
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharged ultrafiltration membranes based on TEMPO-oxidized cellulose nanofibrils/poly(vinyl alcohol) antifouling coating†
Andrea Aguilar-Sanchez a,
Blanca Jalvoa,
Andreas Mautnerb,
Ville Rissanenc,
Katri S. Kontturic,
Hani Nasser Abdelhamida,
Tekla Tammelinc and
Aji P. Mathew*a
a,
Blanca Jalvoa,
Andreas Mautnerb,
Ville Rissanenc,
Katri S. Kontturic,
Hani Nasser Abdelhamida,
Tekla Tammelinc and
Aji P. Mathew*a
aDivision of Materials and Environmental Chemistry, Stockholm University, Frescativägen 8, 10691, Stockholm, Sweden. E-mail: aji.mathew@mmk.su.se
bPolymer and Composite Engineering (PaCE) Group, Institute of Materials Chemistry and Research, Faculty of Chemistry, University of Vienna, Währinger Str. 42, 1090 Wien, Austria
cVTT Technical Research Centre of Finland, Solutions for Natural Resources and Environment, P. O. Box 1000, FI-02044 VTT, Finland
First published on 10th February 2021
Abstract
This study reports the potential of TEMPO-oxidized cellulose nanofibrils (T-CNF)/poly(vinyl alcohol) (PVA) coatings to develop functionalized membranes in the ultrafiltration regime with outstanding antifouling performance and dimensional/pH stability. PVA acts as an anchoring phase interacting with the polyethersulfone (PES) substrate and stabilizing for the hygroscopic T-CNF via crosslinking. The T-CNF/PVA coated PES membranes showed a nano-textured surface, a change in the surface charge, and improved mechanical properties compared to the original PES substrate. A low reduction (4%) in permeance was observed for the coated membranes, attributable to the nanometric coating thickness, surface charge, and hydrophilic nature of the coated layer. The coated membranes exhibited charge specific adsorption driven by electrostatic interaction combined with rejection due to size exclusion (MWCO 530 kDa that correspond to a size of ∼35–40 nm). Furthermore, a significant reduction in organic fouling and biofouling was found for T-CNF/PVA coated membranes when exposed to BSA and E. coli. The results demonstrate the potential of simple modifications using nanocellulose to manipulate the pore structure and surface chemistry of commercially available membranes without compromising on permeability and mechanical stability.
Introduction
Separation using membranes is a common practice on a wide range of feed streams including ions, molecules, and colloids. Membranes for the microfiltration (MF) regime are widely used to retain several micrometers sized particles; while the ultrafiltration (UF) regime is the second-largest pore size type of membranes, separating particles and macromolecules from a fluid feed in the range of 1 and 100 nm.1 By modifying MF membranes to the UF range, size exclusion starts affecting the separation mechanism of the membrane, where small species can pass through and large ones are retained.2 It was shown by Mehta and Zydney,3 through experimental and theoretical methods, that charged UF membranes show a combination of separation and permeability enhancement, similar to what would be achieved with a two-fold reduction in membrane skin thickness in uncharged membranes, but without concerns about protein diffusion, mechanical stability, or defects. Furthermore, electrically charged membranes are a unique class of membranes due to the anionic or cationic group attached to the surface, which provide reduced fouling and separation through rejection of like-charged species or adsorption of oppositely charged species.4 Casting of polymers with appropriate anionic (e.g., carboxylic or sulfonic acid) or cationic (e.g., quaternary amine) groups on various substrates is therefore used to fabricate membranes with increased selectivity towards charged species.3,5,6 Charged membranes are also known to significantly improve the trade-off between selectivity and permeability in the ultrafiltration regime.3Nanocellulose is increasingly being exploited in recent years as a versatile biobased option in charged membrane applications, owing to the nanoscale morphology and surface chemistry, the ability to produce anionic or cationic charged nanometric layers, a high specific surface area (as high as 324 m2 g−1 in the case of TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl radical)-oxidized cellulose nanofibrils (T-CNF))5 and a large number of active sites for selective interaction with charged entities.7–10 Nanocellulose membranes facilitate rejection through mechanisms involving (i) adsorption on the nanoscaled, charged surfaces (ii) size-exclusion filtration by virtue of the nanoscaled porous networks and (iii) a combination of both methods11,12 relying on the principles similar to that of charged membranes. Porous substrates such as paper,13 electrospun mats,14 and commercially available microfiltration membranes15 are successfully used as substrates for nanocellulose-based membrane. Ma et al. demonstrated the potential of polysaccharide nanofibers, including T-CNF for high flux, low-pressure water purification as a barrier layer on polyacrylonitrile (PAN) and polyethylene terephthalate (PET) nonwoven membrane.7 It was shown that an ultrathin nanocellulose layer improves the permeability of membranes due to the nanoscaled porosity of the nanocellulose network and its hydrophilicity that creates water channels throughout the membrane.16 Karim et al. have shown that rejection of charged pollutants as metal ions is typically initiated by electrostatic interaction which switches to size exclusion due to cluster formation.11
While nanocellulose based membranes have several advantages, they are prone to swelling in an aqueous medium and have poor mechanical stability in humid environments due to their intrinsic hygroscopicity.17 Moreover, the nanocellulose layer thickness is crucial in controlling water flux through the membranes. Ultrathin nanocellulose layers on porous substrates are known to provide an active functional layer with water permeability within the ultra to nanofiltration range. UF membranes are categorized by their nominal molecular weight cut-off, which is typically defined as the molecular weight of a solute rejected 90%. Membranes in the UF regime are one of the most common types of separation membranes used and in water treatment, pharmaceutical industry, food, and beverage production, etc. Bai et al. have shown earlier that cellulose nanocrystals have significant advantages over cellulose nanofibers regarding antifouling properties.18 Nevertheless, later studies showed that T-CNF provides not only antifouling properties but also antibacterial properties compared to cellulose nanocrystals.19
The aim of this work was to develop a scalable method to coat polyethersulfone (PES) MF membranes having an average pore size of 0.2 μm with nanocellulose and produce a UF membrane. In the current study poly(vinyl alcohol) (PVA), an inexpensive water-soluble polymer, abundant in hydroxyl groups,20 biodegradable and biocompatible,21–23 was mixed with a aqueous dispersion of T-CNF to develop coating formulations that provide stability17 and adhesion to the substrate. It is well established that PVA can be irreversibly adsorbed by hydrophobic surfaces, such as PES membranes, by hydrophobic interactions.24,25 Furthermore, adequate crosslinking of PVA systems can avoid swelling and dissolution in an aqueous medium.26 Previous studies have also shown that crosslinking of PVA with cellulose improves selectivity and stability of membranes.20 We evaluated the potential of T-CNF/PVA coating as a simple and green route to modify commercial PES membranes to achieve enhanced membrane performance, mechanical properties, and antifouling properties while maintaining high permeance.
Experimental section
Materials
TEMPO-oxidized cellulose nanofibrils from softwood pulp (T-CNF, 1.4 wt%, 1.1 mmol g−1 of carboxyl groups), prepared following the procedure reported by Isogai,27 were provided by Swiss Federal Laboratories for Materials Science and Technology (EMPA, Switzerland). The diameter of the nanofibrils was in the range of 3–5 nm based on AFM data (see ESI Fig. S1†). Polyethersulfone (PES) asymmetric membranes, average pore size 0.2 μm, were given by Sartorius Stedim Biotech (Germany). Poly(vinyl alcohol) (PVA, molecular weight 72 kDa, degree of hydrolysis 85–89%) was purchased from AppliChem (Germany). Glutaraldehyde (GA) solution (50 wt% in water), victoria blue B (VB), bovine serum albumin (BSA) lyophilized powder ≥ 96% with a molecular weight (Mw) of 66 kDa (40 × 140 Å), and phosphate-buffered saline (PBS) at pH 7.2 were purchased from Sigma Aldrich (Sweden). Methyl orange (MO) was purchased from Alfa Aesar (Germany). Hydrochloric acid (HCl) fuming 37% was purchased from AnalaR NORMAPUR (Austria). Branched polyethyleneimine (PEI), with a Mw of 50–100 kDa, was purchased from Polysciences Inc (Germany). Dextran 50 kDa and 150 kDa was purchased from Sigma (Austria) and poly(ethylene glycol) (PEG, 634 kDa) from PolymerLabs (Austria). AT-cut quartz crystal sensors with gold electrodes were purchased from Q-sense AB (Gothenburg, Sweden), with a fundamental resonance frequency of 5 MHz and a sensitivity constant of 0.177 mg m−2 Hz−1 as reported by the supplier.Processing methods
Nanocellulose suspension (T-CNF 1 wt%) was stirred for 24 h and sonicated for 10 min at 75% amplitude. PVA powder was added to cellulose suspension, in order to reach a suspension at 0.5 wt% PVA. The suspension was stirred and heated until it reached a temperature of 60 °C. It was cast coated on the PES membrane at a speed of 5 mm s−1 and a gap distance of 100 μm using an automatic film applicator (Elcometer 4340 motorized). The membrane was immersed in acidic GA solution (5 wt%, pH 2 as set by 1 N HCl) for 1 s and then dried at 70 °C for 20 m. Finally, the membrane was rinsed with de-ionized (DI) water to remove un-reacted GA.Characterization
The membrane surface roughness and morphology were studied using atomic force microscopy (AFM, Multimode Nanoscope V, Bruker) in tapping mode. The PES substrate and T-CNF/PVA coated membranes were attached to metal pugs with double-sided tape and probed with standard TESPA-V2 silicon nitride tips (Bruker, USA) with the spring constant k = 40 N m−1. The collected data were processed with NanoScope Analysis 1.5 (Bruker) software. The roughness of the membranes was measured from the height images at 5 different sections of the membrane, each section with an average area of 0.3 μm2.
The surface chemistry of uncoated and coated substrates was characterized by X-ray photoelectron spectroscopy (XPS). The analysis was performed using a Nexsa XPS system (Thermo-Fisher) with a radiation source gun-type Al Kα operating at 72 W and a pass energy of 200 eV, a spot size of 400 μm, “Standard Lens Mode”, CAE Analyser Mode, an energy step size of 0.1 eV for the survey spectrum, and integrated flood gun. The analysis was done after cleaning the surface with Ar-clusters (1000 atoms, 6000 eV, 1 mm raster size) for 60 s. The high-resolution C 1s spectrum was acquired with 10 passes at pass energy of 50 eV and fitted using Thermo Avantage v5.9914, Build 06617 with Smart background, and Simplex Fitting algorithm using Gauss–Lorentz Product.
The wettability of the uncoated and coated substrates was measured by sessile drop technique. An optical contact angle meter from KSV instruments model CAM 200, equipped with a Basler A602f camera was used. Measurements were performed three times for each type of sample, taking the contact angle value at 0.3 s after the droplet was deposited on the surface. The environmental conditions were controlled, with a temperature of 23 °C and relative humidity (R.H.) of 40 ± 5%.
The surface charge of the membranes was studied by measuring the ζ-potential as a function of pH using a SurPASS electrokinetic analyzer (Anton Paar, Graz, Austria). The specimens were characterized in an adjustable gap cell at a gap width of 130 μm. As an electrolyte, a solution of KCl (1 mmol L−1), was used and the pH value controlled using HCl and KOH (0.05 mol L−1). The ζ-potential was determined from the measured streaming current.
The molecular weight cut off (MWCO) was studied using a feed solution consisting of dextran (50 kDa and 150 kDa) and PEG, (634 kDa) polymer standards to evaluate the rejection of polymer molecules as a function of their molecular weights. The standard solution was filtered through the membranes in a dead-end cell at 0.2 bar (see the schematic representation of the experimental set-up in Fig. S2a†). Gel permeation chromatography (Viscotek TDA302) was used to analyze the fraction of each molecular weight in the filtrate passing the membrane.
Water permeance of the membranes was measured by filtering deionized water using a Convergence Inspector Titan equipment with a constant pressure of 0.2 bar at 20 °C for one hour in dead-end mode. Schematic representation of the set-up is shown in Fig. S2b.† The active filtration area of the membrane was 0.004 m2. The permeate flux data was used for permeability determination.
The mechanical properties of the membranes were characterized using an Instron 5966 Dual Column Tabletop Testing System equipped with a 100 N load cell. Specimens were prepared by cutting rectangular strips of 10 mm × 100 mm from the membranes. Prior to the test, the specimens were conditioned for 40 h at 50 ± 5% R.H. and 23 ± 2 °C. The tensile test was performed at a speed of 25 mm min−1 until failure.
Tensile tests were also performed with the same parameter set under wet conditions using a BioPuls temperature-controlled bath to simulate real usage conditions. Samples were pre-conditioned by placing them underwater, held by the grips for 5 min before starting the test. The temperature of the water-bath was kept at 25 ± 2 °C.
Adsorption/rejection of charged dyes by the membranes was studied to understand the effect of electrostatic interaction on the membrane rejection performance. Uncoated and coated membranes were submerged in a Petri dish containing 2 mg L−1 solution of VB (positive charge at pH 5.6) or MO (negative charge at pH 5.8) for 6 h. Afterward, the dye solution in which the membranes were submerged was analyzed using a UV-vis spectrophotometer (Genesys 150) at a wavelength of 450 nm for MO and 610 nm for VB.
Quartz crystal microbalance with dissipation monitoring (QCM-D E4, Q-sense AB) was used to have a direct assessment of BSA adsorption on model surfaces based on crosslinked T-CNF and T-CNF/PVA based on the procedure reported by Hakalahti et al.28 Briefly, gold sensor crystals were rinsed with MilliQ-water, dried with nitrogen gas, and cleaned with UV/ozone treatment for 10 min. PEI, acting as a cationic anchoring polymer, was allowed to adsorb from 0.1% aqueous solution for 30 min on the surface of the sensor crystal. Subsequently, the crystals were washed with Milli-Q water and dried with nitrogen gas. 200 μL of T-CNF suspension at 0.15 wt% consistency or 0.15 wt% T-CNF/0.075 wt% PVA were spin-coated (WS-400 BZ-6NPP-Lite, Laurell Technologies Corporation, 3000 rpm, 1.5 min) on the sensor. Separately, the pH of a 0.75 wt% GA solution was adjusted to 2 by dropwise addition of 1 N HCl. Afterward, a droplet of this GA solution was placed on the surface and later washed with MilliQ-water. The crystals were dried in an oven (20 min, 70 °C). The crosslinked T-CNF and T-CNF/PVA coated crystals were kept in 0.01 M PBS solution overnight before the adsorption experiments in the QCM-D measurement cell. During the measurements, a continuous flow (0.1 mL min−1) of the PBS (0.01 M) solution was led on the crystal in the QCM-D cell for 30 min. The solution was exchanged to BSA (0.1 wt%) in PBS (0.01 M), and the change in frequency and dissipation was followed for 1 h. Finally, the solution was exchanged back to PBS (0.01 M) solution and rinsed for 30 min. The cell temperature was 37 °C. Data presented was acquired using the fifth overtone (25 MHz, f0 = fundamental resonance frequency = 5 MHz, n = overtone number = 5).
In order to complement the QCM-D analysis, further adsorption of BSA (1 mg mL−1) was performed on PES and T-CNF/PVA coated membranes in PBS (0.01 M, pH 7.2) as a buffer solution. The membranes were equilibrated with PBS overnight at 37 °C and then exposed to a continuous flux of BSA solution for 6 h at 37 °C.
After that, the membranes were rinsed three times with PBS and the adsorbed proteins recovered from the surface by immersing the membranes in dodecyl sulfate (SDS, 1 wt%) for 1 h at 37 °C under slight shaking conditions (200–300 rpm). The SDS solution containing the recovered proteins from the membranes was analyzed using a UV-vis spectrophotometer (Genesys 150) at a wavelength of 280 nm.
For the biofilm formation assessment, exponentially growing cultures of Escherichia coli ATCC 8739 on Nutrient Broth were diluted to an optical density of 0.0138 at a wavelength of 600 nm (OD600), equivalent to 108 cells per mL, and 2 mL of diluted cultures were placed on the surface of the membranes, which were subsequently incubated on polystyrene 24-well plates for 18 h at 37 °C without stirring. Afterward, the membranes were carefully washed with distilled water to remove not adhered cells.
The adhesion of the bacterial cells on the surface of the membranes was visualized by SEM. Cell fixation was carried out in glutaraldehyde 5% (v/v) in 0.2 M sodium cacodylate buffer (pH 7.2) for 1 h at room temperature. Samples were then rinsed in cacodylate buffer and dehydrated in an ascending ethanol series (25%, 50%, 70%, 90%, and 100%) before critical point drying with CO2 and subsequent observation with SEM.
Results and discussion
PES surface functionalization by coating
Fig. 1 shows the schematic representation of the coating process adopted for the surface functionalizing of a PES substrate in the MF regime (0.2 μm). PVA was added to facilitate the interfacial adhesion between hydrophobic PES and hydrophilic T-CNF24,25 and also to crosslink T-CNF. Previous studies suggest that the carboxyl groups in T-CNF can react with the alcohol groups from hydrolyzed PVA to form ester bonds.17 To further improve the stability, mechanical properties and provide insolubility of PVA and T-CNF in aqueous media, GA was used as an additional chemical cross-linker.23,29 The crosslinking generates intramolecular and intermolecular interactions between the hydroxyl groups of PVA and T-CNF, and the aldehyde groups of GA, in presence of strong acids, such as HCl, that catalyzes the reaction20,23,26,30,31 and can further drive the esterification reaction between T-CNF and PVA17 (Fig. 1). It is expected that these ester bonds, the intra- and intermolecular H-bonding between PVA and T-CNF,17 as well as the ability of PVA to interact with hydrophobic polymers, such as PES, by lowering the interfacial free energy, will enhance the stability of the coating.24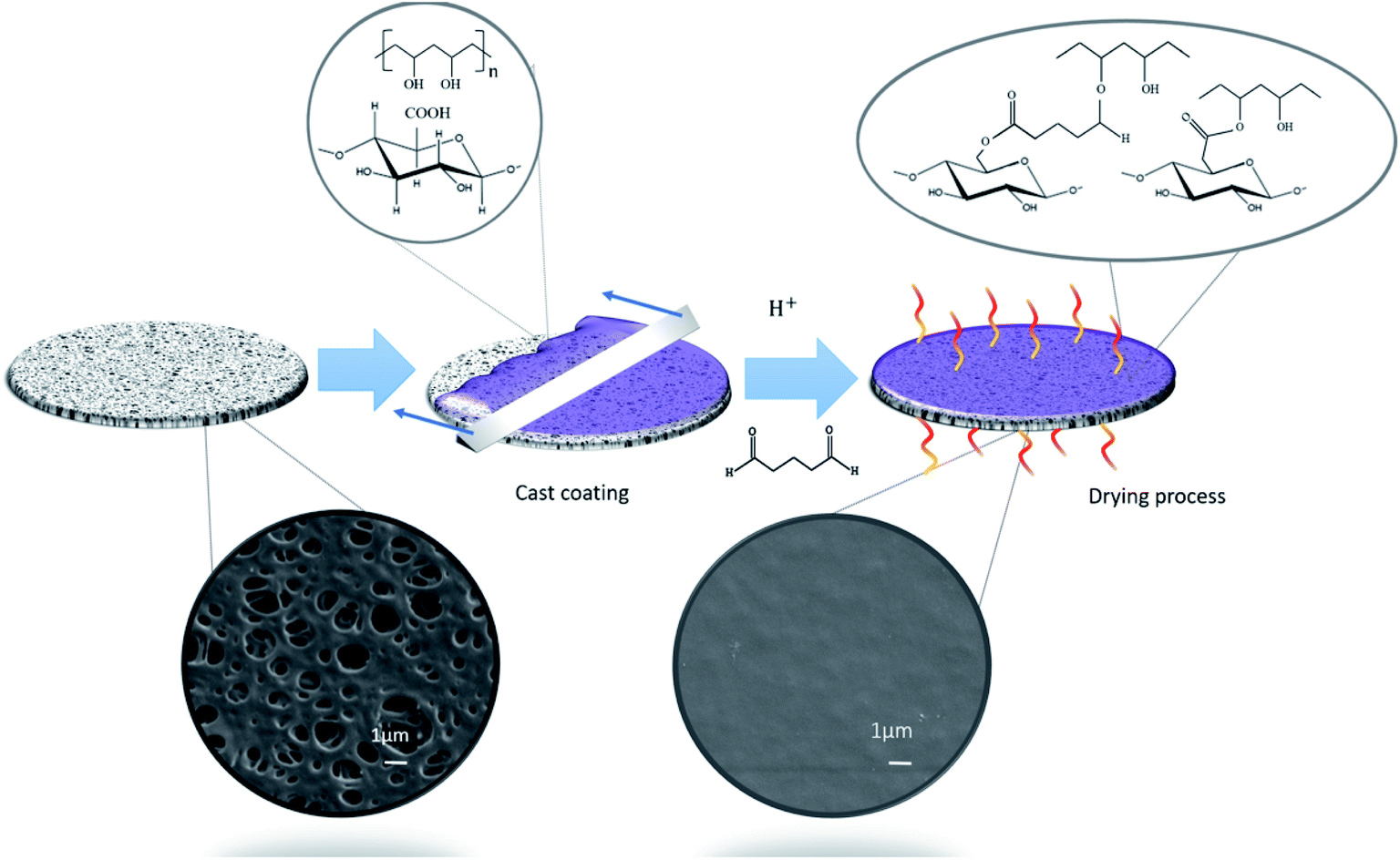 | ||
| Fig. 1 Schematic illustration of the surface modification method of the commercial PES membranes by coating. | ||
Morphology and surface chemistry of the coated membranes
The morphology of the membranes was studied using SEM, from which it was possible to confirm that the T-CNF/PVA layer was deposited homogeneously (Fig. 1) on top of the porous PES substrate. The coating layer thickness was measured from the SEM images to be about 0.47 ± 0.06 μm. (It was also noted that the coated layer remained intact after 24 h of continuous water flux, see ESI Fig. S3a–c†).The AFM topography of the original membrane was compared to the coated membranes and morphology in Fig. 2. The average surface roughness were evaluated from the height images. AFM images also confirmed the microscale porosity of the substrate and the uniform, nanotextured, thin T-CNF/PVA layer on the substrate (Fig. 2a and b). The measured surface roughness for the uncoated membrane was 30.5 ± 13.8 nm, while for the coated membrane it was reduced to 10.8 ± 4.2 nm (Table 1). Higher surface roughness is related to increased surface energy and higher specific surface area, which is expected to increase the adhesion of molecules on the membrane surface.32,33 The decreased surface roughness after coating thus should potentially inhibit surface fouling.
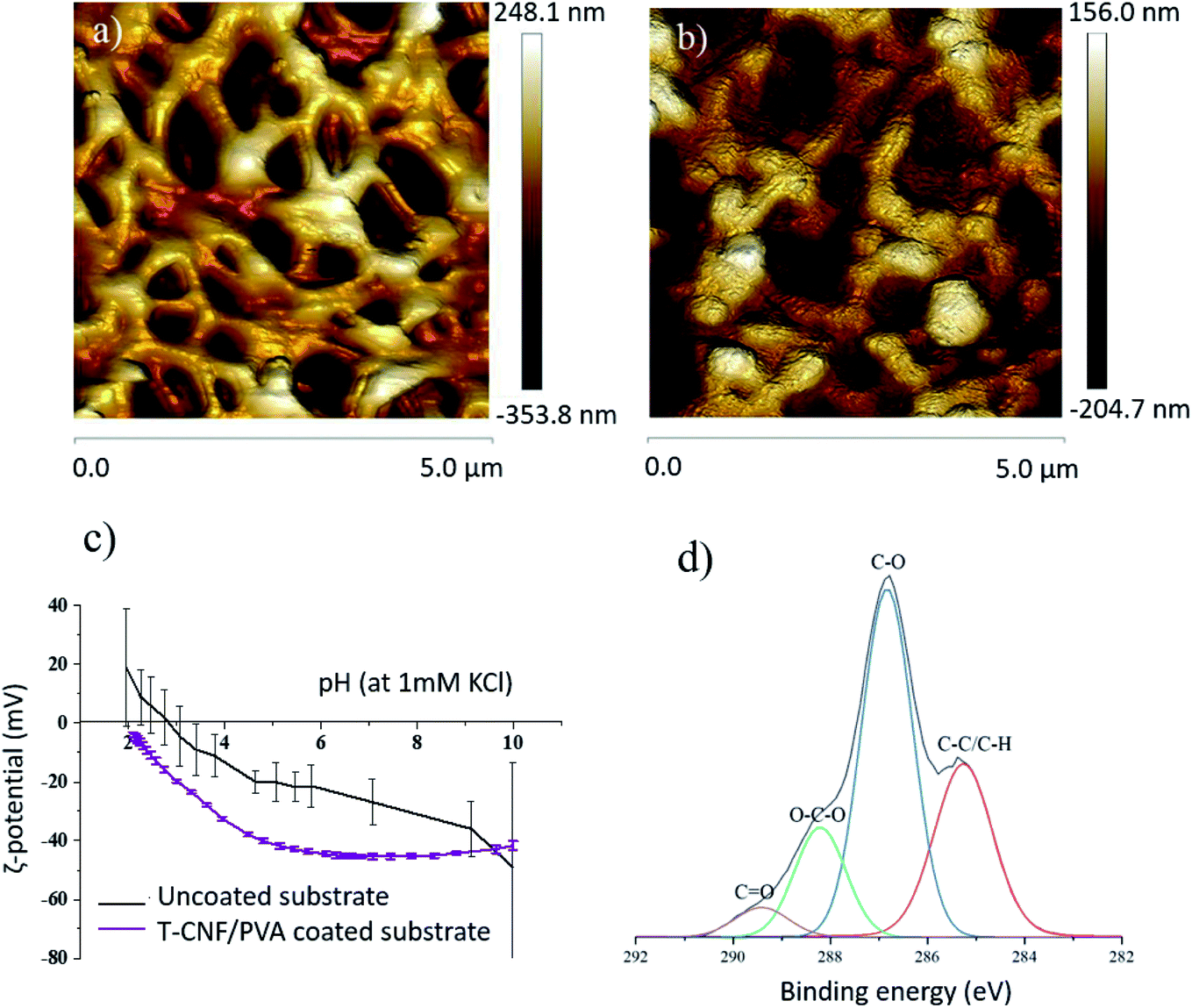 | ||
| Fig. 2 Two dimensional (2D) AFM height images for (a) uncoated PES substrate (b) T-CNF/PVA coated membrane (c) ζ-potential of coated and uncoated membrane in the pH range of 2–10 and (d) deconvoluted C 1s high resolution XPS spectra for the TCNF/PVA coated membrane. | ||
| Sample | Surface roughness Rq, (nm) | Contact angle (°) | Permeance (L m−2 h−1 bar−1) |
|---|---|---|---|
| Uncoated | 30.5 ± 13.8 | 48.8 ± 5.8 | 3230 ± 170 |
| T-CNF/PVA membrane | 10.8 ± 4.2 | 48.6 ± 9.4 | 3110 ± 90 |
The T-CNF/PVA coating had a clear impact on the surface charge of the membranes (Fig. 2c). Over the whole pH range analyzed (pH 2–10), the ζ-potential was lower for the coated membrane compared to the uncoated membrane. The ζ-potential plateau at pH > 5 was between −40 and −45 mV for the coated membrane, reduced from about −20 mV at pH 5 for the uncoated membrane. This indicates the presence of an abundance of negatively charged hydroxyl and aldehyde groups, on the surface of TCNF/PVA membranes. Furthermore, no isoelectric point (IEP) was detected for the coated membranes within the pH range tested, since the functional groups attached have an IEP below pH 2; the IEP of the uncoated membrane was at pH 2.9. This confirmed the presence of the negative surface groups, demonstrating that the surface deposition was done successfully. The surface ζ-potential values were lower for the coated membrane compared to the uncoated membrane indicating a stable coated layer in the whole pH range. These membranes can therefore be successfully used in applications involving a wide pH range.
In addition, the coated membranes were characterized by XPS to analyze the local bonding environment in the functional groups of the different membrane surfaces. Fig. 2d shows the deconvoluted C 1s high-resolution XPS spectra of the T-CNF/PVA coated membrane with a peak at 285.2 eV corresponding to C–C bonds and a second peak at 286.8 eV corresponding to C–O. A peak in 288.2 eV corresponding to O–C–O bonds and a small population of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 289.4 eV were present. Moreover, no peak attributed to –COOH group was found at 290.5 eV19 in the T-CNF/PVA coating which confirms that there are linkages between the reactive C-6 of T-CNF and the PVA and/or glutaraldehyde. Previous studies suggest that the carboxyl groups of T-CNF react with the alcohol groups of PVA, forming covalent ester bonds during the crosslinking.17
O at 289.4 eV were present. Moreover, no peak attributed to –COOH group was found at 290.5 eV19 in the T-CNF/PVA coating which confirms that there are linkages between the reactive C-6 of T-CNF and the PVA and/or glutaraldehyde. Previous studies suggest that the carboxyl groups of T-CNF react with the alcohol groups of PVA, forming covalent ester bonds during the crosslinking.17
The surface modification changed the surface charge but did not have an impact on wettability. The PES substrate had a contact angle of 48.8 ± 5.8° which remained stable after the coating (48.6 ± 9.4°, Table 1). This data agrees with the value reported by Hou et al. for glutaraldehyde crosslinked PVA with regenerated cellulose,20 where the hydroxyl groups of the cellulose are crosslinked with the aldehyde groups of GA and also form hydrogen bonds with the –OH groups in PVA.20
Membrane properties
PES substrates, in microfiltration regime (with a pore size of 0.2 μm) (SEM image in Fig. 1) were converted into ultrafiltration membranes after coating. N2 gas sorption analysis showed DFT pore size in the micro to macropore regime (2.6–74.4 nm), confirming the ultrafiltration regime of the surface layer (see ESI S4† for adsorption–desorption curves). The data analysis of T-CNF/PVA coated membrane shows BET specific surface area (SBET), Langmuir specific surface area (SLang), external surface area (SExt), and total pore volume (Vtotal) of 10 m2 g−1, 12 m2 g−1, 18 m2 g−1, and 0.022 cm3 g−1, respectively.The water flux through the membranes indicated that the nanoscale coating layer did not significantly affect the permeance performance of the PES substrate (Table 1). The permeance of the uncoated membrane in crossflow mode was 3230 ± 170 L m−2 h−1 bar−1, while for the coated one it was 3110 ± 90 L m−2 h−1 bar−1, which corresponds to a reduction in flux by only 4%. This insignificant reduction in permeance was attributed to the nanometer range thickness of the coating layer, the hydrophilic nature of the coated layer that promotes affinity with water, and the high porosity of the substrate.
MWCO of the uncoated and coated membranes are shown in Fig. 3a. The coated membranes exhibited improved rejection with 90% of 530 kDa macromolecules, corresponding to a particle size of 30 nm, confirming the membrane performance in the ultrafiltration region. This improved rejection performance without a significant decrease in permeability is expected to significantly impact the UF membrane application of these membranes.
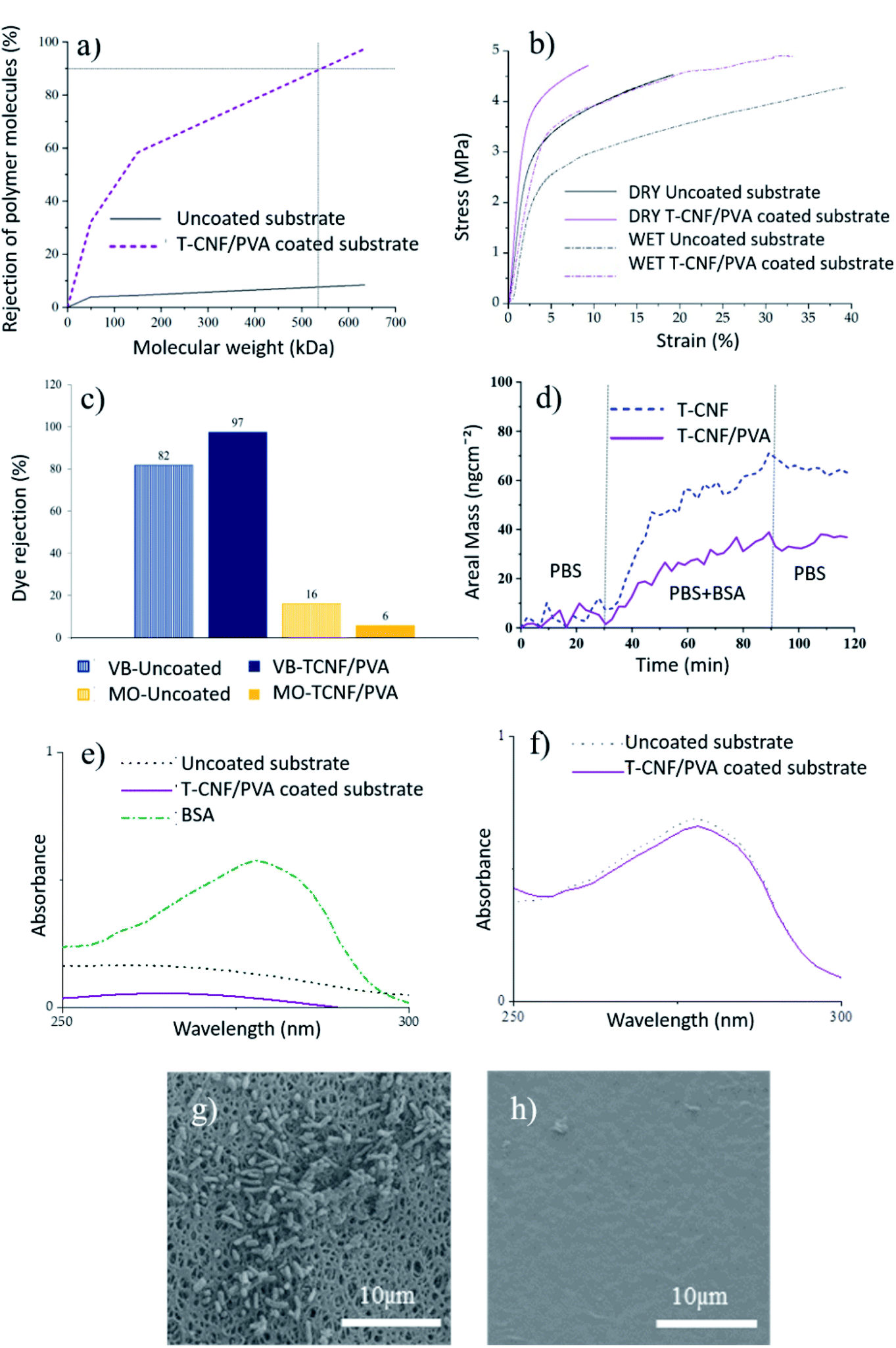 | ||
| Fig. 3 (a) MWCO of T-CNF/PVA coated and uncoated membranes, (b) stress–strain curves of uncoated and coated membranes tested in dry and wet conditions, (c) comparison of dye rejection of uncoated and coated membrane exposed to VB and MO, (d) BSA adsorption on T-CNF/PVA and T-CNF surfaces monitored with QCM-D at 37 °C (f0 = 5 MHz, n = 3, f5/n) represented a change in areal mass as a function of time. (e) UV-vis spectra of BSA solution recovered from uncoated and T-CNF/PVA coated substrate exposed to a continuous flux for 6 hours, (f) UV-vis spectra of BSA solution that passed through the uncoated and T-CNF/PVA coated substrate exposed to a continuous flux for 6 hours. SEM images of E. coli colonization after 18 h of bacterial incubation of: (g) uncoated PES membranes, (h) T-CNF/PVA coated membrane. | ||
Tensile properties of the uncoated and coated membranes were measured, both in dry and wet conditions, to evaluate the membrane integrity in use conditions. The stress–strain curves and the tensile data of the membranes are given in Fig. 3b and Table 2, respectively. Slightly higher maximum tensile strength was obtained for T-CNF/PVA coated membranes compared to the uncoated ones. The coating had a major impact on the Young's-modulus of the membranes, for which in both wet and dry conditions, the stiffness increased by 28% compared to the uncoated ones (Fig. 3b). As expected, the elongation at break decreased. These results agree with improved mechanical properties by crosslinking nanocellulose, and PVA reported in the literature.23,26,34 In wet conditions, both types of samples elongated more than twice than the ones tested in dry conditions, which was attributed to the plasticizing effect of water in hydrophilic materials. Improved mechanical properties were observed for coated membranes compared to uncoated ones, especially in wet conditions which can be attributed to the co-crosslinking of TCNF and PVA in a 3D network using glutaraldehyde. This 3D network formation was explained in the previous section (see Fig. 1), where hydroxyl groups of PVA react with carboxyl groups from T-CNF fibrils, resulting in a covalent ester bonding which stabilize the film structure in aqueous media.17
| Conditions | Sample type | Tensile strength (MPa) | E-Mod (MPa) | Elongation at break (%) |
|---|---|---|---|---|
| Dry | Uncoated | 4.6 ± 0.2 | 144 ± 2 | 20.6 ± 2.4 |
| T-CNF/PVA coated | 4.6 ± 0.4 | 199 ± 3 | 10.2 ± 0.7 | |
| Wet | Uncoated | 4.3 ± 0.2 | 99 ± 3 | 49.9 ± 4.5 |
| T-CNF/PVA coated | 4.9 ± 0.1 | 138 ± 14 | 32.9 ± 2.0 |
Furthermore, the abundant hydroxyl groups in PVA are partially crosslinked by the aldehyde groups of the glutaraldehyde.20 Covalent and H-bonds between the hydrated regions of the glutaraldehyde and the carboxyl group of the T-CNF20 are also expected to mechanically stabilize the membrane in wet conditions. Lower swelling of the crosslinked layer also positively impacted the mechanical stability of the coated membranes.
Dye adsorption/rejection
The coated substrates, being negatively charged, and having a pore structure and MWCO in the ultrafiltration regime, are expected to show rejection performance through electrostatic interaction and size exclusion.Positively and negatively charged dyes were used to evaluate the T-CNF/PVA rejection performance via electrostatic interaction. Positively charged VB (at pH 5) showed a better affinity towards the coated membrane than to the PES substrate attributable to the negative charge on these membranes, as confirmed by streaming potential measurements (Fig. 3c). The rejection was higher (97% vs. 82%) for the T-CNF/PVA coated membrane. In the case of negatively charged MO (at pH 5), the PES substrate and coated membrane showed the opposite trend, with the adsorption being higher for the uncoated membrane (16% compared to 6% for the coated). This can be attributed to the electrostatic repulsions between the negatively charged dye and the negatively charged membrane surface rich in hydroxyl groups from T-CNF/PVA. This data confirms the potential of the membranes for the selective separation of contaminants and particles based on charge. These results agree with the findings reported in the literature that electrically charged membranes have the potential for electrostatic exclusion without significantly reducing permeability and provides a better separation factor vs. permeability combination compared to the uncharged membranes.3
Antifouling performance
The antifouling performance of the uncoated membranes and the T-CNF/PVA coated membranes was analyzed with regards to organic fouling and biofouling (Fig. 3d–h).To reproduce the conditions to which the membranes are exposed in regular use, they were exposed to 6 h of a continuous flux of a BSA solution at neutral pH. After rinsing the samples with SDS solution, the solution was analyzed by UV-vis spectroscopy (Fig. 3e). T-CNF/PVA coated membranes displayed a relative adhesion reduction of 75% compared to the uncoated PES membranes. These reductions in the adhesion of BSA on the membrane were attributed to the abundant hydroxyl groups on the surface of the cellulose nanofibrils and PVA, respectively, which allows a layer of water to bind to the surface, preventing the adhesion of BSA to the surface. This suppresses non-specific interactions with proteins and prevents possible foulants, such as BSA, from attaching to the membrane surface.36 The permeate solution from both uncoated and coated membranes was analyzed by UV-vis spectroscopy (Fig. 3f). Results showed that the BSA passed through the membrane and was neither rejected by size exclusion nor adsorbed by the membrane at pH 7. This was expected as the Mw of BSA is about 66 kDa, and both the membrane and the coating are negatively charged; therefore, there are repulsive interactions responsible for this observation.
Conclusions
This study demonstrates an efficient, water-based, and easy to scale process to coat T-CNF/PVA on microfiltration substrates generating high-flux, negatively charged ultrafiltration membranes. PVA was used to anchor the nanocellulose to the PES substrate and enable the formation of a chemically-crosslinked 3-dimensional network to stabilize the nanocellulose layer. This approach successfully addressed the major challenges while using hygroscopic materials as functional coating viz. the mechanical properties of the membranes in the wet environment, and the stability of the coated layer during water flux as well as pH changes, which confirmed the applicability of the membrane in an aqueous environment. Surface functionalization of the substrate by the addition of chemically-bound T-CNF was confirmed by the change in the membrane charge, surface chemistry, and molecular weight cut off (530 kDa). The charged T-CNF/PVA coated membranes exhibited adsorption driven by electrostatic interaction combined with rejection due to size exclusion without significantly reducing permeability and provides a better separation factor vs. permeability combination compared to the uncharged membranes.The coated membranes exhibited a significant reduction in organic fouling attributable to a decrease in surface roughness compared to the uncoated PES substrates, as well as increased negative surface charge compared to the original membrane over a pH range of 2–10, which generates electrostatic repulsions between negatively charged foulants and the surface of the membrane. Moreover, coated membranes exhibited up to 75% less relative adhesion of BSA detected via spectroscopy and supported by QCM-D studies. The T-CNF coating exhibited not only significantly enhanced antifouling but also credible antibacterial properties against biofouling, such as E. coli, compared to the uncoated substrate, which was assigned to aldehyde groups present on the coated substrate. The data indicate potential use of these multifunctional membranes in single-use applications for e.g. biological sample preparation, sterile filtration of culture media and buffers etc.
Author contributions
The manuscript was written through the contributions of all authors. AA-S prepared the materials and performed most of the characterization and writing of the manuscript. BJ contributed to the planning and analysis of antifouling performance. VR, KSK and TT contributed with the QCM-D studies, AnM performed the MWCO studies and HA performed the N2 adsorption studies. AM contributed to the project planning, AFM data, all data analysis. All authors contributed to the writing and proof reading of the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflict of interests to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 760601. The authors acknowledge Dr Gilberto Siqueira (EMPA Swiss Federal Laboratories for Materials Science and Technology) and Dr Björn Hansmann (Sartorius Stedim Biotech) for providing the materials used in this study and fruitful discussions. Special thanks to Professor Roberto Rosal (University of Alcalá) for the support during the bacterial studies, Dr Luis Valencia (Stockholm University) for the help during the data analysis and Lukas Brandfellner (Universität Wien) for support with the MWCO study.References
- A. Moslehyani, A. F. Ismail, T. Matsuura, M. A. Rahman and P. S. Goh, in Membrane Separation Principles and Applications, Elsevier Inc., 2019, pp. 85–110 Search PubMed.
- W. Eykamp, in Membrane Separation Technology. Principles and Applications, ed. R. D. Noble and A. Stern, Elsevier Science B.V., 1995, pp. 1–43 Search PubMed.
- A. Mehta and A. L. Zydney, J. Membr. Sci., 2005, 249, 245–249 CrossRef CAS.
- D. Breite, M. Went, A. Prager, M. Kuehnert and A. Schulze, Polymers, 2019, 11, 3 CrossRef.
- H. Sehaqui, U. Perez de Larraya, P. Liu, N. Pfenninger, A. P. Mathew, T. Zimmermann and P. Tingaut, Cellulose, 2014, 21, 2831–2844 CrossRef CAS.
- A. L. Zydney, in Encyclopedia of Membranes, ed. E. Drioli and L. Giorno, Springer-Verlag Berlin Heidelberg, 2014, pp. 1–2 Search PubMed.
- H. Ma, C. Burger, B. S. Hsiao and B. Chu, Biomacromolecules, 2011, 12, 970–976 CrossRef CAS.
- P. Liu, K. Oksman and A. P. Mathew, J. Colloid Interface Sci., 2016, 464, 175–182 CrossRef CAS.
- A. Mautner, K.-Y. Lee, P. Lahtinen, M. Hakalahti, T. Tammelin, K. Li and A. Bismarck, Chem. Commun., 2014, 50, 5778–5781 RSC.
- A. Mautner, K.-Y. Lee, T. Tammelin, A. P. Mathew, A. J. Nedoma, K. Li and A. Bismarck, React. Funct. Polym., 2015, 86, 209–214 CrossRef CAS.
- Z. Karim, S. Claudpierre, M. Grahn, K. Oksman and A. P. Mathew, J. Membr. Sci., 2016, 514, 418–428 CrossRef CAS.
- H. Voisin, L. Bergström, P. Liu and A. P. Mathew, nanomaterials, 2017, 7, 1–18 CrossRef.
- Z. Karim, A. P. Mathew, V. Kokol, J. Wei and M. Grahn, RSC Adv., 2016, 6, 20644–20653 RSC.
- L. A. Goetz, N. Naseri, S. S. Nair, Z. Karim and A. P. Mathew, Cellulose, 2018, 25, 3011–3023 CrossRef CAS.
- P. Liu, C. Milletto, S. Monti, C. Zhu and A. P. Mathew, RSC Adv., 2019, 9, 28657–28669 RSC.
- H. Ma, C. Burger, B. S. Hsiao and B. Chu, ACS Macro Lett., 2012, 723–726 CrossRef CAS.
- M. Hakalahti, A. Salminen, J. Seppälä, T. Tammelin and T. Hänninen, Carbohydr. Polym., 2015, 126, 78–82 CrossRef CAS.
- L. Bai, Y. Liu, A. Ding, N. Ren, G. Li and H. Liang, Chemosphere, 2019, 217, 76–84 CrossRef CAS.
- A. Aguilar-Sanchez, B. Jalvo, A. Mautner, S. Nameer, T. Pöhler, T. Tammelin and A. P. Mathew, J. Membr. Sci., 2021, 620, 1–10 CrossRef.
- T. Hou, K. Guo, Z. Wang, X.-F. Zhang, Y. Feng, M. He and J. Yao, Cellulose, 2019, 26, 5065–5074 CrossRef CAS.
- S. Matsumura, H. Kurita and H. Shimokobe, Biotechnol. Lett., 1993, 15, 749–754 CrossRef CAS.
- Q. Zheng, Z. Cai and S. Gong, J. Mater. Chem. A, 2014, 2, 3110–3118 RSC.
- C. Tang, C. D. Saquing, J. R. Harding and S. A. Khan, Macromolecules, 2010, 43, 630–637 CrossRef CAS.
- M. Kozlov, M. Quarmyne, W. Che and T. J. McCarthy, Macromolecules, 2003, 36, 6054–6059 CrossRef CAS.
- X. Ma, Y. Su, Q. Sun, Y. Wang and Z. Jiang, J. Membr. Sci., 2007, 300, 71–78 CrossRef CAS.
- K.-J. Kim, S.-B. Lee and N.-W. Han, Polym. J., 1993, 25, 1295–1302 CrossRef CAS.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- M. Hakalahti, M. Faustini, C. Boissière, E. Kontturi and T. Tammelin, Biomacromolecules, 2017, 18, 2951–2958 CrossRef CAS.
- K.-J. Kim, S.-B. Lee and N.-W. Han, Korean J. Chem. Eng., 1994, 11, 41–47 CrossRef CAS.
- C.-K. Yeom and K.-H. Lee, J. Membr. Sci., 1996, 109, 257–265 CrossRef CAS.
- V. S. Praptowidodo, J. Mol. Struct., 2005, 739, 207–212 CrossRef CAS.
- A. D. Marshall, P. A. Munro and G. Trägårdh, Desalination, 1992, 91, 65–108 CrossRef.
- D. Johnson and N. Hilal, Desalination, 2015, 356, 149–164 CrossRef CAS.
- S. Tanpichai and K. Oksman, Composites, Part A, 2016, 88, 226–233 CrossRef CAS.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- R. Zhang, Y. Liu, M. He, Y. Su, X. Zhao, M. Elimelech and Z. Jiang, Chem. Soc. Rev., 2016, 45, 5888–5924 RSC.
- L. D. Renner and D. B. Weibel, MRS Bull., 2011, 36, 347–355 CrossRef CAS.
- W. Teughels, N. Van Assche, I. Sliepen and M. Quirynen, Clin. Oral Implants Res., 2006, 17, 68–81 CrossRef.
- Y. Zi, M. Zhu, X. Li, Y. Xu, H. Wei, D. Li and C. Mu, Carbohydr. Polym., 2018, 192, 118–125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (S1) AFM image of T-CNF, (S2) experimental set-up for MWCO and permeance measurements, (S3) SEM image of the coated surface after 24 h of continuous flux, a tilted coated sample, and scheme of the membrane's cross-section, (S4) N2 adsorption–desorption curves and pore size distribution determined using DFT model of T-CNF/PVA coated membrane, (S5) experimental details of QCM-D study, and (S6) quantification of bacteria using Image J are given. See DOI: 10.1039/d0ra10220b |
| This journal is © The Royal Society of Chemistry 2021 |