
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
HPLC-UV and UPLC-MS/MS methods for the simultaneous analysis of sildenafil, vardenafil, and tadalafil and their counterfeits dapoxetine, paroxetine, citalopram, tramadol, and yohimbine in aphrodisiac products†
Mohamed A.
Abdelshakour
a,
Randa A.
Abdel Salam
 bc,
Ghada M.
Hadad
b,
Dina M.
Abo-ElMatty
d and
Eman A.
Abdel Hameed
*e
bc,
Ghada M.
Hadad
b,
Dina M.
Abo-ElMatty
d and
Eman A.
Abdel Hameed
*e
aForensic Medicine Administration, Ministry of Justice, Egypt
bDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Suez Canal University, Ismailia, Egypt
cDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Sinai University, Kantara Branch, Egypt
dDepartment of Biochemistry, Faculty of Pharmacy, Suez Canal University, Ismailia, Egypt
eDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Port Said University, Egypt. E-mail: emanali_19@hotmail.com; Fax: +20-64-3561877; Tel: +20-01224448268
First published on 18th February 2021
Abstract
In recent times, the counterfeiting of pharmaceuticals has been considered a serious trouble especially in developing countries that acquire poor inspection programs. Sildenafil, vardenafil and tadalafil (phosphodiesterase type 5 inhibitors) products have gained wide popularity in treating sexual disorders, for which they are subjected to counterfeiting. For this purpose, a simple, rapid, and novel HPLC method with ultraviolet detection has been simply developed for the simultaneous determination of vardenafil, sildenafil, and tadalafil, and their counterfeits (dapoxetine, paroxetine, citalopram, tramadol and yohimbine) in pharmaceutical dosage forms and counterfeit products such as instant coffee and honey. The separation was carried out on a C18 column, with acetonitrile and an aqueous 0.05% formic acid solution as the mobile phase with a gradient program and at a flow rate of 1 mL min−1. UV detection was accurately set at 230 nm. The total run time was 11 min for elution of these eight drugs. A UPLC-MS/MS method was also developed, by which compounds were separated in only 6 min, and it was used as a confirmatory tool for studied compounds by identification of their mass spectra. Proposed methods were validated by following ICH guidelines. Both methods were found to be linear, specific, precise and accurate, and they were efficiently applied to analyze 50 commercial products including honey sachets, instant coffee and pharmaceutical products marketed as aphrodisiacs and suspected to contain PDE5-inhibitors.
1. Introduction
Erectile dysfunction (ED) is really considered the most popular sexual dysfunction. ED is described as the lack of ability to have and/or keep an erection enough for a satisfying sexual act. ED is a serious medical problem that may affect the quality of life and cause anxiety, loss of self-confidence, and sadness. The mental stress because of ED could have many effects on the interaction of patients with others.1 Common causes for ED are diabetes, hypertension, cardiovascular disease, hyperlipidemia, injuries, obesity, anxiety, increased age, stress, smoking, drug use or alcohol use.2 Phosphodiesterase type 5 (PDE-5) inhibitors are broadly taken as a therapy of choice for those patients with ED who do not have a specific contraindication preventing their use.3 In Egypt, sildenafil (SLD), tadalafil (TAD), and vardenafil (VAR) are approved by the Egyptian Ministry of Health for clinical use. Another substance, which is used for the treatment of ED, is yohimbine (YHB). YHB is obtained from the bark and roots of an African yohimbe tree. It is a relatively selective alpha-2-adrenoceptor antagonist, causing raised cholinergic and reduced adrenergic tone. Its extract has long been used as an aphrodisiac and as a remedy for psychogenic erectile insufficiency.4 Premature ejaculation (PME) is another male sexual dysfunction. PME is defined as the inability to control or delay ejaculation, which results in dissatisfaction for the patient.5 Premature ejaculation is classified into four subtypes: lifelong PME, acquired PME, variable PME and subjective PME. There are six major types of treatment for PME: daily use of selective serotonin reuptake inhibitors (SSRIs), the use of dapoxetine (DPX) on-demand, clomipramine, topical local anesthetics, tramadol (TRM) or PDE-5 inhibitors. Only DPX 30 and 60 mg has been registered by European Union agency (EMA) as a rapid-acting SSRI for the treatment of PME on-demand, while other treatments are considered off-label.6 Moreover, some studies suggest that the SSRI paroxetine (PRX) may be administered to treat premature ejaculation.7 Citalopram (CTP) is another SSRI recommended by some studies for the treatment of premature ejaculation.8 Due to the popularity, PDE-5 inhibitors (SLD, VAR and TAD) and products containing these compounds are often subjected to counterfeiting. Moreover, herbal products and dietary supplements adulterated with these compounds have been found in the market.9–17Some clinical trials suggested the use of a combination of SLD with DPX, SLD with PRX,18,19 and SLD with TRM20 for the treatment of PME. In addition, a report in 2010 revealed that counterfeit DPX sold on-line contains unrevealed SLD.21 Some studies showed that SLD and its analogues were found as adulterants in an herbal supplement.22–24 In 2018, SLD and TRM were found in a product sold in herb outlets, in Iran (Tehran).25 Therefore, there was a real need for paying more attention for the analysis of counterfeit pharmaceutical products and herbal and food supplements used for the treatment of sexual dysfunction.
The literature declares that many analytical approaches were reported for simultaneous quantification of PDE-5 inhibitors SLD, TAD, and VAR in counterfeit drugs, pharmaceutical products and dietary supplements using different analytical techniques including TLC and HPLC-PDA-MS methods,13 HPLC-UV-ESI-MS,9 LC-MS,26 LC-MS/MS,27–29 LC/HRMS,30 HPLC-DAD and LC-MS/MS.10 SLD, TAD and YHB was analyzed by pulsed amperometric detection using a gold electrode coupled to HPLC separation.31 SLD, TAD, VAR and YHB were determined by LC-MS/MS12,14 and LC-diode array detector-quadrupole-time-of-flight (DAD-QTOF) system.16 TRM, SLD, and TAD were determined by HPLC using a calixarene stationary phase,32 TRM, SLD, DPX, and YHB were also determined with other compounds by HPLC-UV.33 One article was reported for the determination of SSRIs (DPX and PRX) with the three PDE-5 inhibitors (SLD, TAD, and VAR) by HPLC-DAD.34
The goal of this study was to develop simple and accurate analytical methods for simultaneous quantification of PDE-5 inhibitors (SLD, TAD, and VAR), YHB, TRM and common SSRIs (CTP, PRX and DPX), as this mixture was not separated previously followed by applying them to analyze counterfeited products widely used in the Egyptian market for treatment of male sexual dysfunctions.
2. Experimental
2.1. Instrumentation
2.2. Materials and reagents
Pharmaceutical-grade authentic standards of SLD, TAD, VAR, DPX, PRX, CTP, YHB and TRM were used and verified to acquire a purity of 99.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :99.9% (w/w), on dry weight basis. Methanol and acetonitrile were of HPLC grade (BDH, Poole, UK) and formic acid used was of high analytical grade. Suspected counterfeit samples were obtained from local markets and pharmacies or purchased online.
:99.9% (w/w), on dry weight basis. Methanol and acetonitrile were of HPLC grade (BDH, Poole, UK) and formic acid used was of high analytical grade. Suspected counterfeit samples were obtained from local markets and pharmacies or purchased online.
2.3. Standard solutions
Stock solutions of the studied analytes were prepared by dissolving SLD, TAD, VAR, DPX, PRX, CTP, YHB and TRM separately in methanol to obtain a concentration of 0.5 mg mL−1 and stored at 4 °C till preparation of working solutions.The working solutions were prepared by further dilution of the stock solutions with a specified mobile phase ratio of 0.05% formic acid in water:acetonitrile (50:50) to reach the concentration range stated for HPLC and UPLC-MS/MS methods.
2.4. Sample preparation
:acetonitrile (50:50)) by taking 0.2 mL of the filtered stock sample solution and making up the volume to 10 mL. The general procedures for the HPLC-UV and UPLC-MS/MS approaches explained were followed, and the concentrations of the found drugs were calculated.
:acetonitrile (50:50)) by taking 0.2 mL of the filtered stock sample solution and making up to 10 mL. The general procedures for the HPLC-UV and UPLC-MS/MS approaches explained were followed, and the concentrations of the found drugs were calculated.
:acetonitrile (50:50)) by taking 0.2 mL of the filtered stock sample solution and making up to 10 mL. The general procedures for the HPLC-UV and UPLC-MS/MS approaches explained were followed, and the concentrations of the found drugs were calculated.
2.5. Samples analysis
The developed approaches were efficiently applied for the analysis of 50 different samples labeled to contain SLD, VAR, YHB, and TAD and (or) labeled as totally natural herbal ingredients, traded in the Egyptian market.2.6. Chromatographic conditions
3. Results and discussion
There is no doubt that the spread of social media and the increase in the number of satellite channels, and its connection to every home, have provided platforms for drug counterfeiters and promoters to promote their products of counterfeit drugs. Especially, aphrodisiac drugs often deceive some people with slogans of “natural products, have no side effects and suitable for heart patients and diabetics” and claim that their products treat erectile dysfunction and premature ejaculation. Following the success of inspectors in seizing many of the counterfeits and non-registered drugs in the Egyptian market, there was an urgent need to find a method for the concurrent determination of the most common PDE-5 inhibitors (SLD, VAR, and TAD), and other drugs that are commonly used to manage premature ejaculation (YHB, CTP, PRX, DPX and TRM).3.1. Optimization of chromatographic conditions
It is very important to consider the drugs' acid–base properties first, and hence, the pKa values of the studied drugs were determined: TRM (basic pKa = 9.41, acidic pKa = 13.8), YHB (pKa = 7.65, acidic pKa = 14.68), VAR (basic pKa = 6.2, acidic pKa = 8.01), SLD (basic pKa = 5.99, acidic pKa = 11.14), CTP (pKa = 9.78), PRX (pKa = 9.77), DPX (pKa = 9.04) and TAD (basic pKa = 4.2, acidic pKa = 15.17),35 all of the studied compounds were basic except TAD. In case of ionisable analytes, the mobile phase pH can highly influence the analyte retention time in case of HPLC and sensitivity in case of LC/MS methods. In our study, the mobile phase pH should be acidic, to ionize these basic drugs and consequently decrease their retention times and also enhance their detection in the positive ion mode.Optimization of the chromatographic condition was difficult, and many columns were examined: C8, CN, and C18 columns. Upon trying C8 and CN columns that are more polar stationary phases than C18 and due to the polarity of these studied compounds at acidic pH, DPX and PRX were highly tailed, while TAD was retained on both columns. Therefore, using the C18 column was crucial for such separation offering good resolution for all the studied drugs, as indicated in Table 1. Different column temperatures were examined (25 °C, 30 °C and 35 °C), and it was found that by rising the temperature of the column to 35 °C, all obtained peaks became sharp (Fig. 1). Different mobile phase combinations were tried (acetonitrile with ammonium acetate or ammonium formate buffer, methanol with acetate or formate buffer, or mixture of acetonitrile and methanol as the organic phase with ammonium acetate or ammonium formate), and all these trails gave bad separation even after changing the buffer pH in the range from 3.5 to 5.0. Replacing the buffer with formic acid gave better separation for the critical pairs TRM with YHB and SLD with CTP. Gradient elution is used to simultaneously analyze these eight drugs because of their different structures and physicochemical properties; it helped to push strongly retained compounds, and consequently, shorten the analysis time, improve the quality of separation, and diminish peak tailing. Several time programs with different percentages of acetonitrile were tested to enhance the resolution of TRM with YHB and SLD with CTP and also to decrease the retaining of DPX and TAD on the analytical column. Increasing acetonitrile concentration to more than 30% in the first 5 minutes led to inadequate separation and overlap of TRM, YHB, SLD and CTP peaks. At a lower acetonitrile concentration (<40%) after 8 minutes, separation occurred but excessive tailing for DPX and TAD was observed. For wavelength selection, the spectra of the studied drugs were tested and the maximum wavelengths were noticed at 215 nm, 220 nm, 260 nm, 229 nm, 227 nm, 280 nm, 230 nm and 280 nm for TRM, YHB, VAR, SLD, CTP, PRX, DPX and TAD, respectively. Based on the spectra of the studied compounds, two wavelengths were examined, namely, 214 nm and 230, of which 230 nm showed more selectivity for the studied compounds with minimal noise than the shorter wavelength 214 nm. The separation was satisfactory when 0.05% formic acid in water was used as aqueous phase A and acetonitrile as organic phase B by gradient elution, as previously explained in Section 2.6, which gave very good resolution for the separation of TRM, YHB, VAR, SLD, CTP, PRX, DPX and TAD within 11 minutes (Fig. 1). The selectivity of the HPLC-UV method is demonstrated in Table 1.
| HPLC-UV | |||||
|---|---|---|---|---|---|
| Compound | Retention timea (min) | Capacity factor k | Selectivity factorbα | ResolutionbRs | Tailing factor |
| a The retention time of unretained peak is 0.70 min. b a1, b1 are α and Rs calculated for TRM, and YHB. a2, b2 are α and Rs calculated for YHB and VAR. a3, b3 are α and Rs calculated for VAR, and SLD. a4, b4 are α and Rs calculated for SLD, and CTP. a5, b5 are α and Rs calculated for CTP, and PRX. a6, b6 are α and Rs calculated for PRX, and DPX. a7, b7 are α and Rs calculated for DPX, and TAD. | |||||
| TRM | 3.20 | 3.57 | 1.16 (a1) | 5.30 (b1) | 1.03 |
| YHB | 3.60 | 4.14 | 1.21 (a2) | 6.10 (b2) | 0.96 |
| VAR | 4.20 | 5.00 | 1.21 (a3) | 7.40 (b3) | 0.98 |
| SLD | 4.94 | 6.06 | 1.10 (a4) | 4.90 (b4) | 0.96 |
| CTP | 5.35 | 6.64 | 1.17 (a5) | 8.90 (b5) | 1.04 |
| PRX | 6.14 | 7.77 | 1.22 (a6) | 10.80 (b6) | 1.02 |
| DPX | 7.36 | 9.51 | 1.46 (a7) | 15.30 (b7) | 1.10 |
| TAD | 10.40 | 13.86 | 1.09 | ||
| UPLC-MS/MS | |||
|---|---|---|---|
| Compound | Retention time (min) | Precursor ion [M + H]+ (m/z) | Fragment ions (m/z) |
| TRM | 2.62 | 264.2 | 265.3 |
| YHB | 3.15 | 355.3 | 356.3–357.3 |
| VAR | 3.45 | 489.3 | 490.3 |
| SLD | 3.90 | 475.3 | 476.3–477.3 |
| CTP | 4.12 | 325.3 | 326.3–327.3 |
| PRX | 4.41 | 330.2 | 331.2–332.3 |
| DPX | 4.82 | 306.3 | 307.3–308.3 |
| TAD | 5.81 | 390.2 | 391.2 |
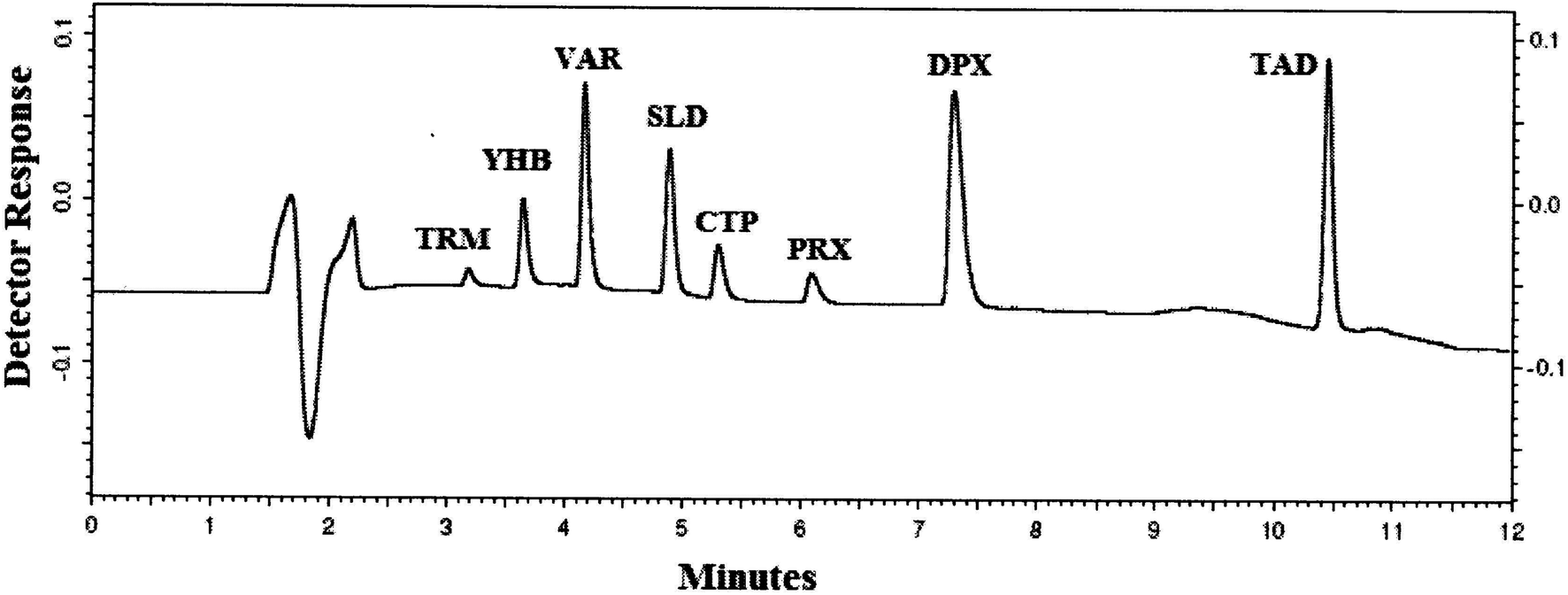 | ||
| Fig. 1 HPLC-UV chromatogram of 5 μL injection of standard prepared mixture containing 10 μg mL−1 of TRM, YHB, VAR, SLD, CTP, PRX, DPX and TAD. | ||
Method transfer to UPLC-MS/MS was easy and optimization was facilitated due to the pre-studied and validated HPLC-UV method, however with little tuning of the separation conditions to fit the specifications of UPLC-MS/MS instrument. Separation was achieved by gradient elution of aqueous 0.1% formic acid as aqueous phase A and acetonitrile as organic phase B. The gradient program was previously explained in Section 2.6. The standard for each analyte was auto-tuned in a positive mode separately according to its masses, namely, 263.200, 354.300, 488.300, 474.300, 324.300, 329.200, 305.300 and 389.200 for TRM, YHB, VAR, SLD, CTP, PRX, DPX and TAD, respectively. The optimum separation for each analyte is presented in Fig. 2. The positive ion mode was selected for MRM analysis, where the protonated precursor ions [M + H]+ of TRM, YHB, VAR, SLD, CTP, PRX, DPX and TAD in the Q1 full-scan mass spectrum were predominant at m/z values (listed in Table 1). The fragmentation pattern obtained in the mass spectra was used for the prediction and identification of studied compounds (Fig. 3).
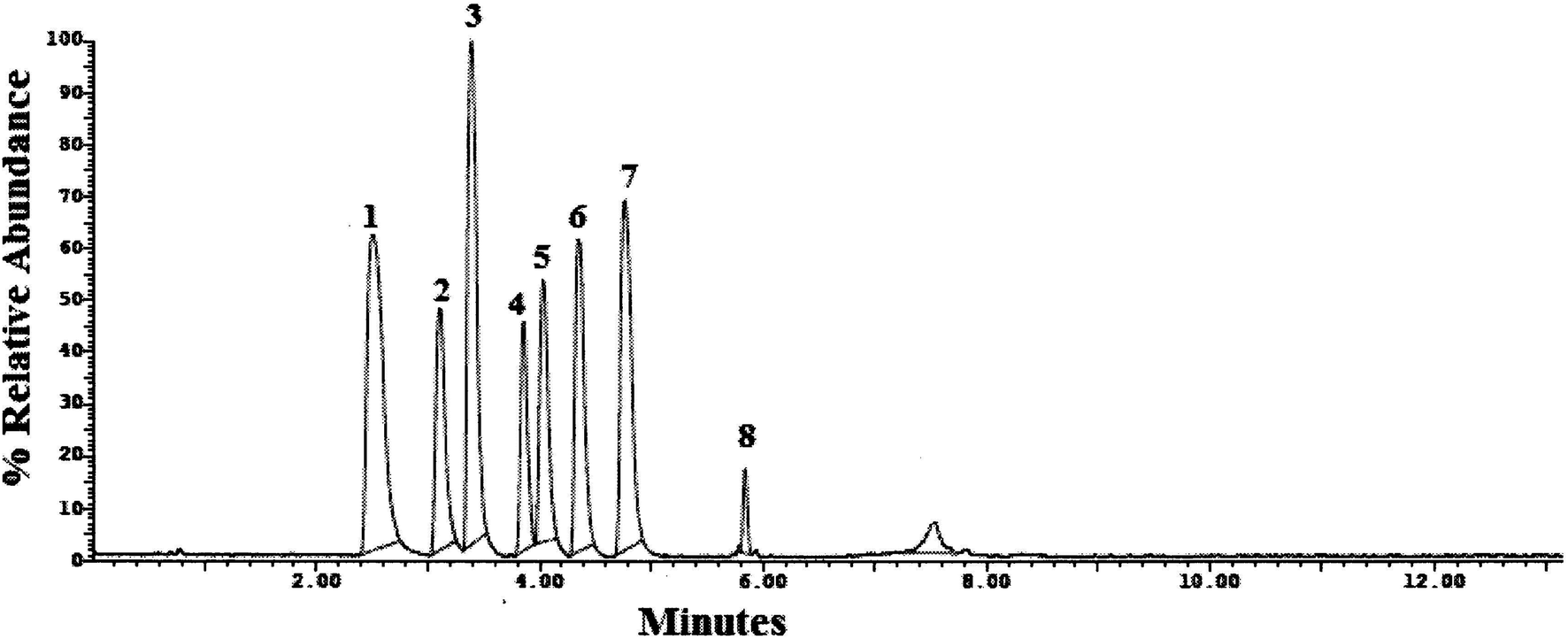 | ||
| Fig. 2 UPLC-MS/MS chromatogram of standard prepared mixture containing (1) TRM, (2) YHB, (3) VAR, (4) SLD, (5) CTP, (6) PRX, (7) DPX and (8) TAD. | ||
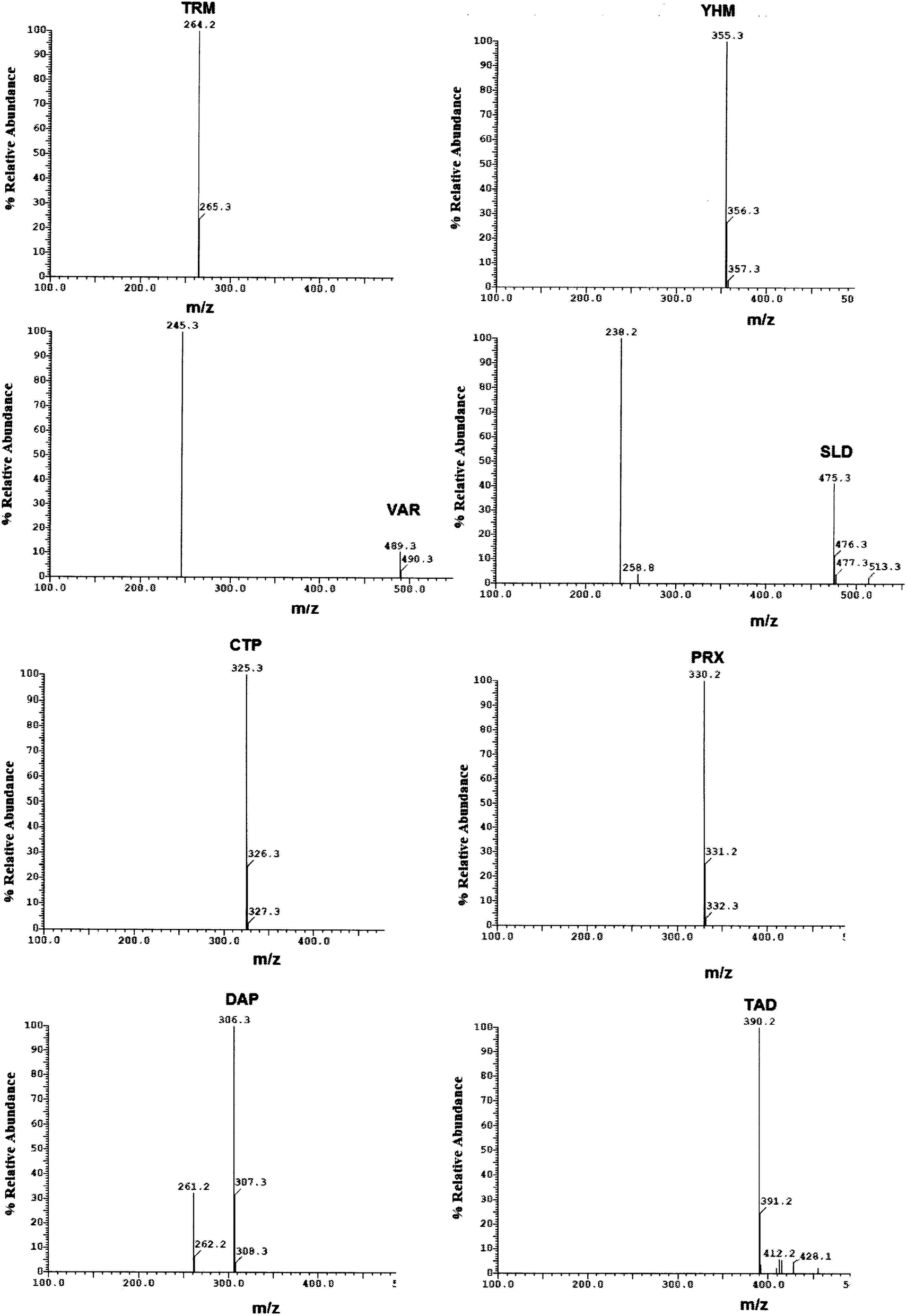 | ||
| Fig. 3 Mass spectra of standard prepared mixture containing TRM, YHB, VAR, SLD, CTP, PRX, DPX and TAD. | ||
3.2. Validation of the methods
| Parameter | TRM | YHB | VAR | SLD | CTP | PRX | DPX | TAD |
|---|---|---|---|---|---|---|---|---|
| a y = a + bC, where C is the concentration in μg mL−1 or ng mL−1 and y is the peak area. b 95% confidence limit. | ||||||||
| HPLC-UV | ||||||||
| Linearity range (μg mL−1) | 0.1–30 | 0.1–30 | 0.1–30 | 0.1–30 | 0.1–30 | 0.1–30 | 0.1–30 | 0.1–30 |
| Determination coefficient (r2) | 0.99996 | 0.99997 | 0.99996 | 0.99996 | 0.99988 | 0.99989 | 0.99998 | 0.99997 |
| LOD (μg mL−1) | 0.0093 | 0.0092 | 0.0130 | 0.0055 | 0.0293 | 0.0280 | 0.0251 | 0.0071 |
| LOQ (μg mL−1) | 0.031 | 0.030 | 0.042 | 0.018 | 0.098 | 0.093 | 0.084 | 0.024 |
| Regression equation(y)a: slope (b) | 2.70 × 105 | 2.10 × 105 | 4.93 × 105 | 4.33 × 105 | 3.29 × 105 | 1.16 × 104 | 2.52 × 105 | 4.41 × 105 |
| Standard deviation of the slope (sb) | 1.19 × 103 | 2.79 × 103 | 2.63 × 103 | 1.11.x103 | 4.12 × 103 | 1.38 × 102 | 2.70 × 103 | 1.32 × 103 |
| Confidence limit of the slopeb | 2.695 × 105 to 2.700 × 105 | 2.089 × 105 to 2.110 × 105 | 4.920 × 105 to 4.940 × 105 | 4.325 × 105 to 4.334 × 105 | 3.274 × 105 to 3.306 × 105 | 1.154 × 104 to 1.165 × 104 | 2.509 × 105 to 2.530 × 105 | 4.405 × 105 to 4.415 × 105 |
| Relative standard deviation of the slope (%) | 0.44 | 0.39 | 0.53 | 0.25 | 1.25 | 1.19 | 1.07 | 0.301 |
| Intercept (a) | 6.03 × 104 | −5.62 × 103 | 6.22 × 103 | 4.51 × 103 | 2.22 × 104 | −1.44 × 102 | −1.70 × 104 | 9.11 × 103 |
| Standard deviation of the intercept (sa) | 1.44 × 104 | 3.38 × 104 | 3.18 × 104 | 1.35 × 104 | 4.99 × 104 | 1.68 × 103 | 3.27 × 104 | 1.61 × 104 |
| Confidence limit of the intercept | 5.97 × 104 to 6.08 × 104 | −5.74 × 103 to −5.49 × 103 | 6.09 × 103 to 6.34 × 103 | 4.45 × 103 to 4.56 × 103 | 2.03 × 104 to 2.41 × 104 | −1.50 × 102 to −1.37 × 102 | −1.82 × 104 to −1.56 × 104 | 9.04 × 103 to 9.17 × 103 |
|
||||||||
| UPLC-MS/MS | ||||||||
| Linearity range (ng mL−1) | 10–100 | 10–100 | 10–100 | 10–100 | 10–100 | 10–100 | 10–100 | 10–100 |
| Determination coefficient (r2) | 0.99997 | 0.99979 | 0.99989 | 0.99997 | 0.99998 | 0.99979 | 0.99969 | 0.99996 |
| LOD (ng mL−1) | 0.008 | 0.042 | 0.033 | 0.011 | 0.011 | 0.041 | 0.044 | 0.027 |
| LOQ (ng mL−1) | 0.028 | 0.140 | 0.109 | 0.036 | 0.036 | 0.137 | 0.0147 | 0.091 |
| Regression equation(y)a: slope (b) | 1.96 × 105 | 8.79 × 104 | 1.73 × 105 | 7.72 × 104 | 7.05 × 104 | 1.09 × 105 | 2.47 × 105 | 3.07 × 105 |
| Standard deviation of the slope (sb) | 6.95 × 102 | 1.58 × 103 | 2.41 × 103 | 3.63 × 102 | 3.62 × 102 | 1.91 × 103 | 4.64 × 103 | 3.59 × 103 |
| Confidence limit of the slopeb | 19.57 × 104 to 19.62 × 104 | 8.73 × 104 to 8.84 × 104 | 17.20 × 104 to 17.39 × 104 | 7.70 × 104 to 7.73 × 104 | 7.03 × 104 to 7.06 × 104 | 10.82 × 104 to 10.97 × 104 | 24.52 × 104 to 24.87 × 104 | 30.56 × 104 to 30.83 × 104 |
| Relative standard deviation of the slope (%) | 0.36 | 1.80 | 1.39 | 0.47 | 0.46 | 1.74 | 1.88 | 1.17 |
| Intercept (a) | −7.54 × 103 | −1.55 × 104 | 8.64 × 104 | 9.28 × 103 | 5.70 × 103 | 1.03 × 104 | 7.07 × 104 | −9.36 × 103 |
| Standard deviation of the intercept (sa) | 3.25 × 104 | 7.39 × 104 | 1.12 × 105 | 1.69 × 104 | 1.52 × 104 | 8.90104 | 2.16 × 105 | 1.67 × 105 |
| Confidence limit of the intercept | −7.66 × 103 to −7.41 × 103 | −18.29 × 103 to −12.70 × 103 | 85.97 × 103 to 88.82 × 103 | 9.21 × 103 to 9.34 × 103 | 5.64 × 103 to 5.75 × 103 | 9.58 × 103 to 11.02 × 103 | 69.88 × 103 to 71.51 × 103 | −9.42 × 103 to −9.29 × 103 |
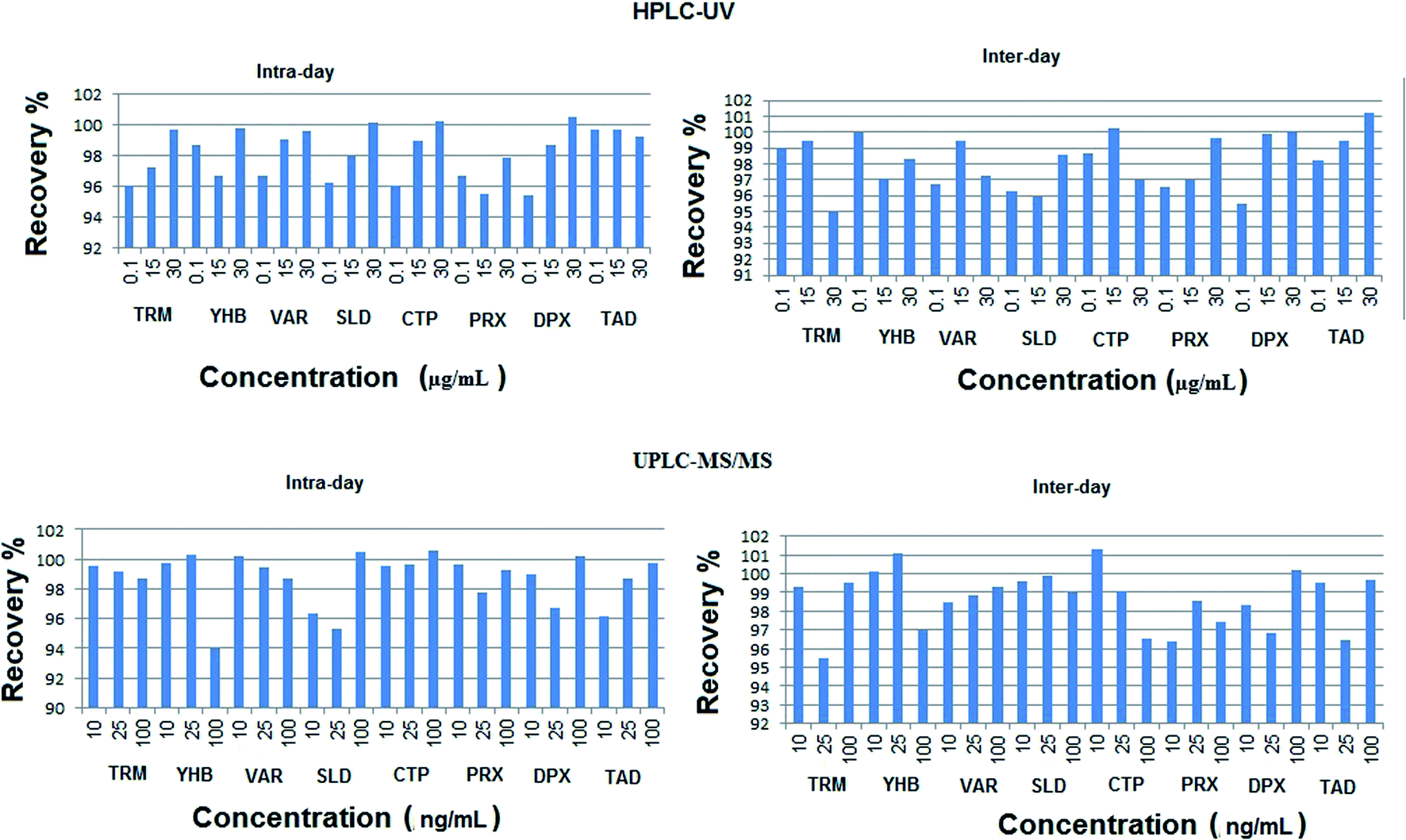 | ||
| Fig. 4 Inter-day and intra-day percentage recoveries of the studied drugs using the proposed HPLC-UV and UPLC-MS/MS methods. | ||
The calculated relative standard deviation of different measurements was below 2% for UPLC-MS/MS and 2.5% for HPLC-UV, which indicates the excellent precision of the proposed analytical methods at both levels of repeatability and intermediate precision.
3.3. Sample analysis
Fifty commercial products marketed as aphrodisiacs and suspected to contain PDE5-inhibitors were analyzed (3 honey sachets, 3 instant coffee, 16 pharmaceutical products labelled to contain natural ingredients, 18 pharmaceutical products labelled to contain SLD, 5 pharmaceutical products labelled to contain TAD, 2 pharmaceutical products labelled to contain VAR, 1 pharmaceutical product that had no content on the label, 1 pharmaceutical product labelled to contain shark extract, 1 pharmaceutical product labelled to contain SLD and DPX and the last sample labelled to contain SLD and TRM). The description, name and ingredients declared on the label of analyzed samples are listed in Table 3. The physical examination of samples showed that samples 5, 6, 15 and 44 in Table 3 were labelled to contain higher concentrations of active ingredients than the average weight of each sample, most of the samples having substandard packaging and printing. Fig. S1† presents some remarks on the packaging that shows a strong and clear indication that these products are counterfeits, sample 37 and 38 having the same name and the same lot number, different expiry dates, different words on the cap and different label colors (Fig. S1a†). Sample 33 the word “distributed” was written as “distribted” (Fig. S1b†). In sample 28, the concentration of the active ingredient was written as “135 mL” instead of “135 mg” (Fig. S1c†). Moreover, Fig. S1d† shows artifacts (cracks, faint imprints) of different tablets in samples 18, 22, 24 and 26.| No. | Name | Description | Content on the label | Found by HPLC | Found by UPLC-MS/MS |
|---|---|---|---|---|---|
| 1 | Cialis | Blue tab | Natural | SLD 103.14 mg | SLD 103.17 mg |
| 2 | Enjoy | Green cap | Natural | SLD 52.46 mg | SLD 51.70 mg |
| 3 | Erectopril | Red cap | Natural | VAR 13.94 mg, DPX 14.34 mg | VAR 13.64 mg, DPX 14.19 mg |
| 4 | GreenValley | Green leaf shape tab | Natural | SLD 46.34 mg | SLD 46.95 mg |
| 5 | Hercules | Yellow cap and white cap | Natural 2000 mg | SLD 114.89 mg, none | SLD 114.99 mg, none |
| 6 | Man's Magic | Yellow kidney shape tab | Natural 3800 mg | SLD 100.92 mg, none | SLD 100.75 mg, none |
| 7 | MAXMAN | Golden capsule | Natural | SLD 95.64 mg, CTP 19.42 mg | SLD 95.99 mg, CTP 19.80 mg |
| 8 | MAXMAN | Black cap and green cap | Natural | SLD 115.52 mg, none | SLD 115.60 mg, none |
| 9 | Plant Viagra | Green leaf shape tab | Natural | SLD 49.04 mg | SLD 49.00 mg |
| 10 | Plant Viagra | Green leaf shape tab | Natural YHB, no conc. | SLD 51.34 mg | SLD 51.39 mg |
| 11 | Super man | Yellow cap, oily cap | Natural | SLD 100.56 mg, none | SLD 100.32 mg, none |
| 12 | Tiger king | Black tab | Natural | SLD 76.23 mg | SLD 76.33 mg |
| 13 | Top man | Black cap and white cap | Natural | SLD 89.78 mg, none | SLD 89.85 mg, none |
| 14 | Vigour 300 | Blue tab | Natural 300 mg | SLD 94.87 mg | SLD 94.99 mg |
| 15 | Vigour 6800 | Blue tab | Natural 6800 mg | SLD 84.43 mg | SLD 84.40 mg |
| 16 | Vigrex | White cap | Natural | SLD 61.67 mg | SLD 61.79 mg |
| 17 | Cajo-150 | Yellow kidney shape tab | SLD 150 mg | SLD 113.04 mg | SLD 113.10 mg |
| 18 | Cobra-125 | Red tab | SLD 125 mg | SLD 96.52 mg | SLD 96.53 mg |
| 19 | DEER-Fox | Red tab | SLD 120 mg | SLD 97.90 mg | SLD 97.94 mg |
| 20 | Erecta Power | Red cap | SLD 140 mg | SLD 96.56 mg | SLD 96.58 mg |
| 21 | Ferrari | Red tab | SLD 130 mg | SLD 127.34 mg | SLD 127.39 mg |
| 22 | FOX | Red tab | SLD 125 mg | SLD 119.43 mg | SLD 119.45 mg |
| 23 | FOX 125 | Red tab | SLD 100 mg | SLD 93.56 mg | SLD 93.57 mg |
| 24 | Goldviagra | Yellow kidney shape tab | SLD 130 mg | SLD 104.12 mg | SLD 104.18 mg |
| 25 | Hard-on | Black tab | SLD 130 mg | SLD 132.46 mg | SLD 132.31 mg |
| 26 | Jaguar 120 | Blue tab | SLD 120 mg | SLD 116.78 mg | SLD 116.80 mg |
| 27 | Jaguar 120 | Red tab | SLD 120 mg | SLD 99.23 mg | SLD 99.31 mg |
| 28 | Plant VIGRA | Green leaf shape tab | SLD 130 mg | SLD 48.12 mg | SLD 48.20 mg |
| 29 | PureGrey | Red tab | SLD 100 mg | SLD 98.40 mg | SLD 98.51 mg |
| 30 | Vega | Blue tab | SLD 50 mg | SLD 49.81 mg | SLD 49.90 mg |
| 31 | Vega b100 | Red tab | SLD 100 mg | SLD 101.54 mg | SLD 101.57 mg |
| 32 | Viag 120 | Red tab | SLD 120 mg | SLD 123.42 mg | SLD 123.46 mg |
| 33 | Viagra 100 | Blue tab | SLD 100 mg | SLD 87.67 mg | SLD 87.79 mg |
| 34 | Fox-125 | Red tab | SLD 125, DPX 20 mg | SLD 122.78 mg | SLD 122.80 mg |
| 35 | Vega b DOL | Red tab | SLD100 mg, TRM 50 mg | SLD 96.47 mg | SLD 96.49 mg |
| 36 | Jaguar Speed | White oblong shape tab | TAD 20 mg | SLD 98.23 mg | SLD 98.20 mg |
| 37 | Cialis | Yellow tab and black cap | TAD 20 mg | None, none | None, none |
| 38 | Cialis | Yellow egg shape tab | TAD 20 mg | None | None |
| 39 | Cialis | Yellow oblong shape tab | TAD 20 mg | None | None |
| 40 | Cialis | Yellow egg shape tab | TAD 20 mg | TAD 18.85 mg | TAD 18.87 mg |
| 41 | Levitra | Yellow tab | VAR 10 mg | VAR 11.4 mg | VAR 11.5 mg |
| 42 | Levitra | Yellow tab | VAR 20 mg | None | None |
| 43 | SuperPower | Black tab | Not labelled | SLD 107.14 mg | SLD 107.15 mg |
| 44 | SHARK Extract | Oblong red tab | Shark extract 3800 mg | SLD 137.33 mg | SLD 137.45 mg |
| 45 | MAXMAN | Coffee sachets | Natural | TAD 16.84 mg | TAD 16.80 mg |
| 46 | Vitamax Power | Coffee sachets | Natural | None | None |
| 47 | Magic coffee | Coffee sachets | Natural | None | None |
| 48 | Vega Honey | Honey sachets | Natural | SLD 98.32 mg | SLD 98.39 mg |
| 49 | Royal Honey | Honey sachets | Natural | SLD 89.50 mg | SLD 89.57 mg |
| 50 | Hard-on | Honey sachets | SLD 130 mg | SLD 94.63 mg | SLD 94.65 mg |
The HPLC-UV analysis revealed that 14 samples labelled to contain natural or herbal ingredients were found to contain SLD (Fig. S2a†); sample 3 was found to contain VAR and DPX (Fig. S2b†), and sample 7 was found to contain SLD and CTP (Fig. S2c†). Instant coffee sample 45 was found to contain TAD without being declared on the package (Fig. S3a†). Coffee samples 46 and 47 were found to be free from any of the studied drugs. Sample 50 that was labelled to contain honey and SLD 130 mg was found to contain SLD as labelled but at a lower concentration (94.63 mg) (Fig. S3b†). Honey samples (No. 48 and 49) were found both with and without SLD, as declared on their packages. Sample 34 was labelled to contain SLD and DPX but was found to contain only SLD; sample 35 that was labelled to contain SLD and TRM was also found to contain only SLD. Sample 36 was found to contain SLD instead of TAD. Samples 37, 38 and 39 were found to be free from any of the studied compounds although they were labelled to contain TAD. For sample 43, the constituents were not declared, and for sample 44, which was labelled to contain shark extract, was found to contain SLD. The proposed UPLC-MS/MS method was used to analyze all samples for more confirmation, and the results are listed in Table 3. The chromatograms and mass spectra of samples 3, 7, 45, and 50 are illustrated in Fig. S4–S7† as an example.
The analysis showed that most of the samples contained concentrations of active substances less than the concentrations declared on their packaging, and these concentrations remained above the permissible therapeutic doses, which requires more attention. Moreover, it confirmed that almost all samples labeled to be natural were adulterated with PDE5 inhibitors without being declared on their packages, as well as advertised and sold as being natural and safe products. This undeclared constituent may really make interaction with nitrates found in some prescription drugs such as nitroglycerin and may also decrease the blood pressure to fatal levels especially for men receiving treatment for diabetes, high blood pressure, hyperlipidemia, or heart disease who often take nitrates.
4. Conclusion
Due to the community heritage and the desire of Egyptians to increase their offspring, the use of sexual stimulants is very frequent, making them vulnerable to commercial fraud and drug counterfeiting. Therefore, simple analytical methods are needed to analyze these drugs to avoid their harmful effects on human health. Chromatographic HPLC/UV and UPLC-MS/MS methods have been successfully developed, optimized and also validated to simultaneously analyze VAR, SLD and TAD and their possible counterfeits TRM, YHB, CTP, PRX, and DPX in different pharmaceutical dosage forms, dietary supplements and herbal products. The HPLC method is considered as a simple, accurate and fast screening tool for counterfeit samples, while the UPLC-MS/MS is considered a highly precise, sensitive confirmatory method, which could be applied successfully to commercial products and suspicious marketed brands.Conflicts of interest
The authors have declared no conflict of interest.References
- L. Fanbin, Y. Mei, Z. Yan, Y. Yirong, Z. Shaoling, C. Yong and X. Peng, Exp. Clin. Transplant., 2014, 12, 184–189 Search PubMed.
- R. ShamLoul and H. Ghanem, Erectile dysfunction, Lancet, 2013, 381, 153–165, DOI:10.1016/s0140-6736(12)60520-0.
- M. Shirai, I. Hiramatsu, Y. Aoki, H. Shimoyama, T. Mizuno, T. Nozaki, S. Fukuhara, A. Iwasa and S. Kageyama, Sex. Med., 2018, 6, 291–296, DOI:10.1016/j.esxm.2018.07.001.
- M. Jahanshahi, E. Nikmahzar, L. Elyasi, F. Babakordi and E. Hooshmand, Int. J. Neurosci., 2018, 128, 404–411, DOI:10.1080/00207454.2017.1389926.
- E. Chung, B. Gilbert, M. Perera and M. Roberts, Aust. Fam. Physician, 2015, 44, 737–743 Search PubMed.
- M. D. Waldinger, Curr. Opin. Psychiatry, 2014, 27, 400–405, DOI:10.1097/yco.0000000000000096.
- D. Zhang, Y. Cheng, K. Wu, Q. Ma, J. Jiang and Z. Yan, BMC Urol., 2019, 19, 2, DOI:10.1186/s12894-018-0431-7.
- T. Karakan, A. Ayyildiz and C. Germiyanoglu, Urol. J., 2009, 5, 41–45 Search PubMed.
- X. Zhu, S. Xiao, B. Chen, F. Zhang, S. Yao, Z. Wan, D. Yang and H. Han, J. Chromatogr. A, 2005, 1066, 89–95, DOI:10.1016/j.chroma.2005.01.038.
- P. Zou, S. S. Oh, P. Hou, M. Y. Low and H. L. Koh, J. Chromatogr. A, 2006, 1104, 113–122, DOI:10.1016/j.chroma.2005.11.103.
- S. Singh, B. Prasad, A. A. Savaliya, R. P. Shah, V. M. Gohil and A. Kaur, TrAC, Trends Anal. Chem., 2009, 28, 13–28, DOI:10.1016/j.trac.2008.09.004.
- Y. Zhang, Z. Huang, L. Ding, H. Yan, M. Wang and S. Zhu, J. Sep. Sci., 2010, 33, 2109–2114, DOI:10.1002/jssc.200900841.
- Y. Cai, T. Cai, Y. Shi, X. Cheng, L. Ma and S. Ma, J. Liq. Chromatogr. Relat. Technol., 2010, 33, 1287–1306, DOI:10.1080/10826076.2010.488979.
- H. M. Lee and B. J. Lee, Food Addit. Contam., Part A, 2011, 28, 396–407, DOI:10.1080/19440049.2011.551947.
- I. Fejos, G. Neumajer, S. Beni and P. Jankovics, J. Pharm. Biomed. Anal., 2014, 98, 327–333, DOI:10.1016/j.jpba.2014.06.010.
- C. Bortolini, A. Pivato, S. Bogialli and P. Pastore, Anal. Bioanal. Chem., 2015, 407, 6207–6216, DOI:10.1007/s00216-015-8801-4.
- Y. d. Jeong, S. Suh and J. Y. Kim, Chromatographia, 2016, 79, 1671–1678, DOI:10.1007/s10337-016-3185-y.
- M. Abu El-Hamd and A. Abdelhamed, Andrologia, 2018, 50(1), 1–6, DOI:10.1111/and.12829.
- K. Sridharan, G. Sivaramakrishnan and R. P. Sequeira, Int. J. Impotence Res., 2018, 30, 215–223, DOI:10.1038/s41443-018-0030-x.
- M. Martyn-St James, K. Cooper, E. Kaltenthaler, K. Dickinson, A. Cantrell, K. Wylie, L. Frodsham and C. Hood, BMC Urol., 2015, 15, 6, DOI:10.1186/1471-2490-15-6.
- J. Dean, R. Klep and J. W. Aquilina, Int. J. Clin. Pract., 2010, 64, 1319–1322, DOI:10.1111/j.1742-1241.2010.02436.x.
- N. M. Reeuwijk, B. J. Venhuis, D. de Kaste, L. A. Hoogenboom, I. M. C. M. Rietjens and M. J. Martena, Food Addit. Contam., Part A, 2013, 30, 2027–2034, DOI:10.1080/19440049.2013.848294.
- N. Campbell, J. P. Clark, V. J. Stecher, J. W. Thomas, A. C. Callanan, B. Donnelly, I. Goldstein and J. C. Kaminetsky, J. Sex. Med., 2013, 10, 1842–1849, DOI:10.1111/jsm.12172.
- Y. C. Huang, H. C. Lee, Y. L. Lin, Y. T. Lin, C. Tsai and H. Cheng, Food Addit. Contam., Part A, 2016, 33, 1637–1642, DOI:10.1080/19440049.2016.1236402.
- N. Saberi, M. Akhgari, L. Bahmanabadi, E. Bazmi and Z. Mousavi, Daru, 2018, 26, 117–127, DOI:10.1007/s40199-018-0216-2.
- T. Tagami, A. Aoyama, A. Takeda, A. Asada, T. Doi, K. Jimura and Y. Sawabe, J. Food Hyg. Soc. Jpn., 2014, 55, 34–40 CrossRef CAS.
- E. S. Lee, J. H. Lee, K. M. Han, J. W. Kim, I. S. Hwang, S. Cho, S. Y. Ham and J. Cheng, J. Pharm. Biomed. Anal., 2013, 83, 171–178, DOI:10.1016/j.jpba.2013.05.009.
- S. Lee, B. Choi, J. Kim, S. In, S. Baeck, S. M. Oh and K. H. Chung, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 978–979, 1–10, DOI:10.1016/j.jchromb.2014.11.024.
- E. Ozturk Er, B. Ozbek and S. Bakirdere, J. Sep. Sci., 2019, 42, 475–483, DOI:10.1002/jssc.201800734.
- S. Strano-Rossi, S. Odoardi, E. Castrignano, G. Serpelloni and M. Chiarotti, J. Pharm. Biomed. Anal., 2015, 106, 144–152, DOI:10.1016/j.jpba.2014.06.011.
- D. T. Muratt, L. S. Müller, T. D. Molin and C. Viana, Anal. Methods, 2018, 10, 2226–2233, 10.1039/C8AY00178B.
- H. Hashem, A. E. Ibrahim and M. Elhenawee, J. Sep. Sci., 2014, 37, 2814–2824, DOI:10.1002/jssc.201400276.
- A. E. Ibrahim, H. Hashem, M. Elhenawee and H. Saleh, RSC Adv., 2020, 10, 1379–1387, DOI:10.1002/jssc.201400276.
- M. Baker, T. Belal, M. S. Mahrous, H. Ahmed and H. Daabees, J. Sep. Sci., 2016, 39, 1656–1665, DOI:10.1002/jssc.201501339.
- D. S. Wishart, Nucleic Acids Res., 2018, 46, D1074–D1082, DOI:10.1093/nar/gkx1037.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guideline—Validation of Analytical Procedures: Text and Methodology Q2(R1), Current Step 4 version, London, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10324a |
| This journal is © The Royal Society of Chemistry 2021 |