
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, crystal structure and antibacterial studies of dihydropyrimidines and their regioselectively oxidized products†
Alakbar E. Huseynzada *a,
Christian Jelchb,
Haji Vahid N. Akhundzadaac,
Sarra Soudanid,
Cherif Ben Nasrd,
Aygun Israyilovae,
Filippo Doriaf,
Ulviyya A. Hasanovaa,
Rana F. Khankishiyevac and
Mauro Freccerof
*a,
Christian Jelchb,
Haji Vahid N. Akhundzadaac,
Sarra Soudanid,
Cherif Ben Nasrd,
Aygun Israyilovae,
Filippo Doriaf,
Ulviyya A. Hasanovaa,
Rana F. Khankishiyevac and
Mauro Freccerof
aBaku State University, ICRL, Z. Khalilov 23, Baku, AZ 1148, Azerbaijan. E-mail: alakbar.huseynzada1117@gmail.com
bUniversité de Lorraine, CNRS, CRM2, 54000 Nancy, France
cInstitute of Radiation Problems of ANAS, B. Vahabzada 9, Baku, AZ 1143, Azerbaijan
dLaboratoire de Chimie des Matériaux, Faculté des Sciences de Bizerte, Université de Carthage, 7021, Zarzouna, Tunisia
eDepartment of Molecular biology and Biotechnology, Baku State University, Z. Khalilov 23, Baku, AZ 1148, Azerbaijan
fUniversita di Pavia, V.le Taramelli 10, 27100 Pavia, Italy
First published on 3rd February 2021
Abstract
The syntheses and investigations of new biologically active derivatives of dihydropyrimidines by Biginelli reaction in the presence of copper triflate are reported. Due to the fact that salicylaldehyde and its derivatives under Biginelli reaction conditions can lead to the formation of 2 types of dihydropyrimidines, the influence of copper triflate on product formation was also investigated. In addition to this, regioselective oxidation of dihydropyrimidines was performed in the presence of cerium ammonium nitrate and novel oxidized dihydropyrimidines were obtained. Single crystals of some of them were obtained and as a result, the structures of them were investigated by X-ray diffraction method, which allows determining the presence of hydrogen bonds in their structures. In addition to this, the presence of hydrogen bonds in their structures affects the formation of the corresponding tautomer during oxidizing of dihydropyrimidines. Since dihydropyrimidines are claimed to be biologically active compounds, activities of the synthesized compounds were studied against Acinetobacter baumanii, Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae and Staphylococcus aureus bacteria.
Introduction
The three-component one-pot Biginelli reaction, which was discovered in 1893 by Pietro Biginelli by condensation of an aldehyde, urea and 1,3-dicarbonyl compound in an acid medium,1 is one of the most important and oldest multicomponent reactions. It is still in trend, due to the fact that this flexible and multilateral tool allows construction of an astonishing class of compounds with a broad spectrum of application area, namely dihydropyrimidines (H2Py).2–8 First of all, it must be mentioned that these marvelous compounds are important heterocyclic systems which play a crucial role as a building block of biomolecules such as DNA and RNA.9 Among other applications of these amazing compounds, the synthesis and development of materials with novel optical properties,10 design of polymers,11 dyes12 and adhesives13 can be mentioned. But the most widespread application area of H2Py is medicine due to their broad spectrum of biological activities. A multicomponent nature of Biginelli reaction provides large product diversity bearing different pharmacophoric groups in the structure of dihydropyrimidines which ensure the optimal supramolecular interactions with a specific biological target (molecular recognition) and trigger or block its biological response.3,14–17 Investigations through molecular manipulations reveal that this class of compounds demonstrate antiviral,3 anti-filarial,18,19 antifungal,3,15 analgesic,20 anti-leishmanial,21 antiproliferative,22 antitumor,23–28 anti-convulsant,29 antibacterial,30–33 anti-inflammatory,34–36 anti-hypertensive37–41 activities, melanin concentrating hormone 1 receptor antagonist,42 anti-HIV,43 antiepileptic,44 antidiabetic,45 anti-SARS,46 anti-malarial,47 mPGES-1 inhibitors,48 anti-hyperglycemic, antitubercular,49 TRPA1 antagonist,50 miscellaneous,51–53 potassium54–56 and calcium channels57 and α1a adrenergic antagonists.58 As a result of these investigations, different drugs based on H2Py such as riboflavin, idoxuridine, aminophylline, emivirine, 5-fluorouracil, methylthiouracil, batzelladine A and B etc. have found their application in medicine.9 Due to the utmost importance of H2Py in pharmaceutics, robust, efficient, cost-effective and “green” chemical synthesis and transformations of dihydropyrimidine scaffolds were developed.3,14,59–63Among important transformations of H2Py, the oxidation reactions are one of the crucial directions due to the fact that the solubility of oxidized products in polar solvents is higher. Moreover, the introduction of various functional groups, as well as the formation of new C–C, S–S and other types of bonds is easier resulting in extending their application area. In addition to this, in some cases, oxidized H2Py are more biologically active than initial H2Py.63–66
Concerning biological activity of H2Py, novel molecules were synthesized by Biginelli reaction in microwave conditions in the presence of copper triflate on the basis of urea, methyl acetoacetate and aldehyde scaffolds. Due to the fact that the Biginelli reaction of salicylaldehyde derivatives can lead to the formation of 2 types of H2Py, we also studied the effect of microwave conditions on product formation. Further, regioselective oxidation of H2Py in the presence of CAN was performed and novel oxidized H2Py were obtained. Moreover, crystals of some H2Py as well as their oxidized products were received and as a result, their structures were investigated by X-ray single crystal diffraction method. Due to the fact that H2Py demonstrate a wide spectrum of biological activities, their activity was analyzed against A. baumanii, E. coli, P. aeruginosa, K. pneumoniae and S. aureus bacteria and promising results were obtained.
Results and discussion
Chemical synthesis
It is known that a lot of protocols were developed for performing Biginelli reaction, leading to the synthesis of H2Py with high yields under “green” conditions and simple work-up procedure. Using catalyst types such as I2/MWI,67 Yb(PFO)3,68 TMSCl/CAN,69 [Hmim]HSO4–NaNO3,70 CAN,71 GaI3,72 HBF4–SiO2,73 Sm(ClO4)3,74 H3PMo12O40,75 La(OTf)3,76 Sc(OTf)3,77 In(OTf)3,78 RuCl3,79 MgCl2·6H2O,80 CeCl3·7H2O,81 acetic acid, NH4Cl17 and others,3–14 it was not possible to perform a Biginelli reaction on the basis of all used aldehyde scaffolds, urea and methylacetoacetate. In addition to this, performing the synthesis in microwave reactor becomes more popular over the conventional method, due to the fact that microwave method increases speed, reproducibility and scalability of the reaction.82,83 Taking into account the positive side of microwave assisted organic synthesis, improving the84 procedure by performing the reaction in the presence of Cu(OTf)2 in microwave conditions allowed us to obtain targeted H2Py 1–7 (Scheme 1 and Table 1). The positive side of this procedure is the almost total absence of work-up stage during the reactions realized for obtaining H2Py (1–7) – the precipitate was just washed with distilled water. Another positive side of using Cu(OTf)2 as a catalyst is that it is an excellent triflate surrogate to other metal triflates because it is cheap, demonstrates high activity and low toxicity making the process more environmentally friendly. The structures of all synthesized novel H2Py's were determined by 1H, 13C NMR, mass spectroscopy and elemental analysis, whereas the spectroscopic data of known compounds were compared with reported in the literature.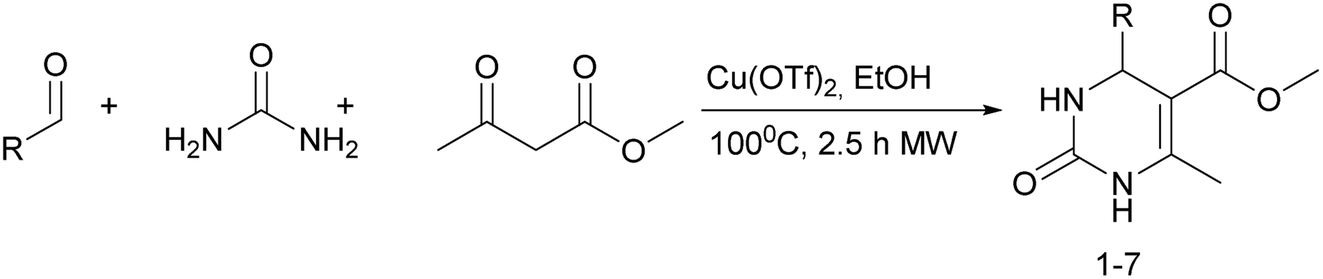 | ||
| Scheme 1 Synthesis of dihydropyrimidines in the presence of Cu(OTf)2. | ||
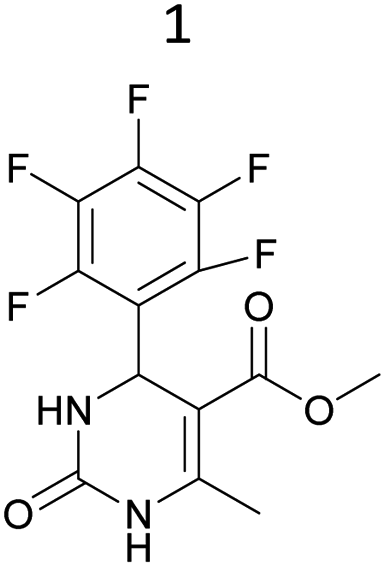
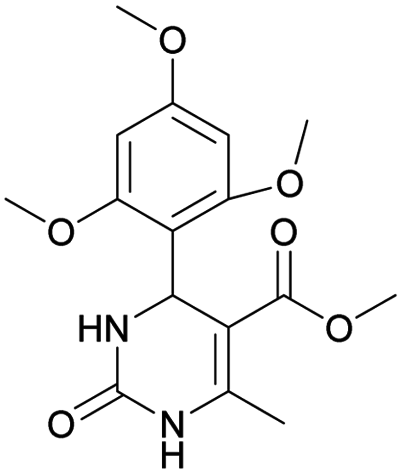
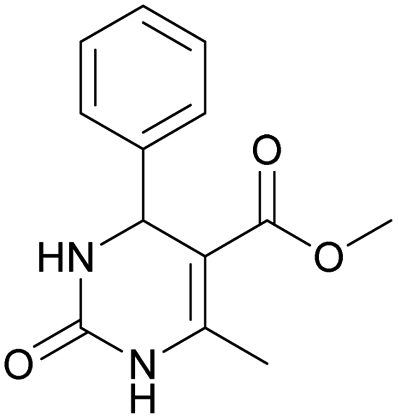
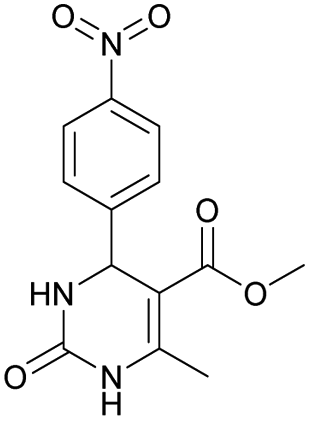
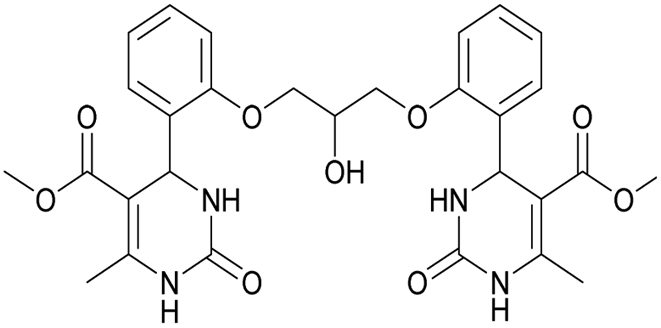
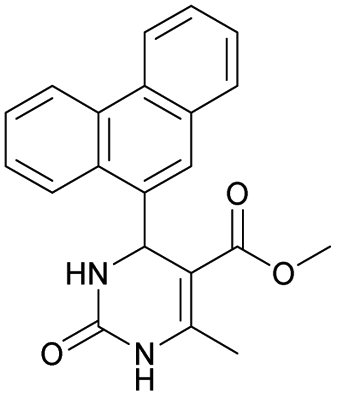
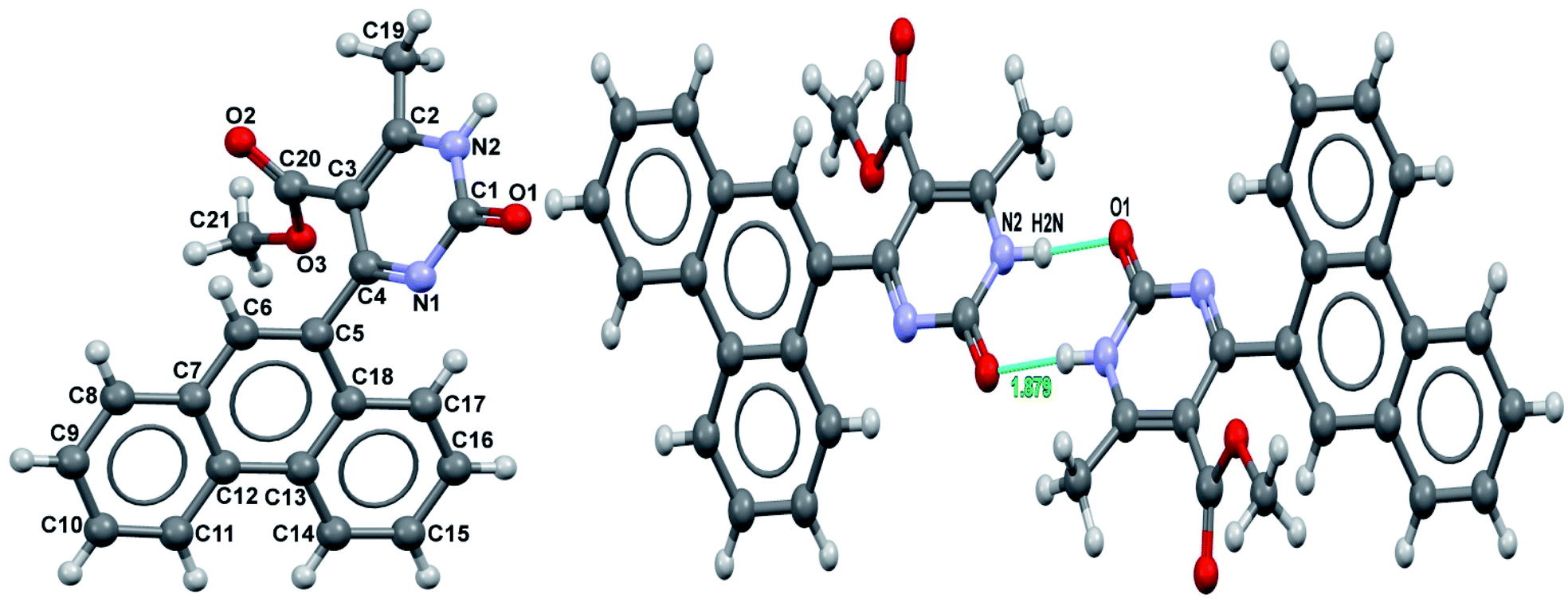
It was also possible to obtain single crystals of compounds 2, 3 and 7 (Fig. 1).
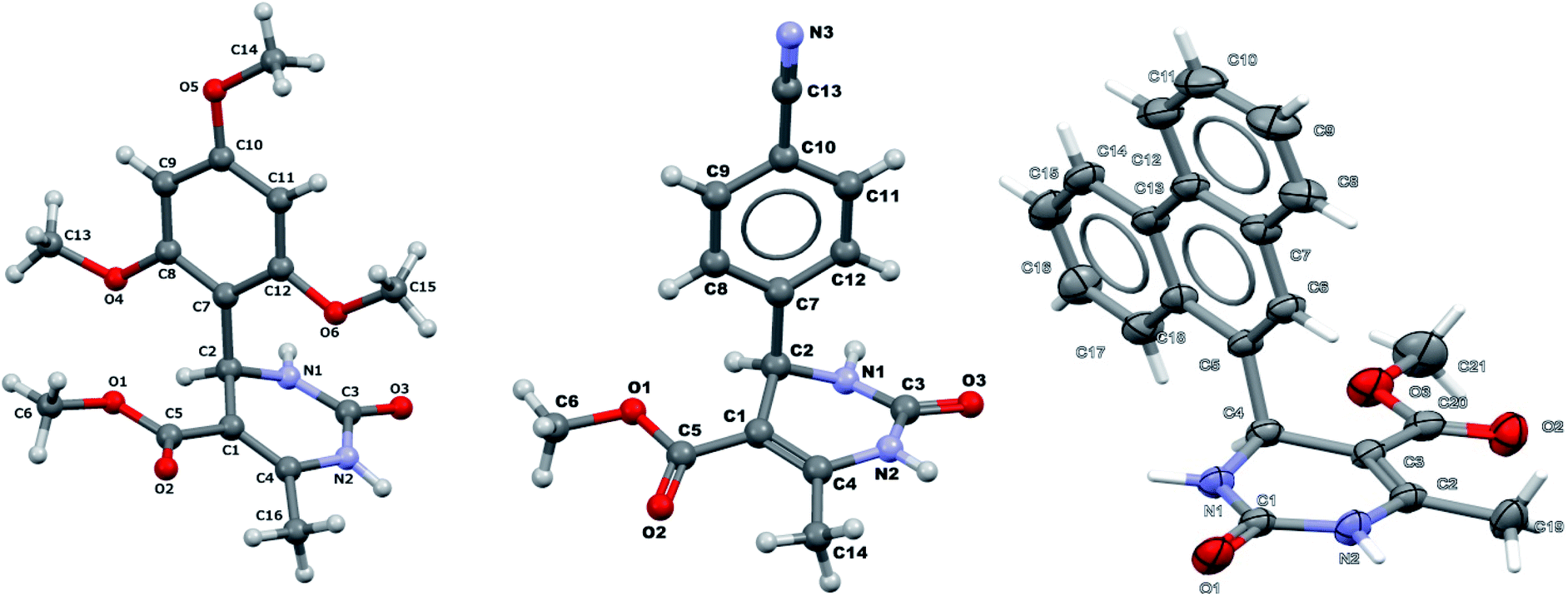 | ||
| Fig. 1 X-ray structure of compounds 2, 3 and 7. | ||
According to the literature data, the participation of 2-hydroxybenzaldehyde (salicylaldehyde) or different substituted 2-hydroxybenzaldehyde derivatives, methylene active compound and urea or thiourea under Biginelli reaction conditions85–97 leads to the formation of oxygen-bridged tricyclic pyrimidine derivative B instead of simple dihydropyrimidine A (Scheme 2).
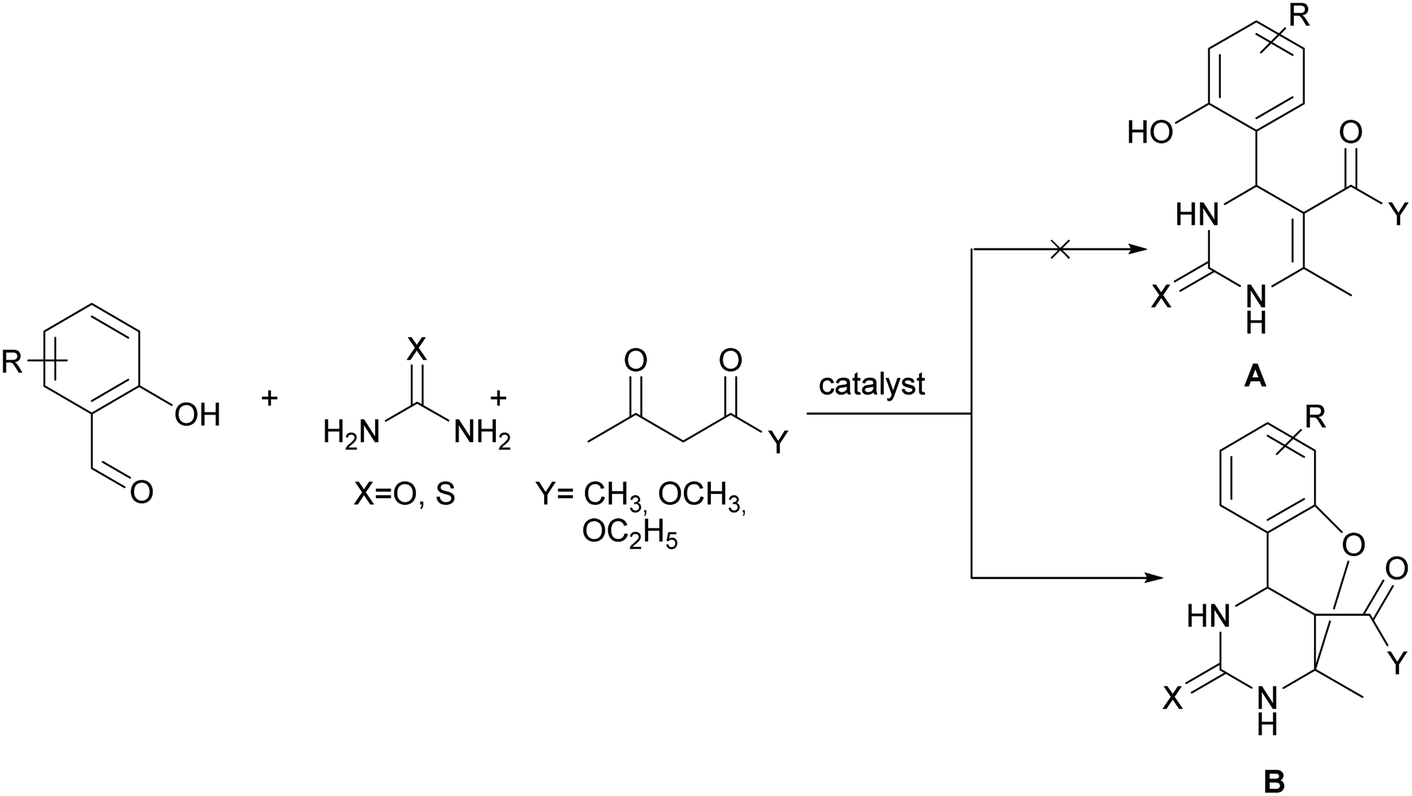 | ||
| Scheme 2 The formation of oxygen-bridged tricyclic pyrimidine derivative. | ||
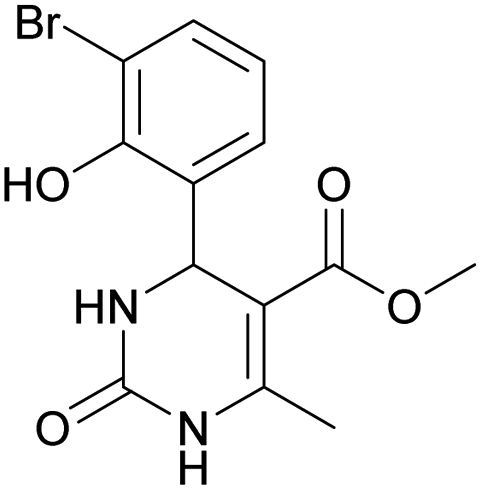
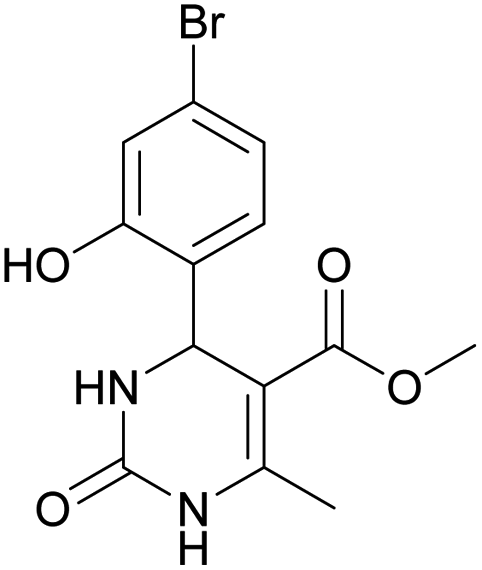
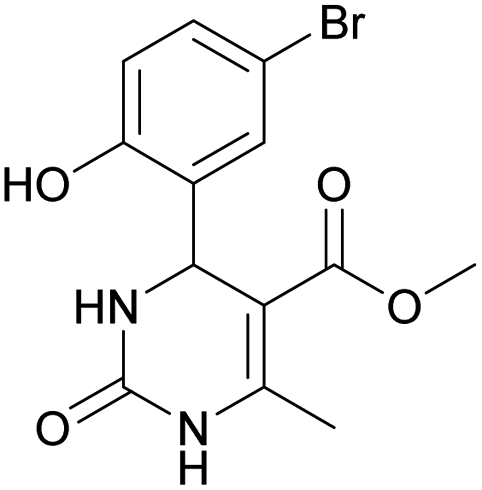
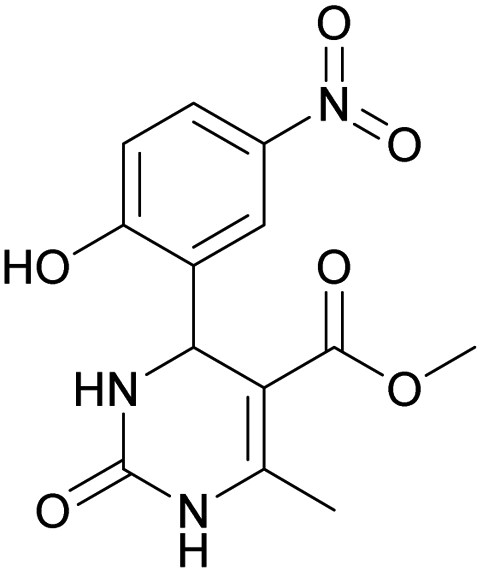
Considering this fact, it was interesting to check the activity of copper triflate in the presence of different substituted 2-hydroxybenzaldehyde derivatives. A small modification of the initial procedure allowed to obtain various A types of H2Py 8–12 (Scheme 3 and Table 2).
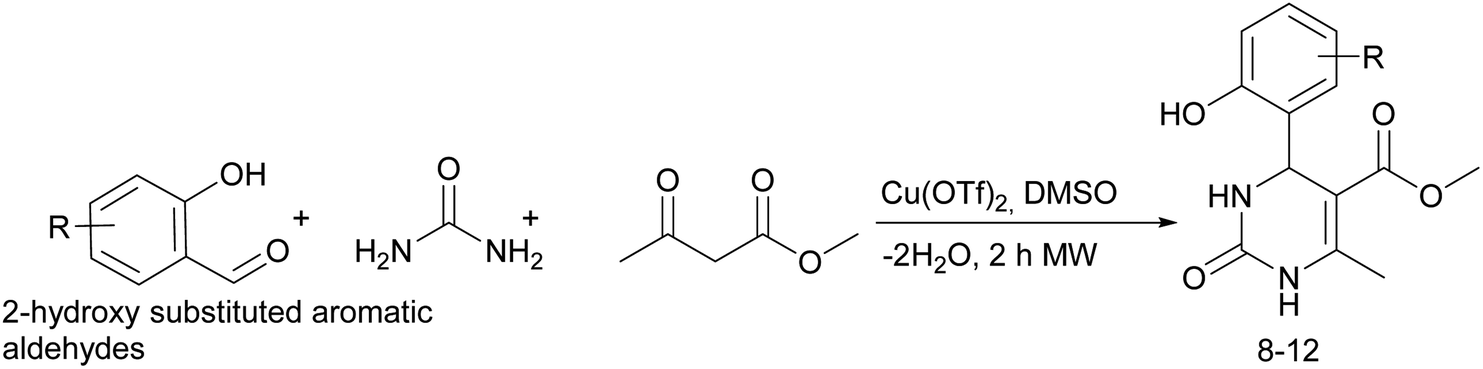 | ||
| Scheme 3 Synthesis of dihydropyrimidines on the basis of 2-hydroxybenzaldehyde derivatives. | ||
| 8 | 9 | 10 | 11 | 12 | |
|---|---|---|---|---|---|
| Product | 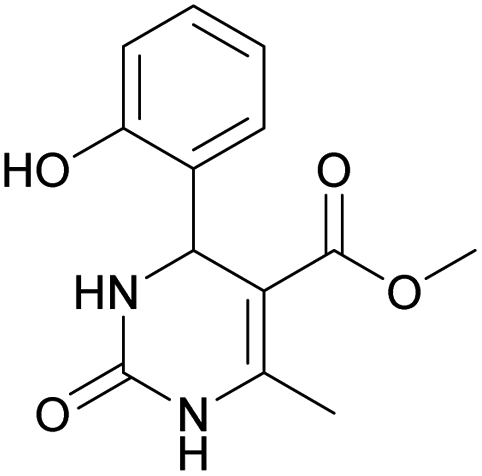 |
 |
 |
 |
 |
| Yield% | 62 | 78 | 77 | 72 | 79 |
The selectivity of copper triflate in the above-mentioned reaction can be enlightened through considering the mechanism of formation of oxygen-bridged tricyclic pyrimidine derivative B (Scheme 4). According to the mechanism of the Biginelli condensation, the formation of oxygen-bridged pyrimidine B occurs through the formation of A. After the formation of molecule A step, the double bond of A must react with the catalyst which will facilitate the attack of oxygen (hydroxyl group) on C-6 position (*). Taking into account the bulkiness of copper triflate molecule as well as the size of –CO–Y and –CH3 groups, this interaction cannot occur because of the steric effect87 (Scheme 4). As a result, the steric effect determines the selectivity of the Biginelli reaction in the presence of copper triflate towards product A. Another factor that supports this idea is the analysis of literature,85–97 according to which the formation of oxygen-bridged tricyclic pyrimidine derivative B in the one-step one-pot process occurs in the presence of Arrhenius acids (H+), which small size facilitates the formation of oxygen-bridged tricyclic compound B.
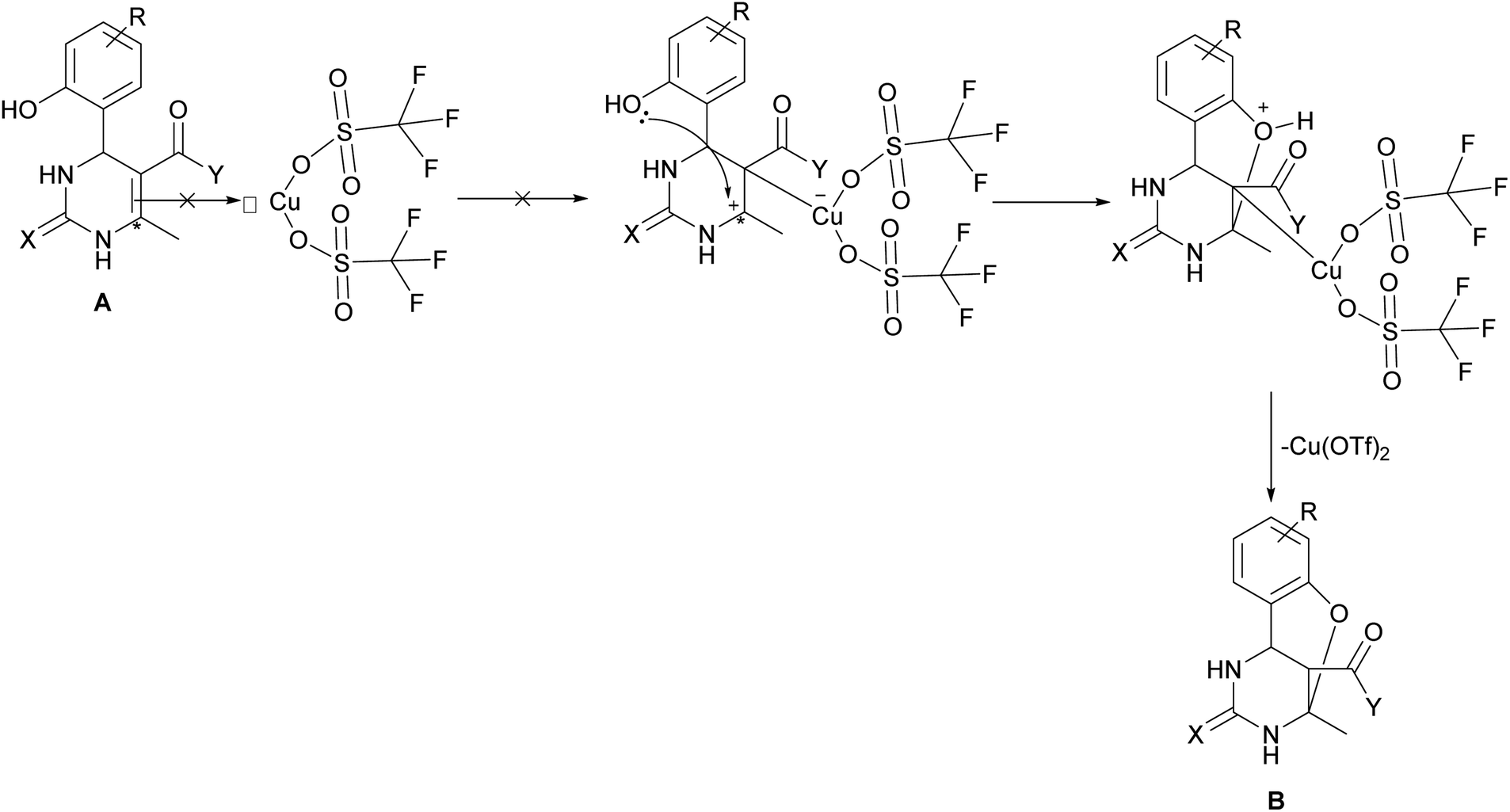 | ||
| Scheme 4 A mechanism of the formation of oxygen-bridged pyrimidine B. | ||
The next stage was performing of regioselective oxidation of H2Py (1–7) in the presence of cerium ammonium nitrate (CAN). Modification of the procedure64 allows us to obtain oxidized H2Py 13–20 (Scheme 5 and Table 3).
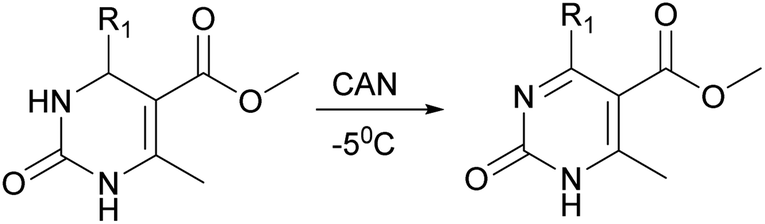 | ||
| Scheme 5 Regioselective oxidation of H2Py in the presence (CAN). | ||
| 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | |
|---|---|---|---|---|---|---|---|---|
| Product | 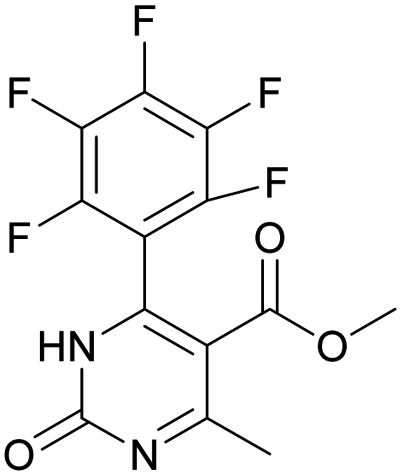 |
 |
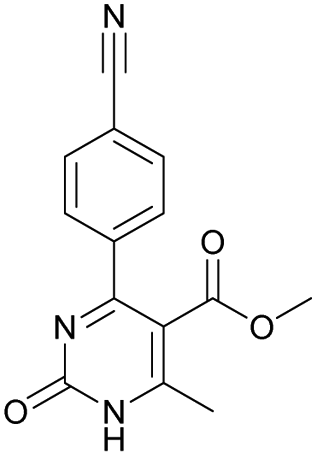 |
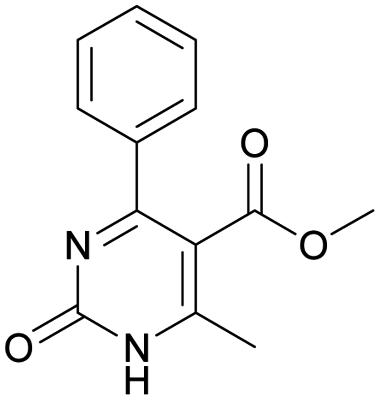 |
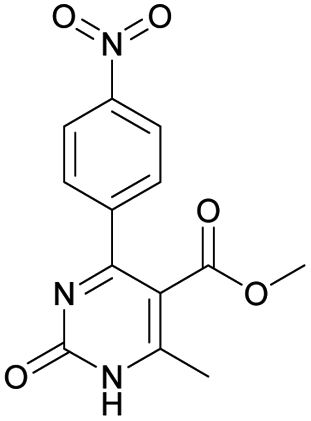 |
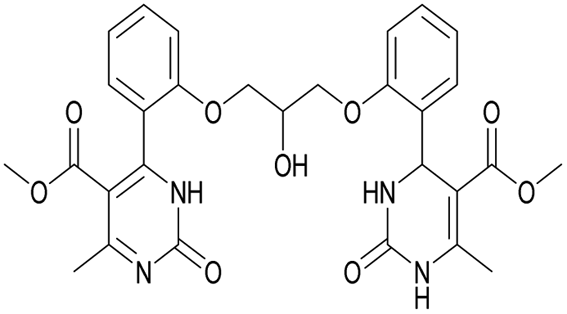 |
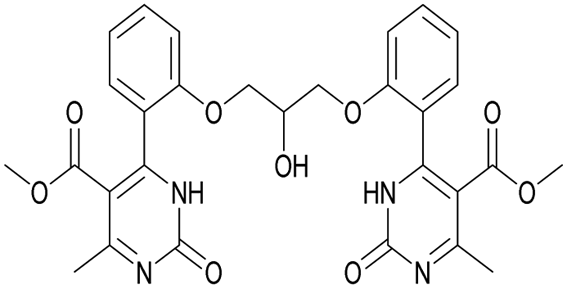 |
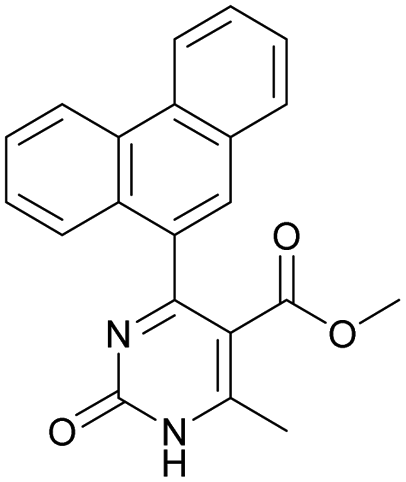 |
| Yield% | 1 | 40 | 27 | 39 | 28 | 15 | 5 | 35 |
The reason of the low yield (1%) of compound 13 can be explained through mechanistic consideration (Scheme 6) of this reaction. According to the mechanism of regioselective oxidation of H2Py in the presence of CAN known in the literature,64 this reaction is a one-electron oxidizing process and, at the first stage, carboradical 1 is formed. In case of compound 13, all five positions in the benzene ring are substituted with fluorine atoms which draw electrons towards themselves resulting in decreasing the stability of the carboradical 1 as well as the subsequently formed carbocation 2 too (Scheme 6). As a result, it leads to the formation of the targeted product 13 with a low yield. This pattern also manifests itself in the case of other oxidation products. The yield of targeted oxidation products with electron enriched aromatic ring (14, 16 and 20) is higher than that of oxidation products with electron-deficient aromatic ring (containing electron-withdrawing –NO2 and –CN groups – 15 and 17). This is due to the fact that the stability of the carboradical (as well as carbocation) in case of electron enriched aromatic ring is higher than that of the electron-deficient aromatic ring.
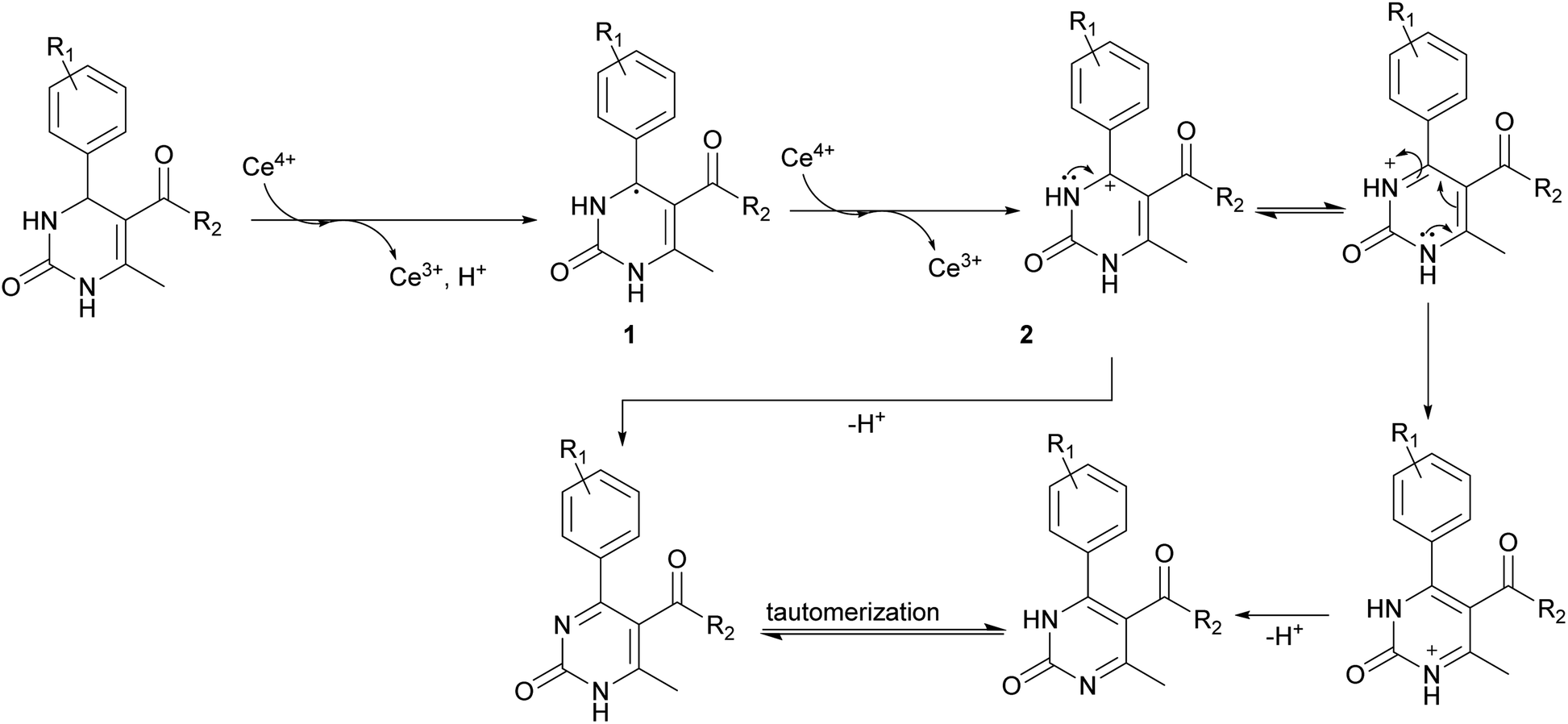 | ||
| Scheme 6 Mechanism of regioselective oxidation of H2Py in the presence of CAN. | ||
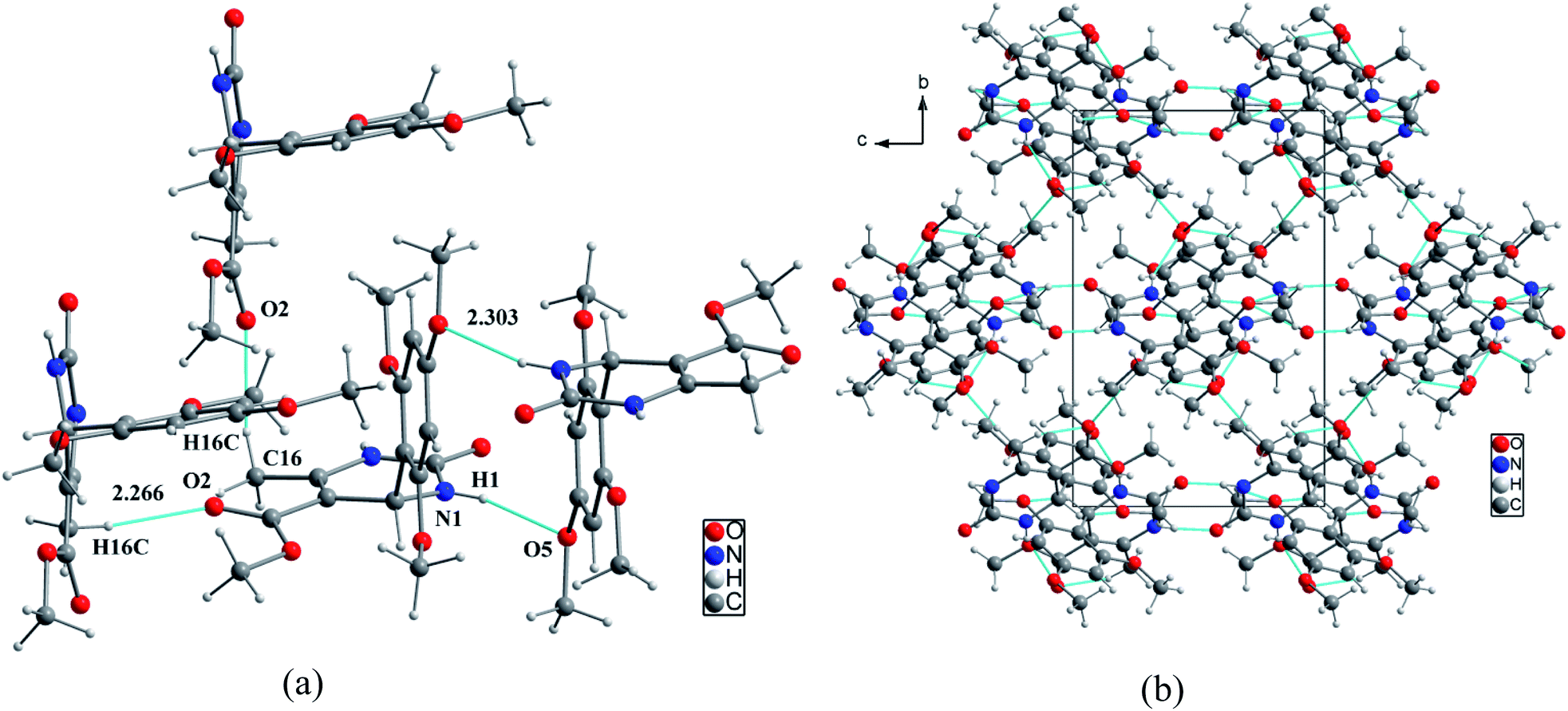
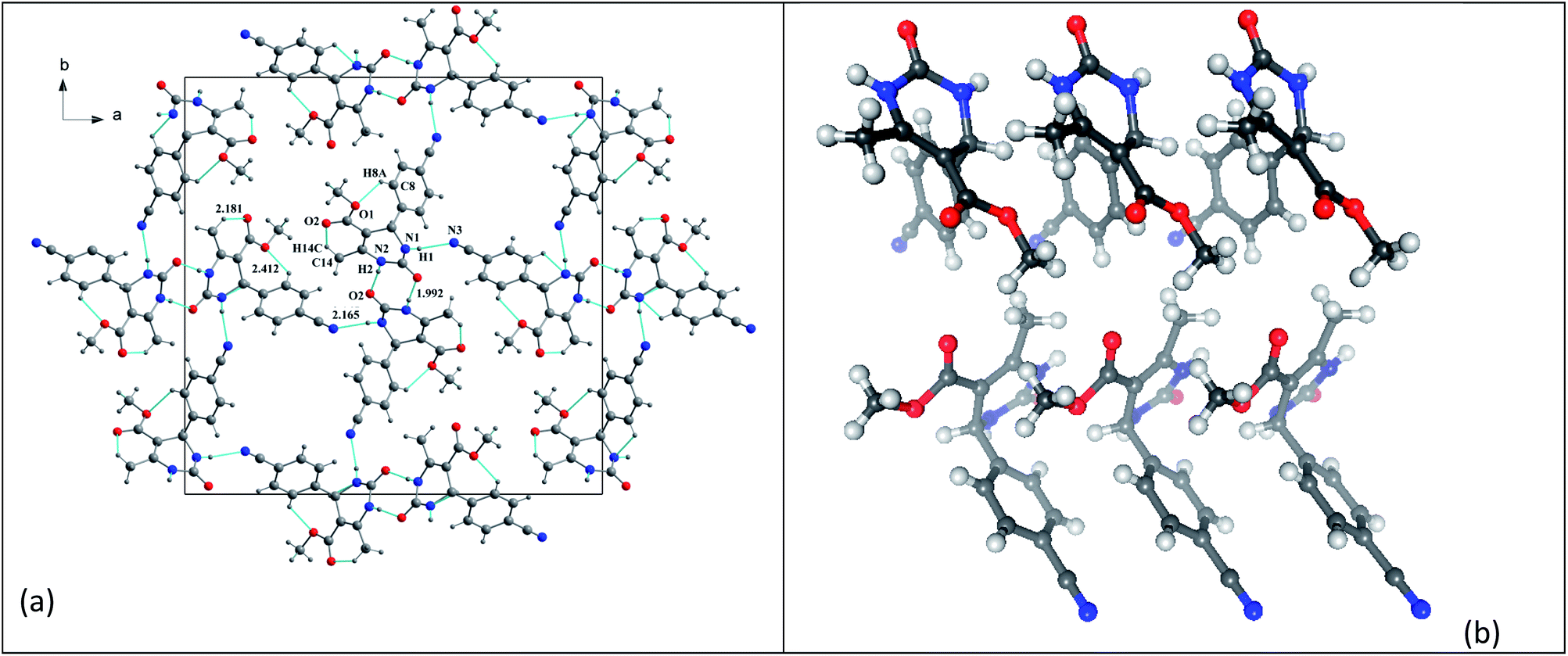
It was possible to obtain single crystals of compounds 14 and 20 (Fig. 2 and 3). According to the literature data,64 oxidized products of H2Py can exist in the solution in the form of various tautomers (Scheme 7).
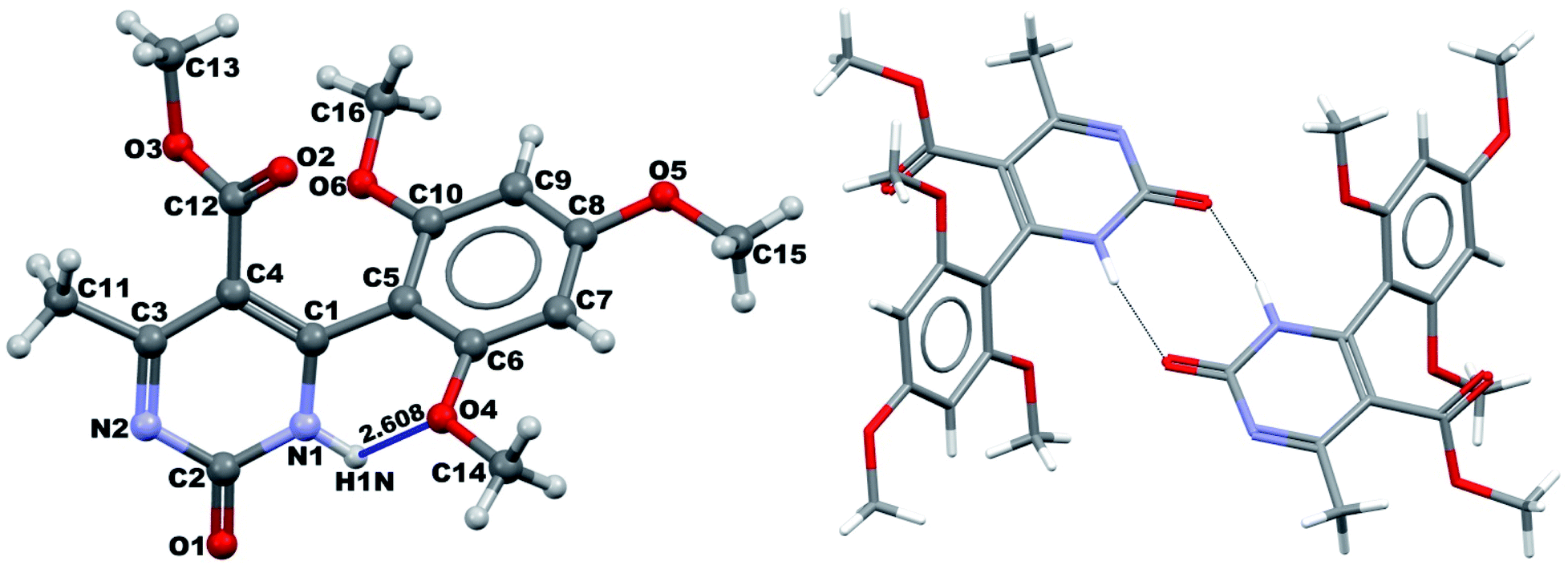 | ||
| Fig. 2 View of the X-ray structure of compound 14 and dimer formed via a double hydrogen bond around an inversion centre. | ||
 | ||
| Fig. 3 View of the X-ray structure of compound 20 and dimer formed via a double hydrogen bond around an inversion centre. | ||
 | ||
| Scheme 7 Two tautomers of pyrimidines. | ||
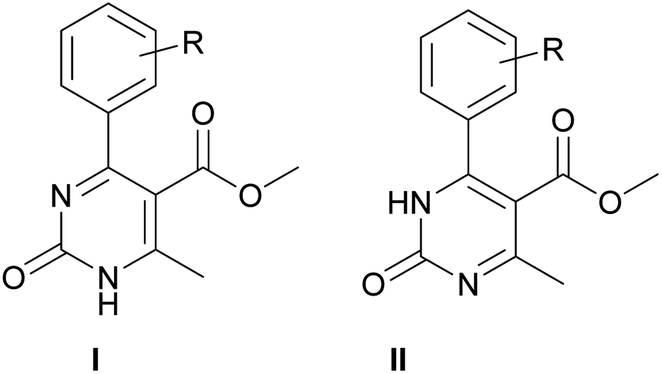
According to the X-ray structure, compound 14 exists in the crystal structure in the form of tautomer II, whereas compound 20 is in the form of tautomer I (Fig. 2 and 3). The reason of existing of compound 14 in the form of tautomer II is caused by the presence of an intramolecular hydrogen bond between H1N and O4 of the methoxy group, which keeps the hydrogen at N1 position and stabilized the presence of tautomer structure II.
X-ray diffraction investigation of compound 20 did not reveal the presence of any intramolecular hydrogen bond in the structure, which makes possible existing of this molecule in the form of tautomer I in crystal. In addition to this, X-ray analysis revealed the presence of compounds 14 and 20 in the form of dimers (Fig. 2 and 3), due to the presence of double intermolecular N–H⋯O hydrogen bonds between O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N–H groups. There is a H1N⋯O1 hydrogen bond in case of compound 14 and H2N⋯O1 in case of compound 20. Considering this fact, we can say that H1N of compound 14 takes part in the formation of both intramolecular and intermolecular hydrogen bonds (bifurcated hydrogen bond with two acceptors) stabilizing the crystal structure of the compound. Furthermore, it is possible to conclude that in case if there is a heteroatom group on the benzene ring which is able to participate in the formation of an intramolecular hydrogen bond with the amine group of the dihydropyrimidine ring, then the oxidized product will be in the form of tautomer II, otherwise in the form of tautomer I.
C–N–H groups. There is a H1N⋯O1 hydrogen bond in case of compound 14 and H2N⋯O1 in case of compound 20. Considering this fact, we can say that H1N of compound 14 takes part in the formation of both intramolecular and intermolecular hydrogen bonds (bifurcated hydrogen bond with two acceptors) stabilizing the crystal structure of the compound. Furthermore, it is possible to conclude that in case if there is a heteroatom group on the benzene ring which is able to participate in the formation of an intramolecular hydrogen bond with the amine group of the dihydropyrimidine ring, then the oxidized product will be in the form of tautomer II, otherwise in the form of tautomer I.
As a result, it is possible to predict that compound 13, as well as 18 and 19, will be in the form of tautomer II.
Structure description
It was also possible to obtain single crystals of compounds 2, 3, 7, 14 and 20, which crystallographic data and details of refinement were given in Table 4.| Compound 2 | Compound 3 | Compound 7 | Compound 14 | Compound 20 | |
|---|---|---|---|---|---|
| a R = ∑||Fo| − |Fc||/∑|Fo|; wR(F2) = [∑w(|Fo|2 − |Fc|2)2/∑w|Fo|4]1/2. | |||||
| Chemical formula | C16H20N2O6 | C14H13N3O3 | C21H18N2O3 | C16H18N2O6 | C21H16N2O3 |
| Formula weight (M) | 336.34 | 271.27 | 346.37 | 343.33 | 344.36 |
| Crystal system | Monoclinic | Tetragonal | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/c | P4/n | C2/c | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a (Å) | 11.6354(5) | 23.7495(15) | 25.515(5) | 8.1493(12) | 9.918(2) |
| b (Å) | 14.5969(5) | 23.7495(15) | 7.710(3) | 9.6420(17) | 13.013(3) |
| c (Å) | 9.3401(3) | 4.8467(3) | 18.240(4) | 11.4398(10) | 12.9630(18) |
| α (°) | 90.00 | 90.00 | 90.00 | 74.396(10) | 90.00 |
| β (°) | 98.173(2) | 90.00 | 108.277(17) | 88.752(10) | 91.984(18) |
| γ (°) | 90.00 | 90.00 | 90.00 | 78.900(17) | 90.00 |
| V (Å3) | 1570.22(10) | 2733.7(4) | 3407.2(17) | 849.1(2) | 1672.0(6) |
| Z | 4 | 8 | 8 | 2 | 4 |
| Temperature (K) | 100(2) | 100(2) | 298(2) | 293(2) | 298(2) |
| Crystal size | 0.320 × 0.290 × 0.145 | 0.320 × 0.054 × 0.040 | 0.650 × 0.360 × 0.220 | 0.580 × 0.430 × 0.320 | 0.300 × 0.220 × 0.060 |
| Density (g cm−3) | 1.423 | 1.318 | 1.350 | 1.343 | 1.368 |
| μ (Mo Kα) (mm−1) | 0.110 | 0.095 | 0.091 | 0.105 | 0.093 |
| F(000) | 712 | 1136 | 1456 | 362 | 720 |
| Goodness of fit on F2 | 1.005 | 1.005 | 1.101 | 1.098 | 1.131 |
| R1, wR2a, [I > 2σ(I)] | 0.0544, 0.1152 | 0.0524, 0.1147 | 0.0637, 0.1320 | 0.0541, 0.1526 | 0.1089, 0.1743 |
| R1, wR2 (all data) | 0.0817, 0.1240 | 0.0663, 0.1201 | 0.0894, 0.1529 | 0.0641, 0.1676 | 0.1932, 0.2282 |
| Residual electron density (max, min) e Å−3 | 0.275, −0.276 | 0.254, −0.230 | 0.199, −0.156 | 0.394, −0.184 | 0.254, −0.262 |
In the structural arrangement of compound 2, the organic molecules are arranged on top of each other to form pillars running along the a-axis direction at (0, 0, 0) and (0, ½, ½) (Fig. 4). Furthermore, each organic molecule is linked to the three nearest neighboring molecules by N–H⋯O and N–H⋯N hydrogen bonds so that the resulting network assumes a three-dimensional form.
 | ||
| Fig. 4 (a) Representation of hydrogen bonds around the organic molecular, (b) projection of the structure of compound 2 along the a-axis. | ||
In the structure of compound 3 (Fig. 5), the organic molecules are linked together by C–H⋯O hydrogen bonds to form a three-dimensional network containing channels centered at (¼, ¼, 0) and (¾, ¾, 0) and extending along the c-axis. This structure also has intramolecular interactions such as C8–H8A⋯O1 and C14–H14C⋯O2.
 | ||
| Fig. 5 Compound 3 (a) projection of the structure of along the c-axis. (b) Crystallographic autostereogram,98 the horizontal translations are along the short c axis. | ||
For compound 7 the organic molecules are interconnected by two hydrogen bonds N1–H1N⋯O2 and N2–H2N⋯O1 generated by the imine group of the pyrimidine ring to form infinite chains running along the b-axis direction (Fig. 6a). This way the pyrimidine rings and the ester groups (–COOCH3) are developed in planes parallel to the plane (b, c) at x = ½n between which the phenanthryl groups are segregated (Fig. 6b).
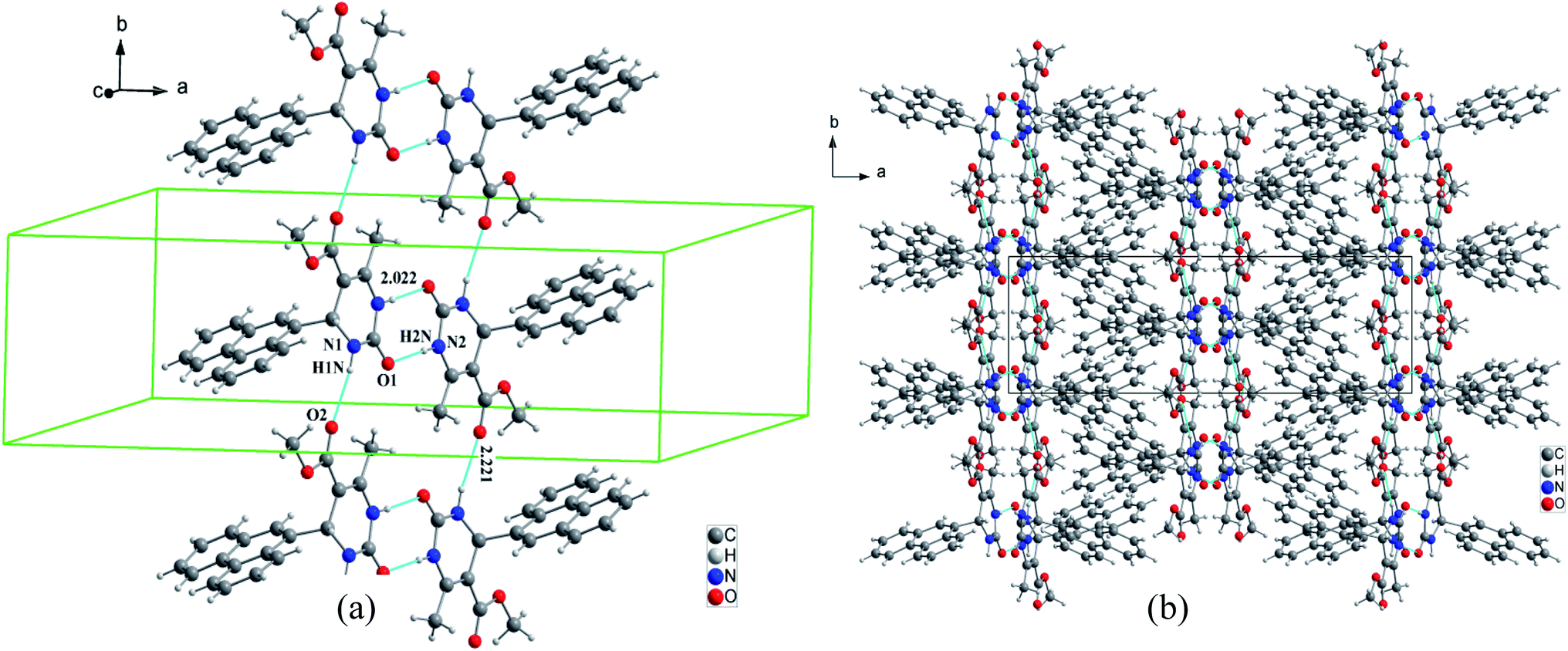 | ||
| Fig. 6 Crystal packing of compound 3 (a) view of the 1D chain parallel to the c-axis for, (b) projection along the b-axis. N–H⋯O hydrogen bonds are represented as light blue lines. | ||
The crystal packing of compound 2 is also stabilized by intermolecular aromatic stacking interactions between neighboring pyrimidine rings, with a face-to-face distance of 3.712 Å (Fig. 7a). For the two compounds 3 and 7, the crystal packing is also stabilized by intermolecular interactions C–H⋯π between hydrogen atoms of the ester groups –COOCH3 and the neighboring aromatic rings, with H⋯π distance of 3.884 Å for compound 3 and 2.802 Å for compound 7 (Fig. 7b and c).
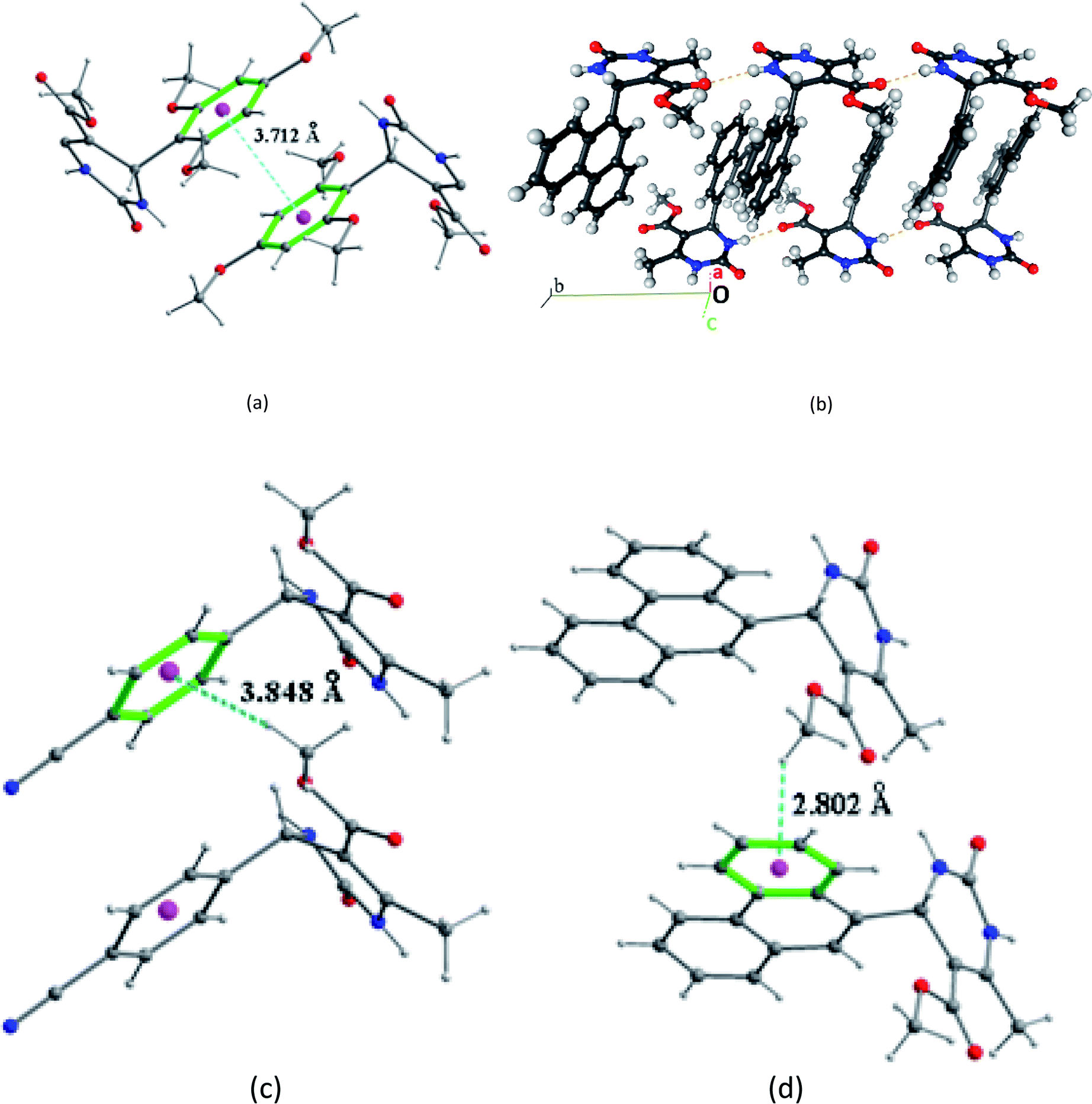 | ||
| Fig. 7 Intermolecular aromatic stacking interactions between the neighboring aromatic rings: (a) in compound 2, (b) in compound 7 (autostereogram with horizontal translation along b). Intermolecular C–H⋯π interactions: (c) in compound 3, (d) compound 7. | ||
Compound 14 is a monohydrate where the water molecule occupies a void which is 74% hydrophobic as surrounded by C and H–C atoms. This more hydrophilic molecule has two strong CO⋯H–N hydrogen bonds and five C–H⋯O/N weak hydrogen bonds.
Compound 20 is characterized by C⋯H, H⋯H and C⋯C contacts which represent up to 71% of the contact surface due to cycle stacking and C–H⋯π interactions. A double CO⋯H–N hydrogen bond occurs within the dimer resulting from an inversion centre.
Hirshfeld surface analysis
Analysis of intermolecular interactions using the Hirshfeld surface was undertaken to gain a better understanding of the three-dimensional crystal packings. The Hirshfeld surface and the decomposed two-dimensional fingerprint plots (ESI Fig. 58–62†) were calculated on the determined crystal structures using Crystal Explorer 3.1 software.99All the five compounds show two spikes at short distances due to the O–H⋯O hydrogen bonds constituting between 17 and 33% of the contact surface. Only compound 2 shows spikes at short distance for H⋯N hydrogen bonds. H⋯H contacts are always the most abundant contacts representing between 30 and 53% of contacts. The amount C⋯C contacts is low except for compound 20 where it reaches 6.2% due to aromatic stacking. In all cases, the most abundant contact is of type H⋯H whose proportion is between 30 and 53%. C⋯H contacts occur in the 12.8–29.1% range, with compound 7 display the highest amount of C–H⋯pi weak hydrogen bonds.
Biological assays
The antimicrobial activity of the synthesized dihydropyrimidines (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 14, 15, 16, 17, 19, 20) was investigated against Gram positive and Gram negative bacterial strains (S. aureus, E. coli, A. baumannii, P. aeruginosa and K. pneumonia). The investigations were started with the determination of the minimum inhibitory concentration (MIC) of the studied compounds by the twofold micro-dilution method.100–102 Obtained results were compared with the results of the pristine antibiotics (cefotaxime and ceftriaxone). As shown in Table 5, compound 10 and 16 showed the highest inhibitory effect against A. baumanii and S. aureus in value of 62.5 μg ml−1. No inhibition was detected for compound 12 against all bacterial strains, while the compound 15 showed the similar activity against all tested cultures. So, the microbial cultures were more susceptible to compound 10, 14, 16 and 20 than other compounds. The lowest MIC in case of E. coli, P. aeruginosa, S. aureus, A. baumannii, K. pneumonia was 125 μg ml−1 (compounds 14), 250 μg ml−1 (compounds 14 and 16), 62.5 μg ml−1 (compounds 10 and 16), 62.5 μg ml−1 (compounds 10 and 16) and 500 μg ml−1 (compound 20) respectively. Moreover, the antibacterial activity of the compound 1 was detected only in case of P. aeruginosa (1000 μg ml−1). Compare with other microbial cultures P. aeruginosa was more susceptible against all of the compounds except 12 and 17. Compound 1, 2 and 12 were not affect the growth of E. coli but the MIC value of the other compounds was identical (1000 μg ml−1) for this strain. S. aureus, A. baumanii, K. pneumonia was not susceptible when tested with the compounds 1, 2, 3 and 6.| Investigated samples | Bacterial strains | ||||
|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. aureus | A. baumannii | K. pneumonia | |
| (1) | — | 1000 | — | — | — |
| (2) | 1000 | 500 | 1000 | 1000 | 1000 |
| (3) | 1000 | 500 | 500 | 1000 | — |
| (4) | 500 | 1000 | 500 | — | — |
| (5) | 500 | 1000 | 250 | 1000 | 1000 |
| (6) | 1000 | 1000 | — | — | — |
| (7) | 1000 | 1000 | 250 | 1000 | — |
| (8) | 500 | 1000 | 250 | 1000 | — |
| (9) | 1000 | 500 | 500 | 500 | 1000 |
| (10) | 1000 | 1000 | 62.5 | 62.5 | 1000 |
| (11) | 1000 | 1000 | 1000 | 500 | 1000 |
| (12) | 1000 | 1000 | 500 | — | — |
| (14) | 125 | 250 | 500 | 1000 | 1000 |
| (15) | 250 | 500 | 250 | 1000 | 1000 |
| (16) | 1000 | 250 | 62.5 | 62.5 | — |
| (17) | — | — | 1000 | — | 1000 |
| (19) | 1000 | 1000 | 1000 | — | 1000 |
| (20) | 250 | 500 | 250 | 500 | 500 |
| Cefotaxime | 250 | 250 | 250 | 250 | 250 |
| Ceftriaxone | 500 | 500 | 500 | 500 | 500 |
The obtaining results revealed that the MICs of compounds 14 in case of E. coli (125 μg ml−1) and 10 and 16 in case of S. aureus and A. baumannii (62.5 μg ml−1) were higher than MICs of both pristine antibiotics (250–500 μg ml−1).
Taking into account the MIC of the compounds and pristine antibiotics, antibacterial activity of compounds 2, 3, 4, 5, 7, 8, 9, 10, 11, 12, 14, 15, 16 and 20 was also investigated by disc-diffusion method.103 Results were compared with the antibacterial activity of pristine antibiotics. As it can be seen from the Fig. 65 (ESI),† the best antibacterial effect against E. coli in comparison with both antibiotics were demonstrated by the compounds 14, 15 and 20, whereas antibacterial effect of compound 8 was equal to ceftriaxone, but lower than cefotaxime. Looking at the inhibition zone in case of P. aeruginosa, compound 14 demonstrated the highest activity in comparison with both antibiotics, whereas the activity of compound 16 was higher than ceftriaxone and equal to cefotaxime (Fig. 63 – ESI†). In case of S. aureus and A. baumannii inhibition zone of compounds 10 and 16 was higher than that of both pristine antibiotics (Fig. 64 and 66 – ESI†). In general, the antibacterial activity of all compounds against K. pneumonia was low; only the effect of compound 20 was higher in comparison with others but lower than that of both cefotaxime and ceftriaxone, equal to 16, 32 and 24 mm respectively (Fig. 67 – ESI†). The pronounced antibacterial effect of the above-mentioned compounds may be related to their ability to enter the bacterial cytoplasm and even to cross the nuclear envelope. In addition to it, they may bind to different classes of enzymes, for example, dihydrofolate reductase, bacterial DNA gyrase, aminoacyl-tRNA synthetases and so on.104
As DMSO was used as a solvent, the record of the results was also carried out with control dishes, without investigated compound. It was determined that DMSO does not influence on the above mentioned Gram-positive and Gram-negative bacteria.
Experimental
Materials and methods
All the solvents and reagents which were purchased from commercial suppliers were of analytical grade and used without further purification. The control of the reactions progress and the determination of the synthesized compounds purity were done by thin layer chromatography (TLC) on Merck silica gel plates (60 F254 aluminum sheets) which were visualized under UV light. Melting points were recorded in open capillary tubes on a Buchi B-540 apparatus and are uncorrected. Elemental analysis was performed on the analyzer Carlo Erba 1108.NMR experiments
The NMR experiments have been performed on a BRUKER FT NMR spectrometer AVANCE 300 (Bruker, Karlsruhe, Germany) (300 MHz for 1H and 75 MHz for 13C) with a BVT 3200 variable temperature unit in 5 mm sample tubes using Bruker standard software (TopSpin 3.1). Chemical shifts were given in ppm (δ) and were referenced to internal tetramethylsilane (TMS). Multiplicities are declared as follow: s (singlet), d (doublet), t (triplet), q (quadruplet), m (multiplet). Coupling constants J are given in Hz. The experimental parameters for 1H are as follows: digital resolution = 0.23 Hz, SWH = 7530 Hz, TD = 32 K, SI = 16 K, 90° pulse-length = 10 ms, PL1 = 3 dB, ns = 1, ds = 0, d1 = 1 s and for 13C as follows: digital resolution = 0.27 Hz, SWH = 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 985 Hz, TD = 64 K, SI = 32 K, 90° pulse length = 9 ms, PL1 = 1.5 dB, ns = 100, ds = 2, d1 = 3 s. The NMR-grade DMSO-d6 (99.7%, containing 0.3% H2O), CDCl3, CD3OD was used for the solutions of synthesized compounds.
985 Hz, TD = 64 K, SI = 32 K, 90° pulse length = 9 ms, PL1 = 1.5 dB, ns = 100, ds = 2, d1 = 3 s. The NMR-grade DMSO-d6 (99.7%, containing 0.3% H2O), CDCl3, CD3OD was used for the solutions of synthesized compounds.
Mass experiments
High-resolution mass spectrometry (HRMS) was performed using electrospray ionization (ESI) in positive-ion or negative-ion detection mode.X-ray analysis
X-ray analyses have been performed on Bruker SMART APEX II Single Crystal X-ray Diffractometer equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The crystal structure was solved by direct methods and refined on F2 by full matrix least-squares using Bruker's SHELXTL-97.105 Crystallographic data for the structural analysis have been deposited to the Cambridge Crystallographic Data Center (CCDC 1998538 for 2, CCDC 1998557 for 3, CCDC 1998539 for 7, CCDC 1998546 for 14 and CCDC 1998547 for 20).Experimental procedures
0.5 mmol of subsequent aldehyde, 0.75 mmol (45 mg) of urea and 0.08 mmol (30 mg) of Cu(OTf)2 were added to a microwave vial with a magnetic stirrer and dissolved in 1 ml of ethanol. Subsequently, 0.46 mmol (50 μl) of methyl acetoacetate was added to a vial, which was sealed and irradiated at 100 °C in a microwave reactor for 2.5 h at a maximum power of 200 W (CEM Discover™ System). At the end of reaction time, the precipitate was formed (in case if precipitate was not formed left it overnight), filtered, washed with distilled water and dried.
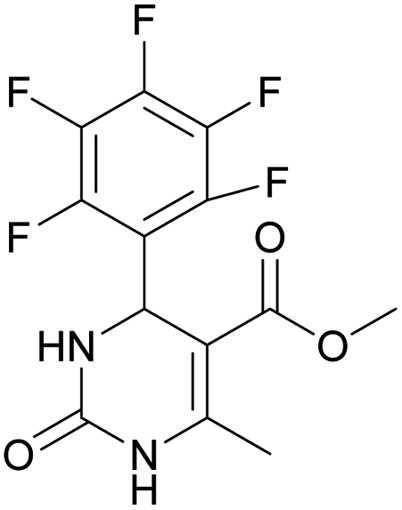
Methyl 6-methyl-2-oxo-4-(perfluorophenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1). The title compound was prepared according to the general procedure using 2,3,4,5,6-pentafluorobenzaldehyde to afford the title compound a colorless precipitate. Yield 56%. Mp 333–335 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.23 s (3H, CH3), 3.5 s (3H, OCH3), 5.69 s (1H, CH), 7.7 s (1H, NH), 9.52 s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 18.26 (CH3), 46.22 (CH), 51.15 (OCH3), 94.08 (C), 118.61 (C), 135.47 (CAr), 143.19 (2CAr), 146.46 (CAr), 150.48 (2CAr), 151.01 (COO), 165.51 (CO). HRMS (ESI-MS): 359.21 [M+ + Na+]. Elemental analysis calcd for C13H9N2O3F5, %: C, 46.44; H, 2.70; N, 8.33. Found, %: C, 46.40; H, 2.76; N, 8.31.
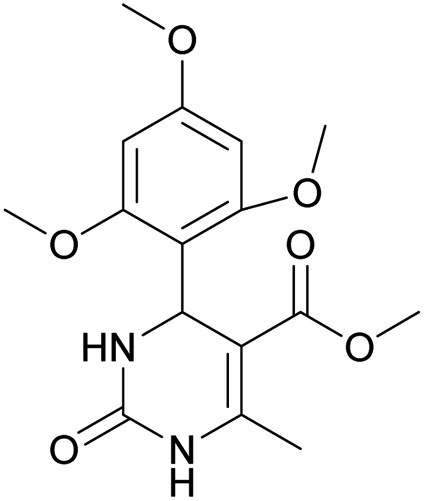
Methyl 6-methyl-2-oxo-4-(2,4,6-trimethoxyphenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (2). The title compound was prepared according to the general procedure using 2,4,6-trimethoxybenzaldehyde to afford the title compound a light yellow precipitate. Yield 74%. Mp 252–254 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.15 s (3H, CH3), 3.39 s (3H, OCH3), 3.72 s (6H, 2OCH3), 3.75 s (3H, OCH3), 5.74 s (1H, CH), 6.18 s (2H, 2CArH), 6.9 s (1H, NH), 8.95 s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 17.78 (CH3), 45.03 (CH), 50.14 (OCH3), 55.1 (OCH3), 55.7 (2OCH3), 91.08 (2CArH), 96.03 (C), 113.71 (C), 147.84 (CAr), 152.12 (CAr), 158.99 (2CAr), 159.91 (COO), 166.25 (CO). HRMS (ESI-MS): 359.17 [M+ + Na+]. Elemental analysis calcd for C16H20N2O6, %: C, 57.14; H, 5.99; N, 8.33. Found, %: C, 57.19; H, 5.96; N, 8.31. This compound can also be obtained by the following procedure: to a solution of 1 mmol of 2,4,6-trimethoxybenzaldehyde in 15 ml of ethanol were added 6.6 mmol of urea, 5.5 mmol of methyl acetoacetate and 4 ml of acetic acid. The reaction mixture was refluxed for 9 h. At the end of reaction time the reaction mixture was cooled with ice and precipitate was formed, which was filtered, washed with distilled water and dried (yield 68%).
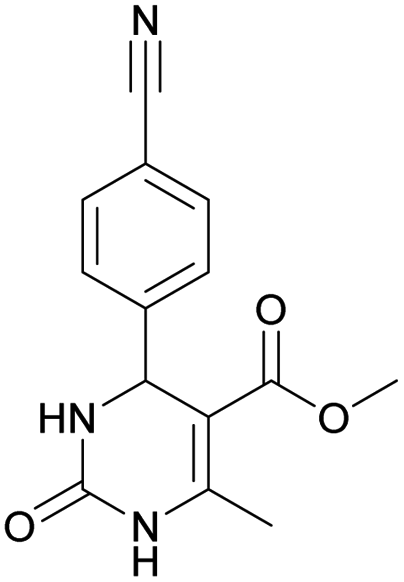
Methyl 4-(4-cyanophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (3). The title compound was prepared according to the general procedure using 4-cyanobenzaldehyde to afford the title compound a colorless precipitate. Yield 74%. Mp 212–214 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.26 s (3H, CH3), 3.54 s (3H, OCH3), 5.21–5.22 d (1H, CH, J = 3 Hz), 7.41–7.44 d (2H, 2CArH, J = 9 Hz), 7.8–7.83 d (2H, 2CArH, J = 9 Hz), 7.86 s (1H, NH), 9.34 s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 18.23 (CH3), 51.23 (CH), 54.03 (OCH3), 98.39 (C), 110.49 (C), 119.08 (CN), 127.61 (2CArH), 132.95 (2CArH), 149.88 (CAr), 150.18 (CAr), 152.2 (COO), 165.95 (CO). Elemental analysis calcd for C14H13N3O3, %: C, 61.99; H, 4.83; N, 15.49. Found, %: C, 61.91; H, 4.87; N, 15.53. The data agrees with the reported literature values.106
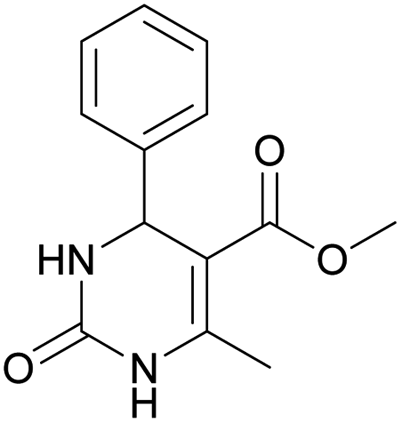
Methyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4). The title compound was prepared according to the general procedure using benzaldehyde to afford the title compound a light yellow precipitate. Yield 79%. Mp 215–217 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.24 s (3H, CH3), 3.52 s (3H, OCH3), 5.14 s (1H, CH), 7.20–7.31 m (5H, 5CArH), 7.77 s (1H, NH), 9.23 s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 17.8 (CH3), 50.7 (CH), 53.8 (OCH3), 99 (C), 126.1 (2CArH), 127.2 (CArH), 128.4 (2CArH), 144.6 (C), 148.6 (CAr), 152.2 (COO), 165.8 (CO). Elemental analysis calcd for C13H14N2O3, %: C, 63.40; H, 5.73; N, 11.38. Found, %: C, 63.47; H, 5.78; N, 11.33. The data agrees with the reported literature values.107 This compound can also be obtained by the following procedure: to a solution of 2.5 mmol of benzaldehyde in 12 ml of ethanol were added 6.6 mmol of urea, 6 mmol of methyl acetoacetate and 2 ml of acetic acid. The reaction mixture was refluxed for 10 h. At the end of reaction time the reaction mixture was cooled with ice and precipitate was formed, which was filtered, washed with distilled water and dried (yield 76%).
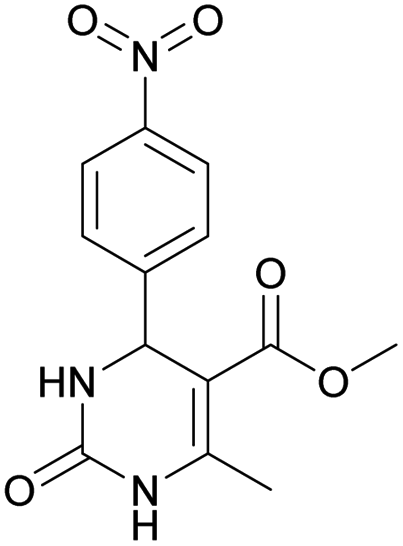
Methyl 6-methyl-4-(4-nitrophenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5). The title compound was prepared according to the general procedure using 4-nitrobenzaldehyde to afford the title compound a colorless precipitate. Yield 72%. Mp 235–237 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.27 s (3H, CH3), 3.54 s (3H, OCH3), 5.28–5.29 d (1H, CH, J = 3 Hz), 7.5–7.53 d (2H, 2CArH, J = 9 Hz), 7.88 s (1H, NH), 8.2–8.22 d (2H, 2CArH, J = 6 Hz), 9.35 s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 18.25 (CH3), 51.23 (CH), 53.91 (OCH3), 98.39 (C), 124.19 (2CArH), 127.93 (2CArH), 147.11 (C), 149.94 (CAr), 152.13 (CAr), 152.16 (COO), 165.92 (CO). Elemental analysis calcd for C13H13N3O5, %: C, 53.61; H, 4.50; N, 14.43. Found, %: C, 53.66; H, 4.54; N, 14.40. The data agrees with the reported literature values.108
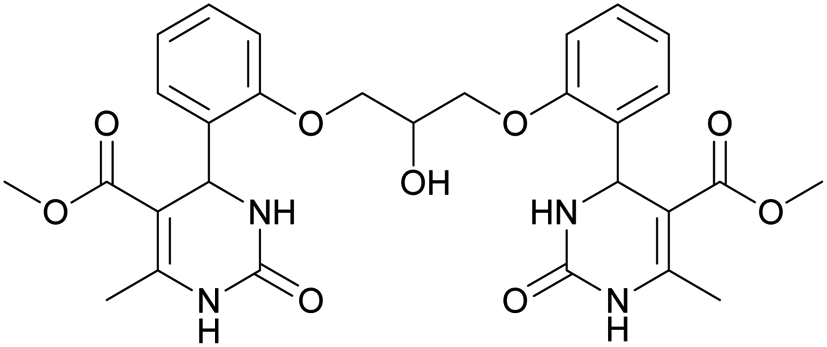
Dimethyl 4,4′-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(2,1-phenylene))bis(6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate) (6). The title compound was prepared according to the general procedure using 2,2′-((2-hydroxypropane-1,3-diyl)bis(oxy))dibenzaldehyde to afford the title compound a colorless precipitate. Yield 60%. Mp 293–295 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.32–2.33 d (6H, 2CH3, J = 3 Hz), 3.38 s (6H, 2OCH3), 4.04–4.1 m (1H, CH), 4.19–4.36 m (4H, 2OCH2), 5.58 s (1H, CH), 5.78–5.8 d (1H, OH, J = 6 Hz), 6.87–6.92 t (2H, 2CArH, J = 9 Hz), 7.01–7.08 q (4H, 4CArH, J = 6 Hz), 7.22–7.28 t (2H, 2CArH, J = 9 Hz), 7.32 s (1H, NH), 7.38 s (1H, NH), 9.2 s (2H, 2NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 17.79 (2CH3), 48.76 (2CH), 50.6 (2OCH3), 67.65 (CH–OH), 69.67 (2CH2O), 96.76 (2C), 112.54 (2CArH), 120.51 (2CArH), 126.55 (2CArH), 128.79 (2CArH), 131.04 (2C), 149.69 (2CAr), 152.27 (2CAr), 155.93 (2COO), 165.75 (2CO). HRMS (ESI-MS): 581.25 [M+ + H+], 579.25 [M+ − H+]. Elemental analysis calcd for C29H32N4O9, %: C, 59.99; H, 5.56; N, 9.65. Found, %: C, 59.94; H, 5.51; N, 9.69.
2,2′-((2-Hydroxypropane-1,3-diyl)bis(oxy))dibenzaldehyde. 2,2′-((2-Hydroxypropane-1,3-diyl)bis(oxy))dibenzaldehyde which was used in above mentioned synthesis, was obtained by following procedure (Scheme 8).109 In details, to the solution of sodium hydroxide (110 mmol, 4.4 g) in 100 ml of water was added 109.4 mmol (11.65 ml) of salicylaldehyde. The solution was stirred and heated till 60 °C under N2, after which 47.1 mmol (3.69 ml) of epichlorohydrin were added during 2 hours. Subsequently, the mixture was stirred at 60 °C for an additional 3 hours. At the end of the reaction time, the mixture was cooled and obtained precipitate was filtered, washed with distilled water and dried. In order to purify obtained dialdehyde, the crude product was recrystallized from methanol–water (8
:1). 2,2′-((2-Hydroxypropane-1,3-diyl)bis(oxy))dibenzaldehyde was finally obtained as white needles. Yield 35%. Mp 109–110 °C. 1H NMR spectrum: (CDCl3, δ, ppm), 3.99–4.01 d (1H, OH, J = 6 Hz), 4.32–4.34 d (4H, 2OCH2, J = 6 Hz), 4.52–4.56 m (1H, CH), 7.02–7.10 m (4H, Ar), 7.53–7.59 m (2H, Ar), 7.78–7.81 m (2H, Ar), 10.41 s (2H, 2COH). 13C NMR spectrum: (CDCl3, δ, ppm), 68.34 (2OCH2), 69.39 (CH), 112.94 (2CArH), 121.43 (2CArH), 124.97 (2CAr), 129.93 (2CArH), 136.16 (2CArH), 160.38 (2CAr), 189.89 (2COH). Elemental analysis calcd for C17H16O5, %: C, 67.99; H, 5.37. Found, %: C, 67.93; H, 5.43. The data agrees with the reported literature values.109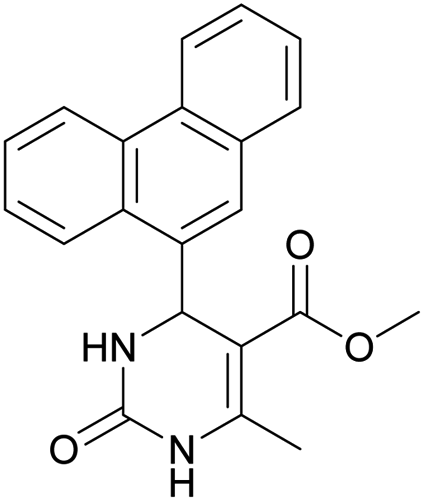
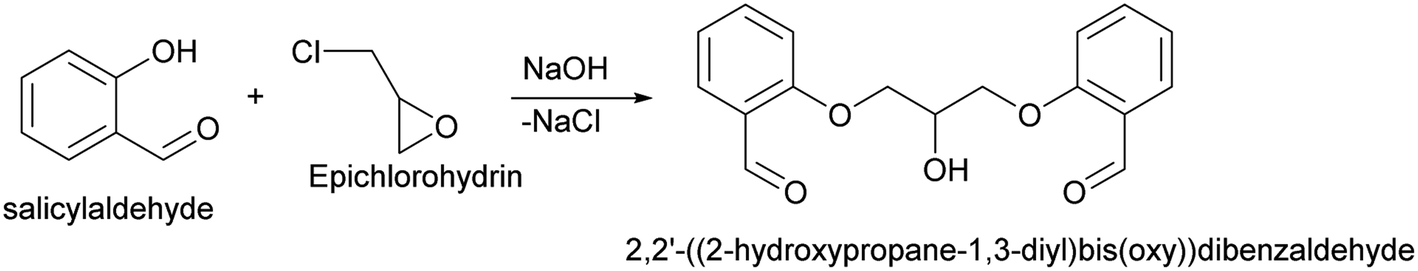 | ||
| Scheme 8 Synthesis of 2,2′-((2-hydroxypropane-1,3-diyl)bis(oxy))dibenzaldehyde. | ||
Methyl 6-methyl-2-oxo-4-(phenanthren-9-yl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7). The title compound was prepared according to the general procedure using phenanthrene-9-carbaldehyde to afford the title compound a colorless precipitate. Yield 54%. Mp 263–265 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.47 s (3H, CH3), 3.39 s (3H, OCH3), 6.1 s (1H, CH), 7.6–7.83 m (6H, 5CArH + NH), 7.96–7.98 d (1H, CArH, J = 6 Hz), 8.38–8.41 m (1H, CArH), 8.8–8.83 d (1H, CArH, J = 9 Hz), 8.89–8.92 m (1H, CArH), 9.34 s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 17.97 (CH3), 50.09 (CH), 50.77 (OCH3), 98.08 (C), 122.7 (CArH), 123.45 (CArH), 124.35 (2CArH), 126.62 (CArH), 126.92 (2CArH), 127.04 (CArH), 128.76 (CArH), 129.25 (C), 129.63 (CAr), 130.49 (CAr), 130.95 (CAr), 137.35 (CAr), 149.8 (CAr), 151.85 (COO), 165.8 (CO). HRMS (ESI-MS): 345.17 [M+ − H+]. Elemental analysis calcd for C21H18N2O3, %: C, 72.82; H, 5.24; N, 8.09. Found, %: C, 72.76; H, 5.29; N, 8.15.
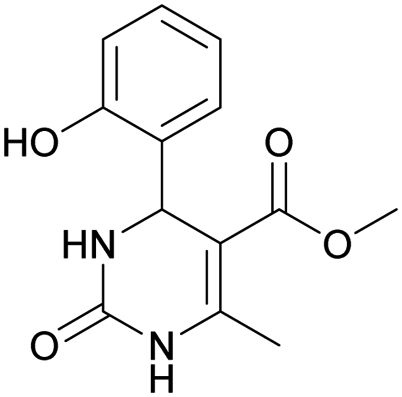
Methyl 4-(2-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (8). The title compound was prepared according to the general procedure using salicylaldehyde to afford the title compound a colorless precipitate. Yield 62%. Mp 230–232 °C. 1H NMR spectrum: (CD3OD, δ, ppm), 2.39 s (3H, CH3), 3.58 s (3H, OCH3), 5.66 s (1H, CH), 6.72–6.82 m (2H, 2CArH), 6.99–7.13 m (2H, 2CArH). Elemental analysis calcd for C13H14N2O4, %: C, 59.54; H, 5.38; N, 10.68. Found, %: C, 59.59; H, 5.31; N, 10.74. The data agrees with the reported literature values.110
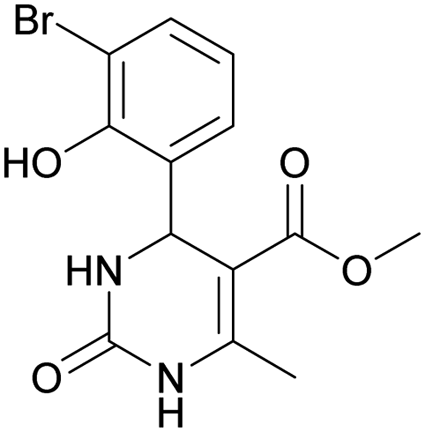
Methyl 4-(3-bromo-2-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (9). The title compound was prepared according to the general procedure using 3-bromo-2-hydroxybenzaldehyde to afford the title compound a yellow-green precipitate. Yield 78%. Mp 224–225 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.29 s (3H, CH3), 3.49 s (3H, OCH3), 5.55 s (1H, CH), 6.75–7.42 m (5H, 3CArH + NH + OH), 9.26 s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 17.87 (CH3), 49.22 (CH), 50.85 (OCH3), 97.53 (C), 111.90 (C), 121.41 (CArH), 126.59 (CArH), 131.75 (CArH), 133.64 (CAr), 149.45 (CAr), 150.70 (CAr), 151.95 (COO), 165.92 (CO). HRMS (ESI-MS): 341.11 [M+ + H+]. Elemental analysis calcd for C13H13N2O4Br, %: C, 45.77; H, 3.84; N, 8.21. Found, %: C, 45.71; H, 3.78; N, 8.28.
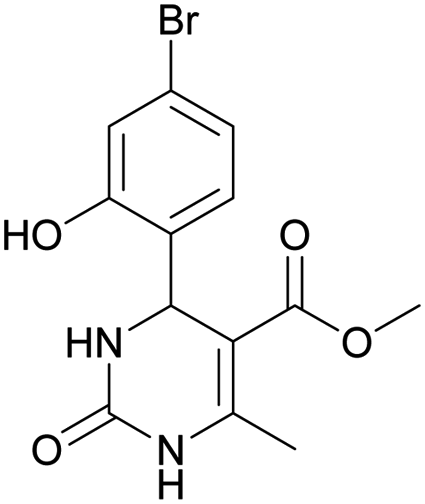
Methyl 4-(4-bromo-2-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (10). The title compound was prepared according to the general procedure using 4-bromo-2-hydroxybenzaldehyde to afford the title compound a yellow-green precipitate. Yield 77%. Mp 231–232 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.27 s (3H, CH3), 3.48 s (3H, OCH3), 5.39 s (1H, CH), 6.91–6.96 m (3H, 3CArH), 7.20 s (1H, NH), 9.17 s (1H, NH), 10.16 s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 17.76 (CH3), 48.78 (CH), 50.70 (OCH3), 96.99 (C), 118.01 (CArH), 120.33 (C), 121.52 (CArH), 128.98 (CAr), 129.37 (CArH), 149.28 (CAr), 152.13 (CAr), 155.89 (COO), 165.74 (CO). HRMS (ESI-MS): 341.11 [M+ + H+]. Elemental analysis calcd for C13H13N2O4Br, %: C, 45.77; H, 3.84; N, 8.21. Found, %: C, 45.70; H, 3.88; N, 8.17.
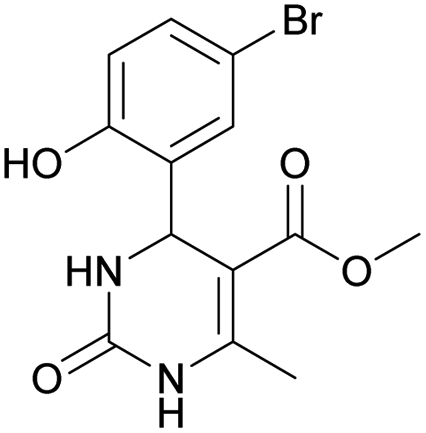
Methyl 4-(5-bromo-2-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (11). The title compound was prepared according to the general procedure using 5-bromo-2-hydroxybenzaldehyde to afford the title compound a yellow-green precipitate. Yield 72%. Mp 215–217 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.27 s (3H, CH3), 3.49 s (3H, OCH3), 5.39 s (1H, CH), 6.74–6.78 d (1H, CArH, J = 12 Hz), 7.0 s (1H, CArH), 7.20–7.27 m (2H, CArH + NH), 9.20 s (1H, NH), 10.01 s (1H, NH). Elemental analysis calcd for C13H13N2O4Br, %: C, 45.77; H, 3.84; N, 8.21. Found, %: C, 45.73; H, 3.81; N, 8.25. The data agrees with the reported literature values.111
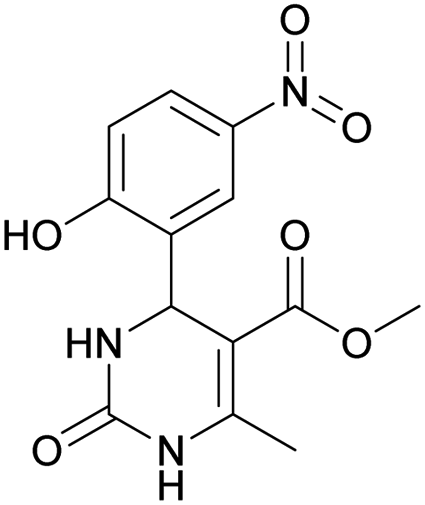
Methyl 4-(2-hydroxy-5-nitrophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (12). The title compound was prepared according to the general procedure using 2-hydroxy-5-nitrobenzaldehyde to afford the title compound a yellow-green precipitate. Yield 79%. Mp 239–240 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.30 s (3H, CH3), 3.49 s (3H, OCH3), 5.48 s (1H, CH), 6.97 s (1H, CArH), 7.44 s (1H, CArH), 7.87 s (1H, CArH), 8.06 s (1H, NH), 9.28 s (1H, NH), 11.36 br s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 17.80 (CH3), 49.38 (CH), 50.78 (OCH3), 96.60 (C), 115.95 (CAr), 123.56 (C), 124.93 (CArH), 139.25 (CArH), 149.60 (CArH), 151.94 (2CAr), 161.68 (COO), 165.63 (CO). HRMS (ESI-MS): 308.08 [M+ + H+], 330.08 [M+ + Na+], 306.08 [M+ − H+]. Elemental analysis calcd for C13H13N3O6, %: C, 50.82; H, 4.26; N, 13.68. Found, %: C, 50.88; H, 4.21; N, 13.61.
:1) allowed obtaining of pure oxidized dihydropyrimidine.
Methyl 6-methyl-2-oxo-4-(perfluorophenyl)-1,2-dihydropyrimidine-5-carboxylate (13). The title compound was prepared according to the general procedure using methyl 6-methyl-2-oxo-4-(perfluorophenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1) to afford the title compound a colorless precipitate. Yield 1%. Mp 311–312 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.31 s (3H, CH3), 3.43 s (3H, OCH3). HRMS (ESI-MS): 357.08 [M+ + Na+]. Elemental analysis calcd for C13H7N2O3F5, %: C, 46.72; H, 2.11; N, 8.38. Found, %: C, 46.78; H, 2.19; N, 8.44.
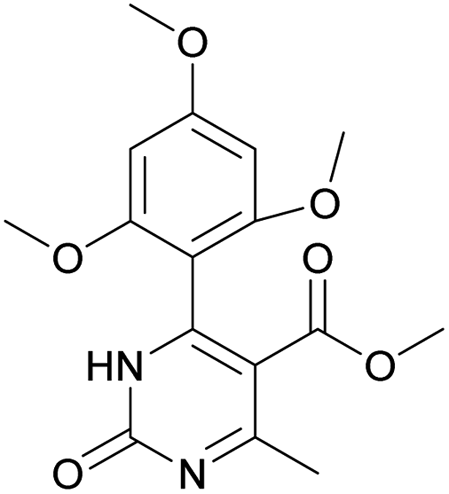
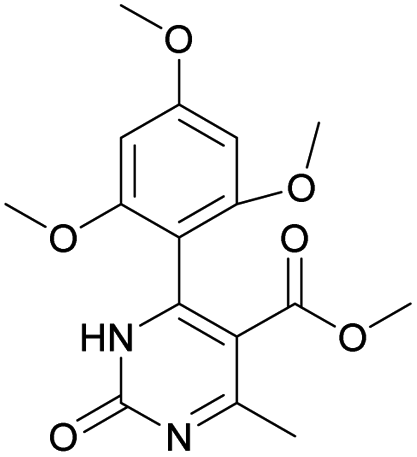
Methyl 6-methyl-2-oxo-4-(2,4,6-trimethoxyphenyl)-1,2-dihydropyrimidine-5-carboxylate (14). The title compound was prepared according to the general procedure using methyl 6-methyl-2-oxo-4-(2,4,6-trimethoxyphenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (2) to afford the title compound a light yellow precipitate. Yield 40%. Mp 220–222 °C. 1H NMR spectrum: (CDCl3, δ, ppm), 2.58 s (3H, CH3), 3.55 s (3H, OCH3), 3.73 s (6H, 2OCH3), 3.84 s (3H, OCH3), 6.13 s (2H, 2CArH). 13C NMR spectrum: (CDCl3, δ, ppm), 24.09 (CH3), 51.54 (OCH3), 55.25 (OCH3), 55.63 (2OCH3), 90.52 (2CArH), 103.72 (C), 112.29 (2C), 157.73 (CAr), 158.2 (3CAr), 163.26 (COO), 165.96 (CO). HRMS (ESI-MS): 335.17 [M+ + H+], 357.17 [M+ + Na+]. Elemental analysis calcd for C16H18N2O6, %: C, 57.48; H, 5.43; N, 8.38. Found, %: C, 57.55; H, 5.36; N, 8.31.
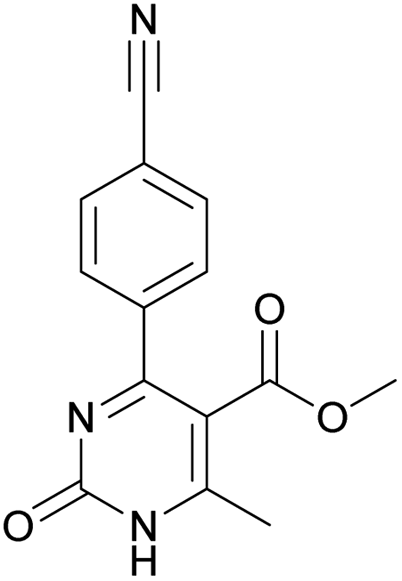
Methyl 4-(4-cyanophenyl)-6-methyl-2-oxo-1,2-dihydropyrimidine-5-carboxylate (15). The title compound was prepared according to the general procedure using methyl 4-(4-cyanophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (3) to afford the title compound a colorless precipitate. Yield 27%. Mp 199–200 °C. 1H NMR spectrum: (CDCl3, δ, ppm), 2.68 s (3H, CH3), 3.62 s (3H, OCH3), 7.72–7.75 m (4H, 4CArH). 13C NMR spectrum: (CDCl3, δ, ppm), 19 (CH3), 52.41 (OCH3), 110.88 (C), 114.35 (2C), 117.93 (2C, CN + CAr), 128.53 (2CArH), 132.00 (2CArH), 141.73 (CAr), 157.94 (COO), 165.50 (CO). HRMS (ESI-MS): 270.08 [M+ + H+], 292.08 [M+ + Na+]. Elemental analysis calcd for C14H11N3O3, %: C, 62.45; H, 4.12; N, 15.61. Found, %: C, 62.51; H, 4.17; N, 15.66.
Methyl 6-methyl-2-oxo-4-phenyl-1,2-dihydropyrimidine-5-carboxylate (16). The title compound was prepared according to the general procedure using methyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4) to afford the title compound a light yellow precipitate. Yield 39%. Mp 193–194 °C. 1H NMR spectrum: (CDCl3, δ, ppm), 2.59 s (3H, CH3), 3.58 s (3H, OCH3), 7.43–7.61 m (6H, 5CArH + NH). Elemental analysis calcd for C13H12N2O3, %: C, 63.93; H, 4.95; N, 11.47. Found, %: C, 63.99; H, 4.89; N, 11.41. The data agrees with the reported literature values.112
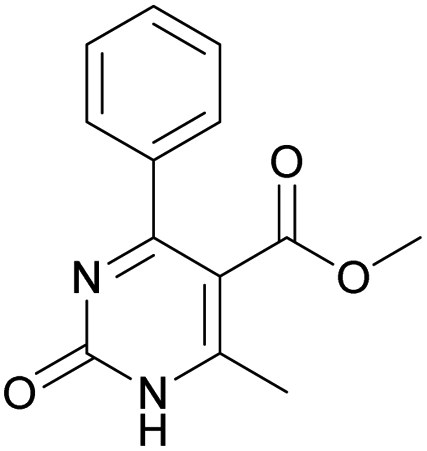
Methyl 6-methyl-4-(4-nitrophenyl)-2-oxo-1,2-dihydropyrimidine-5-carboxylate (17). The title compound was prepared according to the general procedure using methyl 6-methyl-4-(4-nitrophenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5) to afford the title compound a colorless precipitate. Yield 28%. Mp 210–211 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.46 s (3H, CH3), 3.49 s (3H, OCH3), 7.68–7.71 d (2H, 2CArH, J = 9 Hz), 8.30–8.33 d (2H, 2CArH, J = 9 Hz), 12.62 s (1H, NH). 13C NMR spectrum: (DMSO-d6, δ, ppm), 28.99 (CH3), 51.98 (OCH3), 123.42 (2CArH), 128.87 (2CArH), 144.82 (C), 148.12 (C), 154.69 (C), 154.90 (CAr), 161.83 (CAr), 162.12 (COO), 165.60 (CO). Elemental analysis calcd for C13H11N3O5, %: C, 53.98; H, 3.83; N, 14.53. Found, %: C, 53.91; H, 3.89; N, 14.57. The data agrees with the reported literature values.113

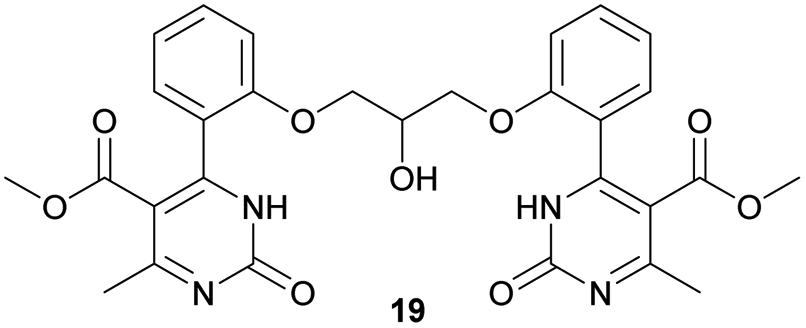
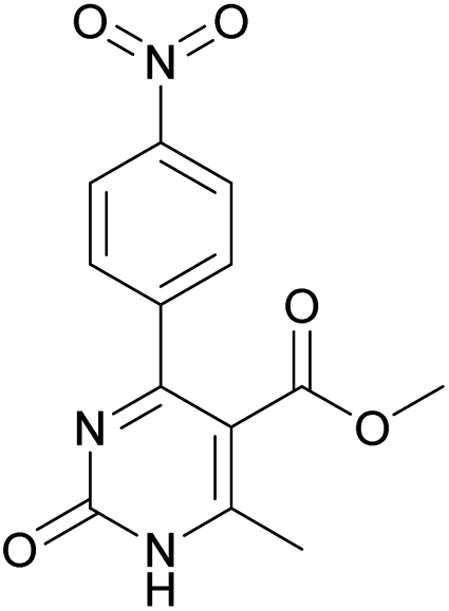
Methyl 4-(2-(2-hydroxy-3-(2-(5-(methoxycarbonyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidin-4-yl)phenoxy)propoxy)phenyl)-6-methyl-2-oxo-1,2-dihydropyrimidine-5-carboxylate (18) and dimethyl 4,4′-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(2,1-phenylene))bis(6-methyl-2-oxo-1,2-dihydropyrimidine-5-carboxylate) (19). The title compounds was prepared according to the general procedure using dimethyl 4,4′-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(2,1-phenylene))bis(6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate) (6) to afford the title compounds a colorless precipitate. Yield 15 and 5% respectively. In case if the amount of sodium bicarbonate and CAN was increased two times in comparison with the general oxidation procedure, then the obtaining of compound 18 was not observed. In this case the yield of compound 19 increased till 22% respectively. Mp 283–285 °C and 267–268 °C accordingly.
1H NMR spectrum of compound 18: (CDCl3, δ, ppm), 1.90 s (1H, NH), 2.33 s (3H, CH3), 2.41–2.43 d (3H, CH3, J = 6 Hz), 3.41–3.42 d (3H, OCH3, J = 3 Hz), 3.46 s (3H, OCH3), 3.87–3.93 t (1H, OH, J = 9 Hz), 4.07–4.20 m (5H, 2OCH2 + CH), 5.56 s (1H, CH), 6.87–6.92 t (1H, CArH, J = 9 Hz), 6.96–7.07 m (3H, 3CArH), 7.11–7.14 m (1H, CArH), 7.23–7.26 m (2H, CArH + NH), 7.32–7.34 d (1H, CArH, J = 6 Hz), 7.40–7.45 t (1H, CArH, J = 9 Hz), 9.19 s (1H, NH). 13C NMR spectrum of compound 18: (CDCl3, δ, ppm), 10.60 (CH3), 17.82 (CH3), 48.49 (CH), 50.70 (OCH3), 51.27 (OCH3), 69.65 (CH–OH), 69.94 (CH2O), 70.48 (CH2O), 96.75 (C), 109.66 (CArH), 111.66 (CArH), 112.30 (CArH), 120.57 (CArH), 126.35 (CArH), 128.75 (CArH), 129.56 (CArH), 131.11 (CArH), 131.31 (C), 149.72 (CAr), 149.77 (CAr), 152.26 (CAr), 152.32 (C), 155.12 (C), 155.16 (C), 155.80 (CAr), 156.06 (COO), 165.65 (COO), 165.67 (CO), 165.76 (CO). HRMS (ESI-MS): 579.33 [M+ + H+], 601.25 [M+ + Na+], 577.33 [M+ − H+]. Elemental analysis calcd for C29H30N4O9, %: C, 60.20; H, 5.23; N, 9.68. Found, %: C, 60.27; H, 5.28; N, 9.61.
1H NMR spectrum of compound 19: (CD3OD, δ, ppm), 3.41 s (6H, 2CH3), 3.54 s (6H, 2OCH3, J = 6 Hz), 3.99–4.02 d (1H, OH, J = 9 Hz), 4.13–4.28 m (5H, 2OCH2 + CH), 7.16–7.21 t (5H, 4CArH + NH, J = 9 Hz), 7.47–7.59 m (5H, 4CArH + NH). 13C NMR spectrum of compound 19: (CDCl3, δ, ppm), 23.88 (2CH3), 52.62 (2OCH3), 69.37 (CH–OH), 71.44 (2CH2O), 113.00 (2C), 113.35 (2CArH), 122.57 (2CArH), 127.78 (2C), 131.19 (2CArH), 133.14 (2C), 133.28 (2CArH), 157.16 (4CAr), 158.96 (2COO), 167.97 (2CO). HRMS (ESI-MS): 577.29 [M+ + H+], 599.27 [M+ + Na+]. Elemental analysis calcd for C29H28N4O9, %: C, 60.41; H, 4.90; N, 9.72. Found, %: C, 60.45; H, 4.83; N, 9.79.
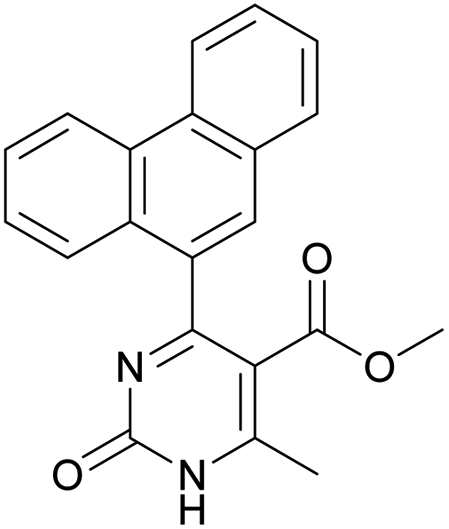
Methyl 6-methyl-2-oxo-4-(phenanthren-9-yl)-1,2-dihydropyrimidine-5-carboxylate (20). The title compound was prepared according to the general procedure using methyl 6-methyl-2-oxo-4-(phenanthren-9-yl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7) to afford the title compound a colorless precipitate. Yield 35%. Mp 239–241 °C. 1H NMR spectrum: (DMSO-d6, δ, ppm), 2.55 s (3H, CH3), 3.07 s (3H, OCH3), 7.61–7.81 m (6H, 6CArH), 8.04–8.07 d (1H, CArH, J = 9 Hz), 8.87–8.93 t (2H, 2CArH, J = 9 Hz). 13C NMR spectrum: (DMSO-d6, δ, ppm), 20.00 (CH3), 51.54 (OCH3), 110.37 (C), 122.92 (CArH), 123.31 (CArH), 125.57 (2CArH), 126.18 (CArH), 127.04 (2CArH), 127.11 (CArH), 127.29 (CArH), 127.88 (C), 128.77 (CAr), 129.07 (CAr), 129.53 (CAr), 129.97 (CAr), 130.28 (CAr), 133.84 (C), 154.62 (COO), 165.10 (CO). HRMS (ESI-MS): 343.17 [M+ − H+]. Elemental analysis calcd for C21H16N2O3, %: C, 73.24; H, 4.68; N, 8.13. Found, %: C, 72.29; H, 4.75; N, 8.17.
Biological assay
The antibacterial activity of the investigated compounds (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 14, 15, 16, 17, 19 and 20) against bacteria A. baumanii, E. coli, P. aeruginosa, K. pneumoniae and S. aureus was assessed determining minimum inhibitory concentration (MIC) by twofold microdilution method as described in elsewhere.100–102 The compounds were prepared according to CLSI guidelines and diluted in U-bottom 96 well microtiter plates which contained Mueller–Hinton Broth (MHB). The freshly prepared bacterial strains about 105 CFU (colony forming unit) in MHB medium were added to each well of the microplate and incubated at 37 °C for 24 hours. At the end of the experiment, the concentration of the tested compounds ranged from 1000 to 7.8 μg ml−1. The growth of the bacterial cells was determined by resazurin method. The solution of the resazurin sodium salt (0.01%) was freshly prepared in sterile distilled water. After incubation, 30 μl of this solution was added in each well of microplate and incubated again at the same condition for about 4 h. MIC was represented as the lowest concentration of the compounds which inhibited the change color from blue to pink since pink color indicated the growth of bacteria. MIC of the studied compound was compared with MICs of pristine antibiotics (cefotaxime and ceftriaxone).In addition to this, the antibacterial activity of the investigated dihydropyrimidines against the above-mentioned bacteria was also studied by disc-diffusion method as described by Mayrhofer.103 In details, the surface of the nutrient medium (meat-peptone agar, potato dextrose agar) was stratified with 1 ml of the diurnal suspension of the test culture (105 CFU ml−1), which was used during 15 min after preparation. Previously prepared discs with certain concentrations were stratified on the surface of the nutrient medium by the sterile tweezers. Dishes were incubated at 37 °C during 24 h. DMSO was used as a solvent. Record of the results was carried out, compared with control dishes without compound and with the known drugs cefotaxime and ceftriaxone.
Conclusion
The new derivatives of dihydropyrimidines were synthesized by Biginelli reaction in microwave conditions in presence of copper triflate. Salicylaldehyde under Biginelli reaction conditions can lead to the formation of 2 types of dihydropyrimidines. This procedure was therefore also used for salicylaldehyde derivatives and the formation of hydroxy substituted dihydropyrimidines was demonstrated. Furthermore regioselective oxidation of dihydropyrimidine ring in the presence of CAN was also done. The structures of all synthesized compounds were investigated by NMR, mass spectroscopy methods and X-ray single crystal diffraction.The crystal structures allowed determining structure-tautomer dependence. The oxidized products of dihydropyrimidines are in the form of tautomer II, in case they have a heteroatom group on the benzene ring that is able to participate in the formation of intramolecular hydrogen bond with amine group in the dihydropyrimidine ring. Otherwise, regioselectively oxidized products are in the form of tautomer I. Along with it, Hirshfeld surface analysis was carried out to gain insight into the crystal packing and molecular interactions. Considering that H2Py can have an ability to act as antibacterial drug, they were tested for the biological activity against A. baumanii, E. coli, P. aeruginosa, K. pneumoniae and S. aureus bacteria. In addition, their activity was also compared with that of pristine antibiotics. The results obtained are promising and suggest that the synthesized compounds are biologically active with antibacterial activity.
Author contributions
Alakbar E. Huseynzada: methodology, investigation, formal analysis, writing – original draft. Christian Jelch: formal analysis, writing – original daft, writing – review & editing. Haji Vahid N. Akhundzada: writing – review & editing. Sarra Soudani: formal analysis, writing – original draft. Cherif Ben Nasr: formal analysis, writing – original draft. Aygun Israyilova: formal analysis, writing – original draft. Filippo Doria: resources, data curation. Ulviyya A. Hasanova: writing – original draft, supervision, funding acquisition. Rana F. Khankishiyeva: writing – review & editing. Mauro Freccero: supervision, project administration.Funding
This work was supported by the Erasmus+ overseas/ICM KA107 programme.Conflicts of interest
The authors declare no conflict of interest.References
- P. Biginelli, Gazz. Chim. Ital., 1893, 23, 360–416 Search PubMed.
- G. C. Tron, A. Minassi and G. Appendino, Eur. J. Org. Chem., 2011, 5541–5550 CrossRef CAS.
- C. O. Kappe, Acc. Chem. Res., 2000, 33, 879–888 CrossRef CAS.
- W. Jie-Ping and Y. Liu, Synthesis, 2010, 23, 3943–3953 Search PubMed.
- M. Kazemi, Synth. Commun., 2020, 50, 1409–1445 CrossRef CAS.
- P. M. Kumar, K. S. Kumar, S. R. Poreddy, P. K. Mohakhud, K. Mukkanti and M. Pal, Tetrahedron Lett., 2011, 52, 1187–1191 CrossRef CAS.
- Y. U. Gadkari, N. T. Hatvate, B. S. Takale and V. N. Telvekar, New J. Chem., 2020, 44, 8167–8170 RSC.
- J. C. M. Willig, et al., RSC Adv., 2020, 10, 3407–3415 RSC.
- R. Kaur, S. Chaudhary, K. Kumar, M. K. Gupta and R. K. Rawal, Eur. J. Med. Chem., 2017, 132, 108–134 CrossRef CAS.
- J. C. Barrow, P. G. Nantermet, H. G. Selnick, K. L. Glass, K. E. Rittle, K. F. Gilbert, T. G. Steele, C. F. Homnick, R. M. Freidinger, R. W. Ransom, P. Kling, D. Reiss, T. P. Broten, T. W. Schorn, R. S. L. Chang, S. S. O'Malley, T. V. Olah, J. D. Ellis, A. Barrish, K. Kassahun, P. Leppert, D. Nagarathnam and C. Forray, J. Med. Chem., 2000, 43, 2703 CrossRef CAS.
- A. C. Boukis, A. Llevot and M. A. R. Meier, Macromol. Rapid Commun., 2016, 37, 643 CrossRef CAS.
- S. R. Patil, A. S. Choudhary, V. S. Patil and N. Sekar, Fibers Polym., 2015, 16, 2349 CrossRef CAS.
- Y. Zhao, Y. Yu, Y. Zhang, X. Wang, B. Yang, Y. Zhang, Q. Zhang, C. Fu, Y. Weia and L. Tao, Polym. Chem., 2015, 6, 4940 RSC.
- S. S. Jagir, Arkivoc, 2012, 66–133 Search PubMed.
- C. G. Wermuth, C. R. Ganellin, P. Lindberg and L. A. Mitscher, Pure Appl. Chem., 1998, 70, 1129–1143 CAS.
- K. S. Atwal, B. N. Swanson, S. E. Unger, D. M. Floyd, S. Moreland, A. Hedberg and B. C. O'Reilly, J. Med. Chem., 1991, 34, 806–811 CrossRef CAS.
- A. M. Maharramov, et al., J. Mol. Struct., 2017, 1141, 39–43 CrossRef CAS.
- P. Murthy and R. Chatterjee, Curr. Sci., 1999, 77, 1084–1089 CAS.
- R. Gaur, S. Dixit, M. Sahoo, M. Khanna, S. Singh and P. Murthy, J. Parasitol., 2007, 134, 537 CrossRef CAS.
- S. M. Sondhi, N. Singh, M. Johar and A. Kumar, Bioorg. Med. Chem., 2005, 13, 6158–6166 CrossRef CAS.
- U. Rashid, et al., Eur. J. Med. Chem., 2016, 115, 230–244 CrossRef CAS.
- (a) X. Zhu, G. Zhao, X. Zhou, X. Xu, G. Xia, Z. Zheng, L. Wang, X. Yang and S. Li, Bioorg. Med. Chem. Lett., 2010, 20, 299 CrossRef CAS; (b) K. L. Dhumaskar, S. N. Meena, S. C. Ghadi and S. G. Tilve, Bioorg. Med. Chem. Lett., 2014, 24, 2897 CrossRef CAS; (c) T. G. M. Treptow, F. Figueir, E. H. F. Jandre, A. M. O. Battastini, C. G. Salbego, J. B. Hoppe, P. S. Taborda, S. B. Rosa, L. A. Piovesan, C. R. M. Doca, D. Russowsky and M. G. M. Doca, Eur. J. Med. Chem., 2015, 95, 552 CrossRef CAS; (d) R. Chikhale, S. Menghani, R. Babu, R. Bansode, G. Bhargavi, N. Karodia, M. V. Rajasekharan, A. Paradkar and P. Khedekar, Eur. J. Med. Chem., 2015, 96, 30 CrossRef CAS; (e) U. Rashid, R. Sultana, N. Shaheen, S. F. Hassan, F. Yaqoob, M. J. Ahmad, F. Iftikhar, N. Sultana, S. Asghar, M. Yasinzai, F. L. Ansari and N. A. Qureshi, Eur. J. Med. Chem., 2016, 115, 230 CrossRef CAS; (f) K. Singh and T. Kaur, RSC Med. Chem., 2016, 7, 749 RSC.
- E. Klein, S. DeBonis, B. Thiede, D. A. Skoufias, F. Kozielskib and L. Lebeaua, Bioorg. Med. Chem., 2007, 15, 6474 CrossRef CAS.
- H. Y. K. Kaan, V. Ulaganathan, O. Rath, H. Prokopcov, D. Dallinger, C. O. Kappe and F. J. Kozielski, Med. Chem., 2010, 53, 5676 CrossRef CAS.
- M. Wright Christine, et al., Bioorg. Med. Chem., 2008, 16, 3291–3301 CrossRef CAS.
- O. C. Agbaje, O. O. Fadeyi, S. A. Fadeyi, L. E. Myles and C. O. Okoro, Bioorg. Med. Chem. Lett., 2011, 21, 989 CrossRef CAS.
- B. R. P. Kumar, G. Sankar, R. B. N. Baig and S. Chandrashekaran, Eur. J. Med. Chem., 2009, 44, 4192 CrossRef.
- D. A. Ibrahim and A. M. El-Metwally, Eur. J. Med. Chem., 2010, 45, 1158 CrossRef CAS.
- S. G. Khanage, S. A. Raju, P. B. Mohite and R. B. Pandhare, Adv. Pharm. Bull., 2012, 2, 213–222 Search PubMed.
- (a) A. Wang, X. Liu, Z. Su and H. Jing, Catal. Sci. Technol., 2014, 4, 71 RSC; (b) B. K. Ghosh, S. Hazra and N. N. Ghosh, Catal. Commun., 2016, 80, 44 CrossRef CAS.
- (a) N. October, N. D. Watermeyer, V. Yardley, T. J. Egan, K. Ncokazi and K. Chibale, ChemMedChem, 2008, 3, 1649 CrossRef CAS; (b) S. Fatima, A. Sharma, R. Saxena, R. Tripathi, S. K. Shukla, S. K. Pandey, R. Tripathi and R. P. Tripathi, Eur. J. Med. Chem., 2012, 55, 195 CrossRef CAS; (c) H. Kaur, M. Machado, C. Kock, P. Smith, K. Chibale, M. Prudencio and K. Singh, Eur. J. Med. Chem., 2015, 101, 266 CrossRef CAS.
- (a) T. N. Akhaja and J. P. Raval, Eur. J. Med. Chem., 2011, 46, 5573 CrossRef CAS; (b) R. K. Yadlapalli, O. P. Chourasia, K. Vemuri, M. Sritharan and R. S. Perali, Bioorg. Med. Chem. Lett., 2012, 22, 2708 CrossRef CAS.
- (a) K. T. Homan, K. M. Larimore, J. M. Elkins, M. Szklarz, S. Knapp and J. J. G. Tesmer, ACS Chem. Biol., 2015, 10, 310 CrossRef CAS; (b) H. V. Waldschmidt, K. T. Homan, O. Cruz-Rodríguez, M. C. Cato, J. Waninger-Saroni, K. M. Larimore, A. Cannavo, J. Song, J. Y. Cheung, P. D. Kirchhoff, W. J. Koch, J. J. G. Tesmer and S. D. Larsen, J. Med. Chem., 2016, 59, 3793 CrossRef CAS.
- S. N. Mokale, S. S. Shinde, R. D. Elgire, J. N. Sangshetti and D. B. Shinde, Bioorg. Med. Chem. Lett., 2010, 20, 4424 CrossRef CAS.
- S. S. Bahekar and D. B. Shinde, Bioorg. Med. Chem. Lett., 2004, 14, 1733 CrossRef CAS.
- S. S. Bahekar and D. B. Shinde, Acta Pharm., 2003, 53, 223 CAS.
- K. S. Atwal, G. C. Rovnyak, J. Schwartz, S. Moreland, A. Hedberg, J. Z. Gougoutas, M. F. Malley and D. M. J. Floyd, Med. Chem., 1990, 33, 1510 CrossRef CAS.
- I. S. Zorkun, S. Sarac, S. Celebib and K. Erolb, Bioorg. Med. Chem., 2006, 14, 8582 CrossRef CAS.
- R. V. Chikhale, R. P. Bhole, P. B. Khedekar and K. P. Bhusari, Eur. J. Med. Chem., 2009, 44, 3645 CrossRef CAS.
- O. Alam, S. A. Khan, N. Siddiqui, W. Ahsan, S. P. Verma and S. J. Gilani, Eur. J. Med. Chem., 2010, 45, 5113 CrossRef CAS.
- C. A. Sehon, G. Z. Wang, A. Q. Viet, K. B. Goodman, S. E. Dowdell, P. A. Elkins, S. F. Semus, C. Evans, L. J. Jolivette, R. B. Kirkpatrick, E. Dul, S. S. Khandekar, T. Yi, L. L. Wright, G. K. Smith, D. J. Behm and R. J. Bentley, Med. Chem., 2008, 51, 6631 CrossRef CAS.
- B. Borowsky, M. M. Durkin, K. Ogozalek, M. R. Marzabadi, J. Deleon, R. Heurich, H. Lichtblau, Z. Shaposhnik, I. Daniewska and T. P. Blackburn, Nat. Med., 2002, 8, 825–830 CrossRef CAS.
- A. D. Patil, N. V. Kumar, W. C. Kokke, M. F. Bean, A. J. Freger, C. Debrossi, S. Mai, A. Truneh, D. J. Faulkner, B. Carte, A. L. Breen, R. P. Hertzbery, R. K. Johnson, J. W. Westley and B. C. M. J. Potts, Org. Chem., 1995, 60, 1182 CrossRef CAS.
- R. W. Lewis, J. Mabry, J. G. Polisar, K. P. Eagen, B. Ganem and G. P. Hess, Biochemistry, 2010, 49, 4841 CrossRef CAS.
- L. Figueroa-Valverde, F. Díaz-Cedillo, M. López-Ramos and E. Garcia-Cervera, Int. J. PharmTech Res., 2010, 2075–2080 CAS.
- R. Ramajayam, K.-P. Tan, H.-G. Liu and P.-H. Liang, Bioorg. Med. Chem. Lett., 2010, 20, 3569–3572 CrossRef CAS.
- (a) K. Folkers and T. B. Johnson, J. Am. Chem. Soc., 1933, 55, 3784 CrossRef CAS; (b) F. S. Sweet and J. D. Fissekis, J. Am. Chem. Soc., 1973, 95, 8741 CrossRef CAS; (c) C. O. Kappe, J. Org. Chem., 1997, 62, 7201 CrossRef CAS; (d) R. De Souza, E. T. Penha, H. M. S. Milagre, S. J. Garden, P. M. Esteves, M. N. Eberlin and O. A. C. Antunes, Chem.–Eur. J., 2009, 15, 9799 CrossRef CAS; (e) L. M. Ramos, A. Y. P. L. Tobio, M. R. Santos, H. C. B. Oliveira, A. F. Gomes, F. C. Gozzo, A. L. Oliveira and B. A. D. J. Neto, Org. Chem., 2012, 77, 10184 CrossRef CAS; (f) M. Puripat, R. Ramozzi, M. Hatanaka, W. Parasuk, V. Parasuk and K. J. Morokuma, Org. Chem., 2015, 80, 6959 CrossRef CAS.
- S. Terracciano, G. Lauro, M. Strocchia, K. Fischer, O. Werz, R. Riccio, I. Bruno and G. Bifulco, ACS Med. Chem. Lett., 2015, 6, 187 CrossRef CAS.
- A. R. Trivedi, V. R. Bhuva, B. H. Dholariya, D. K. Dodiya, V. B. Kataria and V. H. Shah, Bioorg. Med. Chem. Lett., 2010, 20, 6100 CrossRef CAS.
- H. J. Gijsen, D. Berthelot, M. A. De Cleyn, I. Geuens, B. Brone and M. Mercken, Bioorg. Med. Chem. Lett., 2012, 22, 797–800 CrossRef CAS.
- B. K. Singh, M. Mishra, N. Saxena, G. P. Yadav, P. R. Maulik, M. K. Sahoo, R. L. Gaur, P. K. Murthy and R. P. Tripathi, Eur. J. Med. Chem., 2008, 43, 2717 CrossRef CAS.
- J. C. Barrow, P. G. Nantermet, H. G. Selnick, K. L. Glass, K. E. Rittle, K. F. Gilbert, T. G. Steele, C. F. Homnick, R. M. Freidinger, R. W. Ransom, P. Kling, D. Reiss, T. P. Broten, T. W. Schorn, R. S. L. Chang, S. S. O'Malley, T. V. Olah, J. D. Ellis, A. Barrish, K. Kassahun, P. Leppert, D. Nagarathnam and C. Forray, J. Med. Chem., 2000, 43, 2703 CrossRef CAS.
- X. Zhu, G. Zhao, X. Zhou, X. Xu, G. Xia, Z. Zheng, L. Wang, X. Yang and S. Li, Bioorg. Med. Chem. Lett., 2010, 20, 299 CrossRef CAS.
- H. J. Finlay, et al., J. Med. Chem., 2012, 55, 3036–3048 CrossRef CAS.
- J. Lloyd, et al., Bioorg. Med. Chem. Lett., 2009, 19, 5469–5473 CrossRef CAS.
- J. Lloyd, H. J. Finlay, W. Vacarro, T. Hyunh, A. Kover, R. Bhandaru, L. Yan, K. Atwal, M. L. Conder, T. Jenkins-West, H. Shi, C. Huang, D. Li, H. Sun and P. Levesque, Bioorg. Med. Chem. Lett., 2010, 20, 1436 CrossRef CAS.
- G. C. Rovnyak, K. S. Atwal, A. Hedberg, S. D. Kimball, S. Moreland, J. Z. Gougoutas, B. C. O'Reilly, J. Schwartz and M. F. Malley, J. Med. Chem., 1992, 35, 3254–3263 CrossRef CAS.
- S. R. Patil, A. S. Choudhary, V. S. Patil and N. Sekar, Fibers Polym., 2015, 16, 2349 CrossRef CAS.
- M. Heravi, M. Ghavidel and B. Heidari, Curr. Org. Synth., 2016, 13, 569–600 CrossRef CAS.
- J. P. Wan and Y. Liu, Synthesis, 2010, 23, 3943–3953 CrossRef.
- P. Shanmugam and P. T. Perumal, Tetrahedron, 2007, 63, 666–672 CrossRef CAS.
- D. S. Bose, F. Liyakat and H. B. Mereyala, J. Org. Chem., 2003, 68, 587–590 CrossRef CAS.
- A. R. Gholap, K. S. Toti, F. Shirazi, M. V. Deshpande and K. V. Srinivasan, Tetrahedron, 2008, 64, 10214–10223 CrossRef CAS.
- P. Shanmugam and P. T. Perumal, Tetrahedron, 2006, 62, 9726–9734 CrossRef CAS.
- K. Yamamoto, Y. G. Chen and F. G. Buono, Org. Lett., 2005, 7, 4673–4676 CrossRef CAS.
- M. Hayashi, K. I. Okunaga, S. Nishida, K. Kawamura and K. Eda, Tetrahedron Lett., 2010, 51, 6734–6736 CrossRef CAS.
- D. Prajapati, D. Bhuyan, M. Gohain and W. Hu, Mol. Diversity, 2011, 15, 257–261 CrossRef CAS.
- M. Wu, J. Yu, W. Zhao, J. Wu and S. Cao, J. Fluorine Chem., 2011, 132, 155–159 CrossRef CAS.
- J. P. Wan, C. Wang and Y. Pan, Tetrahedron, 2011, 67, 922–926 CrossRef CAS.
- V. P. Srivastava and L. D. S. Yadav, Tetrahedron Lett., 2010, 51, 6436–6438 CrossRef.
- K. U. Sadek, F. Al-Qalaf, M. M. Abdelkhalik and M. H. Elnagdi, J. Heterocycl. Chem., 2010, 47, 284 CAS.
- P. Gupta, S. Gupta, A. Sachar, D. Kour, J. Singh and R. L. Sharma, J. Heterocycl. Chem., 2010, 47, 324–333 CrossRef CAS.
- V. T. Kamble, D. B. Muley, S. T. Atkore and S. D. Dakore, Chin. J. Chem., 2010, 28, 388–392 CrossRef CAS.
- C. J. Liu and J. D. Wang, Molecules, 2010, 15, 2087–2095 CrossRef CAS.
- Z. Pourghobadi and F. Derikvand, Chin. Chem. Lett., 2010, 21, 269–272 CrossRef CAS.
- M. Matache, et al., Tetrahedron, 2009, 65, 5949–5957 CrossRef CAS.
- S. K. De and R. A. Gibbs, Synth. Commun., 2005, 35, 2645–2651 CrossRef CAS.
- M. N. Godoi, H. S. Costenaro, E. Kramer, P. S. Machado, M. G. M. Doca and D. Russowsky, Quim. Nova, 2005, 28, 1010 CrossRef CAS.
- S. K. De and R. A. Gibbs, Synthesis, 2005, 11, 1748–1750 CrossRef.
- G. L. Zhang and X. H. Cai, Synth. Commun., 2005, 35, 829–833 CrossRef CAS.
- A. E. Huseynzada, et al., J. Mol. Struct., 2020, 128581 CrossRef CAS.
- J. Jacob, Int. J. Chem., 2012, 4, 29–43 CAS.
- C. O. Kappe and D. Dallinger, Mol. Diversity, 2009, 13, 71 CrossRef CAS.
- K. K. Pasunooti, H. Chai, C. N. Jensen, B. K. Gorityala, S. Wang and X. W. Liu, Tetrahedron Lett., 2011, 52, 80–84 CrossRef CAS.
- Q. Cheng, Q. Wang, T. Tan, N. Chen and M. Shuaib, J. Heterocycl. Chem., 2012, 49, 1352–1356 CrossRef CAS.
- X. Jing, Z. Li, X. Pan, Q. Wang, C. Yan and H. Zhu, Synth. Commun., 2009, 39, 3796–3803 CrossRef CAS.
- J. Sve, L. Veizerová and V. Kettmann, Tetrahedron Lett., 2008, 49, 3520–3523 CrossRef.
- Q. Liu, J. Xu, F. Teng, A. Chen, N. Pan and W. Zhang, J. Heterocycl. Chem., 2014, 51, 741–746 CrossRef CAS.
- O. Rosati, M. Curini, F. Montanari, M. Nocchetti and S. Genovese, Catal. Lett., 2011, 141, 850–853 CrossRef CAS.
- D. Kumarasamy, et al., Bioorg. Med. Chem. Lett., 2017, 27, 139–142 CrossRef CAS.
- M. M. Ibrahim, H. S. El-Sheshtawy, M. El-Kemary, S. Al-Juaid, M. Youssef and I. H. El-Azab, J. Mol. Struct., 2017, 1137, 714–719 CrossRef CAS.
- R. Zheng, X. Wang, H. Xu and J. Du, Synth. Commun., 2006, 36, 1503–1513 CrossRef CAS.
- N. Y. Fu, Y. F. Yuan, Z. Cao, S. W. Wang, J. T. Wang and C. Peppe, Tetrahedron, 2002, 58, 4801–4807 CrossRef CAS.
- D. S. Bose, M. V. Chary and H. B. Mereyala, Heterocycles, 2006, 68, 1217–1224 CrossRef CAS.
- J. Zhu, M. Zhang, B. Liu and X. Li, Chem. Lett., 2009, 38, 56–57 CrossRef CAS.
- A. Kumar and R. A. Maurya, Tetrahedron Lett., 2007, 48, 4569–4571 CrossRef CAS.
- D. S. Bose, M. Sudharshan and S. W. Chavhan, Arkivoc, 2005, 228, 236 Search PubMed.
- A. Katrusiak, J. Mol. Graphics Modell., 2001, 19, 363–367 CrossRef CAS.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 37, 3814–3816 RSC.
- A. Martin, H. Takiff, P. Vandamme, J. Swings, J. C. Palomino and F. Portaels, J. Antimicrob. Chemother., 2006, 58, 327–331 CrossRef CAS.
- A. Israyilova, S. Buroni, F. Forneris, V. C. Scoffone, N. Q. Shixaliyev, G. Riccardi and L. R. Chiarelli, PLoS One, 2016, 11, 1–17 CrossRef.
- S. Hajiyeva, et al., Turk. J. Chem., 2019, 43, 1711–1721 CrossRef CAS.
- S. Mayrhofer, et al., Appl. Environ. Microbiol., 2008, 74, 3745–3748 CrossRef CAS.
- M. J. Ahmad, et al., Med. Chem. Res., 2016, 25, 1877–1894 CrossRef CAS.
- G. M. Sheldrick, SHELXTL V5. 1, Bruker AXS Inc., 1997 Search PubMed.
- C. Mukhopadhyay and D. Arup, J. Heterocycl. Chem., 2010, 47, 136–146 CAS.
- C. K. Khatri, S. R. Deelip and U. C. Ganesh, New J. Chem., 2016, 40, 10412–10417 RSC.
- J. Safari and S. Gandomi-Ravandi, New J. Chem., 2014, 38, 3514–3521 RSC.
- Z. Bin, et al., Polyhedron, 1996, 15, 1197–1202 CrossRef.
- H. M. Savanur, et al., Tetrahedron Lett., 2016, 57, 3029–3035 CrossRef CAS.
- Q. Cheng, et al., J. Heterocycl. Chem., 2010, 47, 624–628 CrossRef CAS.
- S. Kamaljit Singh and S. Kawaljit, Aust. J. Chem., 2008, 61, 910–913 CrossRef.
- D. S. Kolarović, V. Vladimir and L. Mladen, Arkivoc, 2016, 271–286 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1998538, 1998539, 1998546, 1998547 and 1998557. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra10255e |
| This journal is © The Royal Society of Chemistry 2021 |