DOI:
10.1039/D0RA10227J
(Paper)
RSC Adv., 2021,
11, 12893-12901
Mechanical power driven SPME-SERS ultra-fast detection of illegal additives in aquaculture water†
Received
4th December 2020
, Accepted 26th March 2021
First published on 6th April 2021
Abstract
A dual-function (extraction and detection) porous silver fiber with high enhancement effect was constructed based on a convenient electrochemical etching method. The prepared silver fiber not only had high enrichment capacity and good Surface Enhanced Raman Spectroscopy (SERS) performance but also had good laser stability and uniformity. A strategy combining mechanical power and integration of solid phase extraction (SPME) and SERS detection was used. Driven by mechanical power, the analyte malachite green (MG) was enriched on the prepared silver fiber after 40 seconds, which can realize an ultra-fast and sensitive detection with a detection limit of 8.48 × 10−9 M. At the same time, this fiber can be regenerated after being treated with NaBH4. The silver fiber can be used for the detection of MG and CV after being immersed in NaBH4 solution for a few minutes. After 5 cycles of processing, the measurement signals of the silver fiber can reach 70% of the initial signals. The mechanical power driven SPME-SERS (MPD-SPME-SERS) integrated detection method can be used to analyse aquaculture water within 1 minute with a good linear relationship.
Introduction
SPME is a new type of sample preparation technology and has attracted wide attention since it was first proposed by Canadian professor Pawliszyn.1 SPME does not require organic solvents, which reduces environmental pollution. The solid phase microextraction device is simple, rapid and efficient.2–4 Sampling and enrichment are performed simultaneously by this method. The extraction volume of this method is small, and the loss of matrix can be ignored, which is suitable for on-site sampling and analysis.5,6 Based on the portability and the high efficiency of solvent-free extraction, SPME can be combined with a variety of detection methods to achieve efficient enrichment and detection. SPME has been widely used and combined with many technologies, such as liquid chromatography (LC),7–10 gas chromatography (GC),11–14 mass spectrometry (MS)15,16 and surface enhanced Raman spectroscopy (SERS).17–20 Among them, SERS has the advantages of high sensitivity and fingerprint characteristics. Based on the advantages of SERS and SPME, the two can be combined to realize the integration of sample pretreatment and detection. The SERS substrate provides hot spots and enhances Raman signals. It is also a SPME coating to achieve sampling and enrichment of the analyte. SPME-SERS is an effective in situ detection method for complex systems. Qiu, Lu prepared single-layer graphene-coated Ag nanoparticles on Si fibers and combined SPME and SERS to achieve in situ sensitive determination of BPA in bottled water with a detection limit of 1 μg L−1.21 Chen, Haoxin combined headspace SPME with SERS to achieve sensitive detection of volatile pesticides in apple juice with a detection limit of 5 ppb.22 However, how to enrich the analyte to the extraction fiber faster to shorten the detection time is a key issue that needs to be solved in rapid detection. Hassan Sereshti used a potentiostat to apply a fixed potential to accelerate the enrichment process. The tetracycline in milk can be extracted in 20 minutes.23 Yuan-Ting Li used electrophoresis pre-concentration strategy to complete antibiotic detection within 10 minutes.24 Fast enrichment and detection can be achieved by applying electric driving force.
The factors that affect the extraction effect and time include the thickness of the boundary layer, the thickness of the coating and the distribution constant of the analyte in the coating/solution.25 Similar to electrochemical driving, mechanical driving can also reduce the thickness of the boundary layer. In this study, a mechanical power driven SPME was used in order to solve the problem of excessively large boundary layer thickness in direct diffusion extraction. We realized dynamic and rapid stirring and extraction, and combined with SERS to realize efficient and rapid analysis and determination.
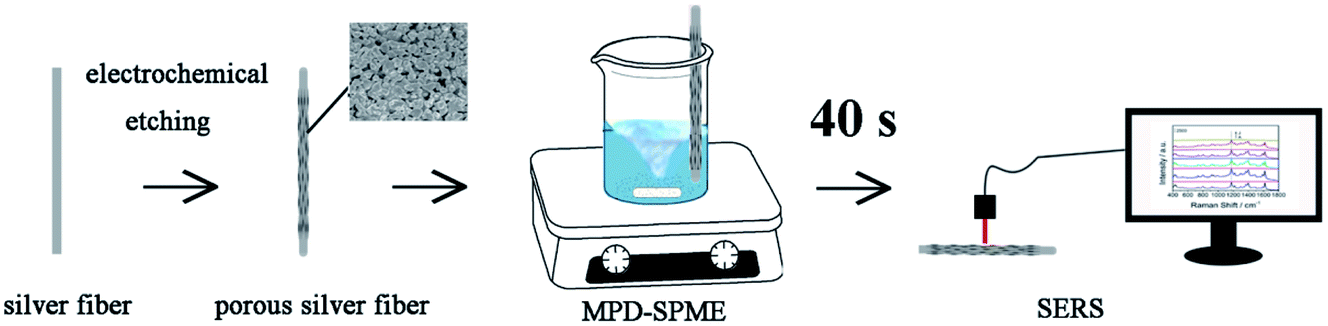
In our work, based on the electrochemical etching method, a three-dimensional silver nanofiber with uniform pores was prepared. And the fiber was used as a dual-function substrate for the integration of extraction and detection. The application of mechanical drive can reduce the thickness of the diffusion layer. We chose the method of stirring with a stir bar to provide mechanical driving force, and SERS signals can be obtained after 40 seconds. Due to the high detection efficiency, this method is expected to play an important role in food safety and environmental monitoring (Fig. 1).
 |
| | Fig. 1 Schematic diagram of MPD-SPME-SERS. | |
Experimental reagent
Malachite green (MG), crystal violet (CV), ethanol, hydrochloric acid, and sodium borohydride (NaBH4, >98%) were purchased from Sinopharm Group, Co., Ltd. Othter reagents used in our experiment were analytical pure. The water used in the experiment was ultrapure water (18.25 MΩ cm). Aquaculture water was taken at a local supermarket.
Preparation of three-dimensional porous silver fiber
Silver wires (diameter 0.4 mm, purity 99.9%, length 2.0 cm) were cleaned with ethanol and ultrapure water for 5 minutes each time by an ultrasonic cleaning machine. The silver fiber was prepared by electrochemical etching. In the electrochemical process, a three-electrode system was used, in which the silver wire was used as the working electrode, Ag/AgCl electrode was used as reference electrode, and Pt electrode was used as counter electrode. 0.1 M hydrochloric acid solution was used as the electrolyte solution, and the silver wire was corroded by electrochemical cyclic voltammetry. The voltage range was from −0.2 V to 0.2 V and the number of cycles was 15. The potential scan rate was 10 mV s−1. The signals of MG collected by porous silver fiber prepared with different potential scan rates were compared in order to select the optimal preparation condition.
Preparation of sample
10−2 M malachite green (MG) and crystal violet (CV) solutions were prepared. The 10−5 to 10−8 M MG and CV solutions were respectively diluted with 10−2 M MG and CV solutions. Actual samples of different concentrations were obtained by adding MG and CV to the aquaculture water.
MPD-SPME-SERS detection
40 mL solution for detection was placed in a 100 mL beaker with a stir bar. We changed the position of the substrate in order to find the best position for sampling. We kept the silver fiber still, and driving force was provided by the stir bar for the solution to improve the extraction efficiency. For a certain substrate at a certain stirring speed, the substrate was taken out for a period of time, and the SERS signals were collected. After finding optimal conditions, the actual sample was added to the beaker and detected under optimal conditions.
Related experimental instruments
Field emission scanning electron microscope (FESEM, JSM-6700F, acceleration voltage 3 kV) was used to characterize the substrate morphology. X-ray energy spectrometer (EDS) was used to characterize the types of elements contained in the substrate. Princeton ParStat 4000 electrochemical workstation was used to prepare porous silver fiber. X-ray Photoelectron Spectroscopy (XPS, ESCALAB 250Xi, ThermoFisher Scientific) was used to judge the valence of Ag. Ocean Optics QE65000 spectrometer was used to collect Raman signals, excitation wavelength: 785 nm, laser power: 150 W, integration time: 1 s.
Results and discussion
Optimization and morphology characterization of silver fiber
Electrochemical etching conditions are very important to obtain a good dual-functional substrate. We explored the conditions of substrates obtained by corrosion in the range of −0.2 V to 0.2 V at different scanning speeds using MG as a probe. The morphology of the obtained silver fiber was characterized. Fig. 2 shows the morphology of the silver fiber obtained at different potential scanning rates. The potential scanning rates of the silver fibers obtained in Fig. 2A–D are 5, 10, 15 and 25 mV s−1, respectively. When the silver fiber prepared under the conditions that the voltage range was from −0.2 V to 0.2 V, the potential scanning rate was 10 mV s−1, and the number of cycles was 15, the highest SERS signals were obtained. When the potential scan rate is slow (for example, Fig. 2A), the electron transmission speed is slow, and the resulting nanoparticle size is large. When the potential scan rate is fast (for example, Fig. 2C and D), the electron transmission speed may exceed the diffusion speed. The oxidation rate of silver exceeds the reduction rate of silver ions, and the reduction is insufficient. Ultimately, the uniformity of the substrate is affected. From the morphology of the prepared sample, when the potential scan rate is 10 mV s−1, the porous silver fiber obtained is the most uniform, and the density of silver nanoparticles on the surface of the silver fiber is the largest, which is important for providing more hot spots and better SERS signals. The SERS enhancement capability is the premise and guarantee for the three-dimensional porous silver to obtain good SERS signals and to be used for the detection of trace samples. In addition, the large surface area of the three-dimensional porous structure is beneficial to increase the enrichment efficiency. The structure of silver fiber determines that it is a good dual-function substrate integrating extraction and detection.
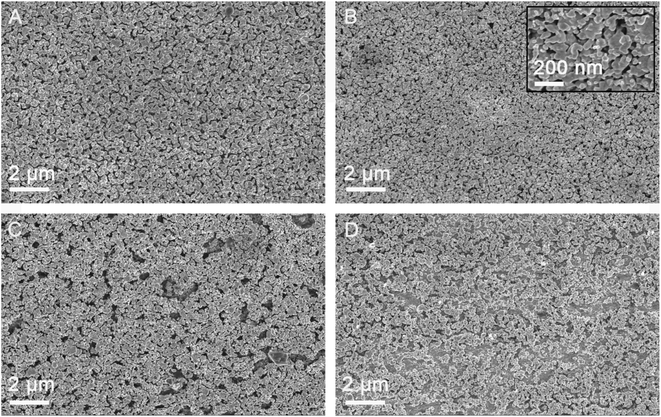 |
| | Fig. 2 SEM images of porous silver fibers prepared by using different potential scan rates (A) 5, (B) 10, (C) 15 and (D) 25 mV s−1, respectively. | |
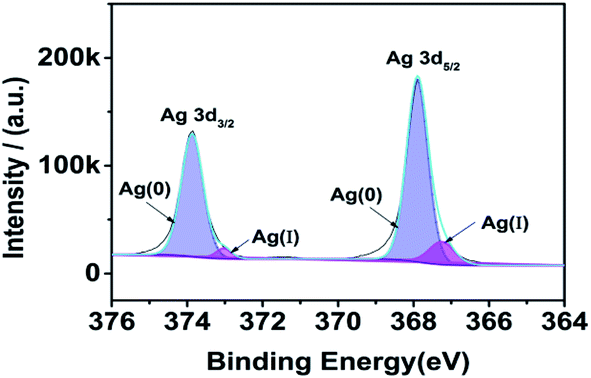
As Fig. 3, we can know that most of the silver element on the surface of the substrate is Ag(0). The peaks observed at binding energy values 367.90 and 373.87 were ascribed to the Ag 3d5/2 and Ag 3d3/2 of Ag(0) state respectively,26,27 with a separation of 5.97 eV.28 The peaks observed at binding energy values 367.25 and 373.03 eV were ascribed to the Ag 3d5/2 and Ag 3d3/2 of Ag(I) state respectively. According to the peak area, the content of Ag(0) is 92.34%.
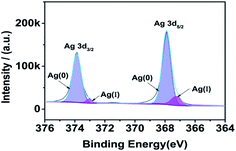 |
| | Fig. 3 XPS result of silver fiber. | |
The sensitivity of the substrate is an important factor for qualitative and quantitative detection. The properties of SERS substrates, such as enhancement factors, uniformity and stability, have important impacts on the application of the substrate. The performance of the prepared porous silver fiber was tested with 10−5 MG as a probe. The characteristic peaks of MG are at 439, 800, 1174, 1398 and 1592 cm−1, respectively. Among them, the peaks at 800 and 1174 cm−1 are attributed to the bending vibration of CH in the radial plane aromatic ring. The peaks at 1398 and 1592 cm−1 are attributed to the in-plane C–C stretching vibration, and the peak at 439 cm−1 is attributed to benzene-C-benzene deforms and vibrates out of the plane.29,30 The peak position is basically the same as the peak position in references, which proves that we have successfully collected the MG signals. SERS signals of MG were collected with silver wires prepared at different potential scan rates. As shown in Fig. 4, as the potential scan rate increased, the SERS signal intensity first increased and then decreased. When the potential scan rate was 10 mV s−1, the signals obtained were the strongest. Therefore, we used a 10 mV s−1 potential scan rate to prepare porous silver fiber in subsequent experiments. We selected the characteristic peak at 1174 cm−1 and calculated the enhancement factor of the substrate according to the formula AEF = (ISERS/CSERS)/(IRaman/CRaman).31 Among them, ISERS: the SERS signal intensity of MG at 1174 cm−1, CSERS: the concentration of MG on the substrate, IRaman: the Raman signal intensity of MG powder at 1174 cm−1, CRaman: the concentration of MG powder. Through calculation, we obtained that the enhancement factor of the prepared porous silver wire to MG can reach 2.68 × 106, and the substrate has a good SERS enhancement effect.
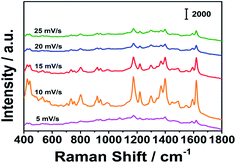 |
| | Fig. 4 SERS signals of MG collected by porous silver fibers prepared by using different potential scan rates. | |
Uniformity and stability
The uniformity and stability of the SERS substrate are also the main parameters we are concerned about. We used MG as a probe molecule, soaked the prepared silver fiber in 10−5 MG solution for 3 hours, and collected the SERS signals of 20 points randomly selected from the substrate. As shown in Fig. 5A, the characteristic peak position and peak intensity are hardly changed. We calculated that the relative standard deviation (RSD) of the peak intensity at 1174 cm−1 is 9.82%, which shows that the prepared SERS substrate has good uniformity. We irradiated the prepared SERS substrate continuously for about 2 minutes under the laser, and collected the signals every 5 s. As shown in Fig. 5B, the characteristic peak position and peak intensity are basically unchanged. We calculated that the relative standard deviation (RSD) of the peak intensity at 1174 cm−1 is 8.55%, which shows that the prepared SERS substrate has good stability. The long-term stability of the substrate is also important. Compared with the signal intensity of MG on the newly prepared substrate, there is almost no change in the signal intensity after 5 days. Therefore, the substrate has long-term stability.
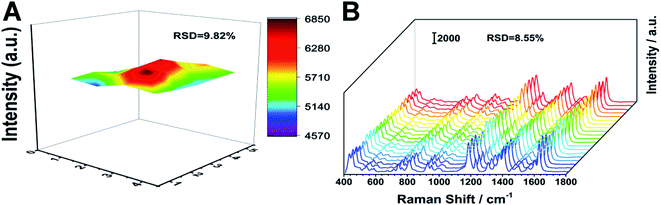 |
| | Fig. 5 (A) Uniformity and (B) laser irradiation stability of the silver fiber. | |
Use of mechanical power
We chose MG and CV as the analytes. We applied different stirring speeds to MG and CV, and took out the substrate at regular intervals to collect SERS signals. We first investigated the position of the characteristic peaks. The characteristic peaks of CV are at 422, 528, 760, 803, 916, 1177, 1394, 1443, 1536, 1590 and 1621 cm−1 respectively. The characteristic peaks at 1443, 1536 and 1621 cm−1 are attributable to the C–C in-plane stretching vibration, the broad peak at 1394 cm−1 is attributable to the in-plane C+–C stretching vibration and the N–benzene ring stretching vibration, the peak at 528 and 916 cm−1 is attributable to the aromatic ring skeleton vibration, the peak at 422 cm−1 is attributable to the out-of-plane deformation vibration of benzene–C–benzene, and the peak at 760, 803, and 1177 cm−1 is attributable to the bending vibration of CH in the aromatic ring hydrocarbon in the radial plane.32,33 The characteristic peaks of signals we collected are basically consistent with the peak positions in the references, which proves that the substrate we prepared can successfully enrich the analytes and achieve qualitative detection. For different analytes, the optimal stirring extraction conditions may be different. We tried different speeds of stirring to MG solution and CV solution. We took out the substrate at 10 second intervals to collect SERS signals on the substrate. The power of the laser was 150 W, and the integration time was 1 s. As shown in Fig. 6, before the extraction equilibrium was reached, the SERS signals increased with stirring time increasing, and after the extraction equilibrium was reached, the SERS signals no longer changed. For MG, when the stirring speed was 500 rpm, the molecular diffusion rate of the analyte was small, so the SERS signal intensity increased slowly with time. When the stirring speed was 1000 rpm and 1500 rpm, the growth rate of SERS signal intensity was greater than 500 rpm, but there was little difference of the kinetic curve between 1000 rpm and 1500 rpm. This shows that as the stirring speed increases, the molecular diffusion rate of the analyte increases. However, because the number of hot spots on the substrate is constant, after the stirring speed increases to the optimal stirring speed, the equilibrium time cannot be shorten by increasing the stirring speed. Therefore, we finally choose the stirring speed of 1000 rpm and the stirring time of 40 s as the optimal stirring conditions for MG. For CV, the experimental result is similar to the result of MG. For CV, we finally choose the stirring speed of 1000 rpm and the stirring time of 40 s as the optimal stirring conditions for CV. In our work, for the analytes, the stirring extraction equilibrium time is less than 1 min.
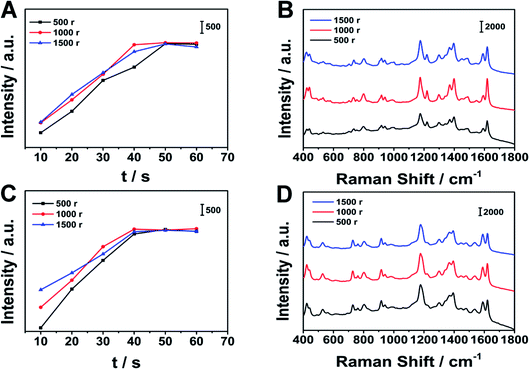 |
| | Fig. 6 SERS signals collected at different stirring speeds. (A), (C) are the changes of the SERS signal intensity of MG at 1174 cm−1 and CV at 1177 cm−1 collected at different stirring speeds over time, (B), (D) are SERS signals collected at different stirring speeds at 40 s of MG and CV respectively. | |
In Fig. 7, we compared the time for different concentrations of MG to reach the equilibrium of extraction by stirring. Under the same stirring conditions, the lower the concentration was, the longer time it took to reach equilibrium. At 1000 r stirring speed, for 10−5 M MG solution, the equilibrium can be reached by stirring for 40 s. When the concentration decreased to 10−6 M, it took 50 s to reach equilibrium. When the concentration decreased to 10−7 M, it took 60 s to reach equilibrium.
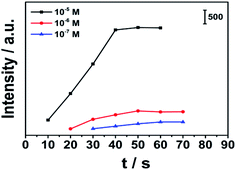 |
| | Fig. 7 The kinetic curve of mechanical power driven SPME of different concentrations (10−5 M, 10−6 M, and 10−7 M MG solution) of MG at 1000 r stirring speed. | |
Principle of MPD-SPME
In SERS detection, if the substrate is immersed in the solution directly, due to the presence of the boundary layer, the analyte diffuses slowly, which is not conducive to rapid detection. It takes a long time for static extraction to reach the equilibrium. We can accelerate this process through stirring. When the liquid sample is ideally stirred, the migration of components in the sample is greatly accelerated, allowing it to enter the substrate quickly. The thickness of the boundary layer can be obtained by the following formula:25| | |
δ = 2.64(b/Re0.50Sc0.43)
| (1) |
b: extraction wire radius; Re: Reynolds number, = 2ub/v; Sc: Schmidt number, Sc = v/Dl; v kinematic viscosity of the medium; Dl: molecular diffusion coefficient. Under the stirring condition of 1000 r and 40 s, we estimated the boundary layer thickness of MG solution. The diffusion coefficient Dl is set to 0.011361 cm2 s−1 and the kinematic viscosity is set to 0.0101 cm2 s−1, and the resulting boundary layer thickness is 2.2 × 10−4 cm. The extraction amount can be obtained by the following formula:25| | |
n = KfsVfC0Vs/(KfsVf + Vs)
| (2) |
Kfs: partition coefficient, Vf: coating volume, C0: initial concentration of the component in the water sample, Vs: water sample volume. From this formula, we know that the extraction volume is related to the partition coefficient, coating volume, concentration, and water sample volume. When the extraction filament and the analyte are determined, the greater the concentration of the analyte, the larger the water sample volume and the greater the extraction volume.
The extraction equilibrium time in this case (defined as the time required for 95% of the components to be extracted from the sample) depends mainly on the diffusion of the analyte on the substrate. The extraction equilibrium time can be obtained by the following formula:25
df is the thickness of the coating and
Df is the diffusion coefficient of the components. However, in reality, there is always a layer of unstirred water fluid around the substrate. Usually we call it the boundary layer. Besides, the fluid on the substrate surface is always stationary under any stirring conditions. As the distance from the coating surface increases, the fluid motion will gradually increase until it is equal to the flow rate of the sample body. If no convection occurs in the boundary layer, the thickness of the boundary layer is determined by the stirring conditions and the viscosity of the fluid. The better we stir, the lower the fluid viscosity and the smaller the boundary layer thickness is. The actual equilibrium time can be obtained by the following formula:
25| | |
te = t95% = 3δKfsds2/2Df
| (4) |
δ is the thickness of the boundary layer, and Kfs is the distribution constant of the component in the substrate/water sample. Combining formula (1), we can know:
| | |
te = t95% = 7.92bv0.50Kfsds2/2Df(2ub)0.50Sc0.43
| (5) |
Comparison of MPD-SPME and direct SPME
We use 10−6 M MG and CV solutions for direct SPME tests. We immersed the substrate in the above three solutions, and collected SERS signals at intervals. We compared the kinetic curves of MPD-SPME and direct SPME. The two extraction kinetic curves of MG and CV are shown in Fig. 8. Under the conditions of direct SPME, it took 155 min to reach the extraction equilibrium of MG. For CV, it took 150 min to reach the extraction equilibrium. In our work, MPD-SERS can help to reach the extraction equilibrium within 1 min. By comparing MPD-SPME with direct SPME, MPD-SPME can greatly shorten the time required to reach the extraction equilibrium, which is conducive to rapid detection.
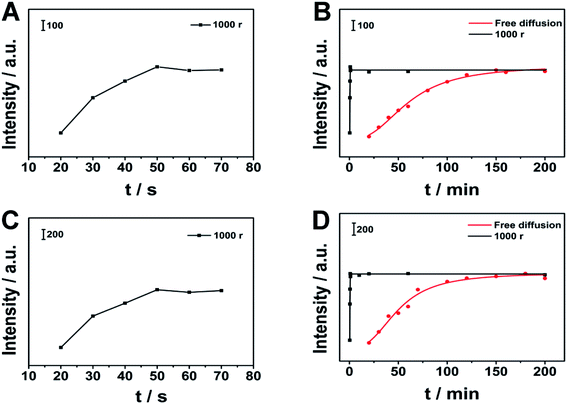 |
| | Fig. 8 Comparison of different extraction methods. (A), (C) are the kinetic curves of MG and CV through MPD-SPME, respectively, (B), (D) are comparison of kinetic curves of MPD-SPME and direct SPME for MG and CV, respectively. | |
Analysis MG and CV by MPD-SPME-SERS
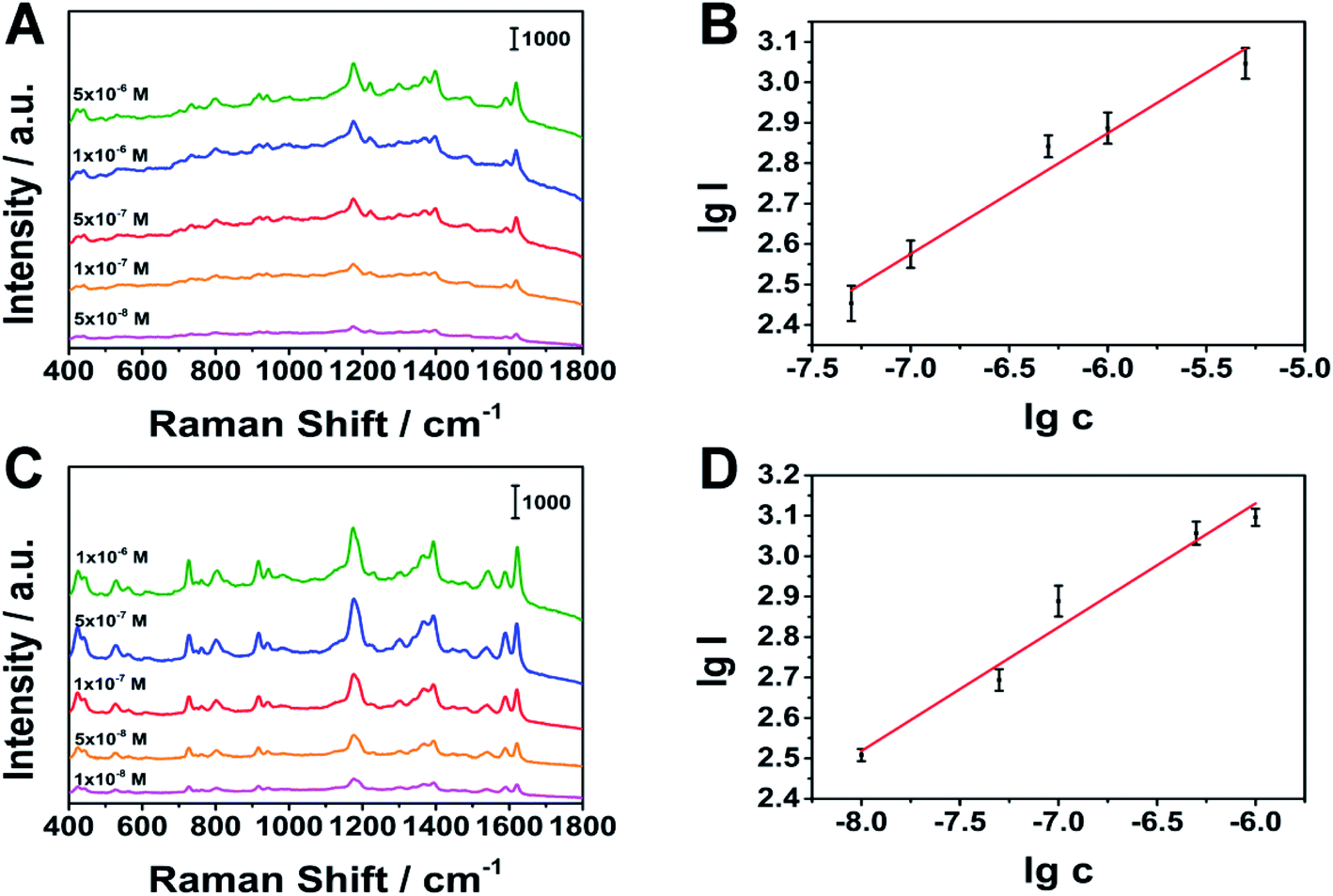
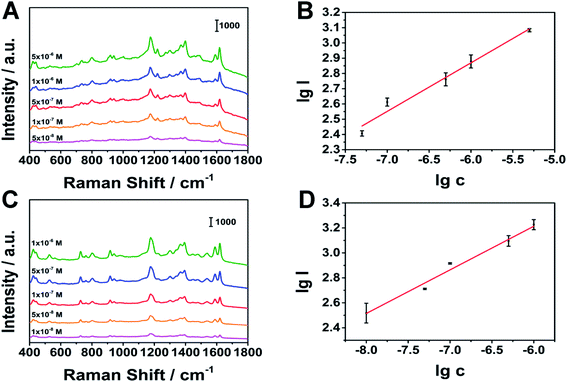
Under optimal stirring conditions, we conducted quantitative tests on MG and CV. As shown in Fig. 9, MG at 1174 cm−1 (linear range: 5 × 10−8 to 5 × 10−6 M), CV at 1177 cm−1 (linear range: 1 × 10−8 to 1 × 10−6 M), have a good linear relationship. The limits of detection (LOD) of MG and CV are 8.48 × 10−9 M, 1.38 × 10−9 M (S/N = 3), respectively. The results show that the porous silver wire prepared by us is expected to achieve rapid qualitative and quantitative detection in the combination of MPD-SPME and SERS.
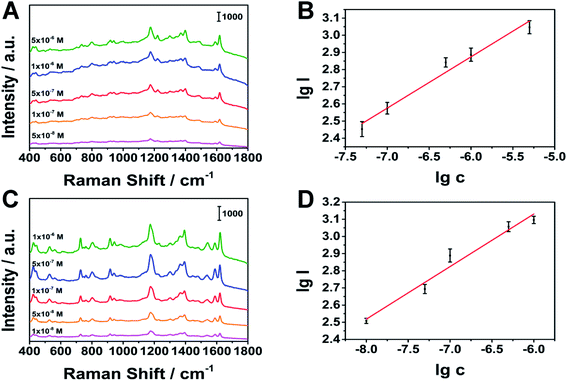 |
| | Fig. 9 Relationship between peak intensity and concentration. (A), (B) MG as the analyte, (C), (D) CV as the analyte, respectively. | |
Recyclability
We verified the recyclability of the substrate. NaBH4 was dissolved in a mixed solution of water and ethanol (water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol = 1:1, volume ratio) as a removal solution. As shown in Fig. 10, the SERS signals collected before and after the NaBH4 solution treatment were recorded separately. When the substrate was immersed in NaBH4 solution for 5 minutes, the characteristic peaks of MG disappeared; when the substrate was immersed for 15 minutes, the characteristic peaks of CV disappeared. The disappearance of the characteristic peaks indicates that the analyte molecule has been removed. After 5 cycles, the position and intensity of the main characteristic peaks did not change significantly. For MG and CV, after 5 cycles, the peak intensity became 70% and 85% of the initial peak intensity, respectively. According to the references, we guess that the mechanism may be that the interaction between Ag fiber and BH4− is strong. This effect is attributed to a decrease in electric potential of the Ag fiber surface on adding BH4− and this could induce desorption of MG and CV from the surface.34–36
:ethanol = 1:1, volume ratio) as a removal solution. As shown in Fig. 10, the SERS signals collected before and after the NaBH4 solution treatment were recorded separately. When the substrate was immersed in NaBH4 solution for 5 minutes, the characteristic peaks of MG disappeared; when the substrate was immersed for 15 minutes, the characteristic peaks of CV disappeared. The disappearance of the characteristic peaks indicates that the analyte molecule has been removed. After 5 cycles, the position and intensity of the main characteristic peaks did not change significantly. For MG and CV, after 5 cycles, the peak intensity became 70% and 85% of the initial peak intensity, respectively. According to the references, we guess that the mechanism may be that the interaction between Ag fiber and BH4− is strong. This effect is attributed to a decrease in electric potential of the Ag fiber surface on adding BH4− and this could induce desorption of MG and CV from the surface.34–36
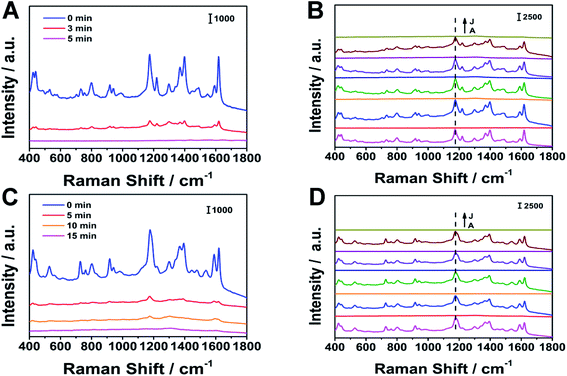 |
| | Fig. 10 The recyclability of the substrate with (A), (B) MG as the analyte and (C), (D) CV as the analyte, respectively. | |
Selectivity
We used nitrofurazone, thiram and atrazine as possible interfering substances in the experiment. In this experiment, we configured 5 × 10−6 M MG, CV, nitrofural, thiram and atrazine solutions, as well as MG and nitrofurazone, MG and thiram, MG and atrazine, CV and nitrofurazone, CV and thiram, CV and atrazine mixed solutions, the concentration of each solute in the mixed solutions was 5 × 10−6 M. When there was only one solute in the solution, as shown in Fig. 11, the characteristic peaks of thiram were observed, but the characteristic peaks of nitrofurazone and atrazine were not observed. In the mixed solutions, the presence of nitrofurazone, thiram and atrazine had little effect on the signals of MG at 1174 cm−1and CV at 1177 cm−1. The position and intensity of characteristic peaks in other positions of MG(439, 800, 1174, 1398 and 1592 cm−1) and CV (422, 528, 760, 803, 916, 1394, 1443, 1536, 1590 and 1621 cm−1)were almost unaffected. This shows that our substrate has good selectivity.
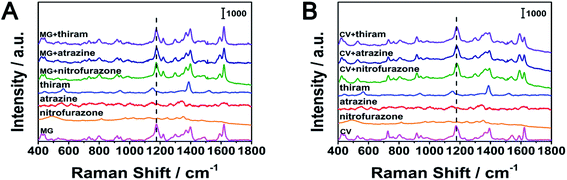 |
| | Fig. 11 The selectivity of the silver fiber. (A) MG as the target analyte (B) CV as the target analyte, respectively. | |
Analysis actual samples by MPD-SPME-SERS
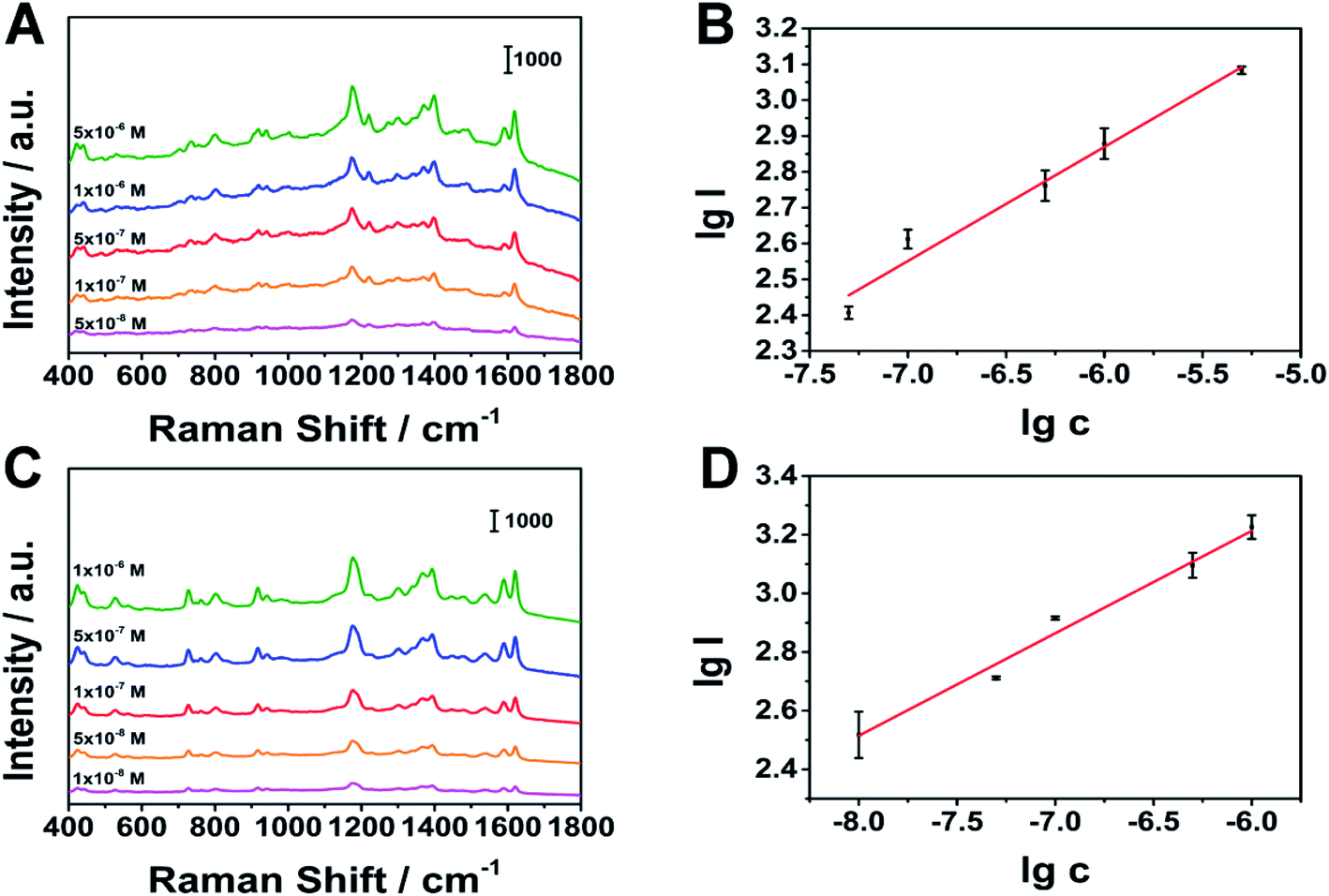
We selected aquaculture water on the market to prepare actual water samples with different concentrations of MG and CV. In actual sample detection, we added malachite green and crystal violet to the aquaculture water to prepare different concentrations of malachite green solutions and different concentrations of crystal violet solutions. Then we took 40 mL of aquaculture water solution in a 100 mL beaker, placed Ag fiber 1.5 cm from the center of the beaker, and placed a stir bar at the bottom of the beaker. We chose the stirring speed of 1000 rpm and the stirring time of 40 s. Finally, we took out the Ag fiber and collected SERS signals. In the actual sample detection, as shown in Fig. 12, there are also good linear relationships. The linear ranges of MG and CV are: 5 × 10−8 to 5 × 10−6 M, 1 × 10−8 to 1 × 10−6 M. The limits of detection are: 1.29 × 10−8 M, 2.11 × 10−9 M. The results show that we can quickly and qualitatively detect the analyte in the actual sample by stirring (Table 1).
 |
| | Fig. 12 SERS spectra of actual samples. (A), (B) SERS spectra of MG in aquaculture water, (C), (D) SERS spectra of CV in aquaculture water, respectively. | |
Table 1 Quantitative analysis results obtained by SERS detection with MPD-SPME
| |
Malachite green (MG) |
Crystal violet (CV) |
| Linear equation |
y = 0.3157x + 4.762 |
y = 0.3571x + 5.358 |
| Linear range/M |
5 × 10−8 to 5 × 10−6 |
1 × 10−8 to 1 × 10−6 |
| LOD/M |
1.27 × 10−8 |
2.24 × 10−9 |
Conclusions
We prepared porous silver fiber by electrochemical etching. The obtained porous silver wire has good enhancement effect, uniformity, stability and recyclability. We accelerated the extraction process by MPD-SPME, and combined with SERS to detect MG and CV. By this method, it is expected to complete the entire analysis and detection process within 1 minute. In summary, the method in this work is expected to play an important role in the fields of rapid food safety detection and environmental monitoring in the future.
Conflicts of interest
There are no conflicts to declare.
References
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS
 .
. - S. Zhu, X. L. Zhang, J. C. Cui, Y. E. Shi, X. H. Jiang, Z. Liu and J. H. Zhan, Analyst, 2015, 140, 2815–2822 RSC .
- S. L. Zhang, Z. Du and G. K. Li, Anal. Chem., 2011, 83, 7531–7541 CrossRef CAS PubMed .
- E. A. Souza-Silva, E. Gionfriddo and J. Pawliszyn, TrAC, Trends Anal. Chem., 2015, 71, 236–248 CrossRef CAS .
- S. Yu, Z. Liu, H. Li, J. Zhang, X. X. Yuan, X. Jia and Y. Wu, Analyst, 2018, 143, 883–890 RSC .
- B. Bojko and J. Pawliszyn, Bioanalysis, 2012, 4, 1263–1265 CrossRef CAS PubMed .
- B. J. Goncalves Silva, R. H. Costa Queiroz and M. E. Costa Queiroz, J. Anal. Toxicol., 2007, 31, 313–320 CrossRef PubMed .
- L. F. da Silva, D. A. Vargas Medina and F. M. Lancas, Talanta, 2021, 221, 121608 CrossRef CAS .
- K. Mejia-Carmona and F. M. Lancas, J. Chromatogr. A, 2020, 1621, 9 CrossRef PubMed .
- F. Saliu, S. Montano, B. W. Hoeksema, M. Lasagni and G. Galli, Anal. Methods, 2020, 12, 1935–1942 RSC .
- P. Rubiolo, F. Belliardo, C. Cordero, E. Liberto, B. Sgorbini and C. Bicchi, Phytochem. Anal., 2006, 17, 217–225 CrossRef CAS PubMed .
- J. Fuller, D. White, H. Yi, J. Colley, Z. Vickery and S. Liu, Chemosphere, 2020, 260 Search PubMed .
- H. Ghosson, D. Raviglione, M.-V. Salvia and C. Bertrand, Anal. Chim. Acta, 2020, 1134, 58–74 CrossRef CAS PubMed .
- Y. Jian, L. Chen, J. Cheng, X. Huang, L. Yan and H. Li, Anal. Chim. Acta, 2020, 1133, 1–10 CrossRef CAS PubMed .
- H. H. Jelen, S. Mildner-Szkudlarz, I. Jasinska and E. Wasowicz, J. Am. Oil Chem. Soc., 2007, 84, 509–517 CrossRef CAS .
- J. H. Kennedy, C. Aurand, R. Shirey, B. C. Laughlin and J. M. Wiseman, Anal. Chem., 2010, 82, 7502–7508 CrossRef CAS PubMed .
- L. Sun, M. Zhang, V. Natarajan, X. Yu, X. Zhang and J. Zhan, RSC Adv., 2017, 7, 23866–23874 RSC .
- B. Li, Y. E. Shi, J. Cui, Z. Liu, X. Zhang and J. Zhan, Anal. Chim. Acta, 2016, 923, 66–73 CrossRef CAS .
- S. H. Yu, Z. G. Liu, W. X. Wang, L. Jin, W. Q. Xu and Y. Q. Wu, Talanta, 2018, 178, 498–506 CrossRef CAS .
- C. C. Liu, X. L. Zhang, L. M. Li, J. C. Cui, Y. E. Shi, L. Wang and J. H. Zhan, Analyst, 2015, 140, 4668–4675 RSC .
- L. Qiu, Q. Liu, X. Zeng, Q. Liu, X. Hou, Y. Tian and L. Wu, Talanta, 2018, 187, 13–18 CrossRef CAS PubMed .
- H. X. Chen, C. R. Wang, Z. Y. Zhang and L. L. He, J. Food Prot., 2018, 81, 1087–1092 CrossRef CAS PubMed .
- H. Sereshti, F. Karami, N. Nouri and A. Farahani, J. Sci. Food Agric., 2021, 101, 2304–2311 CrossRef CAS PubMed .
- Y. T. Li, L. L. Qu, D. W. Li, Q. X. Song, F. Fathi and Y. T. Long, Biosens. Bioelectron., 2013, 43, 94–100 CrossRef CAS PubMed .
- C. Wu, Solid phase microextraction, 2012 Search PubMed .
- G. Hota, S. B. Idage and K. C. Khilar, Colloids Surf., A, 2007, 293, 5–12 CrossRef CAS .
- A. Nigam and S. Kala, AIP Conf. Proc., 2020, 2220, 020116 CrossRef CAS .
- R. Kumar, J. Rashid and M. A. Barakat, Colloids Interface Sci. Commun., 2015, 5, 1–4 CrossRef CAS .
- Y. Y. Zhang, K. Q. Lai, J. L. Zhou, X. C. Wang, B. A. Rasco and Y. Q. Huang, J. Raman Spectrosc., 2012, 43, 1208–1213 CrossRef CAS .
- L. L. He, N. J. Kim, H. Li, Z. Q. Hu and M. S. Lin, J. Agric. Food Chem., 2008, 56, 9843–9847 CrossRef CAS PubMed .
- E. B. E. C. Le Ru, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794–13803 CrossRef CAS .
- E. J. Liang, X. L. Ye and W. Kiefer, J. Phys. Chem. A, 1997, 101, 7330–7335 CrossRef CAS .
- K. R. Strehle, D. Cialla, P. Rosch, T. Henkel, M. Kohler and J. Popp, Anal. Chem., 2007, 79, 1542–1547 CrossRef CAS PubMed .
- S. M. Ansar, F. S. Ameer, W. Hu, S. Zou, C. U. Pittman Jr and D. Zhang, Nano Lett., 2013, 13, 1226–1229 CrossRef CAS PubMed .
- S. S. n.-C. s. a. J. V. Garca-Ramos, J. Raman Spectrosc., 1998, 29, 365–371 CrossRef .
- G. S. Perera, S. M. Ansar, S. Hu, M. Chen, S. Zou, C. U. Pittman and D. Zhang, J. Phys. Chem. C, 2014, 118, 10509–10518 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10227j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*