
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesenchymal stem cells anchored with thymidine phosphorylase for doxifluridine-mediated cancer therapy
Ammar Tarar,
Esmael M. Alyami and
Ching-An Peng*
and
Ching-An Peng*
Department of Chemical & Biological Engineering, University of Idaho, Engineering Physics Building 421, 875 Perimeter Drive, Moscow, ID 83844-0904, USA. E-mail: capeng@uidaho.edu; Tel: +1-208-885-7461
First published on 5th January 2021
Abstract
Many tumors express thymidine phosphorylase (TYMP) with various levels, however due to tumor heterogeneity, the amount of TYMP is usually not enough to convert prodrug doxifluridine (5′-DFUR) to toxic drug 5-fluorouracil (5-FU). Since human mesenchymal stem cells (hMSCs) have unique features of tumor-tropism and low immunogenicity, the purpose of this study is to use mesenchymal stem cells as carriers to deliver TYMP to cancer cells and then trigger their death by administrating doxifluridine. First, the TYMP gene sequence and core streptavidin (core SA) were constructed into pET-30a(+) plasmid. After bacterial transformation and colony screening, TYMP-SA fusion protein was expressed by IPTG induction and purified by immobilized metal affinity chromatography and characterized by SDS-PAGE and western blot with a clear band at 75 kDa. The characterized TYMP-SA was further anchored on the cell membrane of biotinylated hMSCs via biotin–streptavidin binding. hMSCs anchored with TYMP-SA were then co-cultured with adenocarcinoma A549 cells (with different ratios) and treated with 100 μM prodrug doxifluridine over the course of four days. Our results showed that a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio led to the eradication of A549 cells at the end of the experiment with less than 5% confluency, in comparison with the 1:1 and 1:2 ratios which still had about 13% and 20% confluency respectively. In conclusion, harnessing hMSCs as cell carriers for the delivery of TYMP enzyme to cancer cells could lead to significant cell death post-treatment of the prodrug doxifluridine.
:1 ratio led to the eradication of A549 cells at the end of the experiment with less than 5% confluency, in comparison with the 1:1 and 1:2 ratios which still had about 13% and 20% confluency respectively. In conclusion, harnessing hMSCs as cell carriers for the delivery of TYMP enzyme to cancer cells could lead to significant cell death post-treatment of the prodrug doxifluridine.
1. Introduction
According to a recent report of the American Cancer Society, the estimated number of cancer patients will be 1.8 million in the year 2020 which is equal to approximately 5000 new cases each day.1 Several cancer treatments are being practiced clinically including surgery, radiotherapy, immunotherapy, and chemotherapy but all have respective consequences.2 Chemotherapy drugs are widely used for their effectiveness and convenience to eradicate tumors for a long time but have severe side effects i.e. hair loss, anemia, nausea, etc. Targeted chemotherapy using nontoxic prodrug, which can be converted into toxic drugs in the presence of a specific enzyme, is an alternative approach to minimize these side effects.5-Fluorouracil (5-FU) is one of the most commonly used chemotherapy drugs. It was first synthesized by Duschinsky et al. in 1957,3 and its effectiveness as an anti-tumor drug was later recognized by the fundamental clinical studies. Since then, it is approved for various cancer types such as for the treatment of breast, colon, and gastric cancers.4,5 It is usually administrated through continuous infusion to patients because of the short plasma half-life.6 However, to avoid the toxic effect, its prodrug such as capecitabine or doxifluridine (5′-DFUR) are used that are non-toxic. 5′-DFUR is converted into 5-FU by an enzyme named thymidine phosphorylase that is encoded by the gene TYMP. The thymidine phosphorylase (TYMP) is overexpressed in cancer cells and promotes tumor growth, angiogenesis, and inhibits apoptosis.7,8 Several studies have shown the efficacy of 5′-DFUR is linked with the expression and activity of TYMP in tumor sites.4,9 The endogenous expression of TYMP in cancer cells is usually not enough to convert prodrug (5′-DFUR) into a toxic drug (5-FU). The enhanced anti-cancer activity by non-toxic prodrug can be achieved by delivering TYMP on the cancer site to upregulate its expression level.10
Human mesenchymal stem cells (hMSCs) are stromal cells that can be isolated from different sources such as the umbilical cord,11,12 bone marrow,13 and adipose tissues.14,15 But most documented and widely used are bone marrow-derived MSCs.12 The MSCs can differentiate into different lineages that provide great benefits for regenerative medicines.16–19 Their tumor-tropic feature has led to harness MSCs as drug delivery vehicles for cancer therapy.20–26 Moreover, using MSCs as a delivery vehicle has several benefits such as low immunogenicity27 and lesser clinical complication risks.28 Recently, several in vitro experiments using MSCs show promising results to deliver anticancer treatment for various tumor models such as osteosarcoma,29 glioma,30 lung metastatic cancer,31 liver cancer,32 and leukemia.33 In this study, a fusion protein composed of TYMP and core streptavidin (TYMP-SA) was first obtained through bacterial expression and protein purification, and then tethered on the biotinylated cell membrane of hMSCs via robust streptavidin–biotin binding. The hMSCs decorated with exogenous TYMP were co-cultured with A549 lung cancer cells at various hMSC:A549 ratios to mimic TYMP delivery utilizing hMSCs as drug carriers to the tumor site in the body. After administration of prodrug doxifluridine to hMSC/A549 co-cultures up to four days, malignant A549 cells were eradicated significantly by toxic 5-FU converted from 5′-DFUR via the prodrug-activating enzyme (i.e., TYMP) tethered on biotinylated MSCs.
2. Materials and methods
2.1 Materials
Dulbecco's modified eagles' medium (DMEM) culture media, minimum essential medium alpha (αMEM), 0.25% trypsin–EDTA, fetal bovine serum (FBS), L-glutamine, bacterial protein extraction reagent (B-PER), streptavidin PE-Cy5.5, bicinchoninic acid (BCA) protein assay kit, bovine serum albumin (BSA), paraformaldehyde, hematoxylin, biotin-X DHPE (N-((6-biotinoyl amino)hexanoyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt), HisPur™ cobalt-NTA resin, protein concentrator PES (MWCO = 50 K), protease inhibitor EDTA free, phosphate buffer saline (PBS), streptavidin monoclonal antibody, and enhanced chemiluminescence (ECL) substrate were all purchased from Thermo Fisher Scientific (Waltham, MA). Dimethyl sulfoxide (DMSO), anti-TYMP monoclonal antibody, and kanamycin sulfate were obtained from Santa Cruz Biotech (Dallas, TX). Isopropyl-β-D-thiogalactopyranoside (IPTG), penicillin–streptomycin, lysogeny broth (LB) media, imidazole, collagen (bovine achilles tendon, type I), and 5-fluorouracil (5-FU) were purchased from Sigma-Aldrich (St. Louis, MO). Human mesenchymal stem cells (hMSCs) were obtained from RoosterBio (Frederick, MD). Adenocarcinoma A549 cell line was obtained from ATCC (Manassas, VA). Doxifluridine (5′-DFUR) was purchased from APExBIO (Houston, Texas). High fidelity Phusion polymerase, restriction enzymes (EcoRI, EcoRV, and XhoI), Monarch PCR & DNA cleanup kit, T4 DNA ligase, Blunt/TA Ligase Master Mix, T7 Express Lemo21(DE3) competent E. coli, L-rhamnose, and NEB5α competent E. coli (subcloning efficiency) were all purchased from New England BioLabs (NEB) (Ipswich, MA). Plasmid pcDNA3.1+C-eGFP-TYMP and His-Tag monoclonal antibody were acquired from GenScript (Piscataway, NJ). IgG horseradish peroxidase (HRP)-conjugated antibody was purchased from R&D Systems (Minneapolis, MN). Plasmid pET-30a(+) was obtained from Addgene (Watertown, MA). Rapid Coomassie blue stain was obtained from Research Products International (Mount Prospect, IL). Plasmid Miniprep kit was obtained from QIAGEN (Germantown, MD). Forward primer 5′-GCCATGGATATCATGGCAGCCTTGATGACCCC-3′, reverse primer 5′-GATCTCGAATTCTTGCTGCGGCGGCAGAACG-3′, and forward primer 5′-AGATCCGAATTCGGTGCTGCTGAAGCAGGT-3′, reverse primer 5′-ATTATACTCGAGGGAGGCGGCGGACGGCTT-3′ were all obtained from Integrated DNA Technologies (Coralville, Iowa). Laemmli sample buffer, Tris/glycine/SDS buffer, nitrocellulose membrane (0.2 μm), Tris-buffer saline, and Tween-20 were purchased from Bio-Rad (Hercules, CA). 8 μm transwell inserts for 24-well plates were purchased from Corning Inc. (Kennebunk, ME). SDF-1α was purchased from Tonbo Biosciences (San Diego, CA).2.2 Construction of TYMP-SA encoding plasmid
The thymidine phosphorylase (TYMP) cDNA was cloned from pcDNA3.1+C-eGFP-TYMP by polymerase chain reaction (PCR) with forward primer 5′-GCCATGGATATCATGGCAGCCTTGATGACCCC-3′ and reverse primer 5′-GATCTCGAATTCTTGCTGCGGCGGCAGAACG-3′. The TYMP PCR product was achieved with high fidelity Phusion polymerase (NEB) in a T-100 thermocycler (Bio-Rad) with initial denaturation at 98 °C for 30 s, followed by 35 cycles of denaturation at 98 °C for 10 s, annealing at 66 °C for 30 s, and extension at 72 °C for 15 s. The final extension was performed at 72 °C for 5 min. The core-streptavidin (core SA) cDNA was amplified by PCR from pSTE2-215 (yol) plasmid34 using forward primer 5′-AGATCCGAATTCGGTGCTGCTGAAGCAGGT-3′ and reverse primer 5′-ATTATACTCGAGGGAGGCGGCGGACGGCTT-3′. The core streptavidin PCR was performed with an initial denaturation at 98 °C for 30 s, followed by 35 cycles of denaturation at 98 °C for 10 s, annealing at 62 °C for 30 s, and extension at 72 °C for 15 s. Both PCR products (TYMP and core SA) were purified through the Monarch PCR & DNA cleanup kit (NEB) and confirmed by 1% agarose gel electrophoresis. The pET-30a(+) plasmid and core SA PCR product (insert) were cut with restriction enzymes XhoI and EcoRI. The digested pET-30a(+) and core SA were ligated by T4 DNA ligase (NEB) to get the pMT005 plasmid. The ligation product pMT005 was amplified by transformation with NEB5α competent E. coli and purified by Plasmid Miniprep kit. The pMT005 plasmid (vector) and TYMP PCR product (insert) were cut with restriction enzymes EcoRI and EcoRV. The digested pMT005 and TYMP products were ligated by Blunt/TA Ligase Master Mix (NEB) to get the pMT006 plasmid. The pMT006 ligation product was again amplified by transformation with NEB5α competent E. coli and purified by Plasmid Miniprep kit. As illustrated in Fig. 1A, TYMP-SA encoding recombinant plasmid (pMT006) was constructed by inserting the gene sequences of TYMP and core SA into the plasmid vector pET-30a(+). The gene sequences encoding TYMP and core SA from pMT006 cloned by PCR were verified by DNA gel electrophoresis with ∼1.4 kb for TYMP and ∼0.4 kb for core SA (Fig. 1B).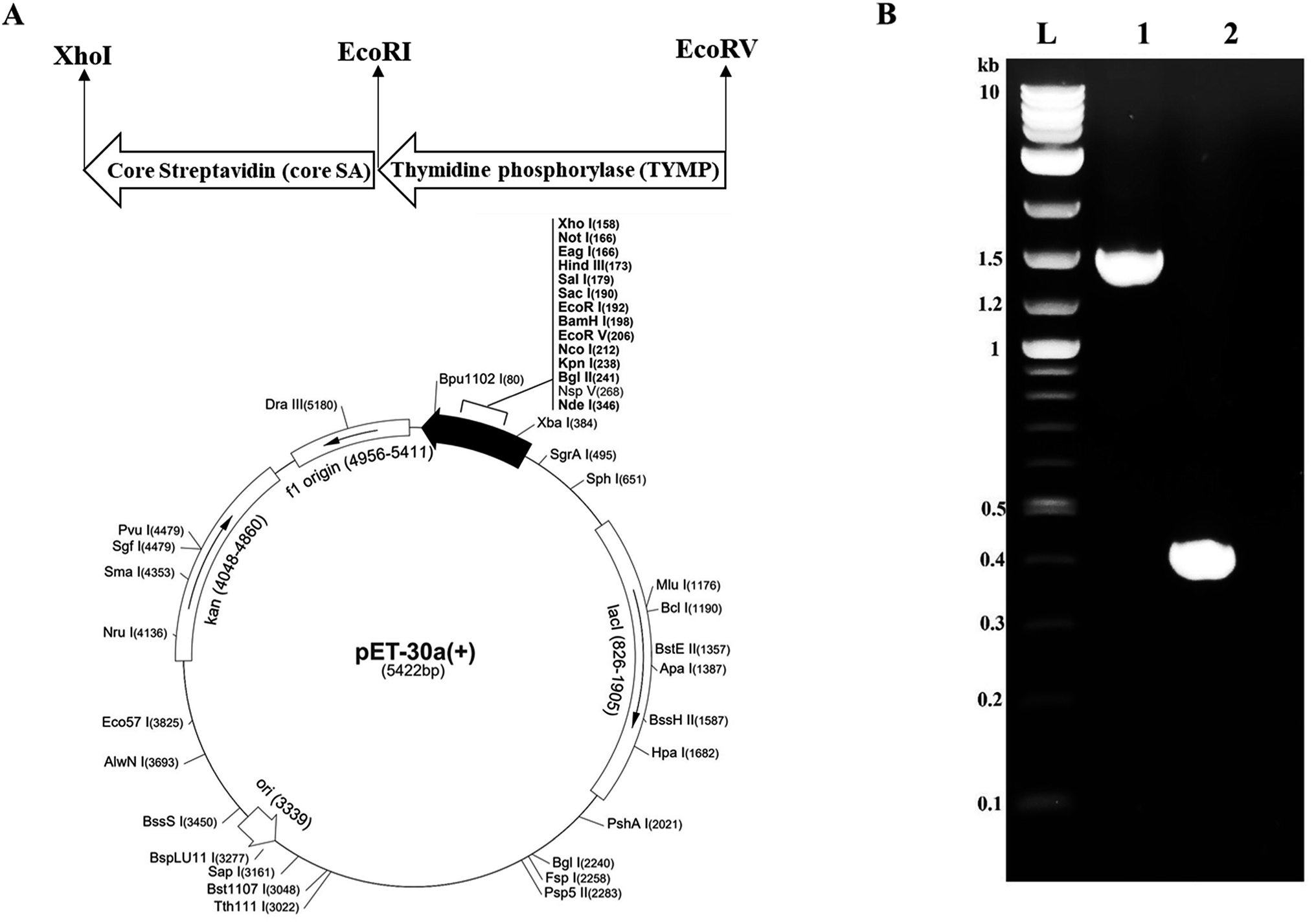 | ||
| Fig. 1 (A) The vector map showing cloning sites of fusion gene TYMP-SA. Core streptavidin was cloned between EcoRI and XhoI to give vector pMT005. The TYMP was cloned between EcoRV and EcoRI to give pMT006; (B) DNA gel electrophoresis of PCR products. L = DNA ladder, 1 = TYMP (∼1.4 kb), and 2 = core streptavidin (∼0.4 kb). | ||
2.3 Expression of TYMP-SA fusion protein
The constructed pMT006 vector was transformed into Lemo21(DE3) competent E. coli (NEB) according to manufacturer standard protocols and was spread on an agar plate (supplemented with 50 μg ml−1 kanamycin) and incubated overnight at 37 °C. Resuspended a single colony in 5 ml of LB media with kanamycin sulfate (50 μg ml−1) and grow again overnight at 37 °C to produce starter culture. The next day 0.2 ml of starter culture was diluted to 200 ml of LB media supplemented with 50 μg ml−1 of kanamycin and 500 μM of L-rhamnose and kept shaking until OD600 reached 0.5, then induced with 400 μM IPTG and kept shaking overnight at 22 °C with 225 rpm. Cells were then harvested by centrifugation at 4500 × g for 15 min. The cell pellets in each expression culture were re-suspended in 2 ml of proprietary nonionic detergent B-PER supplemented with 50 mM Tris–HCl and 1× of protease inhibitor EDTA-free (pH 7.5) to extract bacterial proteins at room temperature. The obtained bacterial lysate was further sonicated on ice with a pulse of 10 s (with 10 s rest in between each pulse) for ten times with power output set at seven (Misonix, Farmingdale, NY) to maximize the protein extraction efficiency. Then, the lysate was centrifuged at 18000 × g, and the supernatant (i.e., crude protein) was collected and proceeded for purification or stored at −20 °C.
2.4 Purification of TYMP-SA fusion protein
Cobalt-NTA affinity chromatography was used to purify TYMP-SA fusion protein from crude fractions. Crude protein was mixed with an equal amount of binding buffer (10 mM imidazole in 1× PBS) and gently shook in HisPur™ cobalt-NTA resin for 1 hour at 4 °C, then washed with five-volume of bed resin with wash buffer (10 mM imidazole in 1× PBS), and finally eluted with 5 bed resin volume with elution buffer (250 mM imidazole in 1× PBS). The optical density at 280 nm for all fractions was measured by a microplate reader (SpectraMax M2e; Molecular Devices, Sunnyvale, CA) and elution fractions were concentrated using protein concentrators PES (MWCO = 50 K). For the control group study using core SA protein only, Lemo21(DE3) was transformed with the constructed pMT005 vector, expressed, and purified by the aforementioned methods.2.5 SDS-PAGE and western blot analysis
Eluted TYMP-SA fusion protein and the crude fraction were diluted separately in 1:1 ratio with 2× Laemmli sample buffer containing 5% 2-mercaptoethanol, 60 mM Tris, 2% sodium dodecyl sulfate (SDS), 10% glycerol, 0.02% bromophenol and heated to 80 °C for 10 min. SDS-PAGE was performed in 12% polyacrylamide gels using 1× Tris/glycine/SDS buffer at 200 V for 40 min. The gel was stained with rapid Coomassie blue staining solution and destained for 1 hour with destain solution (7.5% methanol and 5% acetic acid). For western blot analysis, eluted TYMP-SA was run on SDS-PAGE by the same procedures stated above and then transferred to nitrocellulose membrane by Trans-Blot® semi-dry system (Bio-Rad). The membrane was blocked with 5% BSA in Tris-buffered saline with 0.1% Tween-20 (TBST) for 1 hour at room temperature. Then, washed three times gently with TBST and allowed to react with 1:1000 diluted primary antibody solution prepared in blocking buffer overnight at 4 °C. Two separate western blot experiments were done using different primary antibodies: streptavidin monoclonal antibody and TYMP monoclonal antibody. After overnight shaking in primary antibody solutions, the membranes were washed three times with TBST, followed by incubating in mouse IgG HRP-conjugated antibody for 1 hour at room temperature. Finally, the presence of elution protein was detected by horseradish peroxidase activity using ECL substrate through chemiluminescence imager (PXi Syngene, Frederick, MD).
2.6 Biotinylation of hMSCs
Human mesenchymal stem cells (hMSCs) were cultured in 6-well plates containing αMEM supplemented with 1% penicillin–streptomycin and 16.5% FBS, 2 mM L-glutamine at 37 °C with 5% CO2 and balanced humidifier air. After 24 hours, the culture media were replaced with fresh media supplemented with 0.02 mg ml−1 biotin-lipid (biotin-X DHPE) and incubated up to 48 hours. Cells were washed with 1× PBS and incubated in streptavidin PE-Cy5.5 for 1 hour to allow streptavidin bound with biotin anchored on the outer membrane of MSCs. After that, cells were gently washed with PBS to discard any unbounded streptavidin PE-Cy5.5 and fluorescence intensity was measured using a microplate reader (SpectraMax M2e, Molecular Devices, Sunnyvale, CA). The biotinylation kinetics of MSCs was determined by plotting the detected fluorescence intensity versus the time used for MSCs treated with biotin-lipids.2.7 Enzyme activity of TYMP-SA tethered on biotinylated MSCs
First, BCA protein assay according to the instruction provided by the manufacture was performed to quantify the elution protein. Briefly, BSA stock (2000 μg ml−1) was diluted to 1500, 1000, 750, 500, 250, 125, 25 μg ml−1 in clean vials. Working reagent (WR) was prepared by mixing 50 parts of BCA reagent A and 1 part of BCA reagent B. 200 μl of WR was mixed with 30 μl of each standard and elution protein and incubated for 30 min at 37 °C. All the experiment was done in triplicate and the absorbance at 562 nm was measured using SpectraMax M2e (Molecular Devices). For the anchoring of TYMP-SA on the outer membrane of MSCs, biotinylated cells were cultured in 6-well plates and treated with 20 μl of purified TYMP-SA fusion protein with the concentration determined above for 1 hour. The initial absorbance at 305 nm as a baseline was measured by SpectraMax M2e (Molecular Devices). 100 μM of 5′-DFUR was added into the well and incubated at 37 °C for 2 hours. The final absorbance was measured again at 305 nm, and the amount 5-FU produced was calculated by using the calibration curve of 5-FU at 305 nm.4,8 The calibration curve was obtained by plotting the measured absorbances of 5-FU serial dilutions against its concentrations. The TYMP enzyme activity was then calculated as the amount of 5-FU formed/μg protein per hour using the calibration curve and the concentration of purified protein obtained from the BCA protein assay.2.8 Migration assay of hMSCs tethered with TYMP-SA
The migration capability of hMSCs decorated with TYMP-SA was examined using transwell plates with 8 μm pore size inserts. The transwell inserts were immersed in 10 μg ml−1 collagen solution at 4 °C for 6 to 8 h. The inserts were then rinsed with 1× PBS. 2 × 105 MSCs and MSCs/TYMP-SA were suspended in 200 μl serum-free αMEM and added to the top chamber of each transwell inserts. The lower chamber was loaded with 600 μl αMEM containing 10% FBS and 200 ng ml−1 chemoattractant SDF-1α. CNT-MSCs were allowed to migrate for 24 h at 37 °C in a 5% CO2-balanced and humid culture chamber. The MSCs and MSCs/TYMP-SA that migrated to the bottom of the insert were fixed with 4% paraformaldehyde at room temperature for 15 min. The cells were then stained with hematoxylin at room temperature for 20 min. Cells in the top chamber were removed with a cotton swab. Cells in lower chamber were counted manually by phase contrast microscopy.2.9 Co-culture of hMSC/TYMP-SA and A549 cells with 5′-DFUR prodrug treatment
hMSCs decorated with TYMP-SA (hMSC/TYMP-SA) were co-cultured with A549 cells in 24-well plates with three different hMSC:A549 ratios 1:2, 1:1, and 2:1. hMSCs cell number was fixed at 103 cells per cm2 and the A549 cell number was adjusted according to each ratio and cultured in 50% αMEM and 50% DMEM culture medium followed with the induction of 100 μM prodrug 5′-DFUR. Three sets of control experiments were also performed at the same time. For the first control experiment, co-culture of hMSCs and A549 cells was not treated with 5′-DFUR; for the second one, hMSCs were not biotinylated; for the third one, hMSCs were biotinylated and tethered with core SA (not with TYMP-SA). For the second and third control experiments, cell co-cultures were treated with 100 μM of 5′-DFUR. Cells were observed under a DMi8 microscope equipped with E3 digital color camera (Leica Microsystems, Wetzlar, Germany) over a period of four days and cell viability was quantified using trypan blue and counted by a hemocytometer.
3. Results and discussion
3.1 Expression and purification of recombinant TYMP-SA fusion protein
The pMT006 vector was constructed by cloning TYMP and core SA in between EcoRV/EcoRI and EcoRI/XhoI respectively of pET-30a(+) plasmid as shown in Fig. 1A. The gene coding sequence of TYMP (1458 bp) and core SA (387 bp) in pMT006 were verified by agarose gel shown in Fig. 1B. Given the sequence of TYMP being 1458 bp (∼55 kDa), core SA 387 bp (∼15 kDa), and extras (thrombin site, S tag, 6× His, enterokinase site) being 135 bp (∼5 kDa), the molecular weight of TYPM-SA fusion protein was calculated around 75 kDa. As shown in Fig. 2A, SDS-PAGE revealed enhanced protein band at 75 kDa, verifying the expression of TYMP-SA fusion protein under the induction conditions (i.e., 500 μM L-rhamnose and 400 μM IPTG). The TYMP-SA fusion protein was purified from bacterial lysate by Co-NTA affinity chromatography, as shown in Fig. 2B. The advantage of using cobalt resin is that it binds specifically to His-tagged recombinant protein and can be eluted with mild imidazole treatment. Because higher specificity of cobalt resin to histidine than nickel resin leads to less off-target binding, the washing and binding processes are simplified by using the same buffer (10 mM imidazole in 1× PBS); while for nickel resin the concentration of imidazole in washing buffer is different from the one in binding buffer. However, it should be noted that the yield of TYMP-SA was less using cobalt resin as compared to nickel resin (data not shown) but the purity was much better.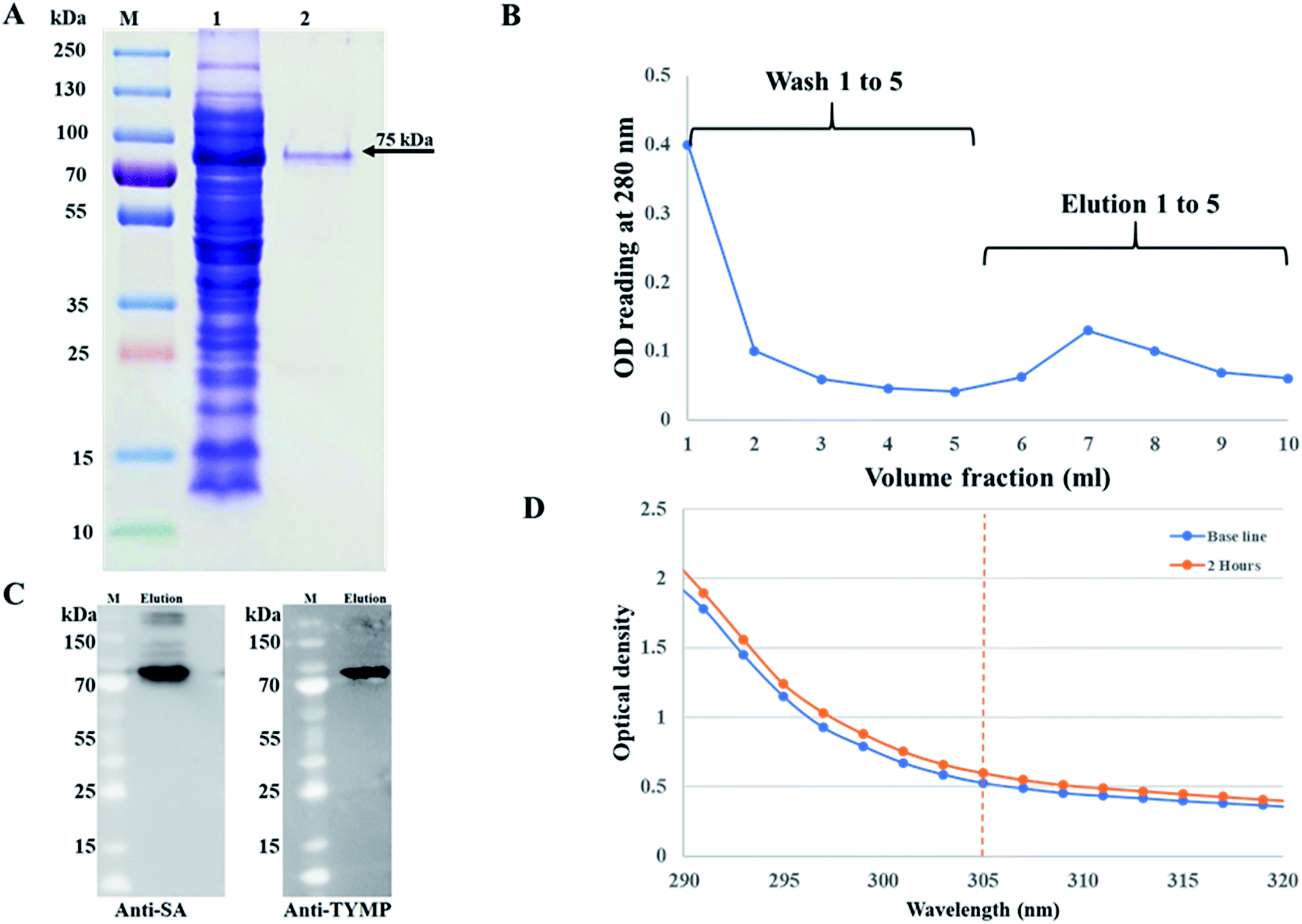 | ||
| Fig. 2 (A) SDS-PAGE of elution protein purified by Co-NTA affinity chromatography. The gel is imaged as (M) protein markers, (1) soluble crude fraction using Lemo21(DE3) competent cells induced with 500 μM L-rhamnose and 400 μM IPTG, and (2) purified TYMP-SA fusion protein; (B) elution profile of TYMP-SA fusion protein using 10 mM imidazole for wash buffer and 250 mM imidazole for elution buffer; (C) western blot analysis of purified TYMP-SA against anti-streptavidin and anti-TYMP monoclonal antibody. M and elution stand for protein markers and eluted TYMP-SA fusion protein, respectively. (D) Spectrometer reading of TYMP decorated hMSC after incubation with 100 μM 5′-DFUR for 2 hours, the difference in OD reading at 305 nm was used to measure the enzyme activity. | ||
3.2 Characterization of TYMP-SA
The purified TYMP-SA fusion protein was characterized by SDS-PAGE. As shown in lane 2 of Fig. 2A, one band with MW around 75 kDa was clearly observed. The eluted protein was further concentrated using a protein concentrator PES (MWCO = 50 K) and the concentration of eluted fusion protein was calculated to be 218 ± 10 μg ml−1 [n = 3] using the BCA assay. The TYMP-SA was further characterized by western blot analysis. As shown in Fig. 2C, the purified TYMP-SA was detected using an anti-streptavidin monoclonal antibody against core SA and anti-TYMP monoclonal antibody against TYMP. The enzyme activity was calculated using spectrometer reading. After 2 h incubation of TYMP decorated on hMSCs with 100 μM 5′-DFUR at 37 °C, the OD reading at 305 nm increased from 0.52 to 0.60 indicating the conversion of 5′-DFUR into 5-FU (Fig. 2D). The enzyme activity of TYMP-SA was calculated to be 1.59 ± 0.06 μmol μg−1 h−1 which is much higher than the reported values.35,36 Such a discrepancy is because the TYMP enzyme activity for current study was calculated by dividing purified protein, yet the ones reported in the literature was divided by total crude protein.3.3 Surface biotinylation and TYMP-SA decoration of hMSCs
The biotinylation of hMSCs was achieved by using biotin-lipid which is a mild approach as compared to conjugating agents such as EZ-link sulfo-NHS-LC-biotin.37 After MSCs attached to the culture plate, the medium was replaced with fresh medium containing 0.02 mg ml−1 of biotin lipid (biotin-X DHPE). It has been reported previously that biotinylation of Vero cells and hMSCs can be biotinylated with the help of biotin lipid without any toxicity effect.28,38 The biotinylated MSCs were then incubated with fluorescent streptavidin PE-Cy5.5 and visualized/imaged under a fluorescence microscope (Fig. 3A). The fluorescent intensity correlated with the biotin level incorporated on MSC cell membrane was measured by a microplate reader. The biotinylation curve was plotted as fluorescence intensity versus the time of biotinylation (Fig. 3B). It has been found that the fluorescence intensity rapidly increased and reached a saturated level after biotinylation for 48 hours.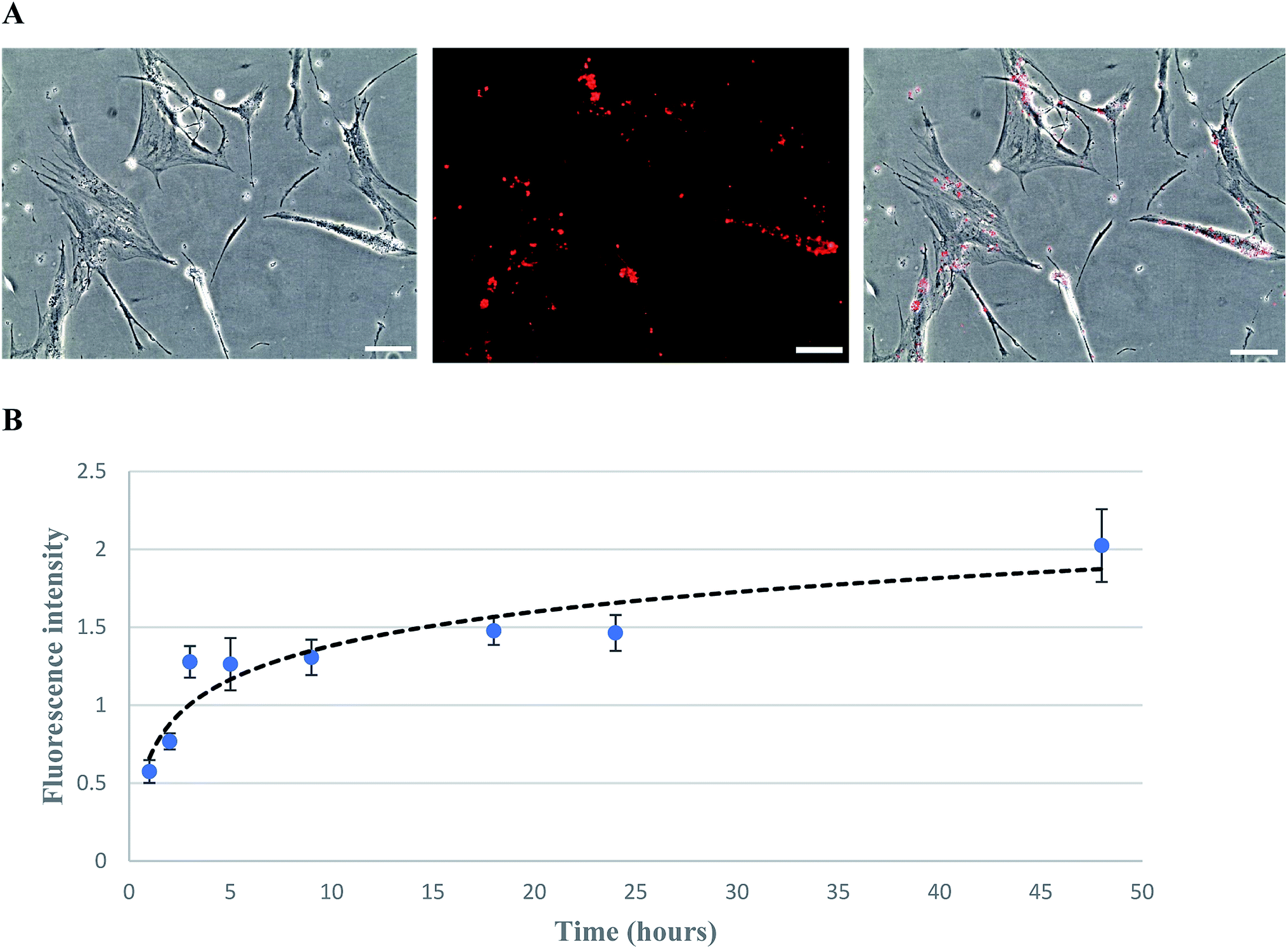 | ||
| Fig. 3 hMSCs treated with 0.02 mg ml−1 biotin X-DHPE for 48 h followed by streptavidin PE-Cy5.5 treatment. (A) Phase-contrast (right), fluorescence (middle), and overlay (left) images were taken with 10× magnification (scale bar denotes 100 μm); (B) the plot of fluorescence intensity versus time of biotinylation (error bars show standard deviation and n = 3). | ||
The biotinylated MSCs after anchored with TYMP-SA by biotin–streptavidin affinity were cultured to obtain the cell growth curve over 7 day cultivation. The growth kinetics revealed that both TYMP-tethered and unmodified MSCs followed a similar proliferation rate (data not shown) which indicates that the processes of biotinylation and decoration with TYMP-SA were not toxic to MSCs. This complies with a previous report indicating that biotinylation of hMSC using biotin-lipid did not affect the cell morphology and its migration abilities.28 For the transwell migration assay, the cell number of migrated MSCs and TYMP-tethered MSCs toward 200 ng ml−1 SDF-1α was about 50 cells per microscopic field for both treated and untreated MSCs (data not shown), indicating the anchoring of TYMP on the outer membrane of MSCs did not alter the migration capacity of MSCs. Moreover, the stability of fusion proteins decorated on cell membrane via streptavidin–biotin binding has been demonstrated to be effective for a long period of time; for instance, CD80-SA decorated splenocyte and human cancer cell lines remained stable and half-life was confirmed to be more than 10 days under in vitro and in vivo conditions.39 Judging from that, our experiment performed for 4 days is expected to be much stable until the end of the experiment.
3.4 Co-culture of hMSC/TYMP-SA and A549 cells with 5′-DFUR prodrug treatment
Prodrug therapy is a novel technique to target cancer cells to avoid the toxic effect of chemotherapy drugs.40 The challenge comes to deliver the specific enzyme required to convert prodrug to chemotherapy drugs at the site of interest. In this study, we targeted A549 lung cancer cells using hMSCs as cell carriers to deliver exogenous TYMP to convert 5′-DFUR into 5-FU. hMSCs have been considered as alternative and safe drug/gene delivery vehicles to target tumor sites due to its intrinsic tumor-tropism and less clinical complications.41,42 We used co-cultures (inoculated with 65 to 75% confluency) in three different ratios i.e. M2:A1, M1:A1, M1:M2 including control of 1:1 (hMSC:A549), and 100 μM of 5′-DFUR was added to each co-culture for four days. The 1:1 control group did not have TYMP-tethered hMSCs, instead just normal hMSCs were used. As shown in Fig. 4, over the cultivation and prodrug treatment period, M2:A1 group revealed the best anticancer effectiveness among all investigated groups.
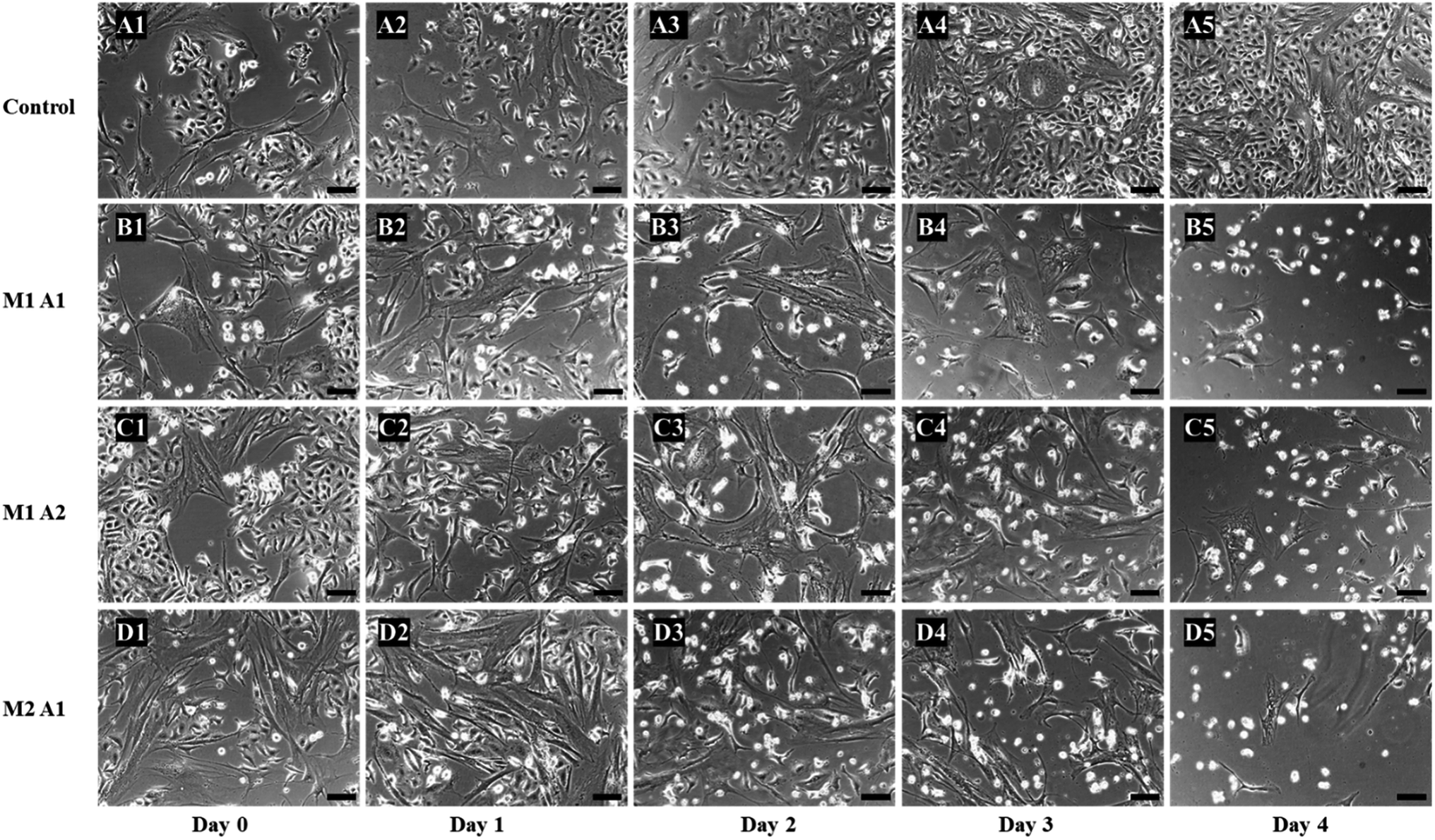 | ||
| Fig. 4 Phase contrast microscopic images of co-culture cytotoxic studies of different ratios of hMSC/TYMP-SA and A549 cells treated with 100 μM of 5′-DFUR. The images of the control co-culture experiment from day 0 to day 4 are from A1 to A5, for M1:A1 (hMSC/TYMP:A549 ratio) is from B1 to B5, for M1:A2 is from C1 to C5, and for M2:A1 is from D1 to D5 (scale bar = 100 μm). Control did not reveal any cell death and reached 100% confluent at day 4 (A5). The rest groups showed significant killing of cancer cells, with maximum efficiency of M2:A1 group on day 4 (D5). | ||
From the images, the death of A549 cancer cells was observed at day 2 and up to day 4 almost no cancer cell survived but few hMSCs are still alive (Fig. 4D1–D5). The confluency decreased by about half in 2 days (from 65% to 33%) and left with less than 5% at the end of the experiment (Fig. 5). The same trends followed for M1:A1 and M1:A2 groups with cell death from day 2 to day 4 (Fig. 4B1–B5 and C1–C5 respectively), but did not kill the cancer cells efficiently at the end of day 4 and final confluency at the end of the experiment were determined to be 20% and 13%, respectively (Fig. 5). It is noteworthy that drug-delivering vehicles (i.e., MSCs) were also killed by the cytotoxic 5-FU converted from 5′-DFUR prodrug via TYMP tethered on the outer membrane of MSCs. This indeed is a desired outcome because the potential for malignant transformation of MSCs under ex vivo production and expansion of cell lines is of concern. In fact, the risk associated with tumorigenesis after stem cell transplantation is widely discussed in the literature.43 On the contrary, the control co-culture group was continuing to grow from day 0 to day 4 (Fig. 4A1–A5) and become 100% confluency at the end of the experiment (Fig. 5). Despite adding the same amount of 5′-DFUR, there was no cell death observed which implies that the endogenous expression level of TYMP (if there is any) in A549 cells was not enough to covert prodrug 5′-DFUR into toxic 5′-FU. Indeed, the reported enzyme activity of endogenous TYMP expressed in A549 was about 475 pmol μg−1 h−1 (ref. 44) which is way less than the one reported in this study.
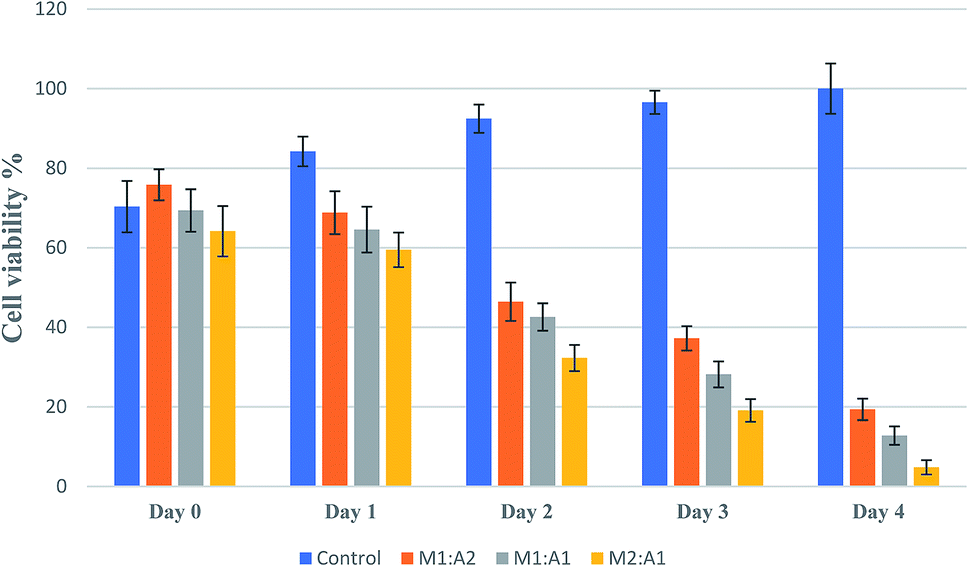 | ||
| Fig. 5 Viable cell confluency (%) of co-culture in different ratios of TYMP-tethered hMSCs and A549 cells (M1:A2, M1:A1, and M2:A1 ratio) when treated with 100 μM 5′-DFUR prodrug for 4 days. All groups had 65% to 75% confluency at the start (day 0) of the experiment. From day 1 to day 4, the cell confluency of control group increased from 84% to 100% (implicating the prodrug has no cytotoxic effect); but for M1:A2, M1:A1, and M2:A1 groups the cell confluency dropped to 20%, 13%, and 5%, respectively, indicating cell death caused by the conversion of prodrug to toxic 5-FU via TYMP-decorated hMSCs. | ||
To further confirm the eradication of cancer cells was only due to 5-FU converted from 5′-DFUR in the presence of TYMP, two more control experiments were performed. First, hMSCs were not biotinylated and loaded with TYMP-SA fusion protein as the procedures given above. After washed with PBS, the co-culture of hMSCs and A549 cells were treated with 100 μM 5′-DFUR. As shown in Fig. 6A1–A4, cells did not show any death or morphological change over 4 day cultivation. For the second control study, hMSCs were biotinylated and incubated with purified core SA protein (no TYMP). After treated with 100 μM 5′-DFUR for 4 days, cells remained healthy (Fig. 6B1–B4). There was no cell death in both control experiments indicating that core SA or 5′-DFUR alone has no effect on cell death. The toxic effect on cells was indeed attributed to 5-FU which is converted from 5′-DFUR by TYMP-anchored on the membrane surface of MSCs.
 | ||
| Fig. 6 Phase-contrast microscopic images of control co-culture experiments. (A1–A4) show that the non-biotinylated hMSCs co-cultured with A549 cells in the presence of 5′-DFUR (100 μM) continued to grow and reached 100% cell confluency on day 4; (B1–B4) show the co-culture of core SA-tethered biotinylated hMSCs and A549 cells treated with 100 μM of 5′-DFUR reached cell confluency on day 4. | ||
Prodrugs are considered as nontoxic and inactive compounds, which can be delivered systematically to the tumor sites and converted into cytotoxic drugs by relevant enzymes.45 The treatment of cancer cells with prodrug enzyme therapy is relatively safe, improving tumor's response to chemotherapeutic and limiting the side effects by sparing the healthy tissues. Because of their low immunogenicity and tumor tropism, hMSCs are emerging as promising cellular vehicles to deliver therapeutic agents to tumor sites. Several in vitro and preclinical animal studies have already proved their safety, efficacy, and have very low side effects as compared to other chemotherapy treatments.46–51 Recently, MSC-driven gene-directed enzyme prodrug therapy clinical trials for the treatment of end-stage gastrointestinal cancer have presented promising results.52 The autologous hMSCs were genetically modified to express herpes simplex virus thymidine kinase that catalyzed prodrug ganciclovir to toxic ganciclovir triphosphate, thereby leading to the death of cancer cells. The treatment was reported safe, feasible, and tolerable with no major side effects. Other ongoing clinical trials (http://ClinicalTrials.gov identifier: NCT03298763 and NCT02530047) using MSCs as drug delivery vehicles to target cancer cells were also reported. However, it should be noted that the results and discussion of this study were made merely at in vitro level. Further study is needed to examine if the anticancer effectiveness of TYMP-tethered hMSCs coupling with 5′-DFUR prodrug will provide promising outcomes using in vivo animal model.
4. Conclusions
Our results showed that TYMP-SA can be successfully anchored on the surface of hMSCs through biotin–streptavidin binding. These TYMP-decorated hMSCs can eradicate A549 cells by converting 5′-DFUR into 5-FU. Our results further revealed that the ratio of TYMP-decorated hMSCs to A549 cancer cells is important, the higher number of hMSCs compared to cancer cells results in greater efficiency of killing over the course of 4 day cultivation. Moreover, from the control experiment, it was concluded that the endogenous TYMP expression of A549 cells was not enough to convert 5′-DFUR into 5-FU as no malignant cell death was observed at the end of the experiment.Conflicts of interest
There are no conflicts to declare.References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2020, 70, 7–30 CrossRef.
- B. W. Stewart and C. P. Wild, World Health Organ., 2014, 1–2 Search PubMed.
- R. Duschinsky, E. Pleven and C. Heidelberger, J. Am. Chem. Soc., 1957, 79, 4560 CrossRef.
- A. J. Sawdon, J. Zhang, X. Wang and C. A. Peng, Nanomaterials, 2018, 8, 1041 CrossRef.
- M. Shida, M. Yasuda, M. Fujita, M. Miyazawa, H. Kajiwara, T. Hirasawa, M. Ikeda, N. Matsui, T. Muramatsu and M. Mikami, Oncol. Lett., 2016, 12, 3215–3223 CrossRef CAS.
- A. De Gramont, G. Louvet, T. André, C. Tournigand and M. Krulik, Eur. J. Cancer, 1998, 34, 619–626 CrossRef CAS.
- S. Akiyama, T. Furukawa, T. Sumizawa, Y. Takebayashi, Y. Nakajima, S. Shimaoka and M. Haraguchi, Cancer Sci., 2004, 95, 851–857 CrossRef CAS.
- A. V. Patterson, H. Zhang, A. Moghaddam, R. Bicknell, D. C. Talbot, I. J. Stratford and A. L. Harris, Br. J. Cancer, 1995, 72, 669–675 CrossRef CAS.
- T. Nishina, I. Hyodo, J. Miyaike, T. Inaba, S. Suzuki and Y. Shiratori, Eur. J. Cancer, 2004, 40, 1566–1571 CrossRef CAS.
- H. W. Lo, C. P. Day and M. C. Hung, Adv. Genet., 2005, 54, 233–255 Search PubMed.
- X. Zhang, M. Hirai, S. Cantero, R. Ciubotariu, L. Dobrila, A. Hirsh, K. Igura, H. Satoh, I. Yokomi, T. Nishimura, S. Yamaguchi, K. Yoshimura, P. Rubinstein and T. A. Takahashi, J. Cell. Biochem., 2011, 112, 1206–1218 CrossRef CAS.
- W. Wagner, F. Wein, A. Seckinger, M. Frankhauser, U. Wirkner, U. Krause, J. Blake, C. Schwager, V. Eckstein, W. Ansorge and A. D. Ho, Exp. Hematol., 2005, 33, 1402–1416 CrossRef CAS.
- S. A. Wexler, C. Donaldson, P. Denning-Kendall, C. Rice, B. Bradley and J. M. Hows, Br. J. Haematol., 2003, 121, 368–374 CrossRef.
- W. J. F. M. Jurgens, M. J. Oedayrajsingh-Varma, M. N. Helder, B. ZandiehDoulabi, T. E. Schouten, D. J. Kuik, M. J. P. F. Ritt and F. J. Van Milligen, Cell Tissue Res., 2008, 332, 415–426 CrossRef.
- J. K. Fraser, I. Wulur, Z. Alfonso, M. Zhu and E. S. Wheeler, Cytotherapy, 2007, 9, 459–467 CrossRef CAS.
- Y. Han, X. Li, Y. Zhang, Y. Han, F. Chang and J. Ding, Cells, 2019, 8, 886 CrossRef CAS.
- R. M. Samsonraj, M. Raghunath, V. Nurcombe, J. H. Hui, A. J. van Wijnen and S. M. Cool, Stem Cells Transl. Med., 2017, 6, 2173–2185 CrossRef.
- F. J. Vizoso, N. Eiro, S. Cid, J. Schneider and R. Perez-Fernandez, Int. J. Mol. Sci., 2017, 18, 1852 CrossRef.
- S. Keshtkar, N. Azarpira and M. H. Ghahremani, Stem Cell Res. Ther., 2018, 9, 63 CrossRef CAS.
- K. Shinagawa, Y. Kitadai, M. Tanaka, T. Sumida, M. Onoyama, M. Ohnishi, E. Ohara, Y. Higashi, S. Tanaka, W. Yasui and K. Chayama, Int. J. Cancer, 2012, 132, 813–823 CrossRef.
- J.-T. Lin, J.-Y. Wang, M.-K. Chen, H.-C. Chen, T.-H. Chang, B.-W. Su and P.-J. Chang, Exp. Cell Res., 2013, 319, 2216–2229 CrossRef CAS.
- K. Shinagawa, Y. Kitadai, M. Tanaka, T. Sumida, M. Kodama, Y. Higashi, S. Tanaka, W. Yasui and K. Chayama, Int. J. Cancer, 2010, 127, 2323–2333 CrossRef CAS.
- M. T. Schweizer, H. Wang, T. J. Bivalacqua, A. W. Partin, S. J. Lim, C. Chapman, R. Abdallah, O. Levy, N. A. Bhowmick, J. M. Karp, A. De Marzo, J. T. Isaacs, W. N. Brennen and S. R. Denmeade, Stem Cells Transl. Med., 2019, 8, 441–449 CrossRef CAS.
- S. Suryaprakash, Y. H. Lao, H. Y. Cho, M. Li, H. Y. Ji, D. Shao, H. Hu, C. H. Quek, D. Huang, R. L. Mintz, J. R. Bagó, S. D. Hingtgen, K. B. Lee and K. W. Leong, Nano Lett., 2019, 19, 1701–1705 CrossRef CAS.
- X. Bin Zheng, X. W. He, L. J. Zhang, H. B. Qin, X. T. Lin, X. H. Liu, C. Zhou, H. S. Liu, T. Hu, H. C. Cheng, X. S. He, X. R. Wu, Y. F. Chen, J. Ke, X. J. Wu and P. Lan, Gastroenterol. Rep., 2019, 7, 127–138 CrossRef.
- E. D. Gomes, J. Vieira de Castro, B. M. Costa and A. J. Salgado, Biochimie, 2018, 155, 59–66 CrossRef CAS.
- M. E. Christensen, B. E. Turner, L. J. Sinfield, K. Kollar, H. Cullup, N. J. Waterhouse, D. N. J. Hart, K. Atkinson and A. M. Rice, Haematologica, 2010, 95, 2102–2110 CrossRef.
- J. Zhang and C.-A. Peng, RSC Adv., 2019, 9, 7156–7164 RSC.
- S. Duchi, G. Sotgiu, E. Lucarelli, M. Ballestri, B. Dozza, S. Santi, A. Guerrini, P. Dambruoso, S. Giannini, D. Donati, C. Ferroni and G. Varchi, J. Controlled Release, 2013, 168, 225–237 CrossRef CAS.
- S. Gunnarsson, D. Bexell, A. Svensson, P. Siesjö, A. Darabi and J. Bengzon, J. Neuroimmunol., 2010, 218, 140–144 CrossRef CAS.
- M. R. Loebinger, A. Eddaoudi, D. Davies and S. M. Janes, Cancer Res., 2009, 69, 4134–4142 CrossRef CAS.
- J. Zhang, L. Hou, X. Wu, D. Zhao, Z. Wang, H. Hu, Y. Fu and J. He, Mol. Cell. Biochem., 2016, 416, 193–203 CrossRef CAS.
- A. Pessina, V. Coccè, L. Pascucci, A. Bonomi, L. Cavicchini, F. Sisto, M. Ferrari, E. Ciusani, A. Crovace, M. L. Falchetti, S. Zicari, A. Caruso, S. Navone, G. Marfia, A. Benetti, P. Ceccarelli, E. Parati and G. Alessandri, Br. J. Haematol., 2013, 160, 766–778 CrossRef CAS.
- S. Dübel, F. Breitling, R. Kontermann, T. Schmidt, A. Skerra and M. Little, J. Immunol. Methods, 1995, 178, 201–209 CrossRef.
- M. Haraguchi, T. Furukawa, T. Sumizawa and S. Akiyama, Cancer Res., 1993, 53, 5680–5682 CAS.
- A. Evrard, P. Cuq, B. Robert, L. Vian, A. Plegrin and J.-P. Cano, Int. J. Cancer, 1999, 80, 465–470 CrossRef CAS.
- A. Knödler and P. Mayinger, BioTechniques, 2005, 38, 858–862 CrossRef.
- B.-H. Huang, Y. Lin, Z.-L. Zhang, F. Zhuan, A.-A. Liu, M. Xie, Z.-Q. Tian, Z. Zhang, H. Wang and D.-W. Pang, ACS Chem. Biol., 2012, 7, 683–688 CrossRef CAS.
- N. P. Singh, E. S. Yolcu, D. D. Taylor, C. Gercel-Taylor, D. S. Metzinger, S. K. Dreisbach and H. Shirwan, Cancer Res., 2003, 63, 4067–4073 CAS.
- R. Walther, J. Rautio and A. N. Zelikin, Adv. Drug Delivery Rev., 2017, 118, 65–77 CrossRef CAS.
- S. Wang, Z. Miao, Q. Yang, Y. Wang and J. Zhang, Can. J. Gastroenterol. Hepatol., 2018, 2018, 7628763 Search PubMed.
- S. Kariminekoo, A. Movassaghpour, A. Rahimzadeh, M. Talebi, K. Shamsasenjan and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 749–757 CrossRef CAS.
- A. Musial-Wysocka, M. Kot and M. Majka, Cell Transplant., 2019, 28, 801–812 Search PubMed.
- S. J. Lee, J. S. Yeo, H. J. Lee, E. J. Lee, S. Y. Kim, S. J. Jang, J. J. Lee, J. S. Ryu and D. H. Moon, Eur. J. Nucl. Med. Mol. Imaging, 2014, 41, 1327–1335 CrossRef CAS.
- C. Zischek, H. Niess, I. Ischenko, C. Conrad, R. Huss, K. W. Jauch, P. J. Nelson and C. Bruns, Ann. Surg., 2009, 250, 747–752 CrossRef.
- L. Kucerova, V. Altanerova, M. Matuskova, S. Tyciakova and C. Altaner, Cancer Res., 2007, 67, 6304–6313 CrossRef CAS.
- I. T. Cavarretta, V. Altanerova, M. Matuskova, L. Kucerova, Z. Culig and C. Altaner, Mol. Ther., 2010, 18, 223–231 CrossRef CAS.
- B. Layek, T. Sadhukha, J. Panyam and S. Prabha, Mol. Cancer Ther., 2018, 17, 1196–1206 CrossRef CAS.
- C. Conrad, Y. Hüsemann, H. Niess, I. Von Luettichau, R. Huss, C. Bauer, K. W. Jauch, C. A. Klein, C. Bruns and P. J. Nelson, Ann. Surg., 2011, 253, 566–571 CrossRef.
- M. R. Loebinger, A. Eddaoudi, D. Davies and S. M. Janes, Cancer Res., 2009, 69, 4134–4142 CrossRef CAS.
- H. Xin, R. Sun, M. Kanehira, T. Takahata, J. Itoh, H. Mizuguchi and Y. Saijo, Mol. Med., 2009, 15, 321–327 CAS.
- J. C. von Einem, C. Güenther, H. D. Volk, G. Grütz, D. Hirsch, C. Salat, O. Stoetzer, P. J. Nelson, M. Michl, D. P. Modest, J. W. Holch, M. Angele, C. Bruns, H. Niess and V. Heinemann, Int. J. Cancer, 2019, 145, 1538–1546 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |