
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu-containing magnetic yolk–shell structured nanocomposite: a powerful, recoverable and highly durable nanocatalyst
Reza Mirbagheria,
Dawood Elhamifar *a and
Shaaker Hajatiab
*a and
Shaaker Hajatiab
aDepartment of Chemistry, Yasouj University, Yasouj 75918-74831, Iran. E-mail: d.elhamifar@yu.ac.ir; dawood.elhamifar@gmail.com; Fax: +98-74-33223048; Tel: +98-74-33223048
bDepartment of Semiconductors, Materials and Energy Research Center (MERC), P.O. Box 31787-316, Tehran, Iran
First published on 10th March 2021
Abstract
A novel method was used to prepare a magnetic phenylene-based periodic mesoporous organosilica nanocomposite with yolk–shell structure (Fe3O4@YSPMO). The Fe3O4@YSPMO nanomaterial was prepared by using easily accessible pluronic-P123 and cetyltrimethylammonium bromide (CTAB) surfactants under basic conditions. This material was employed for effective immobilization of potassium perruthenate to prepare an Fe3O4@YSPMO@Ru nanocatalyst for the aerobic oxidation of alcohols. The physiochemical properties of the designed Fe3O4@YSPMO@Ru nanocomposite were studied using PXRD, FT-IR, TGA, SEM, TEM, ICP, VSM and XPS analyses. Fe3O4@YSPMO@Ru was effectively employed as a highly recoverable nanocatalyst in the selective aerobic oxidation of alcohols.
1. Introduction
On the topic of supported heterogeneous catalysts, the structure and morphology of supports are important properties that can affect the catalyst performance. For example, the surface area of a permeable hallow sphere-supported catalyst is much more than a core–shell one because of its accessible inner layer. Yolk–shell (YS) nanocomposites are known as a new member of the hollow nanoarchitecture family with advantages of high surface area, tunable physiochemical properties, high capacity for adsorption and/or catalyst immobilization and high stability.1–3 The easy functionalization of both cores and shells made the YSs an attractive candidate for different applications such as drug delivery and nanocatalysis.4–9 Especially, YS-structured nanomaterials with mesoporous silica shells are very interesting to chemists owing to their attractive properties such as high stability, non-toxicity, high stability, biocompatibility and biodegradability as well as high surface area and pore volume.10,11 On the other hand, among different cores, the Fe3O4 NPs are more interested in the synthesis of YS-structured materials due to the advantages of low-cost, high magnetic properties, high loading of OH on their surface and also easy preparation.12,13Different methods have been used for the preparation of YS nanocomposites that can be divided into template-assisted and template-free approaches. The well-known template-less methods are Kirkendall14,15 and Ostwald ripening.16–19 Based on the type of surfactant, template-assisted methods are divided into hard20 and soft21 approaches. In the hard approach, the template is located between the core and shell. This template is selectively removed under special conditions to prepare the YS-structure. The disadvantage of tedious processing steps, makes the hard-template method inefficient. The soft-template assisted method has recently been employed as a simple and efficient strategy to prepare YS nanocomposites. To date, a few unusual and/or less-available surfactants have been used as a soft template to prepare YS materials.22–25 Some recently developed surfactants applied in this matter are SDBS and LSB,18 water/oil (W/O) microemulsions such as Igepal CO-520 (NP-5) and cyclohexane in water,26 FC4 with pluronic F-127 (ref. 24) and also FC4 with cetyltrimethylammonium bromide (CTAB).25
As mentioned above, most of these surfactants are expensive and commercially unavailable. Therefore, the preparation of novel yolk–shell nanocomposites using routine, low-cost and easily available soft-templates is a very important progress in this matter.
On the other hand, oxidation is a very important process in chemistry, due to its diverse industrial applications such as ground remediation with trichloroethylene (TCE) or tetrachloroethylene (PCE),27,28 pesticides removal,29 formaldehyde removal30–33 and elimination of chemical oxygen demand (COD) for industrial waste water,34 etc. Especially, the oxidation of alcohols to respective carbonyls is an important subject in organic synthesis. Various reagents and catalysts such as chromium oxide,35–39 manganese oxide,40–42 activated DMSO,43–46 hypervalent iodine,47–49 pyridine-SO3 (ref. 50) and NaOCl with TEMPO51–55 have been used for the oxidation of alcohols. Among different oxidants, molecular oxygen is of more interest due to its availability, low-cost and especially the production of water as only by-product. However, these homogeneous systems suffer from restrictions such as difficulty in catalyst and product separation and waste problems. Therefore, the use of heterogeneous catalysts has been highly developed in this matter. Some of recently developed catalytic systems are based on noble metals such as Pd56–64 and Ru.65–71 A number of reported studies in this matter are hydroxyapatite-supported palladium nanoclusters,72 methyl glycolate-supported gold,73 carbon-supported Pt, Pt–Ru and Pt3Sn,74 graphene-supported Pt and Pt–Ru,75 silica-supported TEMPO,76 PMO-IL@RuO4 (ref. 77) and ILNOS-Ti.78
In view of the above, herein, a novel soft-template assisted strategy has been developed for easy preparation of magnetic phenylene-based ordered mesoporous nanocomposite with yolk–shell structure (Fe3O4@YSPMO). Pleasingly, for the first time, the routine, easily available and low-cost surfactants (pluronic P123 and CTAB) were used as a template to prepare the Fe3O4@YSPMO nanomaterial (Scheme 1).
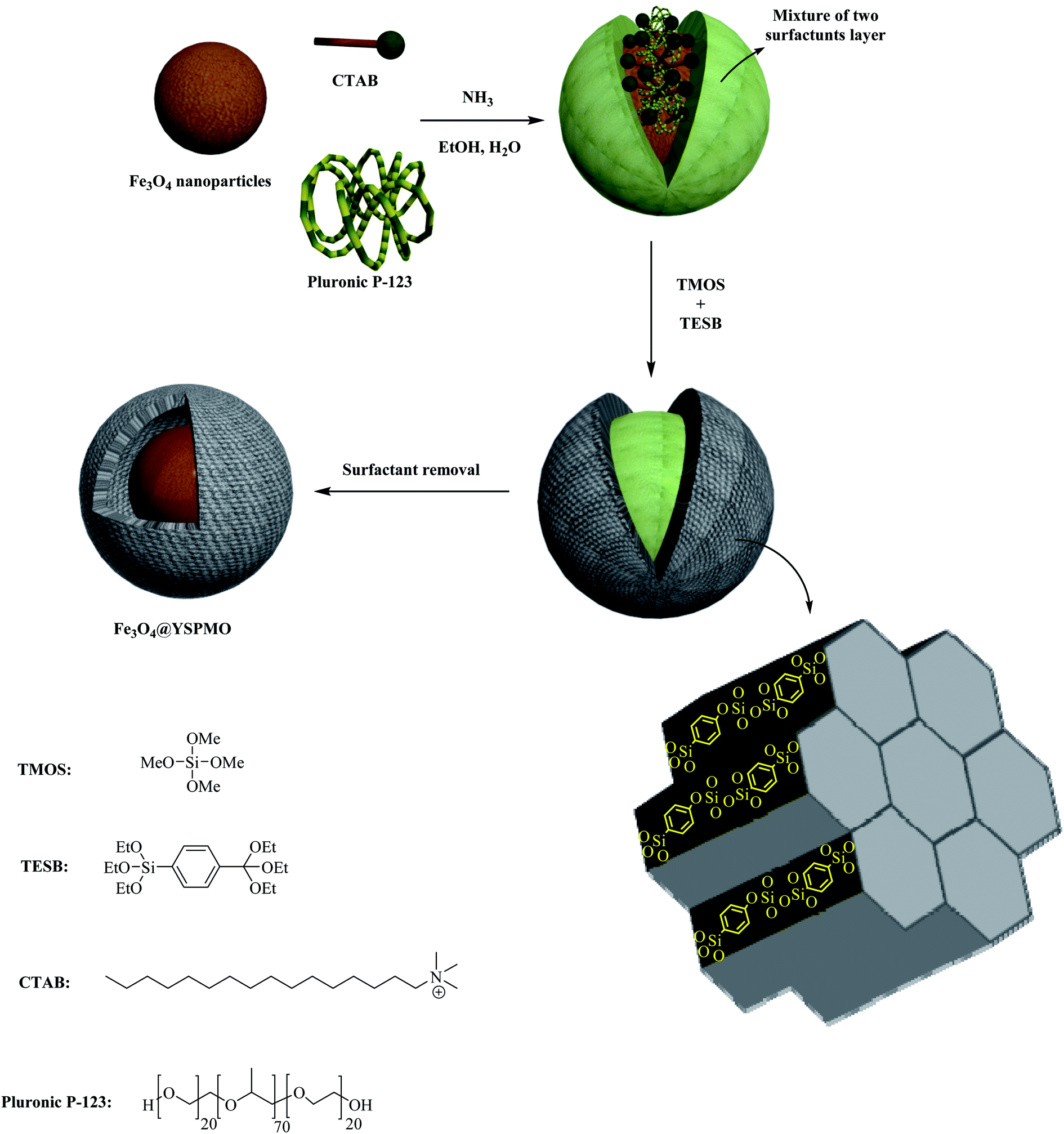 | ||
| Scheme 1 Preparation of Fe3O4@YSPMO nanocomposite. | ||
2. Experimental section
2.1 Preparation of Fe3O4@YSPMO nanocomposite
Magnetic Fe3O4 nanoparticles were synthesized according to our recently reported procedure.79 In order to prepare Fe3O4@YSPMO, the Fe3O4 NP (0.12 g) was first completely dispersed in distilled water (25 mL) and then this was added to a solution of ethanol (16 mL) and water (36 mL) containing CTAB (0.32 g), pluronic P123 (0.52 g) and ammonia (0.4 mL). The resulted mixture was stirred at 35–40 °C. After 30 minutes, TMOS (0.31 mL) and TESB (0.85 mL) were added and the stirring was continued for 1 hour. Next, the obtained combination was heated at 100 °C statically for 17 hours. The final material was collected and washed with H2O and EtOH. The pluronic P123 and CTAB surfactants were removed using a Soxhlet apparatus using acidic ethanol (Scheme 1).2.2 Preparation of Fe3O4@YSPMO@Ru
Firstly, 0.4 g of Fe3O4@YSPMO nanoparticles were dispersed in water (15 mL) under ultrasonic irradiations. Then, potassium perruthenate (0.05 g) was added and the mixture was stirred at RT for 12 h. The resulted material was collected and washed several times with water and acetone to remove unsupported potassium perruthenate. Finally, the product was dried at 50 °C for 8 h and it was denoted as Fe3O4@YSPMO@Ru (Scheme 2).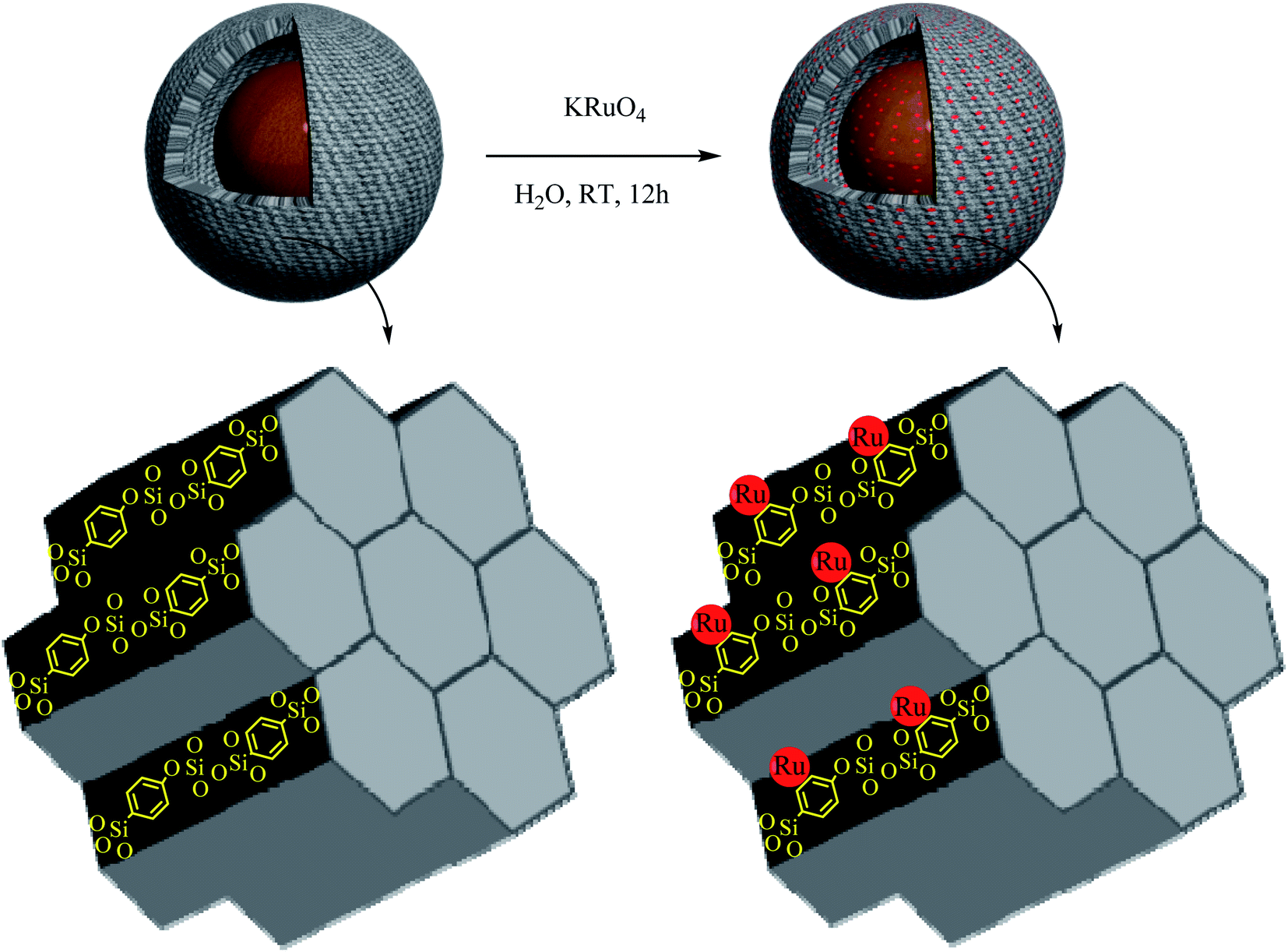 | ||
| Scheme 2 Preparation of Fe3O4@YSPMO@Ru nanocomposite. | ||
2.3 Procedure for the oxidation of alcohols using Fe3O4@YSPMO@Ru
Typically, alcohol (1 mmol) and catalyst (0.3 mol%) were added in a reaction vessel containing TFT (10 mL) and it was stirred at 90 °C under O2 gas (1 atm). The progress of reaction was monitored by GC. After the oxidation process was completed, the catalyst was easily removed using an external magnetic field. The yield and conversion were calculated via GC results.3. Results and discussion
For the preparation of Fe3O4@YSPMO@Ru nanocomposite, firstly, magnetic iron oxide NPs were prepared.80 The Fe3O4 nanoparticles were then modified with periodic mesoporous organosilica shell as follows. The Fe3O4 NPs were completely dispersed in an aqueous ethanol solution containing ammonia, CTAB and P123. After 30 minutes stirring, the TMOS and TESB precursors were added as the source of silica. This mixture was stirred for 1 h at 35–40 °C. The obtained mixture was then statically heated at 100 °C to form mesoporous organosilica shell. Importantly, in this study, for the first time, the routine and easily available surfactants (CTAB and pluronic P123) were used as structure-directing agents under moderate conditions to prepare yolk–shell structured Fe3O4@YSPMO material (Scheme 1). The Fe3O4@YSPMO was used for the immobilization of potassium perruthenate to prepare a powerful nanocatalyst named Fe3O4@YSPMO@Ru (Scheme 2). This nanomaterial was characterized using different techniques and applied as highly recoverable catalyst in the aerobic oxidation of alcohols.The low-angle powder XRD pattern (Fig. 1) demonstrated a sharp d100 peak at 2θ of about 1° confirming the presence of periodicity in a shell with two-dimensional hexagonal structure for the catalyst. According to this peak, it can be concluded that none of the surfactants lonely exists in the outer layer and the final form of the shell is made through a combination of both surfactants.
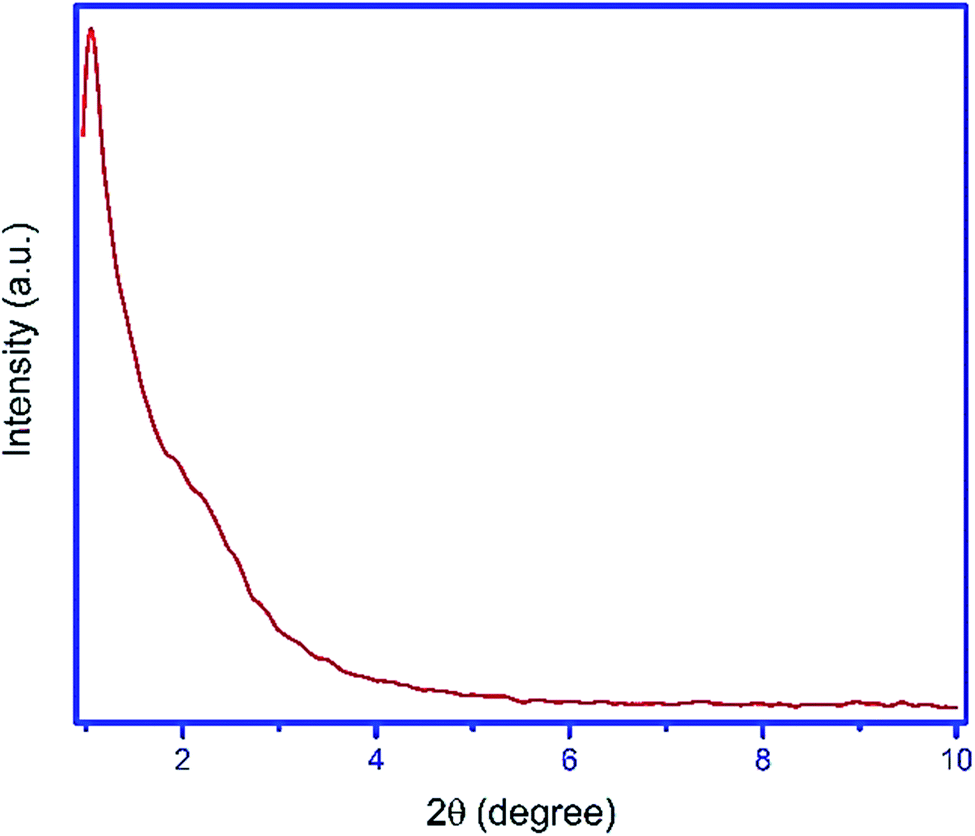 | ||
| Fig. 1 The low-angle powder XRD pattern of Fe3O4@YSPMO@Ru. | ||
Furthermore, the wide-angle PXRD was in agreement with Fe3O4 spinel structure and showed six peaks at 2θs of 30, 35, 43, 54, 57 and 64° corresponding, respectively, to the Miller indices (hkl) values of 220, 311, 400, 422, 440 and 511. The broad peak appeared at 2θ over 20–27 degree is also related to mesoporous silica shell (Fig. 2).
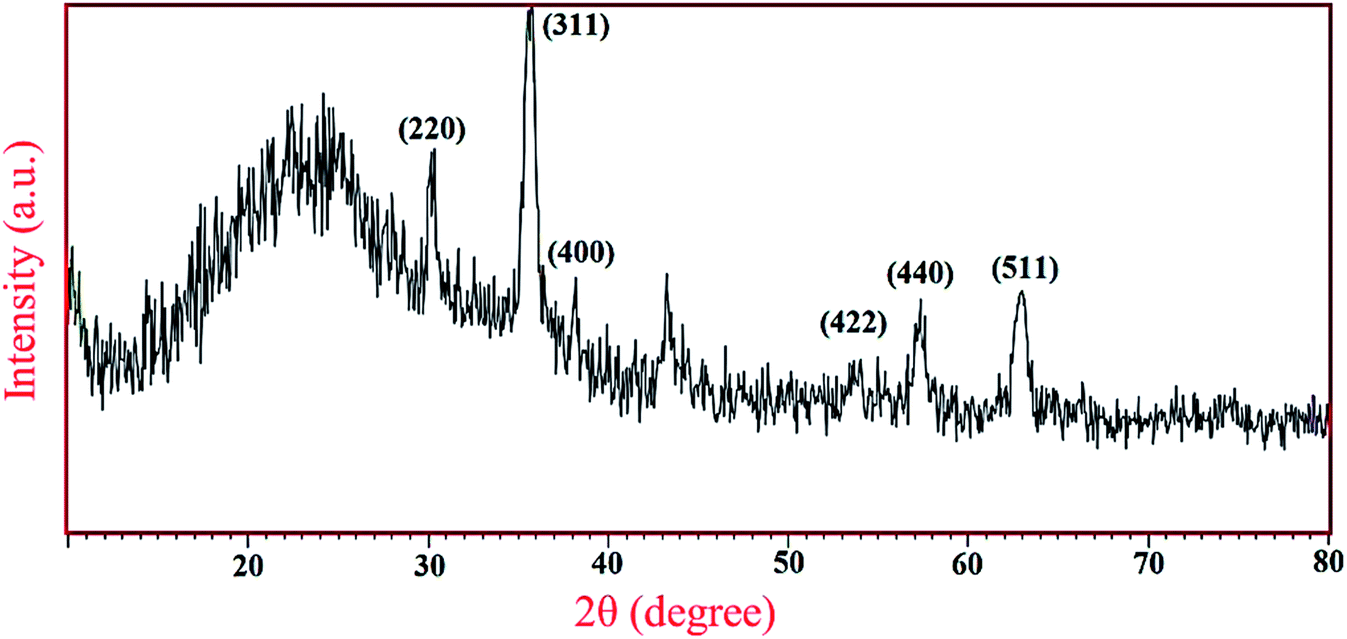 | ||
| Fig. 2 The wide-angle PXRD pattern of Fe3O4@YSPMO@Ru. | ||
The FT-IR spectra of Fe3O4 NPs and also Fe3O4@YSPMO before and after the extraction of surfactants are shown in Fig. 3. These showed a peak at 573 cm−1 corresponding to Fe–O bond for all materials. The O–H bonds of material surface were appeared about 3400 cm−1. Interestingly, new bands were observed for Fe3O4@YSPMO, before and after the surfactant extraction (Fig. 3(B) and (C)). Before the surfactant extraction, the bands observed at 2922 and 2853 cm−1 are due to the aliphatic C–H bonds of P123 and CTAB surfactants (Fig. 3(B)). After the Soxhelet extraction, the latter signals were not seen confirming well removal of surfactants during extraction process (Fig. 3(C)). The peaks at 720–810 cm−1 are attributed to C–Si bonds. Moreover, the bands at 1096 and 932 cm−1 are due to symmetric and asymmetric vibrations of Si–O–Si bonds. The bands appeared around 3100 cm−1 are corresponded to aromatic C–H bonds of PMO shell. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of phenylene rings were also observed at 1630 cm−1. These results confirm successful formation of phenylene-PMO-shell around magnetite NPs.
C bonds of phenylene rings were also observed at 1630 cm−1. These results confirm successful formation of phenylene-PMO-shell around magnetite NPs.
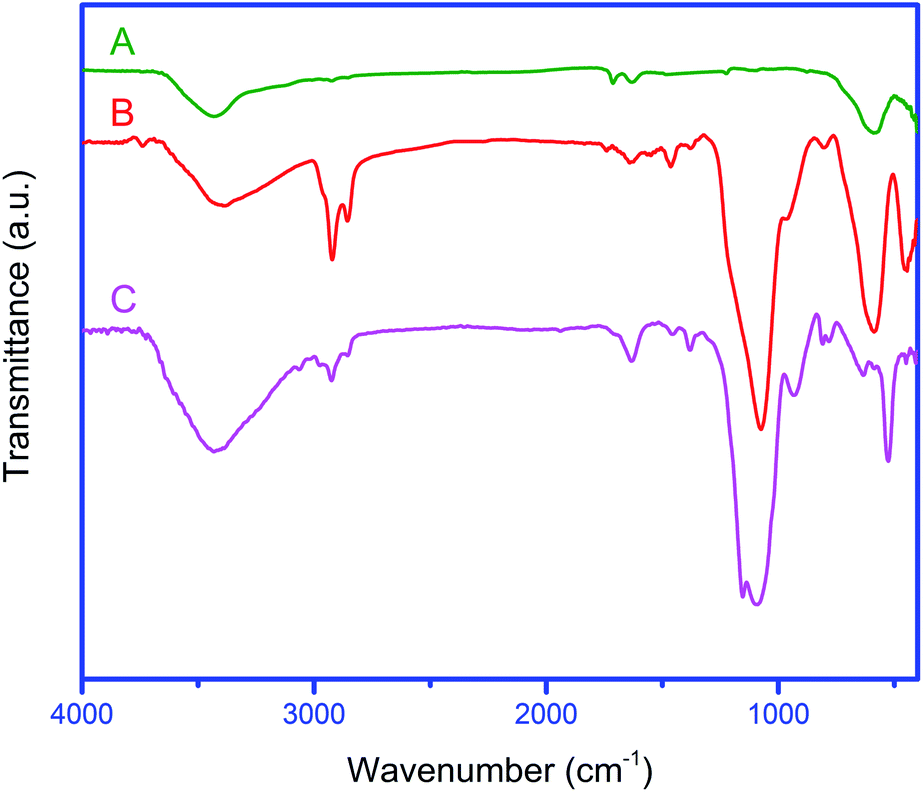 | ||
| Fig. 3 The FT-IR spectra of Fe3O4 (A), Fe3O4@YSPMO nanoparticles before (B), and after (C) surfactant extraction. | ||
Scanning electron microscopy (SEM) image is shown in Fig. 4(a). This image showed uniform spherical particles with an average size of 70 nm for the material. The transmission electron microscopy (TEM) image of Fe3O4@YSPMO is shown in Fig. 4(b). This also showed YS-structured spherical particles with a void space between black cores (Fe3O4 NPs) and gray shells (organosilicas) for the designed material.
 | ||
| Fig. 4 SEM (a) and TEM (b) images of the Fe3O4@YSPMO@Ru nanomaterial. | ||
Thermal stability of Fe3O4@YSPMO@Ru was studied by TGA (Fig. 5). A weight loss of about 6% below 120 °C is attributed to elimination of remained H2O, EtOH and MeOH from the synthetic process. The second weight loss (about 7%) was found at 121–300 °C corresponding to remained surfactants during extraction process. The main weight loss (27%) at 301–800 °C is due to removal of the phenylene moieties incorporated into material shell.
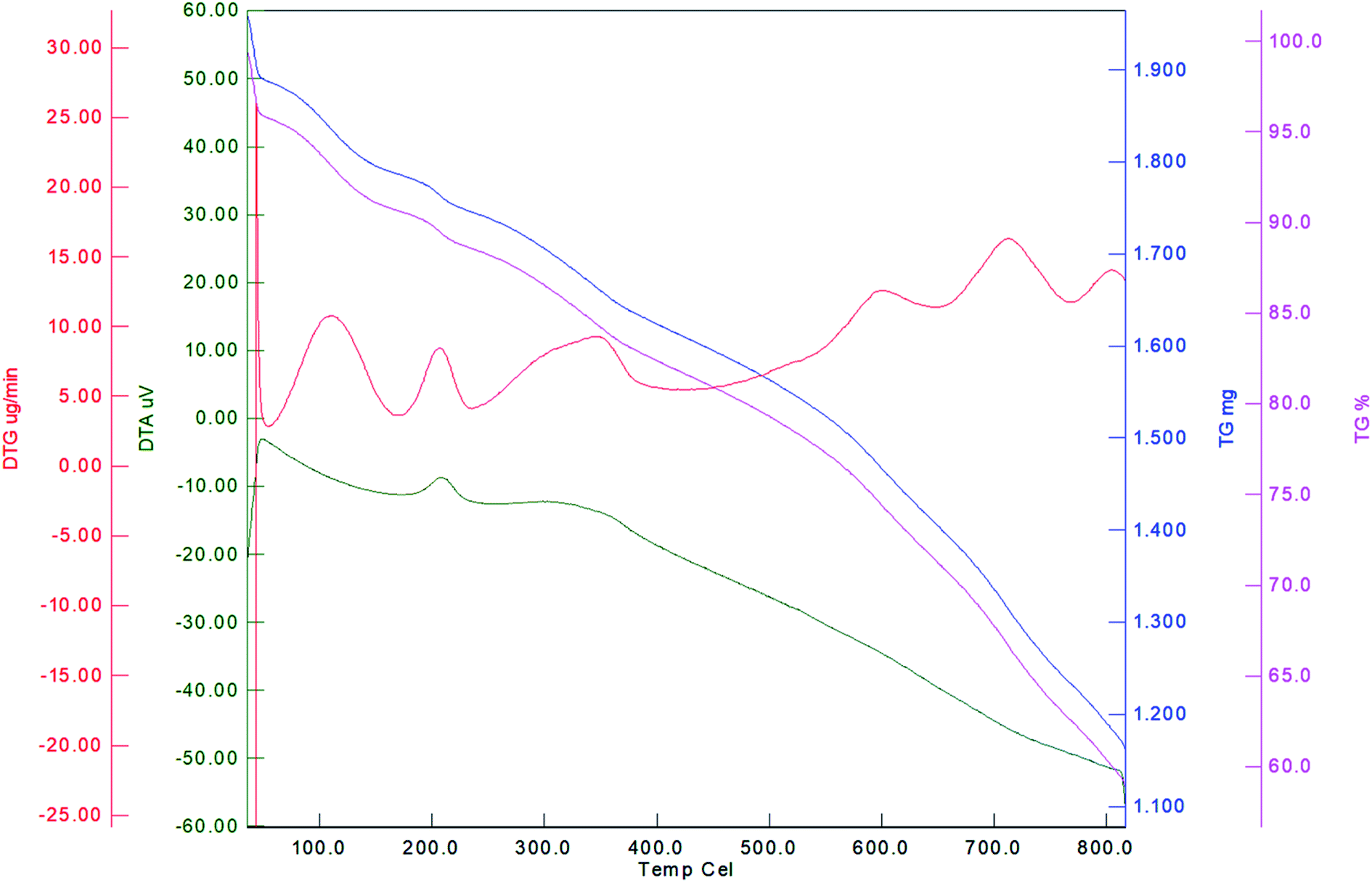 | ||
| Fig. 5 TGA pattern of Fe3O4@YSPMO@Ru. | ||
Immobilization of potassium perruthenate in the prepared material was investigated by using EDS and ICP analyses. The EDS pattern (Fig. 6) proved the presence of Ru, O, Si, Fe and C in the material. The exact amount of ruthenium was calculated by ICP (2.38 mol%). These data were found to be in good agreement with FT-IR and TGA results confirming well incorporation/immobilization of phenylene and perruthenate moieties into/onto material framework.
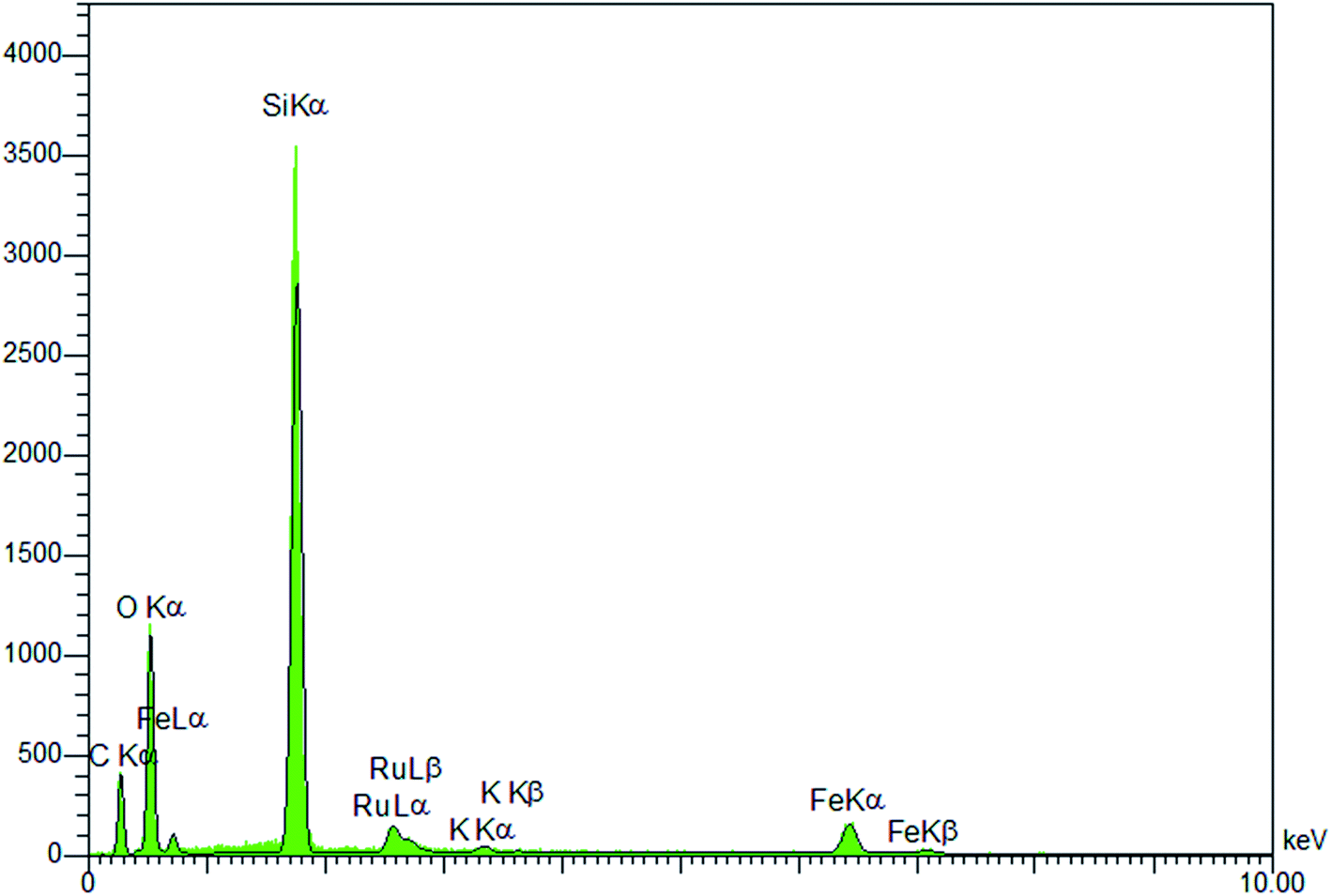 | ||
| Fig. 6 EDS spectrum of Fe3O4@YSPMO@Ru. | ||
The VSM analysis was also performed to investigate the magnetic properties of the designed catalyst (Fig. 7). According to this, the saturation magnetization of Fe3O4@YSPMO@Ru nanocomposite was found to be about 14 emu g−1 confirming good magnetic properties of the catalyst.
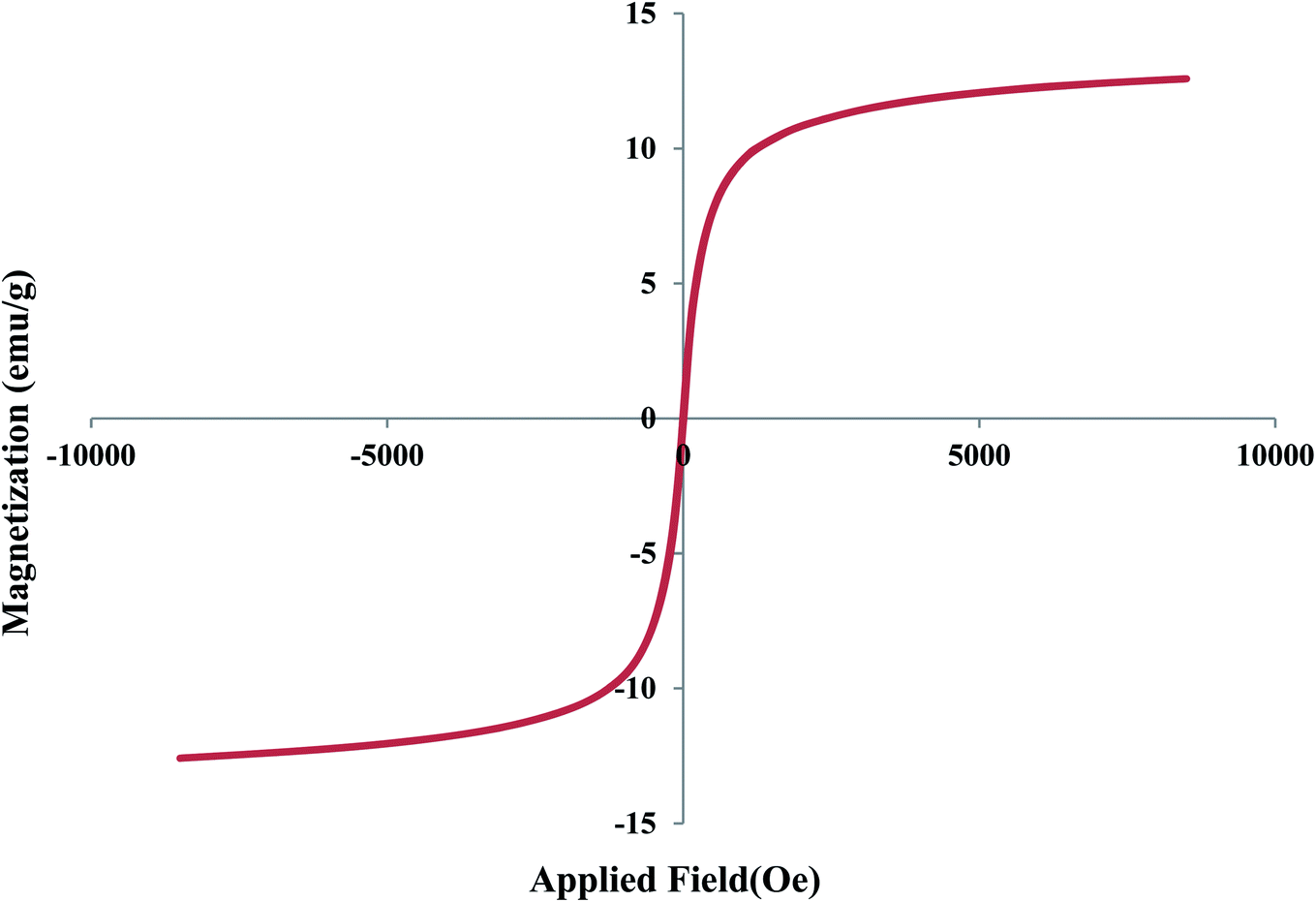 | ||
| Fig. 7 VSM pattern of Fe3O4@YSPMO@Ru. | ||
The XPS survey spectrum (Fig. 8) of the nanocomposite was taken and analyzed to investigate its chemical composition. Before the XPS analysis, Savitzky–Golay filtering was implemented on the XPS spectra to reduce their noise level.81,82 All signals were calibrated to have C 1s at BE of 284.9 eV. As shown in Fig. 8, Si 2p3/2 is observed at 103.27 eV, which confirms its contribution in the PMO part of the material as SiO2. The peak of Ru 3p3/2 appeared at binding energy of 463.5 eV confirming the presence of Ru as Ru (4+). It was also found that the oxygen contributes in the material as SiO2 (533 eV) and RuO2 (530.2 eV). Iron and potassium were not found in XPS spectra though they were shown to be present in the material by using EDS. As known, EDS is a characterization technique for analyzing bulk materials and thus it was able to detect Fe from Fe3O4 as core material. Despite EDS, XPS is a surface sensitive technique with sampling depth of three times of inelastic scattering mean free path of electrons (IMFP) with a given kinetic energy within a given overlayer. For instance, the IMFPs of Fe 2p electrons with kinetic energy of about 543 eV within the overlayers of SiO2 and RuO2 are calculated from Tanuma–Powell–Penn (TPP) formula to be 1.85 and 1.11 nm, respectively. In other words, the escape depth of such electrons within SiO2 and RuO2 are about 5.55 and 3.33 nm. This indicates that the thickness of the RuO2/PMO shell enveloping the core Fe3O4 is larger than the escape depth of Fe 2p electrons. The relative sensitivity factor for Ru 3p is much higher than that of other elements in the prepared core shell that should result in a Ru 3p signal of high intensity. In spite of this, it is of low intensity, which indicates that to the amount of RuO2 is very low. Therefore, the disappearance of Fe signal is mainly attributed to the thick PMO layer.
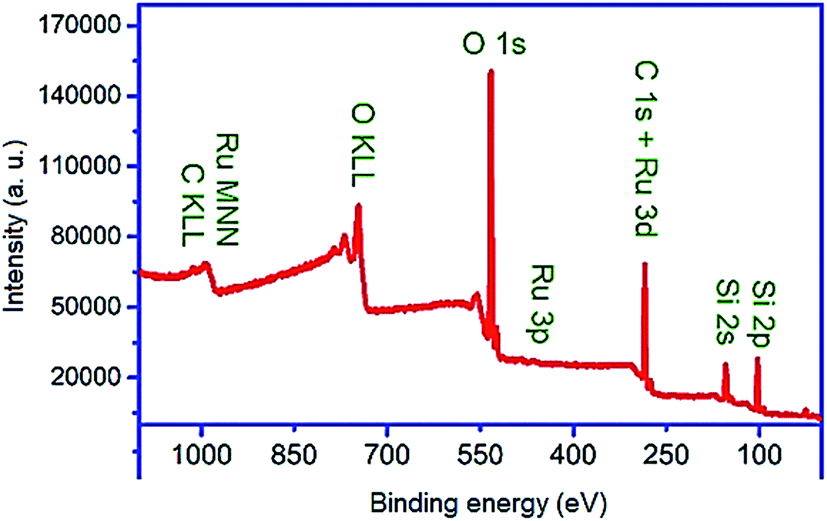 | ||
| Fig. 8 XPS spectrum of Fe3O4@YSPMO@Ru catalyst. | ||
The interaction of aromatic compounds and metal complexes is an important subject because of its importance in the fields of nucleic acids,83,84 proteins,85,86 crystal engineering,87 material science88 and drug design. Especially in ruthenium–arene complexes, an important interaction is between π-electrons of aromatic rings and the Ru cationic sites with high oxidation states.89,90 This kind interaction leads to both immobilization and reduction of Ru species. In the present study, we used potassium perruthenate with the oxidation state of +7 for Ru. However, the XPS analysis showed that the oxidation state of supported Ru is +4 in the designed nanocomposite. This is an evidence of successful immobilization of ruthenium species on the organosilica shell that can be achieved via coordination of aromatic π-electrons with the Ru-centers followed by the reduction of the oxidation state of Ru from +7 to +4.
After the successful preparation and characterization, the Fe3O4@YSPMO@Ru was used as an effective and recoverable catalyst for aerobic oxidation of alcohols. To obtain the best conditions, the oxidation of 1-phenylethanol was selected as a test model. As shown in Table 1, the reaction progress is greatly influenced by the amount of catalyst, solvent and temperature. Accordingly, the best result was obtained using 0.3 mol% of Fe3O4@YSPMO@Ru catalyst in trifluorotoluene (TFT) at 90 °C. In the next study, the catalytic activity of our designed Fe3O4@YSPMO@Ru nanocatalysts was compared with Fe3O4@YSPMO@Pd and Fe3O4@YSPMO@Au nanomaterials (Table 1, entry 7 versus entries 9 and 10). The result showed that the best result is delivered in the presence of Fe3O4@YSPMO@Ru.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Temperature (°C) | Time (h) | Yieldb (%) |
| a Conditions: 1-phenylethanol (1 mmol), O2 (1 atm).b GC yield.c The Fe3O4@YSPMO@Pd was used as catalyst.d The Fe3O4@YSPMO@Au was used as catalyst. | |||||
| 1 | 0.1 | — | R.t. | 12 | — |
| 2 | 0.1 | Toluene | R.t. | 12 | 10 |
| 3 | 0.1 | Toluene | 60 | 12 | 55 |
| 4 | 0.1 | Toluene | 90 | 12 | 67 |
| 5 | 0.2 | Toluene | 90 | 12 | 80 |
| 6 | 0.2 | TFT | 90 | 12 | 96 |
| 7 | 0.3 | TFT | 90 | 6 | 96 |
| 8 | 0.3 | TFT | 75 | 6 | 78 |
| 9c | 0.3 | TFT | 90 | 6 | 94 |
| 10d | 0.3 | TFT | 90 | 6 | 68 |
After that, the applicability of this conditions for other alcohols was examined (Table 2). As shown, different alcohols are effectively converted to their corresponding carbonyls (Table 2, entries 1–8). It is noteworthy that this process was very selective for oxidation of primary benzylic alcohols to aldehydes without the formation of appreciable amounts of carboxylic acids (Table 2, entries 1–3). Moreover, 1-phenylpropanol, 1-phenylethanol and benzhydrol also delivered corresponding ketones in high yields (Table 2, entries 4–6). Interestingly, the Fe3O4@YSPMO@Ru was also effective in the selective oxidation of cinnamyl alcohol and the corresponding α,β-unsaturated carbonyl was produced in a good yield (Table 2, entry 7). The primary and secondary aliphatic alcohols (both acyclic and cyclic) were also reactive in the presence of designed catalytic system and converted to the corresponding carbonyls in good yield and selectivity (Table 2, entries 8–10). These results confirm the high ability of Fe3O4@YSPMO@Ru for selective aerobic oxidation of a broad range of aliphatic and aromatic alcohols.
|
|||||
|---|---|---|---|---|---|
| Entry | Alcohol | Time (h) | Yieldb (%) | TONc | TOFd (h−1) |
| a Conditions: alcohol (1 mmol), O2 (1 atm), catalyst (0.3 mol%) in TFT at 90 °C.b GC yields.c TON: defined as [mmol of product/mmol of catalyst].d TOF: defined as [TON/time].e Conditions: alcohol (1 mmol), O2 (1 atm), catalyst (0.5 mol%) in TFT at 90 °C. | |||||
| 1 | Benzyl alcohol | 5 | 98 | 326.7 | 65.3 |
| 2 | 4-Chlorobenzyl alcohol | 6.5 | 94 | 313.3 | 48.2 |
| 3 | 4-Methoxybenzyl alcohol | 4.5 | 98 | 326.7 | 72.6 |
| 4 | 1-Phenylethanol | 6 | 96 | 320 | 53.3 |
| 5 | 1-Phenylpropanol | 6 | 95 | 316.7 | 52.8 |
| 6 | Benzhydrol | 12 | 92 | 306.7 | 25.6 |
| 7 | Cinnamyl alcohol | 24 | 68 | 226.7 | 9.4 |
| 8e | 2-Octanol | 24 | 68 | 136 | 5.7 |
| 9e | Cycloheptanol | 24 | 73 | 146 | 6.1 |
| 10e | 3-Phenyl-1-propanol | 16 | 56 | 112 | 7 |
Next, the recoverability and reusability of Fe3O4@YSPMO@Ru in 1-phenylethanol oxidation were studied. To do this, after the completion of oxidation process, the catalyst was removed and it was dried and reused in conditions the same as the first run. The result showed that the Fe3O4@YSPMO@Ru can be recovered at least five times with no considerable decrease in performance (Fig. 9).
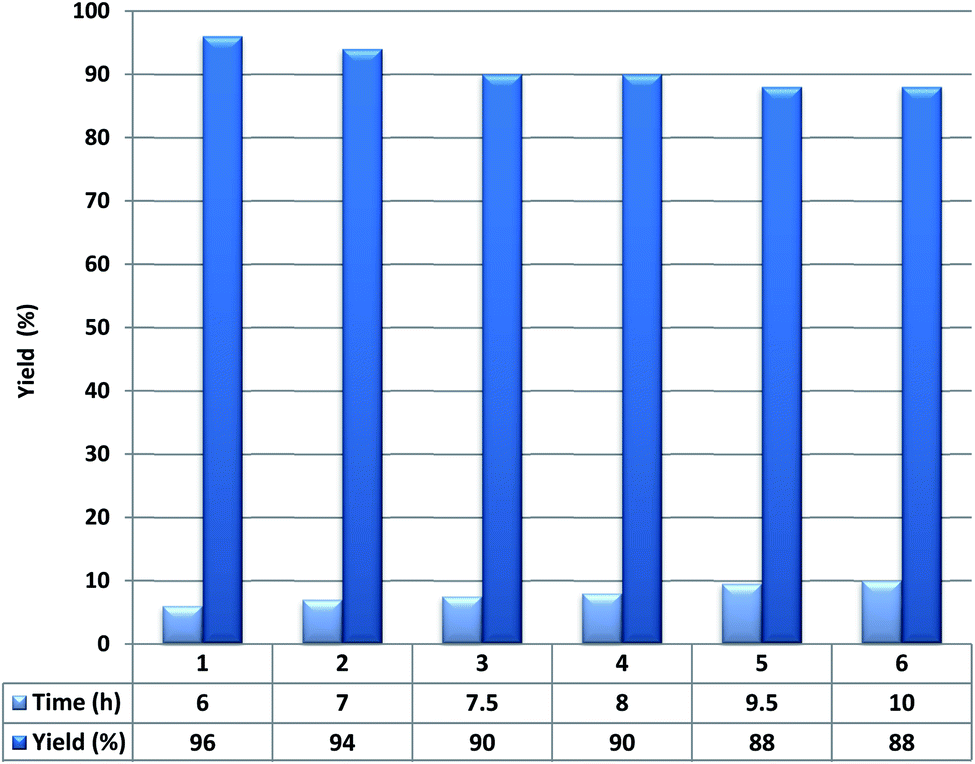 | ||
| Fig. 9 Recoverability of Fe3O4@YSPMO@Ru. | ||
A hot filtration test was also performed on the above-mentioned reaction as a test model. This experiment was performed to make it clear whether the active catalytic species are leaching during the reaction process or not. To this end, after the completion of about 50% of the oxidation process, the catalyst was removed and the reaction progress of residue was monitored. Interestingly, after 10 h, no considerable conversion of alcohol was observed (Fig. 10). Moreover, the atomic absorption analysis also showed no leaching of ruthenium in the filtrate mixture. These results confirm effective immobilization and high stability of active Ru-species during the applied conditions.
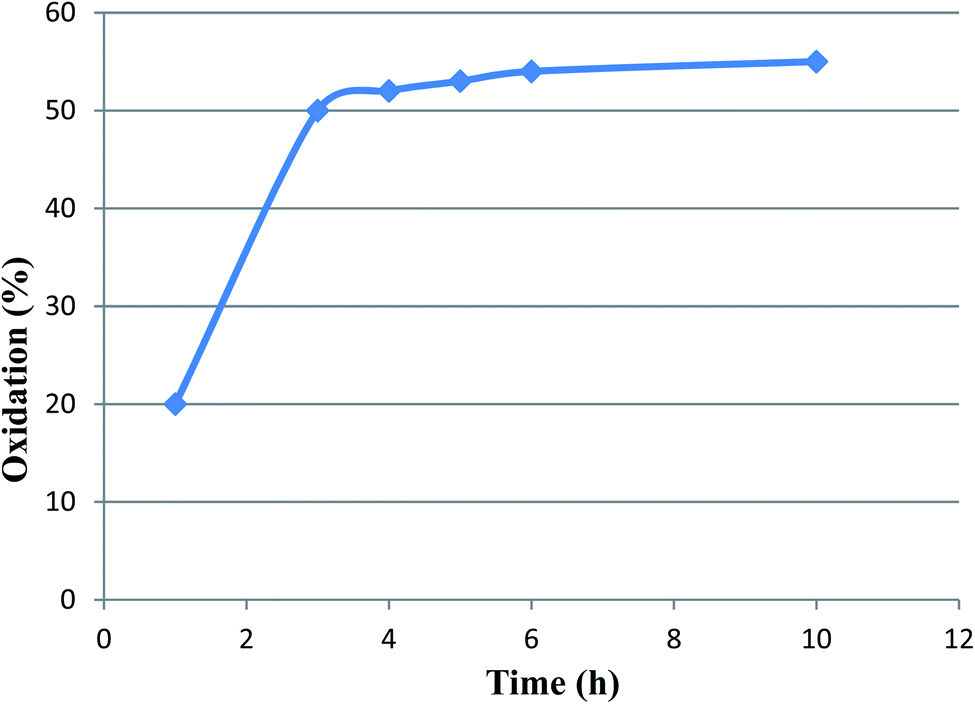 | ||
| Fig. 10 The effect of hot filtration test in the oxidation process. | ||
Finally, the efficiency of Fe3O4@YSPMO@Ru was compared with some of recently reported ruthenium based heterogeneous catalysts in the oxidation of 1-phenylethanol (Table 3). As shown, the amount of applied catalyst in our study is much lower than previous studies. Moreover, the recovery times for Fe3O4@YSPMO@Ru is more than the former catalysts. These observations clearly confirm high catalytic efficiency and also high durability of the Fe3O4@YSPMO@Ru in comparison to reported catalysts.
| Entry | Catalyst | Conditions | Time (h) | Recovery times | Ref. |
|---|---|---|---|---|---|
| 1 | Ru(OH)x/Fe3O4 | Toluene, cat. (3.8 mol%) O2 (1 atm), 105 °C | 2 | 1 | 91 |
| 2 | Fe3O4@SiO2/Ru(OH)x | Toluene, cat. (12.6 mol%) O2 (10 atm), 80 °C | 7 | 2 | 92 |
| 3 | RuO4@MCM-41 | Toluene, cat. (1.3 mol%) O2 (10 atm), 80 °C | 24 | 1 | 93 |
| 4 | Ru@PMO-IL | TFT, cat. (2.5 mol%) O2 (1 atm), 70 °C | 5.5 | 4 | 77 |
| 5 | Fe3O4@YSPMO@Ru | TFT, cat. (0.3 mol%) O2 (1 atm), 90 °C | 6 | 5 | This work |
4. Conclusion
In summary, the routine and commercially available CTAB and pluronic P123 surfactants were firstly used as efficient structure directing agents to prepare a yolk–shell nanocomposite with magnetite core and periodic mesoporous organosilica shell. This nanomaterial was effectively used for immobilization of potassium perruthenate species to deliver a novel catalyst named Fe3O4@YSPMO@Ru. The low angle PXRD analysis of Fe3O4@YSPMO@Ru confirmed the presence of a mesoporous shell for the material. The TG, FT-IR, EDX and XPS analyses showed well immobilization/incorporation and high stability of expected moieties into/onto material framework. The Fe3O4@YSPMO@Ru was successfully used as catalyst in the green oxidation of alcohols and gave the corresponding carbonyls in high selectivity and yield. Moreover, this catalyst was recovered and reused several times with keeping its efficiency. Therefore, this powerful and cost-effective catalyst can be promisingly used for practical applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Yasouj University and the Iran National Science Foundation (INSF) for supporting this work.References
- Q. Wen, M. Zhang, J. Zheng and J. Xu, CrystEngComm, 2018, 20, 5377–5386 RSC.
- J. Hu, M. Zhang, L. Liu, J. Zheng, H. Alsulami, M. A. Kutbi and J. Xu, Inorg. Chem., 2020, 59, 9356–9363 CrossRef CAS PubMed.
- W. He, X. Guo, J. Zheng, J. Xu, T. Hayat, N. S. Alharbi and M. Zhang, Inorg. Chem., 2019, 58, 7255–7266 CrossRef CAS PubMed.
- X. W. D. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- Y. Zhao and L. Jiang, Adv. Mater., 2009, 21, 3621–3638 CrossRef CAS.
- J. Liu, S. Z. Qiao and Q. H. Hu, Small, 2011, 7, 425–443 CrossRef CAS PubMed.
- J. C. Park and H. Song, Nano Res., 2011, 4, 33–49 CrossRef CAS.
- Q. Zhang, W. Wang, J. Goebl and Y. Yin, Nano Today, 2009, 4, 494–507 CrossRef CAS.
- A. Cao, R. Lu and G. Veser, Phys. Chem. Chem. Phys., 2010, 12, 13499–13510 RSC.
- J. Fan, G. Fang, X. Wang, F. Zeng, Y. Xiang and S. Wu, Nanotechnology, 2011, 22, 455102 CrossRef PubMed.
- C. Bharti, U. Nagaich, A. K. Pal and N. Gulati, Int. J. Pharm. Invest., 2015, 5, 124 CrossRef CAS PubMed.
- M. Zhang, L. Ding, J. Zheng, L. Liu, H. Alsulami, M. A. Kutbi and J. Xu, Appl. Surf. Sci., 2020, 509, 145348 CrossRef CAS.
- N. Lu, M. Zhang, L. Ding, J. Zheng, C. Zeng, Y. Wen, G. Liu, A. Aldalbahi, J. Shi and S. Song, Nanoscale, 2017, 9, 4508–4515 RSC.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- J. Gao, G. Liang, B. Zhang, Y. Kuang, X. Zhang and B. Xu, J. Am. Chem. Soc., 2007, 129, 1428–1433 CrossRef CAS PubMed.
- J. Liu, H. Xia, D. Xue and L. Lu, J. Am. Chem. Soc., 2009, 131, 12086–12087 CrossRef CAS PubMed.
- H. Li, Z. Bian, J. Zhu, D. Zhang, G. Li, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 8406–8407 CrossRef CAS PubMed.
- X.-J. Wu and D. Xu, J. Am. Chem. Soc., 2009, 131, 2774–2775 CrossRef CAS PubMed.
- G. Li, Q. Shi, S. Yuan, K. Neoh, E. Kang and X. Yang, Chem. Mater., 2010, 22, 1309–1317 CrossRef CAS.
- J. Dai, H. Zou, R. Wang, Y. Wang, Z. Shi and S. Qiu, Green Chem., 2017, 19, 1336–1344 RSC.
- J. Liu, S. Z. Qiao, S. Budi Hartono and G. Q. Lu, Angew. Chem., Int. Ed., 2010, 49, 4981–4985 CrossRef CAS PubMed.
- J. Liu, H. Q. Yang, F. Kleitz, Z. G. Chen, T. Yang, E. Strounina, G. Q. M. Lu and S. Z. Qiao, Adv. Funct. Mater., 2012, 22, 591–599 CrossRef CAS.
- J. Liu, B. Wang, S. B. Hartono, T. Liu, P. Kantharidis, A. P. Middelberg, G. Q. M. Lu, L. He and S. Z. Qiao, Biomaterials, 2012, 33, 970–978 CrossRef CAS PubMed.
- J. Liu, S. Z. Qiao, S. Budi Hartono and G. Q. M. Lu, Angew. Chem., 2010, 122, 5101–5105 CrossRef.
- L. Zhang, S. Qiao, Y. Jin, Z. Chen, H. Gu and G. Q. Lu, Adv. Mater., 2008, 20, 805–809 CrossRef CAS.
- S.-H. Wu, C.-T. Tseng, Y.-S. Lin, C.-H. Lin, Y. Hung and C.-Y. Mou, J. Mater. Chem., 2011, 21, 789–794 RSC.
- Y. E. Yan and F. W. Schwartz, Environ. Sci. Technol., 2000, 34, 2535–2541 CrossRef CAS.
- Z. Miao, X. Gu, S. Lu, X. Zang, X. Wu, M. Xu, L. B. B. Ndong, Z. Qiu, Q. Sui and G. Y. Fu, Chemosphere, 2015, 119, 1120–1125 CrossRef CAS.
- R. Broséus, S. Vincent, K. Aboulfadl, A. Daneshvar, S. Sauvé, B. Barbeau and M. Prévost, Water Res., 2009, 43, 4707–4717 CrossRef.
- G. Moussavi, A. Yazdanbakhsh and M. Heidarizad, J. Hazard. Mater., 2009, 171, 907–913 CrossRef CAS PubMed.
- W. Kujawski, A. Warszawski, W. Ratajczak, T. Porebski, W. Capała and I. Ostrowska, Desalination, 2004, 163, 287–296 CrossRef CAS.
- W. Zhang, L. Chen, L. Xu, H. Dong, H. Hu, Y. Xiao, M. Zheng, Y. Liu and Y. Liang, J. Colloid Interface Sci., 2019, 537, 562–568 CrossRef CAS PubMed.
- C. Yao, A. Yuan, H. Zhang, B. Li, J. Liu, F. Xi and X. Dong, J. Colloid Interface Sci., 2019, 533, 144–153 CrossRef CAS PubMed.
- U. Kurt, O. Apaydin and M. T. Gonullu, J. Hazard. Mater., 2007, 143, 33–40 CrossRef CAS PubMed.
- K. Bowden, I. Heilbron, E. Jones and B. Weedon, J. Chem. Soc., 1946, 39–45 RSC.
- E. J. Corey and J. W. Suggs, Tetrahedron Lett., 1975, 16, 2647–2650 CrossRef.
- G. Piancatelli, A. Scettri and M. D'auria, Synthesis, 1982, 1982, 245–258 CrossRef.
- A. De, J. Sci. Ind. Res., 1982, 41, 484–494 CAS.
- F. A. Luzzio and F. S. Guziec Jr, Org. Prep. Proced. Int., 1988, 20, 533–584 CrossRef CAS.
- J. Ladbury and C. Cullis, Chem. Rev., 1958, 58, 403–438 CrossRef CAS.
- A. J. Fatiadi, Synthesis, 1976, 1976, 65–104 CrossRef.
- R. J. Taylor, M. Reid, J. Foot and S. A. Raw, Acc. Chem. Res., 2005, 38, 851–869 CrossRef CAS.
- K. Pfitzner and J. Moffatt, J. Am. Chem. Soc., 1963, 85, 3027–3028 Search PubMed.
- A. J. Mancuso, S.-L. Huang and D. Swern, J. Org. Chem., 1978, 43, 2480–2482 CrossRef CAS.
- A. J. Mancuso, D. S. Brownfain and D. Swern, J. Org. Chem., 1979, 44, 4148–4150 CrossRef CAS.
- T. T. Tidwell, Synthesis, 1990, 1990, 857–870 CrossRef.
- D. Dess and J. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- M. Frigerio and M. Santagostino, Tetrahedron Lett., 1994, 35, 8019–8022 CrossRef CAS.
- M. Uyanik and K. Ishihara, Chem. Commun., 2009, 2086–2099 RSC.
- J. R. Parikh and W. v. E. Doering, J. Am. Chem. Soc., 1967, 89, 5505–5507 CrossRef CAS.
- P. Lucio Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559–2562 CrossRef CAS.
- A. E. De Nooy, A. C. Besemer and H. van Bekkum, Synthesis, 1996, 1996, 1153–1176 CrossRef.
- M. V. De Souza, Mini-Rev. Org. Chem., 2006, 3, 155–165 CrossRef CAS.
- T. Vogler and A. Studer, Synthesis, 2008, 2008, 1979–1993 CrossRef.
- R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2009, 14, 245–251 CrossRef.
- S. Patnaik, D. P. Sahoo and K. Parida, J. Colloid Interface Sci., 2020, 560, 519–535 CrossRef CAS.
- S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400–3420 CrossRef CAS PubMed.
- T. Nishimura, T. Onoue, K. Ohe and S. Uemura, J. Org. Chem., 1999, 64, 6750–6755 CrossRef CAS PubMed.
- M. J. Schultz, C. C. Park and M. S. Sigman, Chem. Commun., 2002, 3034–3035 RSC.
- G. J. t. Brink, I. W. Arends, M. Hoogenraad, G. Verspui and R. A. Sheldon, Adv. Synth. Catal., 2003, 345, 1341–1352 CrossRef.
- J. A. Mueller, C. P. Goller and M. S. Sigman, J. Am. Chem. Soc., 2004, 126, 9724–9734 CrossRef CAS PubMed.
- M. J. Schultz, S. S. Hamilton, D. R. Jensen and M. S. Sigman, J. Org. Chem., 2005, 70, 3343–3352 CrossRef CAS PubMed.
- M. Grßinne, Chem. Commun., 2010, 46, 7238–7240 RSC.
- P. R. Kasturi, R. Harivignesh, Y. S. Lee and R. K. Selvan, J. Colloid Interface Sci., 2020, 561, 358–371 CrossRef PubMed.
- A. Dijksman, A. Marino-González, A. Mairata i Payeras, I. W. Arends and R. A. Sheldon, J. Am. Chem. Soc., 2001, 123, 6826–6833 CrossRef CAS.
- R. Lenz and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 1997, 3291–3292 RSC.
- M. Hasan, M. Musawir, P. N. Davey and I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 2002, 180, 77–84 CrossRef CAS.
- A. Goti and M. Romani, Tetrahedron Lett., 1994, 35, 6567–6570 CrossRef CAS.
- A. Goti, F. De Sarlo and M. Romani, Tetrahedron Lett., 1994, 35, 6571–6574 CrossRef CAS.
- S. V. Ley, C. Ramarao and M. D. Smith, Chem. Commun., 2001, 2278–2279 RSC.
- J. Li, Z. Liu and R. Wang, J. Colloid Interface Sci., 2018, 531, 204–215 CrossRef CAS PubMed.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS PubMed.
- T. Hayashi, T. Inagaki, N. Itayama and H. Baba, Catal. Today, 2006, 117, 210–213 CrossRef CAS.
- H. Wang, Z. Jusys and R. Behm, J. Power Sources, 2006, 154, 351–359 CrossRef CAS.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS.
- T. Fey, H. Fischer, S. Bachmann, K. Albert and C. Bolm, J. Org. Chem., 2001, 66, 8154–8159 CrossRef CAS.
- B. Karimi, D. Elhamifar, O. Yari, M. Khorasani, H. Vali, J. H. Clark and A. J. Hunt, Chem.–Eur. J., 2012, 18, 13520–13530 CrossRef CAS.
- D. Elhamifar, O. Yari and B. Karimi, J. Colloid Interface Sci., 2017, 500, 212–219 CrossRef CAS PubMed.
- D. Elhamifar, Z. Ramazani, M. Norouzi and R. Mirbagheri, J. Colloid Interface Sci., 2018, 511, 392–401 CrossRef CAS PubMed.
- Y. S. Kang, S. Risbud, J. F. Rabolt and P. Stroeve, Chem. Mater., 1996, 8, 2209–2211 CrossRef CAS.
- S. Hajati, S. Tougaard, J. Walton and N. Fairley, Surf. Sci., 2008, 602, 3064–3070 CrossRef CAS.
- S. Hajati, V. Zaporojtchenko, F. Faupel and S. Tougaard, Surf. Sci., 2007, 601, 3261–3267 CrossRef CAS.
- V. L. Malinovskii, F. Samain and R. Häner, Angew. Chem., 2007, 119, 4548–4551 CrossRef.
- A. Christopher and K. Jeremy, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef.
- S. Balakrishnan and S. P. Sarma, Biochemistry, 2017, 56, 4346–4359 CrossRef CAS PubMed.
- Q. Hou, R. Bourgeas, F. Pucci and M. Rooman, Sci. Rep., 2018, 8, 1–13 CrossRef PubMed.
- N. J. Vickers, Curr. Biol., 2017, 27, R713–R715 CrossRef CAS PubMed.
- T. Chen, M. Li and J. Liu, Cryst. Growth Des., 2018, 18, 2765–2783 CrossRef CAS.
- D. P. Malenov and S. D. Zarić, Coord. Chem. Rev., 2020, 419, 213338 CrossRef CAS.
- D. P. Malenov and S. D. Zarić, CrystEngComm, 2019, 21, 7204–7210 RSC.
- Y. Zhao, J. Li, S. Zhang and X. Wang, RSC Adv., 2014, 4, 32710–32717 RSC.
- Y. Zhao, X. Wang, J. Li and X. Wang, Polym. Chem., 2015, 6, 5376–5384 RSC.
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |