
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-based design, molecular dynamics and ADMET studies of suggested SARS-CoV-2 Mpro inhibitors
Nada M. Mohamed *a,
Eslam M. H. Aliab and
Asmaa M. AboulMagdc
*a,
Eslam M. H. Aliab and
Asmaa M. AboulMagdc
aPharmaceutical Chemistry Department, Faculty of Pharmacy, Modern University for Technology and Information (MTI), Egypt. E-mail: nada.mostafa@pharm.mti.edu.eg
bUniversity of Science & Technology (UST), Yuseong-gu, Daejeon, 34113, Republic of Korea
cPharmaceutical Chemistry Department, Faculty of Pharmacy, Nahda University (NUB), Beni-Suef, Egypt
First published on 22nd January 2021
Abstract
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has been the choice of recent studies worldwide to control its pandemic. Given the similarity with the earlier SARS-CoV, it is possible to use the previously reported inhibitors to develop a new treatment for the current attack of SARS-CoV-2. This study used the formerly published SARS-CoV Mpro small-molecule protease inhibitors to develop a pharmacophore model in order to design new ligands. Several strategies and scaffolds were evaluated in silico giving rise to ten newly designed compounds. Molecular docking and dynamics simulations were performed on Mpro enzyme in its active site to evaluate the newly designed ligands I–X. The results obtained from this work showed that compounds III–VI had a better molecular docking score than the co-crystallized ligand baicalein (3WL) giving −5.99, −5.94, −6.31, −6.56 and −5.74 kcal mol−1, respectively. Moreover, they could bind to the Mpro binding site better than I, II and VII–X. The most promising chromen-2-one based compounds V–VI had sufficiently acceptable physicochemical and ADMET properties to be considered new leads for further investigations. This new understanding should help to improve predictions of the impact of new treatments on COVID-19.
1. Introduction
The World Health Organization (WHO) announced the pandemic alert for Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in March 2020. The newly-discovered virus has over 40 million confirmed cases worldwide including over one million deaths as reported by the WHO in October 2020 and the numbers are still rising.1 Since then, many researchers have been studying its phylogenic similarity with other Coronaviridae family members in order to find or design a suitable treatment. Scientists found that SARS-CoV-2 was ∼80% similar to SARS-CoV in its primary structure, however there are some differences in the occurrence of the accessory proteins.2,3 Both SARS viruses use the human angiotensin II receptor (ACE-II) as their port to conquer the human cell and control its machinery. Several studies have been undertaken to meet the need to find a suitable treatment for SARS-CoV-2 symptoms and control its complications. To the date of commencing this study, no small molecule drug has been approved for SARS-CoV-2 treatment, however, only remdesivir (ribonucleotide analogue inhibitor of viral RNA-dependent RNA polymerase) has been approved by the Food and Drug Administration (FDA) for emergency use in hospitalized patients.4,5The Coronaviridae family shared the positive-sense single stranded RNA genome but with variable sizes between 26–32 Kb. This genome encoded 4 structural (spike, envelop, membrane and nucleocapsid), 15 non-structural proteins (nsps) one of which is nsp5 that encodes chymotrypsin-like cysteine major protease enzyme (Mpro) and 8 accessory proteins as presented in Fig. 1.6,7 The Mpro is found to be a crucial enzyme in the viral gene expression and replication. This enzyme is responsible for the major proteolysis of the large polyproteins 1a (PP1a) and 1b (PP1b) to yield many functional proteins like RNA-dependent RNA polymerase. Consequently, inhibiting Mpro stopped the viral replication inside the host cell.8,9
 | ||
| Fig. 1 Graphical presentation of SARS-CoV-2 viral composition showing the binding pocket of Mpro where the white arrows pointed to the catalytic dyad. | ||
Unlike other cysteine and serine proteases; Mpro has only two residues forming the catalytic dyad (His 41 and Cys 145) and one molecule of water located close to His 41 which supposedly acts as the third residue for its catalytic activity.10,11 The proteolytic mechanism of Mpro involves several steps initiated by the imidazole ring of His 41 deprotonates the sulfhydryl group of Cys 145. Then the resulting nucleophilic thiolate ion attacks the peptide linkage of the viral polyproteins at 11 different sites with the recognition sequence of Leu-Gln↓-(Ser/Ala/Gly) (↓ meant the cleavage site) as explained in Fig. 2.12,13
 | ||
| Fig. 2 Steps of the proteolytic mechanism of SARS-CoV-2 Mpro enzyme.12,13 | ||
Structurally, Mpro enzyme is a homodimer in which each monomer is almost perpendicular on one another and its monomeric form is enzymatically inactive.14 Each monomer consists of three main domains involving the amino acid residues 1–101, 102–184 and 201–301, respectively. Considering their spatial conformation; the first two domains are in the form of antiparallel β-barrel while the third is formed of five α-helices where the substrate binding site is located at the cleft between the first two domains as shown in Fig. 3.15 The substrate binding site consists of three main regions namely; catalytic dyad, deeply buried subunits S1, S2 and shallow buried subunits S3–S5. The catalytic dyad that is responsible for the proteolytic activity of the Mpro which is formed of His 41 as proton acceptor and Cys 145 as the nucleophile as explained earlier. The deeply buried subunits S1 and S2 are the main site for hydrophobic and electrostatic interaction between Mpro and the substrate or the inhibitor agent. It consists of His 163, Glu 166, Cys 145, Gly 143, His 172, Phe 140 and Cys 145, His 41, Thr 25 as S1 and S2, respectively. On the other hand, the shallow subunits S3–S5 are formed from Met 165, Glu 166, Gln 189, Met 49 and His 41.16 The dimerization formed by the interaction of the N-terminus of one monomer with Glu 166 of the other is crucial to keep the correct conformation of the substrate binding pocket in S1 subunit.13,17 Therefore, if an agent could bind to the catalytic dyad, dimerization essential amino acids or other residues that blocked the access of the natural substrate then it might inhibit with great possibility the proteolytic action of the enzyme and hinder the viral replication.
 | ||
| Fig. 3 SARS-CoV-2 Mpro protein structure (PDB: 6M2N) where (A): the full structure of the enzyme showing β-barrel of both domain I and II (blue), the helical shape of domain III (magenta) and the binding site of ligand (green spheres) (B): the binding site structure showing the ligand (green spheres) was surrounding with the crucial amino acid residues for interaction. | ||
Recent homology studies have revealed that the structure of Mpro enzyme of both SARS-CoV and SARS-CoV-2 have 96% similarity in their amino acid composition. However, they differ in only 12 amino acid residues with one residue (A46S) present in the proximity of the substrate binding site and other considerable mutations of T285A and I286L. This mutation was reported to increase the SARS-CoV Mpro catalytic activity by 3.6 folds which might explain the observed higher proteolytic activity of SARS-CoV-2 Mpro.14,18 Nonetheless; none of the 12 variant residues practically affect its enzymatic activity.19,20 Therefore, inhibitors of SARS-CoV Mpro enzyme are considered the milestone in designing new inhibitors for its SARS-CoV-2 Mpro sibling enzyme. In addition, the human cells have no proteases with a matching cleavage specificity of Mpro which offers an excellent drug target with minimal toxicity level.
Several publications had been screened for potent small molecule inhibitors of SARS-CoV-2 Mpro either of natural or synthetic nature. Amongst the natural ones; Sekiou et al. had reported many natural products with in silico inhibitory activity on CoV-2 Mpro namely; quercetin 1, hispidulin 2, cirsimaritin 3, artemisin 4, curcumin 5, thymoquinone 6 and eugenol 7 as shown in Fig. 4.21 It was reported that the ability of 1–7 to bind to the crucial amino acid residues of Mpro active site using PDB: 6LU7 with binding affinities −7.5, −7.3, −7.2, −6.8, −6.8, 5.1 and −4.9 kcal mol−1, respectively. On the other hand, Yao et al. had proven in vitro the capability of baicalein 8, a flavonoid found in traditional Chinese medicine, and its glycosylated form baicalin 9 to inhibit Mpro proteolytic activity with IC50 0.94 and 6.41 μM, respectively. Moreover, both 8 and 9 had shown free binding affinity ΔGITC of −30.78 and −28.2 kJ mol−1, respectively.22 Compounds with chromen-4-one scaffold 1–3 and 8–9 had demonstrated remarkable inhibitory activity on SARS-CoV-2 Mpro which was used as the core of this study.
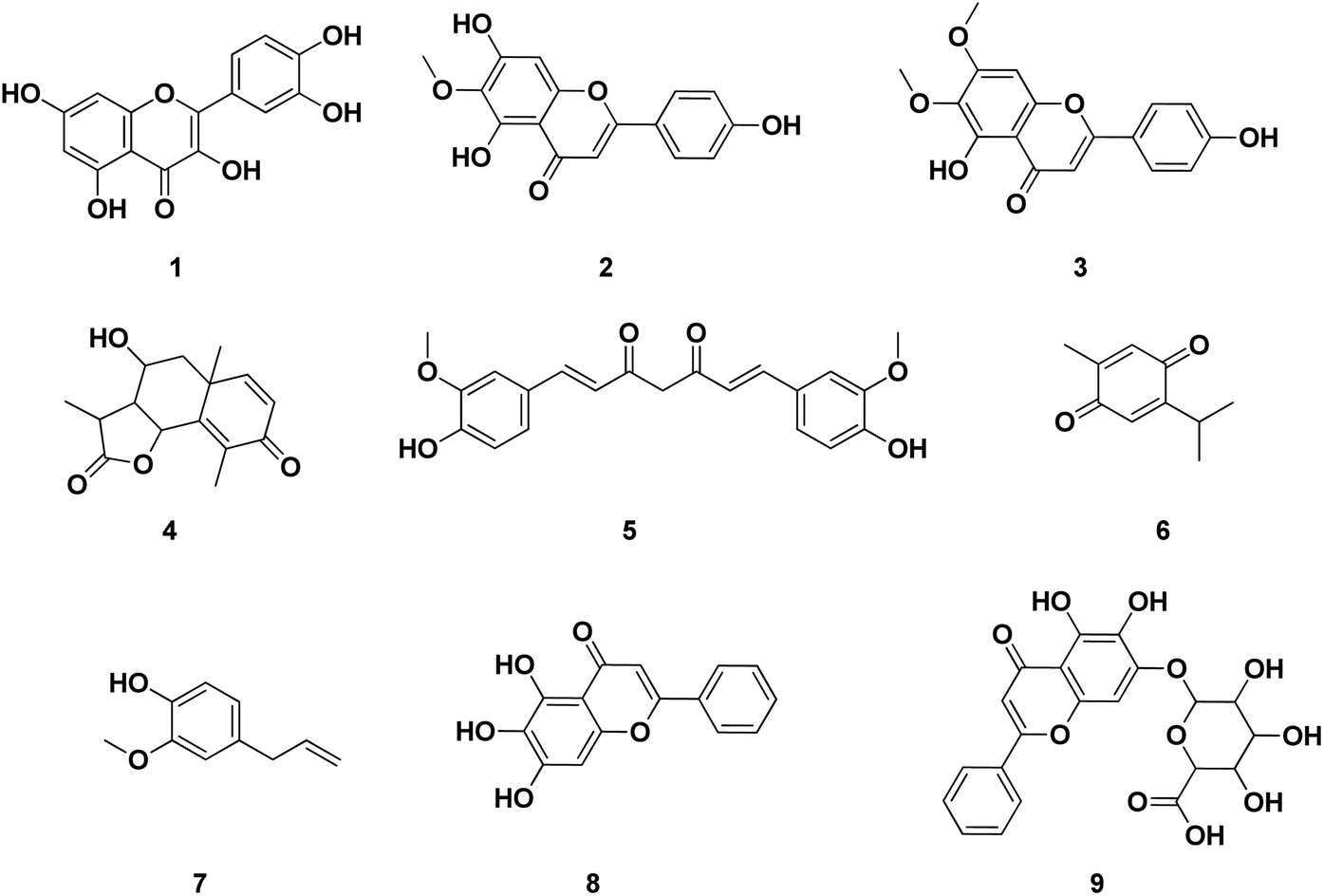 | ||
| Fig. 4 Reported structures with inhibitory activities on SARS-CoV-2 Mpro enzyme. | ||
2. Rational and design
Literature review of the SARS-CoV Mpro small molecule inhibitors revealed common structural features. Almost all inhibitors have two hydrophobic centers; with one has one or more H-bond acceptor atoms, both linked with 2 to 3 H-bond acceptor/donor atoms as detailed in Fig. 5 and 6. Consequently, pharmacophore mapping was formed to screen the newly suggested structures followed by docking on SARS-CoV-2 Mpro and molecular dynamic simulation to predict their activity.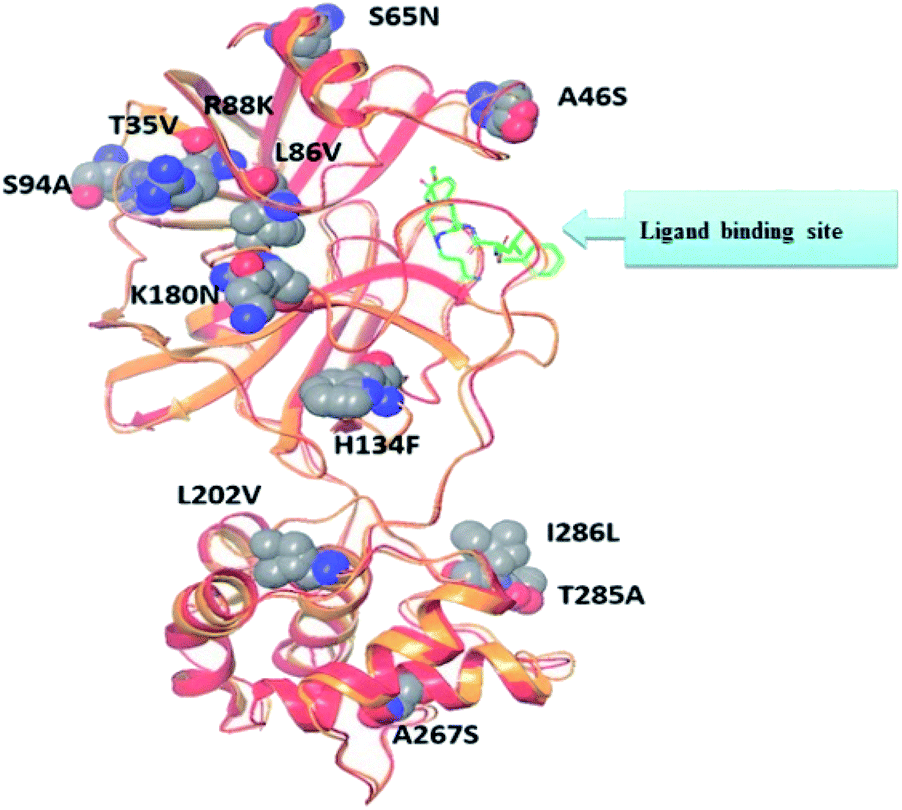 | ||
| Fig. 5 Alignment of SARS-CoV Mpro (PDB ID: 2A5I, red ribbon) and SARS-CoV-2's Mpro (PDB ID: 6LU7, orange ribbon) X-ray structures. As a result of pairwise alignment, sequence identity showed 96%. The green stick indicates the inhibitor binding site, and sphere model indicates residues that are not conserved between both sequences.28 | ||
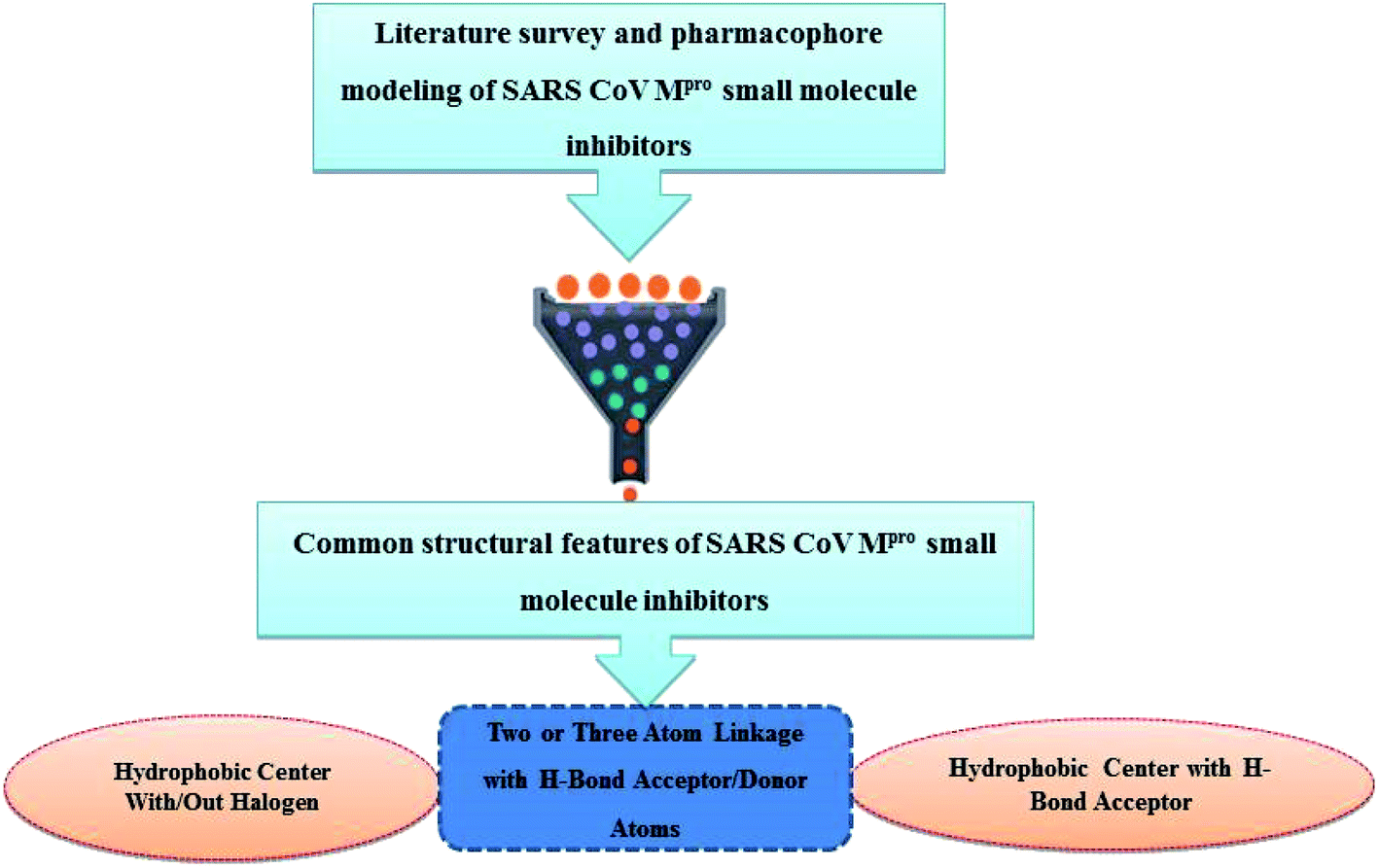 | ||
| Fig. 6 Common structural features of SARS-Mpro small molecule inhibitors. | ||
Herein, the newly designed molecules reserved the common structural features of SARS-CoV Mpro inhibitors with modification on one of the hydrophobic centers using bioisostere rings either five- or six membered. This study focused on benzotriazole, pyridine and coumarin scaffolds with proven inhibitory activity on SARS-CoV Mpro.23–27 Considering the linkage atoms; other alternatives of the ester linkage were to be evaluated. In this study, amide as the ester bioisostere and guanidine linkers were evaluated as detailed in Fig. 7.
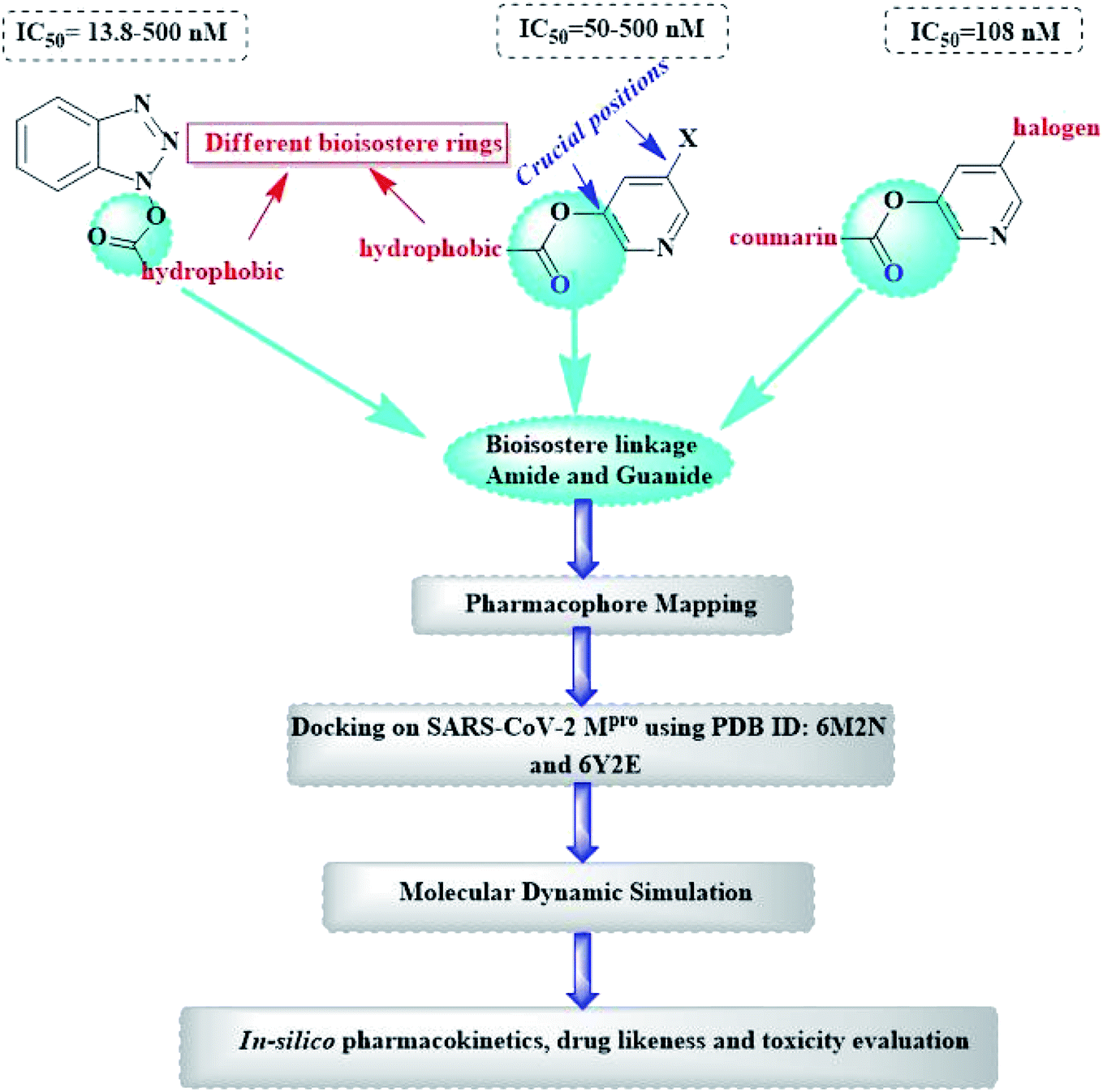 | ||
| Fig. 7 Design of newly suggested SARS-CoV-2 Mpro small molecule inhibitors. | ||
Computational techniques have long been of value in rational drug design and discovering new hits. This study focused in using ligand-based drug design theories as a source to get the required pharmacophore features to inhibit SARS-CoV-2 Mpro enzyme. Moreover, molecular dynamic simulation (MD) would be used to evaluate the interaction between the Mpro enzyme and its newly-designed inhibitors where MD studies the dynamics between the enzyme in solution with the inhibitors at the atomic level according Newton's equation of motion.29,30
3. Experimental methods
All in silico methods were conducted and visualized using MOE® 2020 (Molecular Operating Environment) software through Windows® 10 operating system.3.1. SARS-CoV and SARS-CoV-2 Mpro alignment
As stated earlier, the binding site of both SARS-CoV and SARS-CoV-2 Mpro shared very close amino acid residues and spatial conformation therefore in this current work, protein alignment protocol was conducted at first to demonstrate both enzymes binding sites superimposition and assess their similarity using Protein Data Bank (PDB) files 2A5K and 6M2N for SARS-CoV and SARS-COV-2 Mpro, respectively. This step was essential to prove that both files used for the upcoming in silico study were of high similarity and matched the literature.The two crystal structures were downloaded where their energies were minimized. The protein chains were designated to unique protein unit chain tag and were subjected to flexible alignment. The superposition algorithm started with alignment of the sequences using a modified version of Needleman and Wunsch algorithm.31 The alignment began with building up the initial pairwise similarity matrix using either a progressive or a tree-based method. Then round-robin realignment was performed, followed by a randomized iterative refinement. Finally, structure based realignment was performed using the existing or predicted protein secondary element information. After this, 3D superposition would be done on the best aligned model.
3.2. Pharmacophore modeling
3.3. Molecular docking
Molecular docking was performed using MOE® 2020 and MMFF94x force field. The X-ray crystal structures of SARS-CoV-2 Mpro enzyme were downloaded for the Protein Data Bank (PDB: 6M2N co-crystallized with 3WL and 6Y2E as free enzyme). Before implementing the docking procedure; both protein amino acid sequences were corrected and protonated at the default pH and 293 K temperature as mentioned in the experimental section of the PDB file. Visualization and generation of the 3D figures were performed using MOE® 2020 software.Chain a of PDB: 6M2N was selected for docking and the method was validated giving RMSD of 0.35 using triangle matcher as placement algorithm, London dG as rescoring function 1 and GBVI/WSA dG as rescoring function 2 algorithm. Similarly; the free enzyme crystal PDB: 6Y2E was docked using dummy atoms algorithm allocated to the critical interacting amino acid residues with the human ACE-II receptor using the same force field, placement and rescoring functions.
3.4. Molecular dynamics simulation
The molecular dynamics (MD) simulation study was performed for the suggested compounds to target SARS-CoV-2 Mpro complex (PDB: 6M2N) using standard default parameter setting in the MOE® 2020 software. There are four algorithms implemented in MOE® software for MD simulations; the Nos_e-Poincar_e-Andesen (NPA), the Nos_e-Hoover-Andersen (NHA), Berendsen velocity/position (BER) and Nanoscale Molecular Dynamics (NAMD). In this study, the NPA which is the most precise and sensitive algorithm, was used to study the molecular dynamics of ligands.32During the MD calculations, the system was optimized by selecting MMFF94x force field, water as a solvent, six margins and delete far existing solvent with distance greater than 4 Å. Also, the trajectories were stored in the traj.trr file for the models and structural analysis was done at every picosecond.
4. Discussion
Establishing a full understanding of the close similarity of both amino acid sequence and structure between SARS-CoV and SARS-CoV-2 Mpro enabled to study several small molecule inhibitors of SARS-CoV Mpro to propose a pharmacophore model which in turn would be used for developing new inhibitors for SARS-CoV-2 Mpro.4.1. SARS-CoV and SARS-CoV-2 Mpro alignment
The presented comparison of both SARS-CoV and SARS-CoV-2 Mpro binding sites effectively defined the ligand molecular binding to provide useful visions into the molecular interaction pattern of the co-crystalized ligands. As demonstrated in Fig. 8a and b, a similarity of 95.8–96.1% was observed between both binding sites of SARS-CoV and SARS-CoV-2 Mpro where the two chains of 2A5K were identical. These achieved data complied with the reported data of high similarity between both Coronae viruses Mpro active sites and the fact that the majority of the dissimilarity was slightly far from the targeted site. The difference in their amino acid sequence was found to be as follow where the first letter belongs to SARS-CoV and the last belongs to SARS-CoV-2; T35V, A46S, S65N, L86V, R88K, S94A, H134F, K180N, L202V, A267S, T285A, I286L which matched the literature showing only A46S was at the vicinity of the active site without affecting its proteolytic activity.33 Furthermore; this alignment displayed that whether the inhibitor was small molecule as 3WL or peptide – like as AZP, they should bind to the same residues to achieve a respectful inhibitory activity. Consequently, the design of small molecule SARS-CoV-2 Mpro inhibitors based on the previously published SARS-CoV Mpro enzyme inhibitors was of appreciated value during this study. The superposition of the two active sites with and without ligands illustrated a high similarity between the corresponding enzymes sequence that could clearly appear in their ligands (AZP and 3WL) high alignment as displayed in Fig. 8c and d.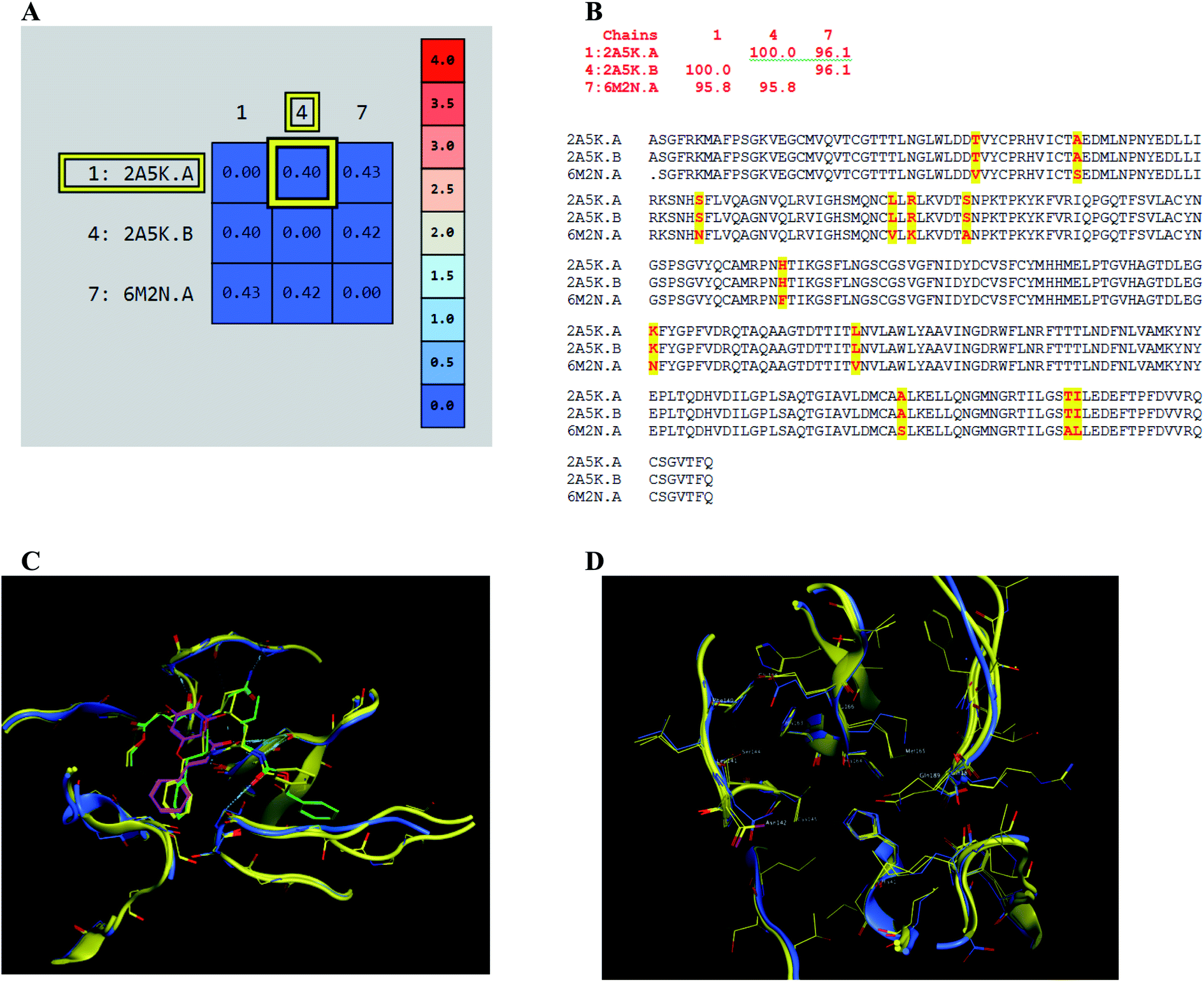 | ||
| Fig. 8 Similarity of both SARS-CoV and SARS-CoV-2 Mpro as amino acid sequence and spatial conformation where (A) was pairwise similarity matrix, (B) was amino acid sequence of both chains of 2A5K and one chain of 6M2N with highlighted differences. (C and D) Were both pockets alignment where 2A5K chains appeared in yellow and 6M2N chain in blue with and without ligands, respectively where 3WL showed in magenta color and AZP in green. | ||
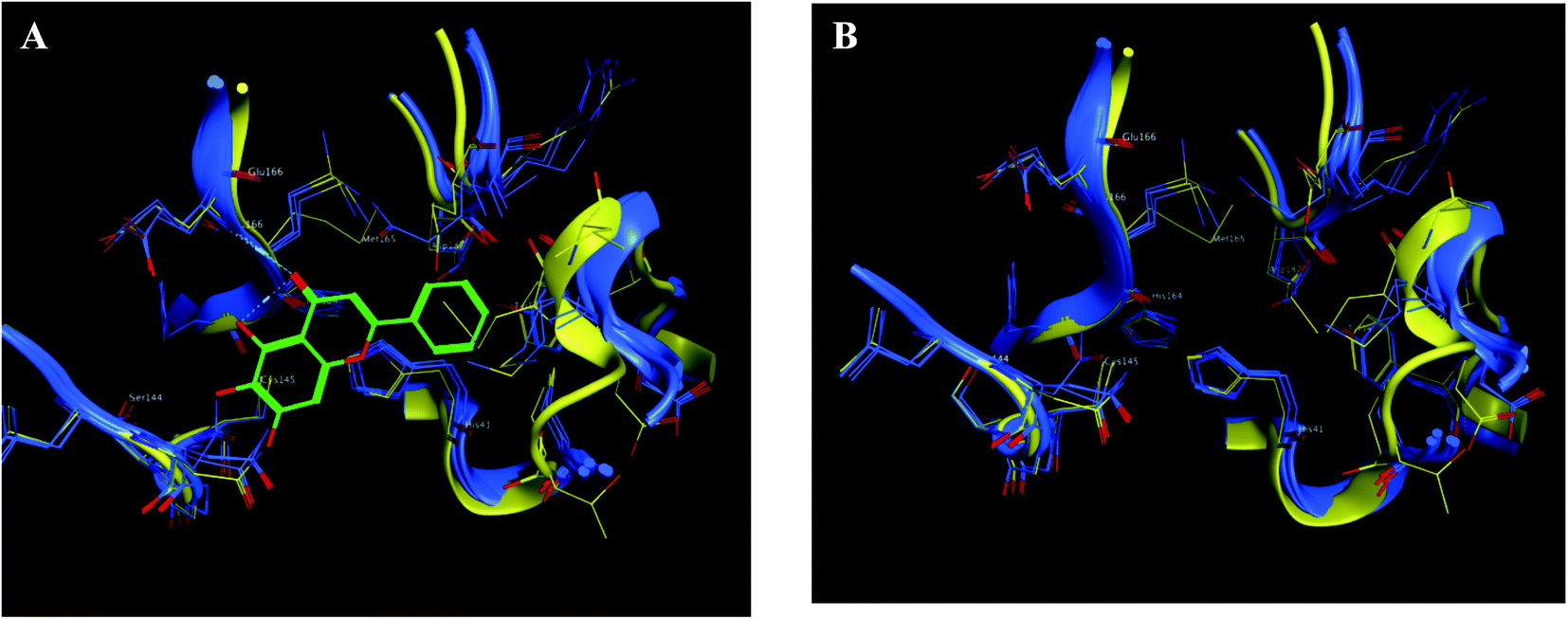 | ||
| Fig. 9 The 2D representation of SARS-CoV-2 Mpro conformations both the free PDB: 6Y2E and the bound PDB: 6M2N. (A) and (B) Showed the conformation similarity between both PDB crystals with and without ligand 3WL, respectively where 6M2N chains appeared blue, 6Y2E chain in yellow and 3WL in green sticks. | ||
4.2. SARS-CoV Mpro pharmacophore model development
The 3D pharmacophore model was built using the previously reported CoV Mpro small molecule inhibitors which exhibited a broad range of structural variability and biological activity.24–26,34,35 Those inhibitors training set could properly detect the essential moieties of the existing SARS-CoV Mpro inhibitors having the same mechanism of inhibition. Afterward, the model was used as 3D search query for predicting the structural requirements of each compound to identify SARS-CoV-2 Mpro new inhibitors. In this study, a training set of 43 reported SARS-CoV Mpro inhibitors were collected and their chemical structures are shown in Fig. 10. | ||
| Fig. 10 Structures of the used reported small molecule SARS-CoV Mpro inhibitors to design the pharmacophore model. | ||
The selected pharmacophore model represented by two aromatic hydrophobic centers (F1: Aro/Hyd and F2: Aro/Hyd) and one H-bond acceptor–metal ligature center (F3: ACC/ML) was shown in Fig. 11.
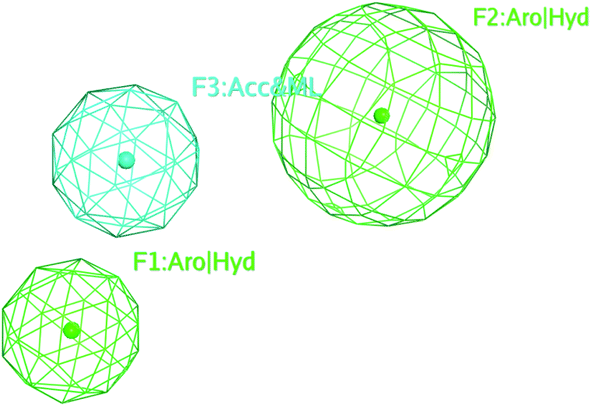 | ||
| Fig. 11 The common features of the pharmacophore model generated from training set alignment of SARS-CoV Mpro inhibitors. | ||
4.3. Structural consideration to design new SARS-CoV-2 Mpro inhibitors
Based on the literature survey of many active SARS-CoV Mpro small molecule inhibitors with benzotriazole, pyridine and coumarin scaffolds and its proven similarity with SARS-CoV-2 Mpro; several new compounds were proposed aiming to identify a potent lead for the current SARS-CoV-2 outbreak control. Designing these new compounds considered the previously discussed structural considerations which in turn were submitted for pharmacophore mapping and molecular docking to choose the best lead. Compounds I–X showed in Fig. 12 were the best regarding their pharmacophore RMSD and molecular docking binding energy score as detailed later.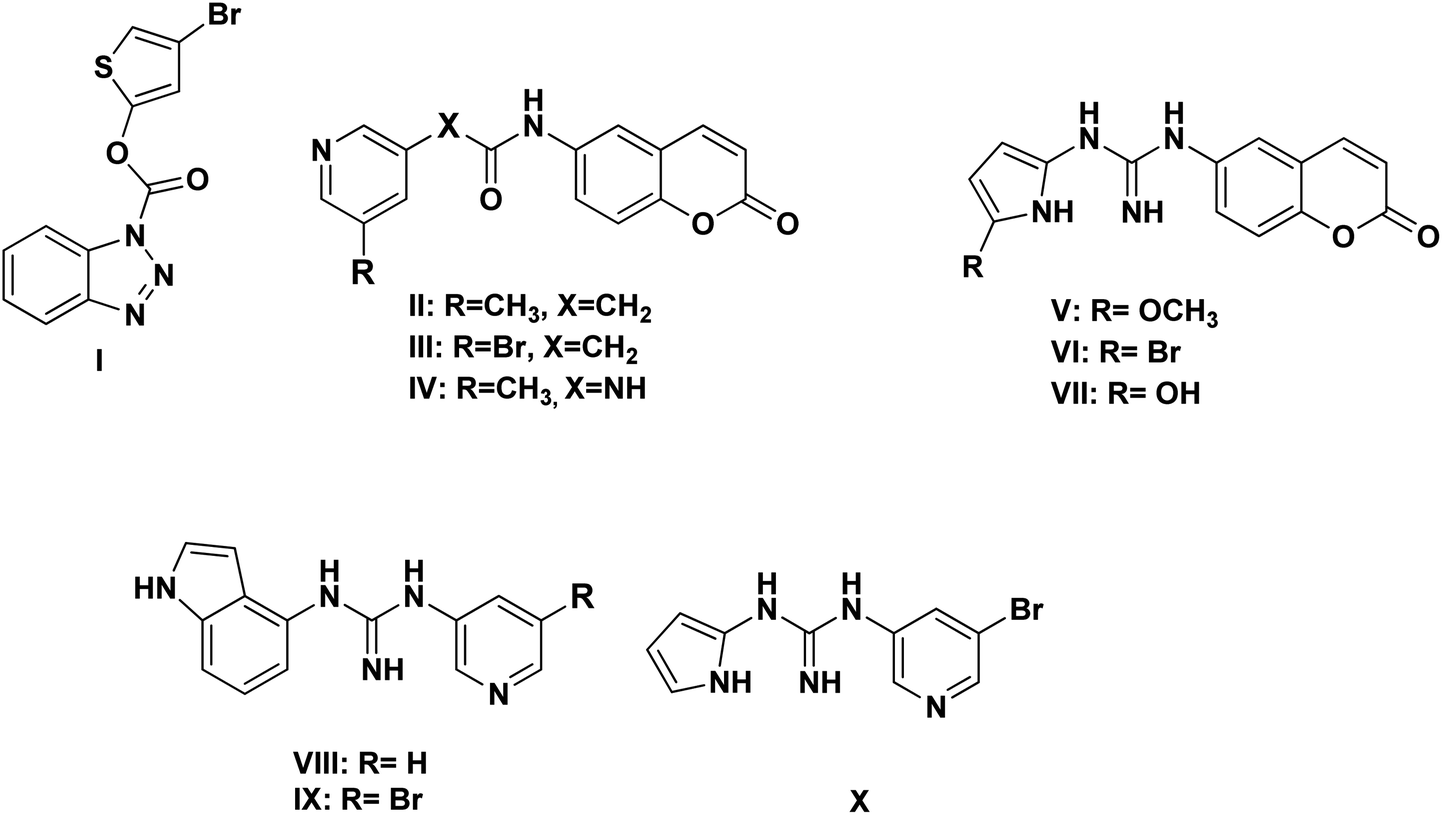 | ||
| Fig. 12 Chemical structures of the newly proposed compounds I–X as SARS-CoV-2 Mpro inhibitors. | ||
Upon screening the best benzotriazole structural features; Wu et al. mentioned that the position of the ester linkage was critical versus the nitrogen and was superior to ether, carbonyl or methylene linkage.24,26 Moreover, the importance of adding a halogen group to the meta position of the other aromatic ring was described.
Considering the other reported SARS-CoV Mpro inhibitors with pyridine center; the published data stated the importance of adding a halogen atom to the meta position of the pyridine ring which was linked to the other hydrophobic center at the other meta position. Interestingly, adding bromide group to the pyridine ring was superior to chloride. Furthermore, changing the ester linkage with an amide abolished their inhibitory activity which was considered during the design of the new compounds VIII–X using guanidine linkage instead.25,34
4.4. Pharmacophore mapping
The obtained data illustrated the high affinity of the tested compounds I–X in the selected pharmacophore model with RMSD values less than one. Hence; they all preserved the main structural features generated in the pharmacophore model as shown in Fig. 13.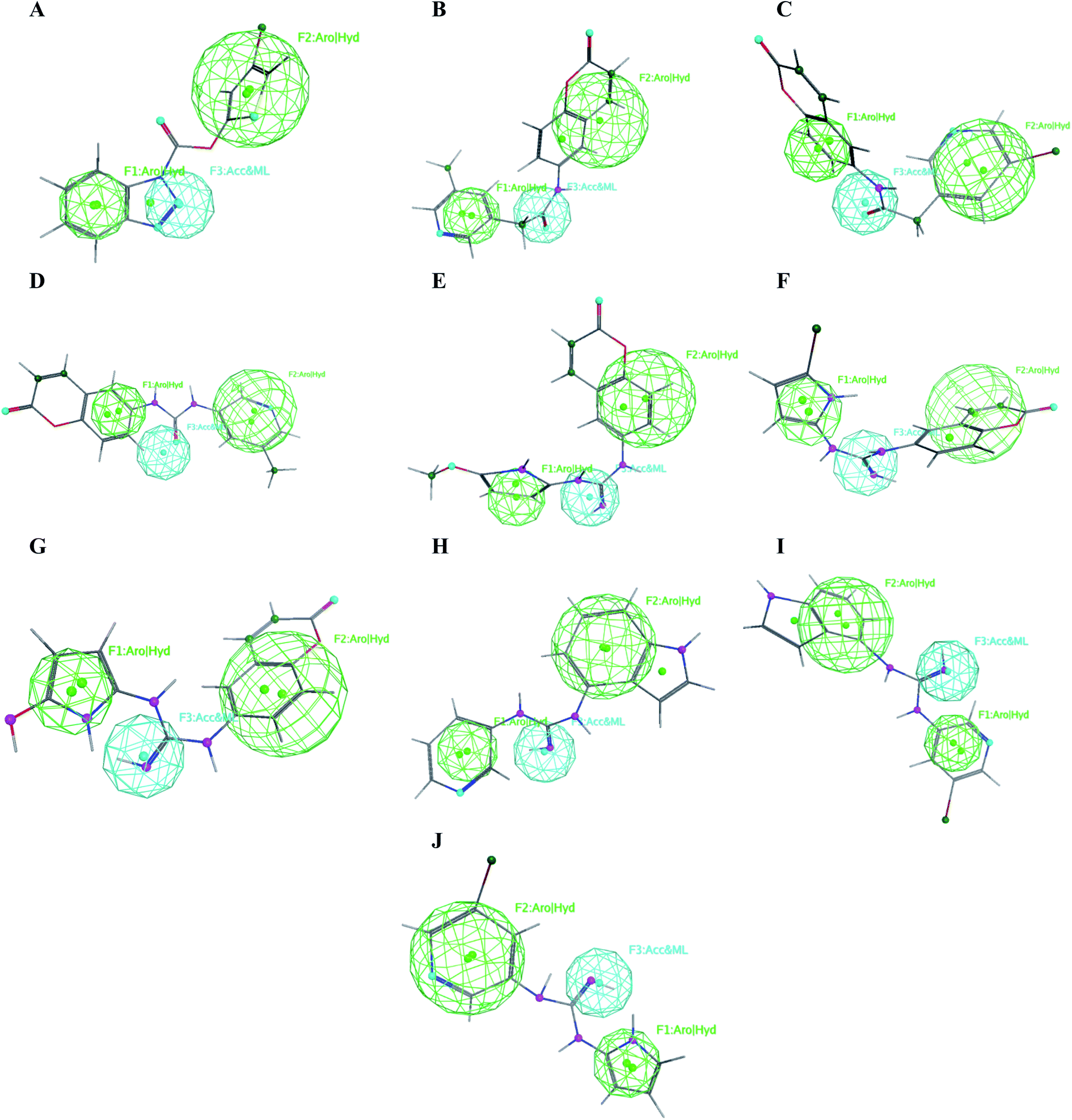 | ||
| Fig. 13 Mapping of the target compounds I (A), II (B), III (C), IV (D), V (E), VI (F), VII (G), VIII (H), IX (I) and X (J) on the generated pharmacophore model. | ||
It was observed that compounds II–IV fitted the model where the H-bond acceptor–metal ligature center was characterized by the acetamide or urea moieties and both pyridine and chromen rings represented the required aromatic hydrophobic centers F1 and F2-Aro/Hyd, respectively. In a quite similar manner, V–X showed similar pharmacophore fitting behavior where the central guanidine moiety represented the H-bond acceptor–metal ligature center, while the lateral aromatic rings represented the two aromatic hydrophobic centers F1 and F2-Aro/Hyd, respectively. However; compound I behaved differently, the H-bond acceptor–metal ligature center was represented by the three nitrogen atoms of the triazole ring. In the mean while the terminal thiophen ring indicated one of the aromatic hydrophobic centers and the other center represented by the phenyl part of benzo[d][1,2,3]triazole moiety as declared in Fig. 13a.
4.5. Molecular docking
In order to get a reliable docking result; two X-ray crystals namely, PDB: 6M2N and 6Y2E were used to computationally evaluate the proposed compounds I–X binding energies and their interactions with the crucial amino acids inside SARS-CoV-2 Mpro binding site. Consequently; to validate the used docking method using PDB: 6M2N the cocrystallized ligand baicalein 8 (3WL) was redocked using several docking protocols and algorithms untill reaching RMSD 0.35 and close 3D alignment as showed in Fig. 14. Meanwhile docking using dummies allocated by MOE® site finder algorithm showing the crucial amino acids was used while docking using PDB: 6Y2E.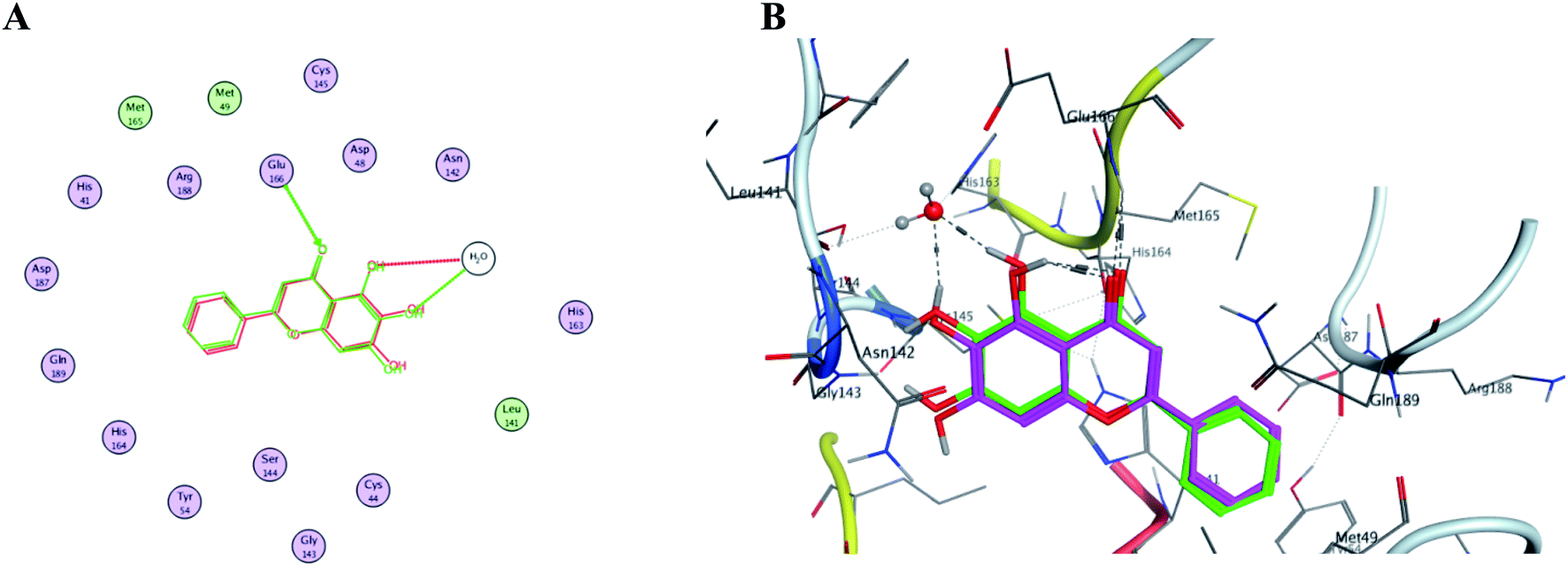 | ||
| Fig. 14 The overlaid co-crystallized ligand 3WL of SARS-CoV-2 Mpro (PDB: 6M2N) whilst docking method validation. (A) Shows 2D structures of the co-crystallized ligand (red) and the re-docked ligand (green) showing the interacting amino acid residues. (B) 3D visualization of overlay co-crystallized ligand (green sticks) and re-docked ligand (magenta sticks). | ||
Interestingly; all the ten proposed compounds showed either better energy score or number of interaction with the crucial residues than 3WL as summarized in Table 1 and presented in Fig. 15 and 16. The co-crystallized ligand 3WL was docked to 6Y2E as a reference to assess I–X using same docking protocol.
| # | Pharmacophore RMSD | Docking using 6M2N | Docking using 6Y2E | ||||
|---|---|---|---|---|---|---|---|
| Score (kcal mol−1) | No. of interacting residues | Interacting residues | Score | No. of interacting residues | Interacting residues | ||
| a NA is not available. | |||||||
| 3WL | NAa | −5.77 | 2 | Glu 166, His 163 | −4.66 | 2 | Cys 145, Glu 166 |
| VI | 0.57 | −6.56 | 3 | His 41, Arg188, Thr 190 | −5.16 | 2 | His 41, Cys 145 |
| V | 0.43 | −6.31 | 1 | His 41 | −5.69 | 2 | Cys 145, Asn 142 |
| III | 0.65 | −5.99 | 2 | Cys 145, Glu 166 | −5.73 | 1 | Cys 145 |
| IV | 0.70 | −5.94 | 6 | His 41, Cys 145, Gly 143, Asn 142, Thr 25, Thr 26 | −5.66 | 5 | His 41, Glu 166, Asn 142, Met 49 |
| II | 0.21 | −5.82 | 2 | Cys 145, Glu 166 | −5.72 | 2 | His 41, Cys 145 |
| VII | 0.52 | −5.75 | 1 | Cys 145 | −5.68 | 2 | Thr 26, Asn 142 |
| I | 0.20 | −5.64 | 1 | His 41 | −5.16 | 3 | His 41, Cys 145, Asn 142 |
| V | 0.67 | −5.49 | 2 | Thr 26, Asn 142 | −5.50 | 1 | Met 165 |
| VI | 0.58 | −5.33 | 2 | His 41, Gly 143 | −5.01 | 1 | Glu 166 |
| IV | 0.54 | −5.25 | 3 | Asn 142, Thr 25, Cys 145 | −5.27 | 1 | Met 165 |
 | ||
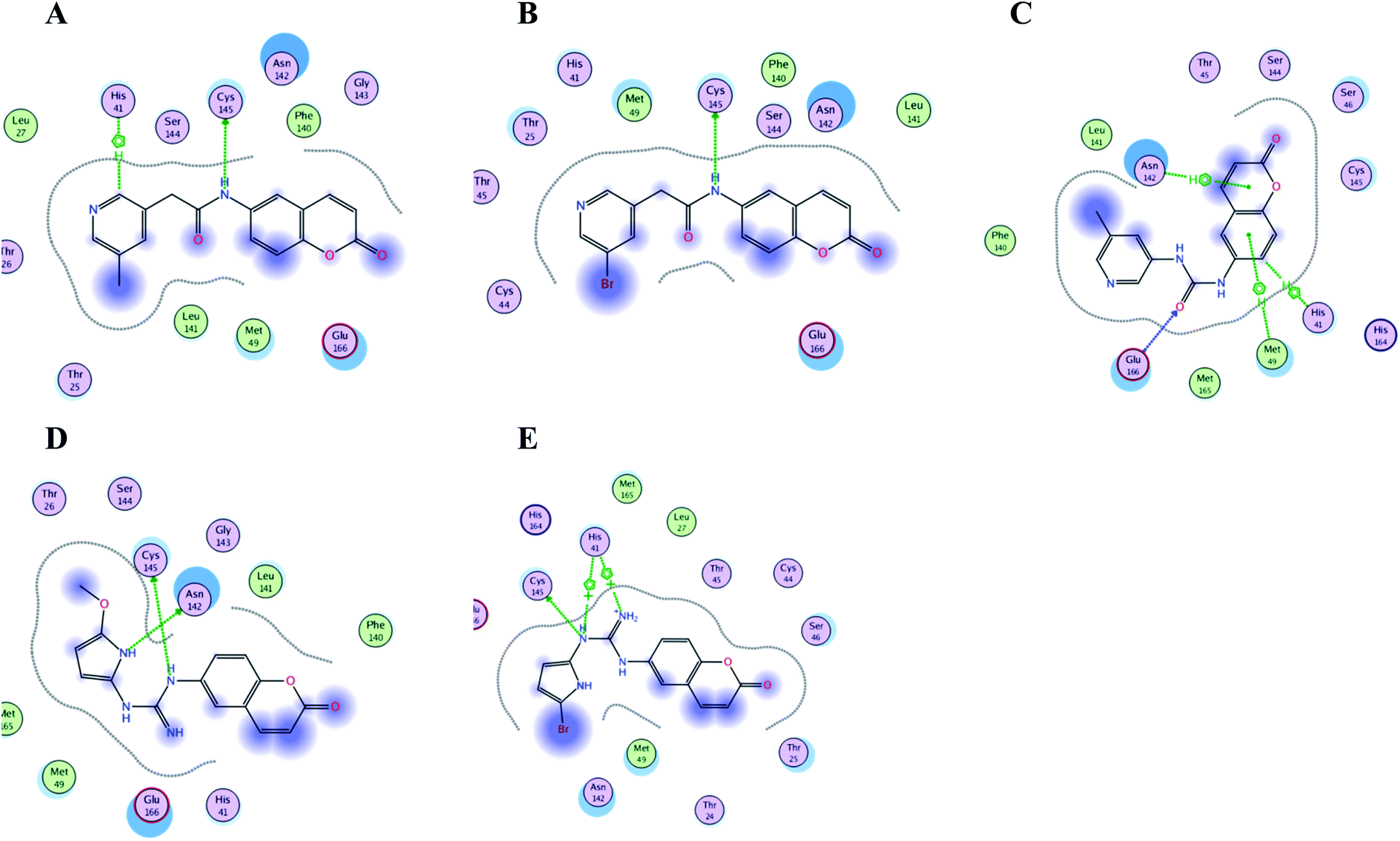
| Fig. 15 Crucial amino acids 2D interaction between SARS-CoV-2 Mpro (PDB: 6M2N) and the proposed compounds II–VI where (A) is for II, (B) for III, (C) for IV, (D) for V and (E) for VI. | ||
 | ||
| Fig. 16 Crucial amino acids 2D interaction between SARS-CoV-2 Mpro (PDB: 6Y2E) and the proposed compounds II–VI where (A) is for II, (B) for III, (C) for IV, (D) for V and (E) for VI. | ||
It was reported that baicalein 8 (3WL) demonstrated 99.4% and 87% inhibition against SARS-CoV-2 Mpro when used in 100 and 10 μM, respectively with an IC50 of 0.94 and 1.18 μM against SARS-CoV-2 and SARS-CoV Mpro, respectively.22 Thereby considering the ability of the proposed compounds I–X to interact with the SARS-CoV-2 Mpro binding site with higher binding score and/or number of residues interaction using two different X-ray crystals might open the way to discover a potent antiviral Mpro inhibitor.
It was noticed that the best computational outcome of I–X were the coumarin containing scaffold II–VI. The mentioned compounds showed binding energy scores of −6.56, −6.31, −5.99, −5.94 and −5.82 kcal mol−1 for VI, V, III, IV and II, respectively which preceded the co-crystallized ligand 3WL that showed −5.77 kcal mol−1. Moreover unlike 3WL; these compounds managed to interact with at least one of the two critical catalytic residues of Mpro, His 41 and Cys 145, as declared in Table 1. The most promising compound VI bound by its coumarin scaffold to 6M2N His 41 through arene–arene interaction while acted as H- bond donor to engage with both Arg 188 and Thr 190 through guanine –NH linkage and pyrrole –NH, respectively. On the other hand, it succeeded to bind to both 6Y2E catalytic residues through two arene–cation linkages and two H-bonds interactions with His 41 and Cys 145, respectively. Likewise, the second promising compound V bound through its coumarin scaffold to His 41 by arene–arene bond and to Cys 145 and Asn 142 through H-bond in 6M2N and 6Y2E, respectively. In a similar manner, compound III served as H-bond donor via its amide –NH to anchor Cys 145 in both 6M2N and 6Y2E. However; it managed to form another H-bond with 6M2N Glu 166, the essential residue for Mpro dimerization, through its amide carbonyl. Interestingly; out of the ten compounds, compound IV showed the highest number of interactions with Mpro crucial residues in both PDB: 6M2N and 6Y2E with binding energy score −5.94 and −5.66 kcal mol−1, respectively which indeed surpassed 3WL score and contact with Mpro binding site. The graphical representation of III, IV and VI interaction with the 6M2N Mpro substrate binding site is shown in Fig. 17.
 | ||
| Fig. 17 A graphical interaction between III, IV and VI (in magenta sticks) and the crucial residues of 6M2N Mpro where (A) was VI showing two hydrogen bonds (black dotted line) with distance 1.56 and 2.56 Å, (B) was III forming two H bonds (black dotted lines) with distance 2.24 and 3.51 Å, and (C) was IV forming many bonds (green and black dotted lines). | ||
Even though compounds II–IV and V–VII shared close chemical structure, yet changing few chemical groups altered their binding scores in different ways. Considering their chemical structures, the addition of a bromide group to the pyridine ring of II or changing its amide linkage to urea in both III and IV, respectively caused a mild improvement of its score value from −5.82 to −5.99 and −5.94 kcal mol−1, respectively. In the same aspect; changing the methoxy group of V to a bromide in VI resulted in a moderate enhancement of its binding energy score value from −6.31 to −6.56 kcal mol−1, respectively. However, changing the methoxy group of V to a hydroxyl group in VII dropped its binding score from −6.31 to −5.75 kcal mol−1, respectively.
Regarding the other four compounds I and VIII–X; despite displaying less score values than 3WL in 6M2N, they showed appreciated number of interactions with the crucial residues in the catalytic center and substrate binding site in both 6M2N and 6Y2E unlike 3WL as explained earlier. Moreover, those compounds showed better binding energy score values in molecular docking using 6Y2E than 3WL as previously stated in Table 1.
4.6. Molecular dynamics simulation
The conformational flexibilities of the proposed docked compounds with the Mpro enzyme were examined via MD process to attain dependable drug receptor – binding affinities. MD process calculations were run for 600 ps on the most promising compounds III–VI to target Mpro enzyme (PDB: 6M2N). Fig. 18 showed the results of the atomic potential energy function (U) during dynamic study calculation for the proposed compounds III–VI in the 6M2N at its binding site. To explore the dynamic stability of the 6M2N/inhibitor drugs complexes, the time-dependent potential energy of the complex were calculated during MD trajectories. It was apparent in Fig. 18, that complex (A) (6M2N/compound V) achieved equilibrium around 400 ps. Meanwhile complex (B) (6M2N/compound VI) achieved the equilibrium around 300 ps. On the other hand, complex (C) and complex (D) (6M2N/compound IV and III, respectively) achieved the equilibrium stability around 400 ps. | ||
| Fig. 18 The evaluation of potential energy of complex of (A) compound V, (B) compound VI, (C) compound IV, and (D) compound III with PDB: 6M2N receptor site as function of time. | ||
4.7. In silico physicochemical, drug likeliness and pharmacokinetics evaluation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P should not exceed 5.37 While Veber's declared that the compound to be accepted as drug potential should have no more than 10 rotatable bonds and its topological polar surface area should not exceed 120 Å to pass through the cell membrane.38,39
P should not exceed 5.37 While Veber's declared that the compound to be accepted as drug potential should have no more than 10 rotatable bonds and its topological polar surface area should not exceed 120 Å to pass through the cell membrane.38,39The calculated physicochemical properties of I–X were shown in Table 2 stating no violation and full compliance to both drug-likeliness rules except for VII that had six H-bond donor groups. However, VII is still considered as a good theoretical drug candidate as explained by Lipinski's. Moreover all compounds I–X had bioavailability score of 0.55 and complied Ghose's, Egan's and Mugge's rules of drug likeness as calculated from SwissADME®.
| # | lip_acc | lip_don | lip_drug like | logP(o/w) |
Weight (dalton) | lip_violation | opr_nrot | TPSA (Å2) | Mutagen groups |
|---|---|---|---|---|---|---|---|---|---|
| I | 5.00 | 0.00 | 1.00 | 3.79 | 324.16 | 0.00 | 2.00 | 57.01 | 0.00 |
| II | 5.00 | 1.00 | 1.00 | 2.35 | 294.31 | 0.00 | 3.00 | 68.29 | 0.00 |
| III | 7.00 | 4.00 | 1.00 | 3.40 | 298.30 | 0.00 | 5.00 | 99.23 | 0.00 |
| IV | 5.00 | 4.00 | 1.00 | 2.53 | 251.29 | 0.00 | 4.00 | 76.59 | 0.00 |
| V | 5.00 | 4.00 | 1.00 | 3.37 | 330.19 | 0.00 | 4.00 | 76.59 | 0.00 |
| VI | 5.00 | 4.00 | 1.00 | 2.34 | 280.13 | 0.00 | 4.00 | 76.59 | 0.00 |
| VII | 7.00 | 6.00 | 1.00 | 2.93 | 285.28 | 1.00 | 4.00 | 111.97 | 0.00 |
| VIII | 6.00 | 5.00 | 1.00 | 4.03 | 348.74 | 0.00 | 4.00 | 91.74 | 0.00 |
| IX | 6.00 | 1.00 | 1.00 | 1.90 | 295.29 | 0.00 | 2.00 | 80.32 | 0.00 |
| X | 5.00 | 1.00 | 1.00 | 2.85 | 359.18 | 0.00 | 3.00 | 68.29 | 0.00 |
On the other hand, I–X displayed the complete absence of any possible mutagenic groups consequently; they could be submitted for further chemical and biological in vitro investigation as the second phase.
| # | GI absorption | BBB permeability | P-glycoprotein substrate | Inhibitor of CYP P450 |
|---|---|---|---|---|
| I | High | No | No | Yes |
| II | High | Yes | No | Yes |
| III | High | Yes | No | Yes |
| IV | High | No | No | No except CYP 1A2 |
| V | High | No | No | No except CYP 1A2 |
| VI | High | No | No | No except CYP 1A2 |
| VII | High | No | No | No except CYP 1A2 |
| VIII | High | Yes | Yes | No except CYP 1A2, 2D6 |
| IX | High | Yes | Yes | Yes |
| X | High | Yes | No | No except CYP 1A2 |
Additionally; SwissADME® could also predict the possibility of the compounds to be affected by P-glycoprotein, a cellular efflux pump that pumps out drugs to extracellular fluid, where only VIII and IX were predicted to be substrate for it that might affect their plasma concentration and biological effect. Moreover; cytochrome P450 (CYP P450) enzymes family might be affected with different degrees by I–X as assumed according to their chemical structures that detailed in Table 3.
The top docking scored V and VI were presumed to be neither BBB permeable nor susceptible to P-glycoprotein. In addition, they expectedly did not inhibit CYP family expect CYP1A2 that might be affected to certain degree.
5. Conclusion
The most influential finding to emerge from this work was that compounds I–X revealed good molecular docking results compared with baicalein 8 in terms of binding energy scores and their interactions with the crucial amino acids of SARS-CoV-2 Mpro binding site. The best in silico outcome was achieved by derivatives V and VI regarding the pharmacophore RMSD, docking scores to SARS-CoV-2 Mpro PDB files and molecular dynamic simulation. Both V and VI showed better binding scores versus the crystallized ligand 8 giving −6.31, −6.56 and −5.77 kcal mol−1, respectively. They matched the calculated pharmacophore model with RMSD 0.43 and 0.57 for V and VI, respectively. Also, the mentioned compounds showed encouraging binding affinity to Mpro enzyme and good compliance with both Lipinisk's and Veber's rules. Further investigation and experimentation to these derivatives is highly recommended which may become promising candidates for COVID-19 therapy.Contributors
NMM initiated and developed the scope of the study with conceptualization, formal analysis, project administration, methodology in molecular docking and ADMET data generation and analysis. EMHA contributed with the protein alignment and pharmacophore generation part of the study with their corresponding data analysis and figures elucidation. AMA designed the molecular dynamics study with its corresponding data analysis and figures elucidation. All authors contributed jointly with the manuscript original draft writing where NMM generated the final revised manuscript that was read and approved by the other authors.Conflicts of interests
We declare that we have no conflicts of interest.Acknowledgements
The authors would gratefully thank the staff members of Nahda and MTI Universities for their collaborations. Sincere appreciation from all the authors to the spirit of our mentor Professor Dr Aly M Taha, professor of medicinla chmistry at MTI University.References
- W. H. Organization, WHO Coronavirus Disease (COVID-19) Dashboard, https://covid19.who.int/?gclid=Cj0KCQjw-O35BRDVARIsAJU5mQWekRZOAT2nMi7M_-aCZ0knit3e5R3pblHmDIUZ_W8OtC1oGSnDjOEaApjZEALw_wcB, accessed 22 October 2020 Search PubMed.
- A. Wu, Y. Peng and B. Huang, Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China, Cell Host Microbe, 2020, 27, 325–328 CrossRef CAS.
- F. Giorgi and C. Ceraolo, Genomic Variance of the 2019-nCoV Coronavirus, J. Med. Virol., 2020, 92(5), 522–528 CrossRef.
- F. Organization, FDA Approves First Treatment for COVID-19, https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19, accessed 31 October 2020 Search PubMed.
- F. Organization, Fact Sheet for Healthcare Providers Emergency Use Authorization (EUA) of Veklury® (Remdesivir) for Hospitalized Pediatric Patients Weighing 3.5 kg to Less Than 40 kg or Hospitalized Pediatric Patients Less Than 12 Years of Age Weighing at Least 3.5 kg, https://www.fda.gov/media/137566/download, accessed 01 Nov. 2020 Search PubMed.
- P. C. Y. Woo, Y. Huang, S. K. P. Lau and K.-Y. Yuen, Coronavirus Genomics and Bioinformatics Analysis, Viruses, 2010, 2, 1804–1820 CrossRef CAS.
- A. Wu, Y. Peng and B. Huang, Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China, Cell Host Microbe, 2020, 27, 325–328 CrossRef CAS.
- J. Ziebuhr, E. J. Snijder and A. E. Gorbalenya, Virus-Encoded Proteinases and Proteolytic Processing in the Nidovirales, J. Gen. Virol., 2000, 81(4), 853–879 CrossRef CAS.
- J. Zeibuhr and A. Hegyi, Conservation of Substrate Specificities among Coronavirus Main Proteases, J. Gen. Virol., 2002, 83(3), 595–599 CrossRef.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Coronavirus Main Proteinase (3CLPro) Structure: Basis for Design of Anti-Sars Drugs, Science, 2003, 300(5626), 1763–1767 CrossRef CAS.
- J. Zeibuhr, Molecular Biology of Severe Acute Respiratory Syndrome Coronavirus, Curr. Opin. Microbiol., 2004, 7(4), 412–419 CrossRef.
- K. Ś. A. V. Moliner, Revealing the Molecular Mechanisms of Proteolysis of SARS-CoV-2 Mpro by Qm/Mm Computational Methods, Chem. Sci., 2020, 11(39), 10626–10630 RSC.
- B. G. A. D. Goyal, Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy, ACS Comb. Sci., 2020, 22(6), 297–305 CrossRef CAS.
- L. Zhang, D. Lin and X. Sun, Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved Α-Ketoamide Inhibitors, Science, 2020, 368, 409–412 CrossRef CAS.
- U. Bacha, J. Barrila and A. Velazquez-Campoy, Identification of Novel Inhibitors of the SARS Coronavirus Main Protease 3CLPro, Biochemistry, 2004, 43, 4906–4912 CrossRef CAS.
- L. Lu, N. Mahindroo and P. Liang, Structure-Based Drug Design and Structural Biology Study of Novel Nonpeptide Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Main Protease, J. Med. Chem., 2006, 49(17), 5154–5161 CrossRef.
- N. Z. Zhong and S. Zou, Without Its N-Finger, the Main Protease of Severe Acute Respiratory Syndrome Coronavirus Can Form a Novel Dimer through Its C-Terminal Domain, J. Virol., 2008, 82, 4227–4234 CrossRef CAS.
- J. S. L. Lim, Y. Mu and J. Song, Dynamically-Driven Enhancement of the Catalytic Machinery of the SARS 3C-Like Protease by the S284–T285-I286/a Mutations on the Extra Domain, PLoS One, 2014, 9(7), e101941 CrossRef.
- M. Stoermer, Homology Models of Coronavirus 2019-Ncov 3CLpro Protease, ChemRxiv, 2020 DOI:10.26434/chemrxiv.11637294.v3.
- J. S. Morse, T. Lalonde, S. Xu and W. R. Liu, Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV, ChemBioChem, 2020, 21, 730–738 CrossRef CAS.
- O. Sekiou, B. Ismail, B. Zihad and D. Abdelhak, In Silico Identification of Potent Inhibitors of COVID-19 Main Protease (Mpro) and Angiotensin Converting Enzyme 2 (ACE-2) from Natural Products: Quercetin, Hispidulin, and Cirsimaritin Exhibited Better Potential Inhibition Than Hydroxy-Chloroquine against COVID-19 Main Protease Active Site and ACE-2, 2020 Search PubMed.
- H. Su, S. Yao and W. Zhao, Anti-SARS-CoV-2 Activities in vitro of Shuanghuanglian Preparations and Bioactive Ingredients, Acta Pharmacol. Sin., 2020, 1–11 CAS.
- A. S.-H. J. Thanigaimalai Pillaiyar, An Overview of Severe Acute Respiratory Syndrome–Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy, J. Med. Chem., 2016, 59, 6595–6628 CrossRef.
- K. Pumpor, S. Anemüller and K. Verschueren, A Structural View of the Inactivation of the SARS Coronavirus Main Proteinase by Benzotriazole Esters, Chem. Biol., 2008, 15, 597–606 CrossRef.
- J. Yin, J. Zhang and C. Niu, Molecular Docking Identifies the Binding of 3-Chloropyridine Moieties Specifically to the S1 Pocket of SARS-CoV Mpro, Bioorg. Med. Chem., 2008, 16, 293–302 CrossRef.
- K. King, C. Kuo and C. Wu, Stable Benzotriazole Esters as Mechanism-Based Inactivators of the Severe Acute Respiratory Syndrome 3CL Protease, Chem. Biol., 2006, 13, 261–268 CrossRef.
- L. Eltis, H. Pettersson and J. Zhang, Design, Synthesis, and Evaluation of Inhibitors for Severe Acute Respiratory Syndrome 3C-Like Protease Based on Phthalhydrazide Ketones or Heteroaromatic Esters, J. Med. Chem., 2007, 50, 1850–1864 CrossRef.
- N. Yasuo, M. Sekijima and R. Yoshino, Identification of Key Interactions between SARS-CoV-2 Main Protease and Inhibitor Drug Candidates, Sci. Rep., 2020, 10, 12493 CrossRef.
- T. Giorgino, G. Fabritiis and I. Buch, Complete Reconstruction of an Enzyme-Inhibitor Binding Process by Molecular Dynamics Simulations, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10184–10189 CrossRef.
- Y. E. A. Shan, How Does a Drug Molecule Find Its Target Binding Site, J. Am. Chem. Soc., 2011, 133, 9181–9183 CrossRef CAS.
- S. B. Needleman and C. D. Wunsch, A General Method Applicable to the Search for Similarities in the Amino Acid Sequences of Two Proteins, J. Mol. Biol., 1970, 48, 443–453 CrossRef CAS.
- J. B. Sturgeon and B. B. Laird, Symplectic Algorithm for Constant pressure Molecular Dynamics Using a Nos_E-Poincar_E Thermostat, J. Chem. Phys., 2000, 112(8), 3474–3482 CrossRef CAS.
- B. Goyal and D. Goyal, Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy, ACS Comb. Sci., 2020, 22(6), 297–305 CrossRef CAS.
- H. Pettersson, C. Huitema and J. Zhange, Design, Synthesis, and Evaluation of Inhibitors for Severe Acute Respiratory Syndrome 3C-Like Protease Based on Phthalhydrazide Ketones or Heteroaromatic Esters, J. Med. Chem., 2007, 50(8), 1850–1864 CrossRef.
- Y. Wang, Y. Lin and L. Chen, Synthesis and Evaluation of Isatin Derivatives as Effective SARS Coronavirus 3CL Protease Inhibitors, Bioorg. Med. Chem. Lett., 2005, 15(12), 3058–3062 CrossRef.
- A. Daina, O. Michielin and V. Zoete, SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules, Sci. Rep., 2017, 7, 42717 CrossRef.
- C. Lipinski, Lead- and Drug Like Compounds: The Rule of Five Revolution, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS.
- S. J. D. Veber and H. Cheng, Molecular Properties That Influence the Oral Bioavailability of Drug Candidates, J. Med. Chem., 2002, 45(12), 2615–2623 CrossRef.
- J. Kelder, Polar Molecular Surface as a Dominating Determinant for Oral Absorption and Brain Penetration of Drugs, Pharm. Res., 1999, 16(10), 1514–1519 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |