
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmino-1H-tetrazole-regulated high-density nitrogen-doped hollow carbon nanospheres for long-life Zn–air batteries†
Shizhu Song‡
a,
Tao Yang‡a,
Rongwei Shi‡ b and
Qi Li*ac
b and
Qi Li*ac
aDepartment of Materials Science, School of Chemistry and Chemical Engineering, Nantong University, Nantong, Jiangsu 226019, P. R. China. E-mail: zhuiqiuzhizhuo@163.com
bDepartment of Materials Science, School of Material and Chemical Engineering, Tongren University, Tongren, Guizhou 554300, P. R. China
cNantong Key Lab of Intelligent and New Energy Materials, Nantong University, Nantong, Jiangsu 226019, P. R. China
First published on 24th December 2020
Abstract
High-density nitrogen-doped porous carbon catalysts have been regarded as promising alternatives to precious metals in proton-exchange membrane fuel cells (PEMFC) and metal–air batteries based on the oxygen reduction reaction (ORR). We herein synthesized high-density pyridinic and graphitic N-doped hollow carbon nanospheres (G&P N-HCS) using a high-yield amino-1H-tetrazole (ATTZ) via a self-sacrificial-template method. The synthesized G&P N-HCS shows a high N content (15.2 at%), in which pyridinic (Pr) and graphitic (Gr) N are highly reactive for the ORR catalysis. We found that the half-wave potential and limiting current density of G&P N-HCS are comparable to the state-of-the-art Pt/C, whereas its cyclic durability is much superior to that of Pt/C. Experimental results indicate that an optimal ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) between Gr N and Pr N in G&P N-HCS exhibits the highest ORR performances, rather than Gr N-dominated N-HCS or Pr N-dominated N-HCS. Notably, N-HCS containing only Gr N and Pr N has poor catalytic performance for ORR in alkaline electrolytes. Density functional theory (DFT) simulations untangle the catalytic nature of Pr and Gr N and decipher the relations between the N type(s) and total N content required for the ORR catalysis. This study provides a new way to design efficient N-doped porous carbon-enriched active sites, and solves the cathode catalyst in the commercialization of PEMFC and metal–air batteries.
:1) between Gr N and Pr N in G&P N-HCS exhibits the highest ORR performances, rather than Gr N-dominated N-HCS or Pr N-dominated N-HCS. Notably, N-HCS containing only Gr N and Pr N has poor catalytic performance for ORR in alkaline electrolytes. Density functional theory (DFT) simulations untangle the catalytic nature of Pr and Gr N and decipher the relations between the N type(s) and total N content required for the ORR catalysis. This study provides a new way to design efficient N-doped porous carbon-enriched active sites, and solves the cathode catalyst in the commercialization of PEMFC and metal–air batteries.
Introduction
To promote the commercialization of Zn–air batteries, it is required to solve problems relating to the cathode catalyst, for example its high usage cost, low activity, and short lifetime. Rational design and exploration of metal-free catalysts to achieve a high-efficiency oxygen reduction reaction (ORR) is among the most promising strategies proposed as an alternative to precious metals.1–4 Over the past decades, heteroatom-doped carbons, particularly N-doped porous carbons, have formed the subject of intensive research.5–8 Nitrogen-doped carbons, particularly graphitic (Gr) and pyridinic (Pr) N significantly improve ORR activity by enhancing the positivity of carbon atoms adjacent to Gr N and Pr N.9–11 Consequently, the synthesis of high-density Gr N- and Pr N-doped carbon catalysts and elucidating their role as ORR catalysis has become a hot research topic.Recent advances have been made regarding the influence of total N content and/or different N types (e.g., graphitic, pyridinic, and pyrrolic N) on ORR activity.12–20 When located in the edge area of carbon frameworks, N-doped carbons containing only effective N species provide plentiful active sites for the catalysis of ORR.21 However, conflicting reports have been published with respect to the active site afforded by N species. For example, Wei and co-workers demonstrated that pyridinic and pyrrolic N with planar sp2 hybridization are active in the ORR, whereas graphitic N with a 3D sp3 structure is responsible for poor catalysis.22 Zhang, Kwak, Yan, and co-workers also verified the important role played by pyridinic N in the catalysis of ORR.23–26 However, Haque and co-workers concluded from experimental results and computational simulation that the total content of N has no influence on ORR, whereas graphitic N (or quaternary N) was observed to be effective in the ORR via four-electron transfer pathways.22 Colavita et al., Zhao et al., and Huang et al. determined that the synergistic effect between Gr N and Pr N improves the activity and selection of ORR by facilitating the dissociative adsorption of O2 molecules along with the desorption of intermediates.27–29 Amidst these ongoing research efforts, established wisdom suggests that both Gr N and Pr N play an indispensable role in catalysis of the ORR. Thus, it is of significance to uncover the influence of relationships between Gr N and Pr N on this reaction.
To achieve maximum usage efficiency of catalysts, the four-electron transfer pathways of N-doped carbon nanostructures are the most desirable alternatives to Pt catalysts.30–34 Many efforts have been devoted to developing high-level N-doping (mainly using Gr N and Pr N) porous carbons able to catalyze the ORR via direct, four-electron transfer pathways.35–38 For better evaluating the catalyst containing Gr N and Pr N, the following two factors must be ensured: (1) high-level N-doping guarantees catalytic activity for the ORR; (2) precisely tunable content of Gr N and Pr N is used to explore well-defined doped active sites in carbon frameworks. Among the various N-doped porous carbons studied, the selection of C and N sources directly influences the total content and hybrid types of N, which has a substantive impact on the performance of the ORR,39,40 however, difficulties in accurately controlling the content of one of the two N types while keeping another type constant have not yet been resolved via a facile synthetic strategy. In addition, mechanism for the catalysis of ORR in Gr N and/or Pr N active sites require to be verified in both alkaline and acidic electrolytes.
In this work, protonated graphitic carbon nitride (P-g-C3N4) and 5-aminotetrazole (ATTZ) were explored as N precursors for the synthesis of high-density Gr N- and Pr N-doped hollow carbon nanospheres (G&P N-HCS) via a facile self-sacrificing template method followed by heat treatment. We investigated the effects of the proportion of effectual N in the total N content as well as the interplay between Gr N and Pr N on the overall activity of as-prepared G&P N-HCS electrodes in the ORR using an alkaline electrolyte. The content of effectual N at the surface of G&P N-HCS determined using X-ray photoelectron spectroscopy (XPS) exceeded 12 at%. Careful optimization of pyrolysis temperatures guarantees the conversion of pyrrolic N into Gr N and/or Pr N while excluding the influence of pyrrolic N. Notably, the content of Pr N enhances with increasing ATTZ, which enables convenient comparison between the activities of Gr N and Pr N. Based the experimental results and density functional theory, we show that both Gr N and Pr N are effective in the ORR, and that optimal proportions of Gr N and Pr N possess the highest ORR activity. To the best of our knowledge, few previous works have investigated the effects of high-density Gr N and Pr N, and their proportions, on the ORR. These results thus provide robust evidence and new insights into the design and synthesis of efficient ORR catalysts.
Results and discussion
Synthesis and morphology characterization of G&P N-HCS. Polystyrene (PS) nanospheres with a homogeneous dispersion state were synthesized via emulsion polymerization according to a slightly modified version of our previous method.41 Meanwhile, we successfully prepared P-g-C3N4 nanosheets from bulk g-C3N4 based on our previous results (see details in Experimental section). For the present work, high-level N-doped HCS with high-density Gr N and Pr N using efficient precursors of P-g-C3N4 and ATTZ was synthesized via a self-sacrificing template strategy, follow by controlled pyrolysis to obtain G&P N-HCSx (where x refers to the ratios of ATTZ and P-g-C3N4; see details in ESI†) as depicted in Scheme 1.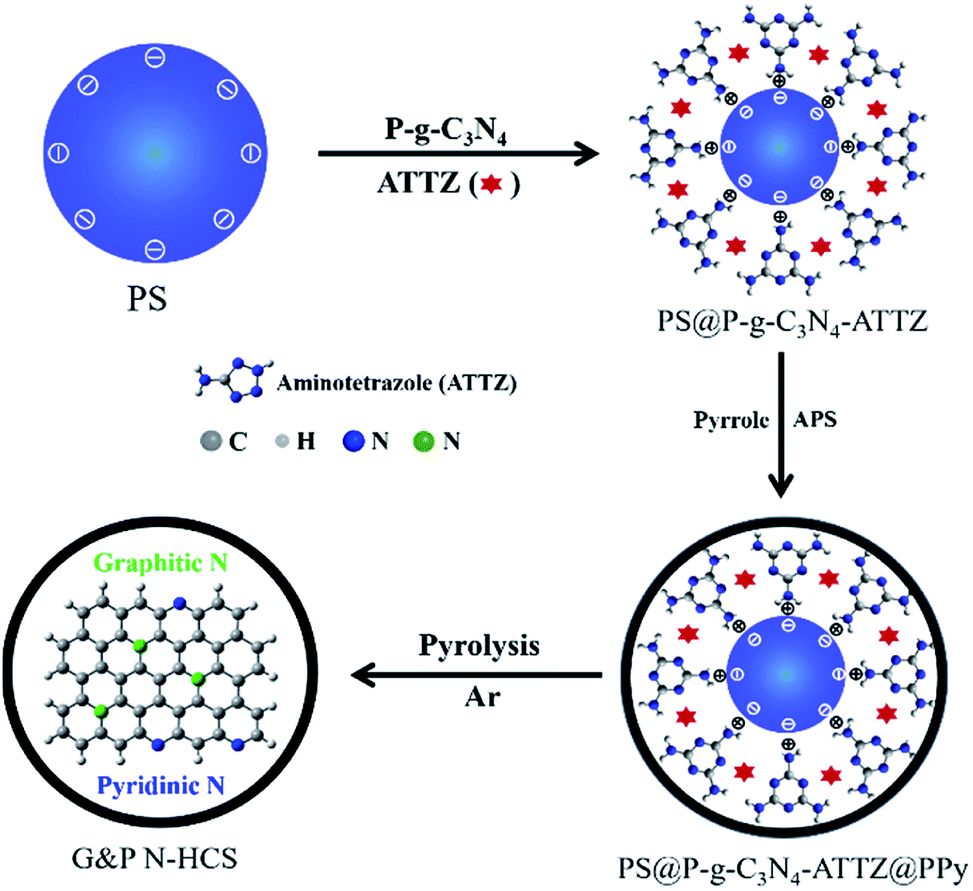 | ||
| Scheme 1 The schematic illustration of the graphitic and pyridinic nitrogen-doped hollow carbon nanospheres (G&P N-HCS). (PS refers to the negatively charged polystyrene nanospheres; P-g-C3N4 refers to the protonated graphitic carbon nitride; APS represents ammonium persulfate.) | ||
Negatively charged PS nanospheres with a uniform diameter of 220 ± 10 nm were used directly as the self-sacrificing template, as demonstrated by TEM and SEM images (Fig. 1a and S1†). These PS nanospheres exhibited a uniform dispersed state with a smooth surface, but also possessed a stable spherical structure. Protonated g-C3N4 nanosheets (P-g-C3N4) played a key role in linking PS and ATTZ (the N source) via electrostatic interactions and hydrogen bonding, respectively.5 In contrast to bulk g-C3N4, characterized by a large size, smaller positively charged P-g-C3N4 nanosheets were observed, as verified in Fig. S2.† After coating these materials with a layer of polypyrrole (PPy), we obtained the N-containing precursor of PS@P-g-C3N4-ATTZ@PPy with a unique core–shell structure. As indicated in Fig. 1b and S3,† this spherical PS@P-g-C3N4-ATTZ@PPy exhibited a diameter of 250 ± 10 nm and its rough surface remained after polymerization, suggesting that the PPy coating successfully covered the PS nanospheres. Controlled carbonization at 950 °C under Ar flux produced high-density Gr- and Pr-N-doped hollow carbon nanospheres (G&P N-HCS4). The high temperature used guaranteed the conversion of pyrrolic N into graphitic N, thereby enabling our investigation of the effects of Gr and Pr N on ORR using efficient nitrogen sources of ATTZ. Fig. 1c and S4† display TEM and SEM images of G&P N-HCS4, respectively. A hollow carbon shell with a uniform diameter of 230 ± 10 nm was observed, confirming the feasibility of our experimental plan. Our as-prepared G&P N-HCS4 exhibited a clear shell thickness of 8 ± 1 nm, indicating good structural stability and robust stiffness (Fig. 1d). High-resolution TEM images (Fig. 1e) show the crystalline region of G&P N-HCS4, which corresponds to the graphitic phase (yellow dotted circle); the presence of this phase was as also verified by Raman spectroscopy (Fig. S5†).
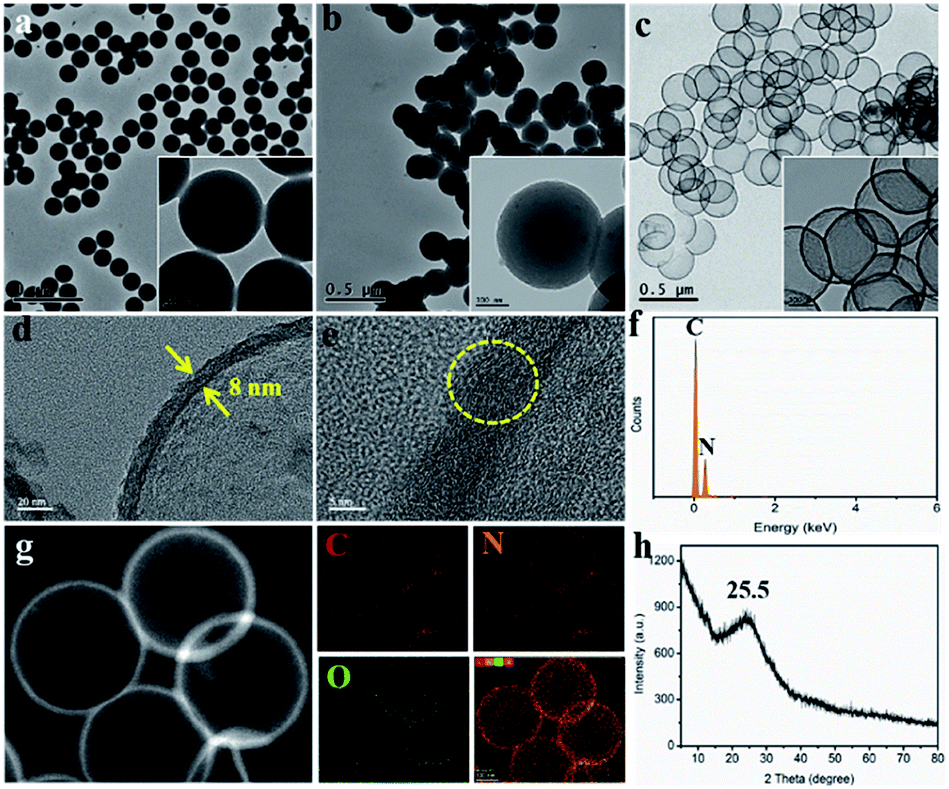 | ||
| Fig. 1 (a)–(c) TEM images of PS nanospheres, PS@P-g-C3N4-ATTZ@PPy, and G&P N-HCS4, respectively, the inserts are corresponding high-magnification TEM images. (d) and (e) High-resolution TEM images of G&P N-HCS4 (the yellow dotted circle refers to the crystalline region). (f) EDS curve of G&P N-HCS4. (g) Scanning transmission electron microscopy (STEM) image of G&P N-HCS4 and the corresponding elements mapping and overlapping image of C, N, O elements. (h) XRD profile of G&P N-HCS4. | ||
The content and dispersed state of nitrogen have a significant influence on the performance of carbon frameworks. High-level N-doping was demonstrated by energy dispersive X-ray spectroscopy (EDS) (Fig. 1f), whereas element analysis (EA) technology further gave further details of N element (Table S1†). The total N content in G&P N-HCS samples were all greater than 12.0 at% (with a maximum value of 15.2 at%), which exceeds the corresponding value in most N-doped carbons. Fig. 1g shows a high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) micrograph of G&P N-HCS4 together with the corresponding elemental maps. N was homogeneously distributed throughout the skeleton of G&P N-HCS4, together with a small quantity of O arising from the adsorption of sodium dodecyl sulfate and O2 onto the PS surface. The powder X-ray diffraction (XRD) pattern of this material, displayed in Fig. 1h, exhibits a broad peak at 25.5° ascribed to the (002) plane of the graphite.
The high specific surface area (SBET) and rational pore size distribution (PSD) measured by N2 adsorption/desorption isotherms at 77 K allow improved catalytic activity of G&P N-HCS. As shown in Fig. S6,† a typical H-4 isotherm corresponding to an obvious hysteresis loop was observed, indicating the characteristics of micropores/macropores derived from capillary condensation in all samples.42,43 Notably, G&P N-HCS exhibited the highest adsorption capability, such that the resulting SBET and PSD were superior to that of either HCS or N-HCS. Detailed textural parameters are summarized and displayed in Fig. S7 and Table S2.† Benefiting from the pyrolysis of P-g-C3N4 and ATTZ, G&P N-HCS4 also displayed a high SBET of 258 m2 g−1, which is much higher than the corresponding value in either HCS (85 m2 g−1) or N-HCS (154 m2 g−1). The total volume of G&P N-HCS4 was determined to be 0.81 cm3 g−1 according to the Barrett–Joyner–Halenda model at specific pressure (P/P0 = 0.99); this value was the highest among all samples, revealing that ATTZ can greatly enhance pore volume. Moreover, the micropore-dominated G&P N-HCS samples, with total micropore volume of 0.73 cm3 g−1 and mean pore size of 0.65 nm are advantageous for absorbing oxygen. Based on the results discussed above, our G&P N-HCS samples possess optimal physical structures and thus efficiently promote the ORR.
X-ray photoelectron spectroscopy (XPS) studies
The chemical environment of our samples was investigated using X-ray photoelectron spectroscopy (XPS). As shown in Fig. 2a, signals centered at 285, 400, and 530 eV were verified, corresponding to C 1s, N 1s, and O 1s, respectively. This is consistent with result from XRD and element mapping. Notably, signals of O were observed in all samples, which is ascribed to the adsorption of O on the surface of our samples. In addition, XPS survey spectra (Fig. 2a) revealed that the surface N contents of our samples were 4.2, 6.5, 8.7, 9.1, 10.8, 13.2, and 13.4 at%, respectively, consistent with EDS and EA measurements. Fig. 2b shows the high-resolution C 1s XPS spectra of all samples, which can be deconvoluted into three peaks centered around 284.6, 285.5, and 287.2 eV, corresponding to the C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N/CN, and C–O bonds, respectively. The appearance of the C–N/CN peak at 285.5 eV demonstrates successful N-doping of the C frameworks. A significant effect of total N content and N types (Gr N and Pr N) on the ORR of N-doped carbons has been demonstrated by pioneering works. N species were identified using high-resolution N 1s spectra (Fig. 2c). The high-resolution N 1s spectrum of HCS derived from PPy at 750 °C was deconvoluted into three peaks centered around 398.5, 400.1, and 401.1 eV, corresponding to Pr N, pyrrolic N, and Gr N, respectively. The proportion of pyrrolic N is highest among these species, indicating the low-efficiency conversion of N originating from PPy into pyridinic and graphitic N at 750 °C. Following the addition of P-g-C3N4 and a temperature increase to 950 °C, the high-resolution N 1s spectrum of N-HCS exhibited a high level of Gr and Pr N, revealing that increased temperatures significantly facilitate the conversion of pyrrolic N into Gr and Pr N.21 Compared to mainstream N-doped carbon catalysts, N-HCS possesses a low total N content such that it fails to further improve the performance of the ORR. Increasing the total N content for the enhancement of the ORR can be achieved by using an efficient N source of ATTZ. As expected, the total N content, including Gr N and Pr N, exhibited a rising trend, with the highest value reaching 13.4 at% i.e., higher than that of the majority N-doped carbon catalysts reported previously. As shown in Fig. 2c and S8,† both the ratio and percentage of Gr N and Pr N in G&P N-HCSx samples changed significantly following the increase of ATTZ amount. This is very beneficial for investigating the influence of Gr N and Pr N on ORR performance and for the design of an efficient N-doped carbon catalyst.
C, C–N/CN, and C–O bonds, respectively. The appearance of the C–N/CN peak at 285.5 eV demonstrates successful N-doping of the C frameworks. A significant effect of total N content and N types (Gr N and Pr N) on the ORR of N-doped carbons has been demonstrated by pioneering works. N species were identified using high-resolution N 1s spectra (Fig. 2c). The high-resolution N 1s spectrum of HCS derived from PPy at 750 °C was deconvoluted into three peaks centered around 398.5, 400.1, and 401.1 eV, corresponding to Pr N, pyrrolic N, and Gr N, respectively. The proportion of pyrrolic N is highest among these species, indicating the low-efficiency conversion of N originating from PPy into pyridinic and graphitic N at 750 °C. Following the addition of P-g-C3N4 and a temperature increase to 950 °C, the high-resolution N 1s spectrum of N-HCS exhibited a high level of Gr and Pr N, revealing that increased temperatures significantly facilitate the conversion of pyrrolic N into Gr and Pr N.21 Compared to mainstream N-doped carbon catalysts, N-HCS possesses a low total N content such that it fails to further improve the performance of the ORR. Increasing the total N content for the enhancement of the ORR can be achieved by using an efficient N source of ATTZ. As expected, the total N content, including Gr N and Pr N, exhibited a rising trend, with the highest value reaching 13.4 at% i.e., higher than that of the majority N-doped carbon catalysts reported previously. As shown in Fig. 2c and S8,† both the ratio and percentage of Gr N and Pr N in G&P N-HCSx samples changed significantly following the increase of ATTZ amount. This is very beneficial for investigating the influence of Gr N and Pr N on ORR performance and for the design of an efficient N-doped carbon catalyst.
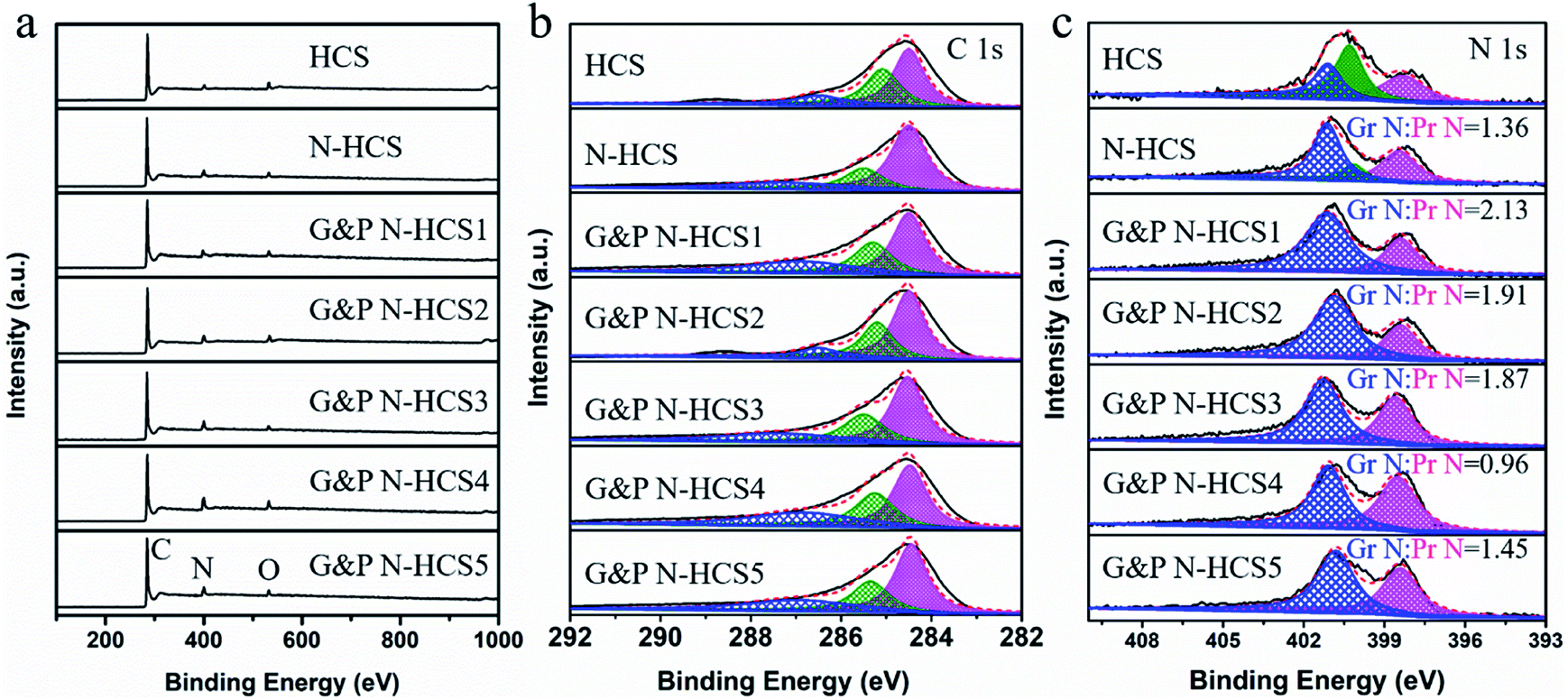 | ||
| Fig. 2 (a) X-ray photoelectron spectroscopy (XPS) survey spectra, (b) deconvoluted XPS spectra for high-resolution C 1s and (c) N 1s of HCS, N-HCS, G&P N-HCSx (x = 1, 2, 3, 4, and 5), respectively. The ratio of Gr N and Pr N refers to the proportion between peak area of graphitic N and peak area of pyridinic N. | ||
Electrocatalytic performance for ORR
In order to decipher the influence of structural stability and the catalytic activity of Gr N and Pr N on the ORR, electrochemical measurements based on a three-electrode workstation were performed at 0.1 M KOH (Fig. 3). The reversibility of the glass carbon electrode (working electrode) was demonstrated using a standard cyclic voltammetry (CV) test of the [Fe(CN)6]3−/[Fe(CN)6]4− redox couple (Fig. S9†) before each electrochemical measurement. Potential differences between the cathodic and anodic peaks were approximately 72 mV, suggesting that our glass carbon electrode is highly reversible. Fig. 3a shows the CV curves of G&P N-HCS4. Compared to the CV curve under the N2 flow, an obvious O2 cathodic peak was observed under the O2 flow, indicating that G&P N-HCS4 possesses outstanding ORR activity. Fig. S10† demonstrates the influence of N content on ORR, which was initially evaluated by linear sweep voltammetry (LSV) technology at 1600 rpm with a scanning rate of 10 mV s−1. The HCS sample showed a low half-wave potential (0.58 V) and on-set potential (0.68 V), i.e., inferior to that of N-HCS with increasing N content. As the N content continues to increase, the ORR performance of G&P N-HCS4 including half-wave potential (0.81 V), on-set potential at which current density reaches 0.5 mA cm−2 (0.91 V),21 and diffusion-limited current density (4.5 mA cm−2) was observed to increase (Fig. S11†), revealing that N content has a positive effect on ORR in alkaline media.44 Moreover, the high-level N (13.2 at%) in G&P N-HCS4 demonstrates the applicability of our design ideas and preparing process.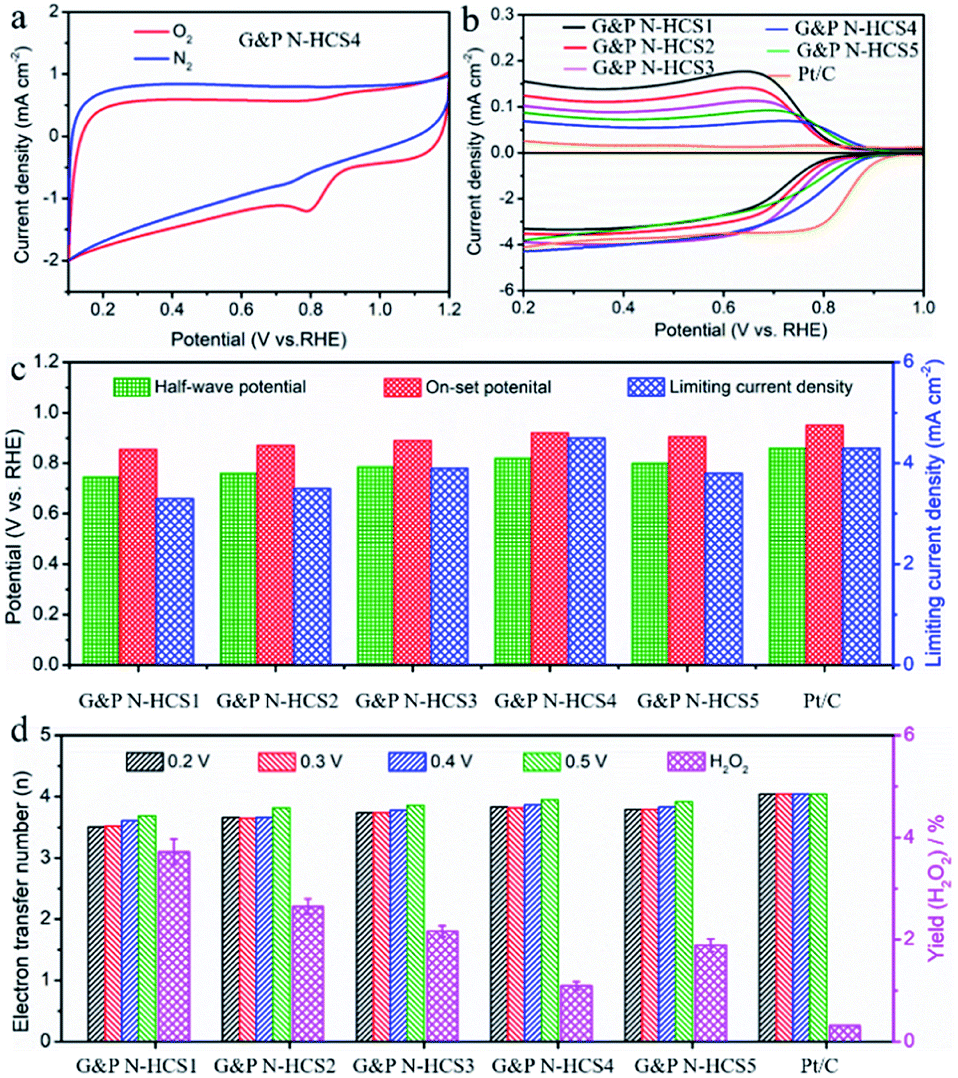 | ||
| Fig. 3 Evaluation of ORR performance of these samples and the commercial JM 40% Pt/C in 0.1 M KOH solution. (a) Cyclic voltammetry (CV) curves of G&P N-HCS4 in O2 and N2-saturated 0.1 M KOH electrolyte with a scan rate of 20 mV s−1. (b) RRDE measurement of G&P N-HCSx (x = 1, 2, 3, 4, and 5), and Pt/C samples under 1600 rpm with a scan rate of 10 mV s−1. (c) The half-wave potential, on-set potential, and diffusion-limited current density of G&P N-HCSx (x = 1, 2, 3, 4, and 5), and Pt/C samples, respectively. (d) Electron transfer number (n) obtained from RRDE tests and the H2O2 yield of G&P N-HCSx (x = 1, 2, 3, 4, and 5), and Pt/C samples, respectively. | ||
The influence of the proportion of Gr N and Pr N on ORR performance was also investigated by using rotating disk electrode and rotating ring-disk electrode (RRDE) tests in 0.1 M KOH solution (Fig. S12–S17†). As indicated in Fig. 3b, G&P N-HCSx (x = 1, 2, 3, 4, and 5) samples exhibited outstanding ORR performance. The LSV plot of G&P N-HCS1 (Gr N:Pr N = 2.13) exhibited the lowest ORR on-set potential (Eon = 0.85 V), determined as the potential at which current density reaches 0.5 mA cm−2. As the proportion of Pr N continues to increase, G&P N-HCS4 with a ratio of Pr N to Gr N close to 1 (Gr N:Pr N = 0.98) displayed the most positive on-set potential (Eon = 0.91 V), indicating the strong catalytic capability of Pr N. In addition, the increase of the Gr N proportion facilitated the ORR, such as in the on-set potential: G&P N-HCS3 > G&P N-HCS2 > G&P N-HCS1 (Table 1). However, G&P N-HCS5 with the highest Gr N proportion (7.9 at%) exhibited the second highest on-set potential (Eon = 0.90 V), revealing that Gr N is slightly inferior to Pr N in term of catalytic capability. Detailed electrochemical parameters, including half-wave potential, on-set potential, and limiting current density, are summarized in Fig. 3c. RRDE tests (Fig. 3b) also demonstrated that G&P N-HCS4 exhibits the lowest ring current, which is superior to G&P N-HCS5. The corresponding H2O2 yield and transfer electron number calculated at different potentials are summarized in Fig. 3d, S18 and S19.† Electrochemical impedance spectroscopy (EIS) results indicated that the electrochemical resistances of our samples are as follows: G&P N-HCS5 > G&P N-HCS4 > G&P N-HCS3 > G&P N-HCS2 > G&P N-HCS1 (Fig. S20†). This reflect the fact that G&P N-HCS5 endowed with the highest Gr N proportion (7.9 at%) improved the conductivity of carbon frameworks. Notably, the ORR performance of G&P N-HCS4 is comparable to that of commercial Pt/C, particularly in accelerated durability test, methanol tolerance test, and in terms of transfer electron number (Table 1, Fig. S21 and S22†). The above mentioned discussion verified that Pr N plays an indispensable role in catalyzing ORR, whereas Gr is important but not decisive.
| Samples | Total N content (at%) | Gr N percentage (%) | Pr N percentage (%) | Gr N content (at%) | Pr N content (at%) | Ring currenta (mA cm−2) | Transfer electron numberb (n) |
|---|---|---|---|---|---|---|---|
| a Ring current is measured at 0.2 V (vs. RHE).b The n refers to the mean of transfer electron number between 0.2 and 0.4 V (vs. RHE). | |||||||
| G&P N-HCS1 | 8.7 | 68.1 | 31.9 | 5.9 | 2.8 | 0.14 | 3.92 |
| G&P N-HCS2 | 9.1 | 65.6 | 34.4 | 6.1 | 3.0 | 0.11 | 3.94 |
| G&P N-HCS3 | 10.8 | 65.1 | 34.9 | 7.0 | 3.1 | 0.09 | 3.95 |
| G&P N-HCS4 | 13.2 | 48.9 | 51.1 | 6.5 | 6.7 | 0.06 | 3.97 |
| G&P N-HCS5 | 13.4. | 59.2 | 40.8 | 7.9 | 5.5 | 0.07 | 3.96 |
| Pt/C | — | — | — | — | — | 0.02 | 4.06 |
Theoretical study of G&P N-HCS for ORR
Density function theory analyse were performed in order to elucidate ORR performance by investigating the structural stability and catalytic activity of Gr N and Pr N, respectively. Pure graphene (Gr) and seven N-doped graphene models (denoted as 0PrN_6GrN, 1PrN_5GrN, 2PrN_4GrN, 3PrN_3GrN, 4PrN_2GrN, 5PrN_1GrN and 6PrN_0GrN) with different Pr N/Gr N ratio were optimized for this purpose (Fig. 4).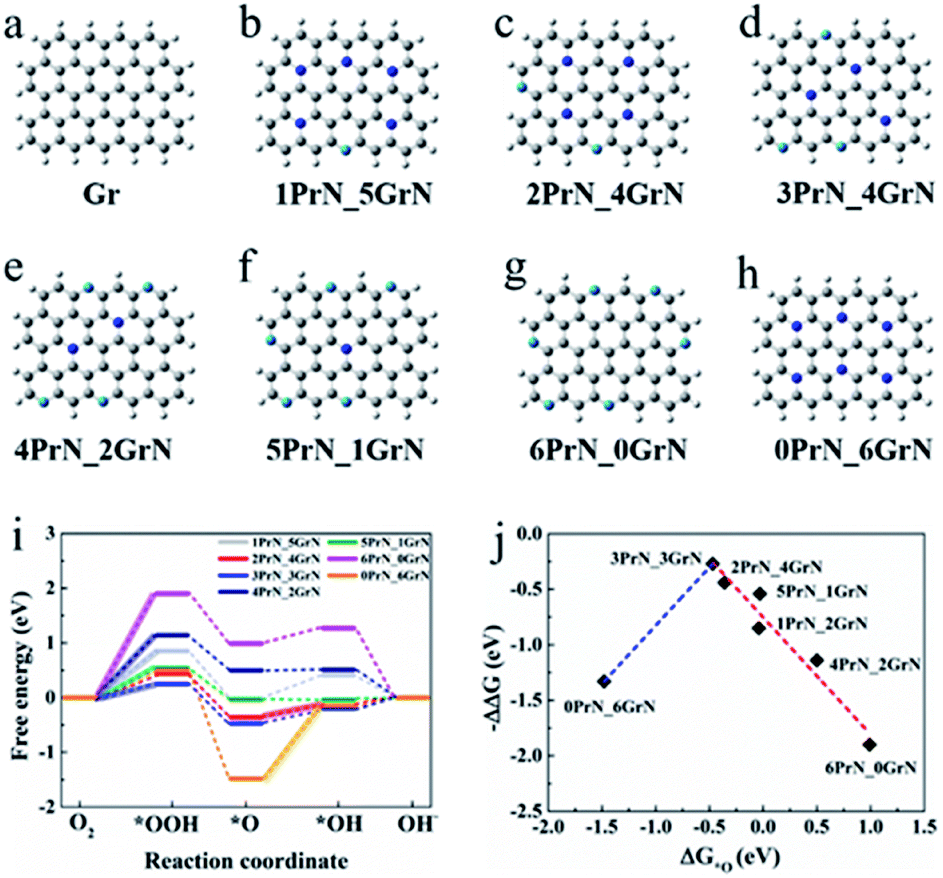 | ||
Fig. 4 Structural models and DFT calculations. (a–h) Optimized configurations for graphene and seven N-doped models. (C atom: gray, Gr N atom: blue, Pr N atom: light blue, H atom: white). (i) Gibbs free energy diagrams at 0.460 V for ORR over seven nitrogen doped graphene models in alkaline media. The highlights indicate the rate-determining step with the values of the limiting energy barrier labelled. (j) The “volcanic curve” relationship between 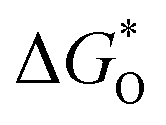 and negative energy barrier (−ΔΔG). and negative energy barrier (−ΔΔG). | ||
The energy separation between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), a simple indicator of kinetic stability, was used to conduct an initial investigation of the catalytic ability of each graphene modal, with or without nitrogen doping. A small HOMO–LUMO gap can be considered to reflect low kinetic stability and high chemical reactivity, thus easing the catalytic reaction.45 Values of the HOMO–LUMO energy gaps for these graphene models are listed in Table S3.† Compared to pure graphene (1.90 eV), the substitution of C by N leads to a significant decrease in energy gap; however, overdoping by Pr N, as in the case of 6PrN_0GrN, results in a remarkable increase in the energy gap. As a result, 6PrN_0GrN has the highest energy gap (1.93 eV), which is similar to that of Gr. The energy gap gradually increased from 1.12 to 1.93 eV when the number of Pr N in doped models rose from 0 to 6, except in the case of Pr N:Gr N = 5, suggesting that the catalytic performance of doped Gr, except 5PrN_1GrN (1.26 eV), falls the increase of Pr N:Gr N. Indeed, energy gap analysis is not always strictly related to ORR catalytic ability, for example, ORR free energy calculations showed that 0PrN_6GrN (1.12 eV) exhibits low catalytic reactivity, verifying that the addition of Pr N leads to improved catalytic capacity. Overall, energy gap calculations demonstrated that either Pr N or Gr N plays an important role in ORR catalytic activity related to doped graphene, consistent with the above-mentioned experimental results.
To illustrate in detail, the effect of the ratio of Pr N and Gr N on the catalytic performance of the ORR, the variation of ORR free energy was analyzed. The optimal atomic configurations of ORR intermediate states for the doped models are shown in Fig. S29† and their adsorption free energy changes are listed in Table S4.† From these optimized intermediates, O2 molecules could not be directly adsorbed on pyridinic N-graphene, as is the case of 1PrN_5GrN, 2PrN_4GrN, 3PrN_3GrN and 6PrN_0GrN, consistent with previous reports.46–48 OH and OOH underwent top site adsorption, while O was adsorbed on either a top site or a “bridge” site (Fig. S26 and S27†). Free energy diagrams revealed an uphill energy (highlighted in Fig. 4i) increase in the following order: 3PrN_3GrN (0.27 eV) < 2PrN_4GrN (0.44 eV) < 5PrN_1GrN (0.54 eV) < 1PrN_5GrN (0.85 eV) < 4PrN_2GrN (1.14 eV) < 0PrN_6GrN (1.33 eV) < 6PrN_0GrN (1.93 eV) for ORR with the adsorption of *OOH or *OH as the rate-limiting step (Fig. 4i and Table S5†) at U = 0.46 V in alkaline media. 3PrN_3GrN with an optimum Gr N:Pr N ratio of 1 and the lowest energy barrier (ΔΔG = 0.25 eV) exhibited the highest ORR catalytic activity, confirming the above experimental descriptions suggesting that G&P N-HCS4 exhibited the most positive on-set potential with a Gr N:Pr N ratio of 0.98. Moreover, our theoretical calculations indicated that the energy barrier for 2PrN_4GrN, 1PrN_5GrN and 0PrN_6GrN increased gradually from 0.59 to 1.33 eV with increasing Gr N:Pr N ratio, denoting that the catalytic activity for these three N-doped models declined slowly and would be very weak when not Pr N-doped, such as in the case of 0PrN_6GrN. However, the over-doping of Pr N, such as in the case of 6PrN_0GrN (1.93 eV), led to dramatic drops in catalytic performance, suggesting that Pr N is indispensable for ORR catalysis; this is in good agreement with experimental evidence. Furthermore, the adsorption of the *O intermediate appears to be a reliable descriptor for ORR activity and a “volcanic curve” relationship between 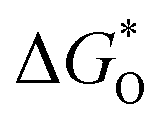 and negative energy barrier (−ΔΔG) was obtained (Fig. 4j). Closer to the vertex of the “volcanic curve”, energy barriers of the rate-limiting steps are lower. A similar trend in ORR activity was observed in 3PrN_3GrN with a lower *O adsorption energy of −0.47 eV and in the lowest energy barrier of 0.25 eV, which also corroborated experimental results.
and negative energy barrier (−ΔΔG) was obtained (Fig. 4j). Closer to the vertex of the “volcanic curve”, energy barriers of the rate-limiting steps are lower. A similar trend in ORR activity was observed in 3PrN_3GrN with a lower *O adsorption energy of −0.47 eV and in the lowest energy barrier of 0.25 eV, which also corroborated experimental results.
Electrocatalytic performance for Zn–air battery
The practical applications of our G&P N-HCS catalysts for ORR were examined using a home-made Zn–air battery device. As indicated in Fig. 5a, the open-circuit voltage (OCV) of the G&P N-HCS4-based Zn–air battery reaches 1.42 V even after 2000 s, which is closed to the Zn–air battery fabricated with the state-of-the-art Pt/C catalyst. This stable OCV verifies the high catalytic activity of the G&P N-HCS4 catalyst for ORR. The insert in Fig. 5a (left) reveals that the Zn–air battery based on G&P N-HCS4 catalyst delivered a OCV of 1.43 V (two batteries connected in series). Moreover, two Zn–air batteries assembled with G&P N-HCS4 catalyst could light up a pattern (NTU represents the Nantong University) composed of many LEDs (2.5 V). Fig. 5b displays the discharge polarization curves as well as the corresponding power density plots of G&P N-HCS4 and Pt/C catalysts, respectively. The Zn–air battery assembled with G&P N-HCS4 catalyst delivered a high power density of 236 mW cm−2 at a current density of 350 mA cm−2, which is comparable to the Pt/C catalyst. The cyclic stability of Zn–air batteries fabricated with the as-prepared catalysts was evaluated by a galvanostatic discharge–charge test at 10 mA cm−2 for both discharging (5 min) and charging (5 min). As shown in Fig. 5c, the cyclic stability of G&P N-HCS4 is far superior to Pt/C after 500 cycles (84.2 h), as demonstrated by the stable discharge plateau at about 1.1 V. Moreover, such the remarkable cyclic durability outperforms a number of N-doped catalysts.49–54 The specific capacity of the Zn–air battery based on the G&P N-HCS4 reaches 696 mA h g−1 at a discharge current density of 5 mA cm−2 (Fig. 5d). Moreover, the practical application of solid state Zn–air battery assembled with G&P N-HCS4 catalyst was confirmed by powering a pattern composed of many 2.5 V LEDs (Fig. S30†). These results firmly suggest that the promising application of our G&P N-HCS catalysts in energy storage devices.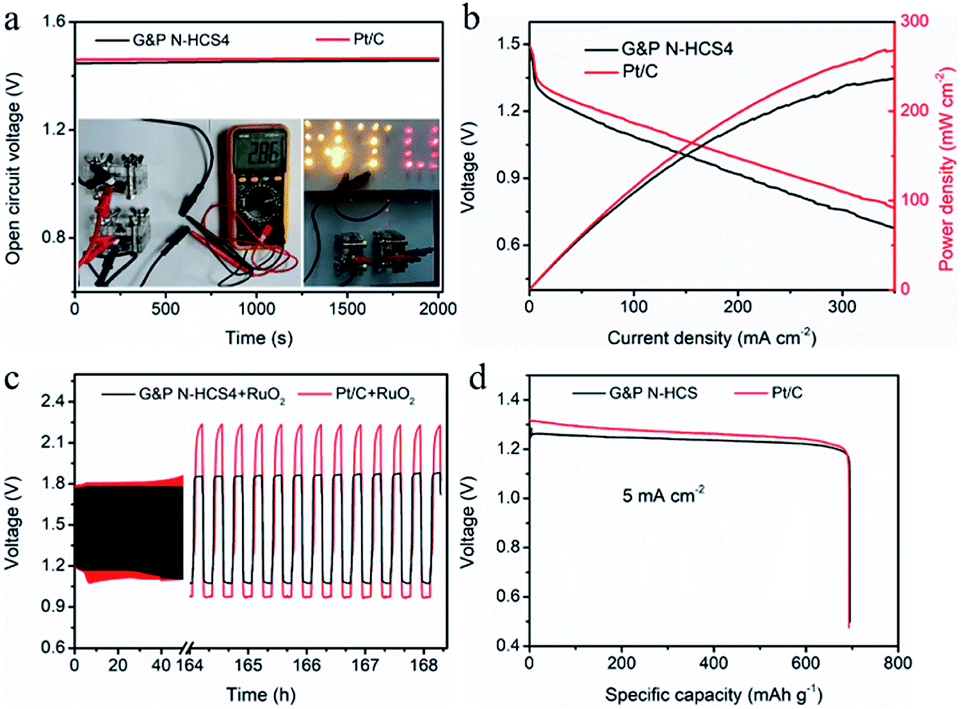 | ||
| Fig. 5 The performances of Zn–air battery fabricated with the G&P N-HCS4 and commercial Pt/C. (a) Open-circuit voltage time-plots, insert is a photograph of two Zn–air batteries connected in series showing an open potential of 2.86 V (Left), a pattern (NTU refer to the abbreviation of Nantong University) composed of LEDs being powered two Zn–air batteries connected in series (Right). (b) Discharge polarization curves and corresponding power–density profiles. (c) Galvanostatic discharge–charge (10 mA cm−2) cycling performance of a rechargeable battery based on G&P N-HCS4 and Pt/C catalysts, 5 min for discharging and 5 min for charging. (d) Specific capacity of Zn–air batteries based on G&P N-HCS4 and Pt/C catalysts at a constant discharge density of 5mA cm−2. | ||
Conclusions
In summary, a series of G&P N-HCS metal-free catalysts, exhibiting specific nitrogen sites and the most efficient structure model, were studied in order to untangle the effect of the structural stability and catalytic activity of different N sites on the ORR, this series was rationally constructed from P-g-C3N4. As-prepared G&P N-HCS, featuring high-density Gr N and Pr N, were explored as ideal model catalyst to decipher the intrinsic properties of N-doped carbon catalysts. We carried out a range of electrochemical measurements including linear sweep voltammetry and an accelerated durability test, on these catalysts and the corresponding results provide direct evidence that both the structural stability and catalytic activity of Gr N and Pr N contribute to the superior catalytic performance of our catalysts for ORR via the 4e− pathway in G&P N-HCS catalysts. Gr N-dominated and Pr N-dominated N-HCS catalysts were observed to possess inferior catalytic activity toward the ORR. Our findings were supported by density functional theory calculations, wherein the carbon skeleton adjacent to the Gr N and Pr N sites displays low Gibbs free energy and high catalytic activity, when compared with the catalyst containing only Gr N and Pr N sites. Taken together, this study provides an insightful statement for ORR catalysis, unambiguously revealing the superior intrinsic catalytic behaviour of Gr N and Pr N.Experimental section
Chemicals and regents
Pyrrole (C4H5N, CP), styrene (C8H8, >99.5%), amino-1H-tetrazole (CH3N5, >99.0%), sodium dodecyl sulfate (SDS, >99.0%), were purchased from Aladdin Chemistry Co., Ltd. Pyrrole and styrene monomer were distilled under reduced pressure before use. Pt/C (40%, JM) powders (HiSPEC 3000) were obtained from Alfa Aesar. Thiourea (CH4N2S, >99.0%), sodium carbonate (Na2CO3, >98.0%), potassium persulfate (K2S2O8, >99.0%), sodium persulfate ((NH4)2S2O8, >98.0%), polyvinyl alcohol (PVA) were purchased from Sinopharm Chemicals Co., Ltd. Potassium hydroxide (KOH, >95.0%), sulfuric acid (H2SO4, >98.0%), methanol (CH3OH, >99.5%), ethanol (C2H5OH, >99.0%) were provided by Xilong Chemical Reagent Co., Ltd. Distilled deionized water (>18.2 MΩ) was produced by reverse osmosis followed by ion-exchange and filtration (HBI, Shenzhen Honbo Water Treatment Equipment Co., Ltd., China). All chemicals and regents were used without further purification apart from pyrrole and styrene.Synthesis of negatively charged polystyrene (PS) nanospheres
PS dispersion was synthesized via a modified emulsion polymerization method according to our previous work. In brief, anhydrous Na2CO3 (0.1 g, 0.94 mmol) and SDS (0.2 g, 0.69 mmol) were dissolved into 300 mL distilled water, then transferred them into a round-bottom flask and purging with N2 for 30 min. Subsequently, styrene (30 mL) was added rapidly, keeping intense agitation for 30 min at 60 °C. Finally, pouring the K2S2O8 aqueous solution (10 mL, 1 mmol) into the above reaction system, heated to 75 °C, and keeping for 20 h. After cooling down to room temperature, the products were centrifuged at 15000 rpm for 30 min for several times with distilled water. The as-obtained PS nanospheres were re-dispersed in 90 mL distilled water, and was denoted PS mother solution.
Synthesis of protonated graphitic carbon nitride (P-g-C3N4) nanosheets
Bulk g-C3N4 powder was used to synthesize the P-g-C3N4 nanosheets. In brief, thiourea (5.0 g) was encapsulated into the corundum crucible with a lid, then the crucible was placed in tubular furnace, followed by a heat treatment from room temperature to 550 °C (keeping for 2 h) with a heating rate of 2.3 °C min−1 in N2 flow. After cooling down to room temperature, the products were grinded thoroughly with an agate mortar, and was denoted as bulk g-C3N4. For the synthesis of P-g-C3N4, bulk g-C3N4 (200 mg) powder was added into a 500 mL round flask containing the mixed concentrated sulfuric acid and nitric acid (40 mL) with a volume ratio of 1:3. Afterwards, the round flask was sonicated severely until the solution is clear. Pouring deionized water (400 mL) into the clear solution to form a dispersion of P-g-C3N4 nanosheets. After the centrifugation by using deionized water at 10000 rpm for several times, the as-obtained product was collected, and was denoted P-g-C3N4.
Synthesis of PS@P-g-C3N4-ATTZ@PPy precursor
Briefly, PS mother solution (2 mL) was dispersed into a beaker containing 100 mL deionized water, then P-g-C3N4 (100 mg) and ATTZ (50 mg) were added to generate homogeneous dispersion. Subsequently, 0.1 mL pyrrole monomer was added to the above system under stirring, accompanied by a polymerization process via adding the potassium persulfate solution (20 mmol L−1, 20 mL) drop by drop. After four hours of polymerization, the mixture was filtered and washed with deionized water and ethanol several times. Finally, the product was dried at 60 °C vacuum oven over the night, which denotes as PS@P-g-C3N4-ATTZ@PPy. For better understanding the effect of nitrogen content on ORR, a series of PS@P-g-C3N4-ATTZ@PPy precursors were prepared in the same way excepting for different amounts of ATTZ (100, 150, 200, 250 mg).Synthesis of high-density graphitic and pyridinic nitrogen-doped hollow carbon nanospheres (G&P N-HCS): the as-obtained several precursors was carbonized at 950 °C with a heating rate of 5 °C min−1 for 2 h under N2 flowing, which denotes as G&P N-HCS. Notably, different amounts of ATTZ derived G&P N-HCS products were denoted G&P N-HCSx (x refer to the weight ratio of P-g-C3N4 and ATTZ). For example, as the amount of ATTZ to be 50 mg (ATTZ:P-g-C3N4 = 1:2), the as-prepared product was denoted G&P N-HCS1. Other products were denoted G&P N-HCS2, G&P N-HCS3, G&P N-HCS4, and G&P N-HCS5 successively. In particular, when the ATTZ is absent, the corresponding product was denoted N-HCS. The product derived from PPy denoted as HCS.
Characterization
Field emission scanning electron microscopy (FESEM) images were performed by a Gemini SEM 300. Transmission electron microscopic (TEM) images and energy dispersive spectroscopy (EDS) mappings were analyzed using a Tecnai G2 F20 at 200 kV. Powder X-ray diffraction (XRD) patterns were collected on a Philips X'pert PRO with nickel-fleered Cu Kα radiation. Raman spectra were carried out using a LabRAM HR800 with an excitation wavelength of 532 nm. The chemical composition and environment were conducted on an X-ray photoelectron spectroscopic (XPS, Shimadzu ESCA-3400) using an Mg Kα source. N2 adsorption/desorption isotherms were recorded on a Micromeritics ASAP 2020 instrument using a Brunauer–Emmett–Teller (BET) method at 77 K. Pore-size distributions (PSD) were measured by using the non-localized density functional theory (NLDFT) method. Mesopore volumes were calculated by Barrett, Joyner and Halenda (BJH) method. The elemental contents of catalysts were performed by elemental analyzer (EA).Electrochemical measurements
The ORR performances were conducted by using an electrochemical workstation (CHI760E, Shanghai, Chenhua, Co., Ltd.) equipped with a three-electrode system in 0.1 M KOH electrolyte at 25 °C. The ORR activity was measured by using rotation disk electrode (RDE) and rotation ring-disk electrode (RRDE) technologies. The catalysts inks were prepared as follows. The as-prepared catalyst (16 mg) was added to a Schering bottles (20 mL) containing 5 wt% Nafion solution (160 μL), deionized water (3 mL), and isopropanol (1 mL), then the reactor was sonicated for at least 30 min to form a homogenous ink. The catalyst ink (15 μL) was drop-casted onto a glass carbon electrode (5 mm in diameter) and dried at 30 °C for 12 h successively, then denote as working electrode. The loading of catalysts is controlled to be 0.3 mg cm−2 for all samples in the same way. For comparison, the JM Pt/C (40 wt%) benchmark catalyst with the same loading amount was also fabricated. An Pt ring used as the counter electrode and a Ag/AgCl electrode (saturated KCl) acted as the reference electrode. All the potentials were calibrated to the reversible hydrogen electrode (RHE) scale according to Evs. RHE = Evs. Ag/AgCl +0.0591pH + 0.197.The activity of glass carbon electrode was verified by using cyclic voltammetry (CV) test with a scanning rate of 50 mV s−1 in the electrolyte containing K3 [Fe(CN)6] (1 mM) and KNO3 (0.5 M) until the potential difference between oxidation and reduction peaks of [Fe(CN)6]3−/[Fe(CN)6]4− redox couple less than 70 mV. O2 and/or N2 was ventilated in 0.1 M KOH and/or 0.5 M H2SO4 solution at least 30 min to guarantee the saturation of electrolyte before each test. For ORR, CV tests were conducted at different scanning rate (10, 20, 50, 100, and 200 mV s−1) over a potential window of 0.1–1.1 V (vs. RHE). The kinetics tests were recorded on a linear sweep voltammetry (LSV) technology with a rotation speed range from 225 to 2025 rpm at a scanning rate of 10 mV s−1 over a potential window of 0.1–1.1 V (vs. RHE). Tafel slopes were calculated from the Tafel equation:
|
η = a + blogj
| (1) |
For RDE method, the kinetic-limiting current and electron transfer numbers were calculated according to the Koutecky–Levich equation.
| 1/j = 1/jK +1/jL = 1/jK + 1/(Bω1/2) | (2) |
|
B = 0.62nFC0(D0)2/3ν−1/6
| (3) |
485C mol−1), C0 is the concentration of O2 (1.2 × 10−6 mol cm−3), D0 represents the diffusion coefficient of O2 in the electrolyte (1.9 × 10−5 cm2 s−1), and ν is the kinetic viscosity (0.01 cm2 s−1).
For RRDE method, the LSV tests were carried out in O2 and/or N2-saturated electrolyte at a scanning rate of 10 mV s−1 under 1600 rpm. The production of H2O2 was detected by using LSV measured at 0.5 V vs. RHE. The yield of H2O2 and electron transfer number were calculated by the following equations.
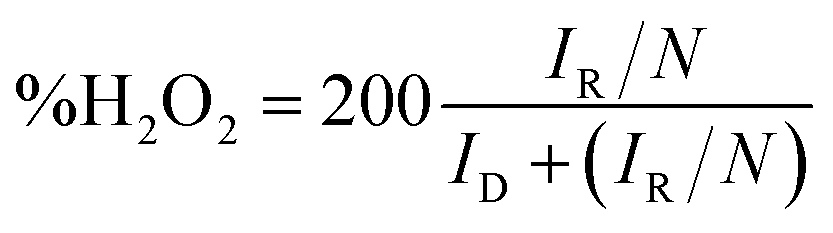 | (4) |
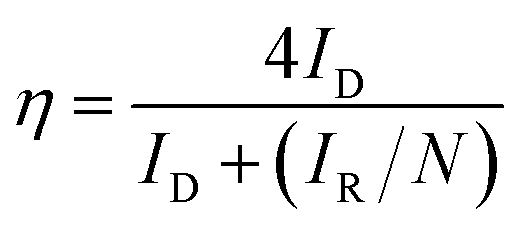 | (5) |
Aqueous Zn–air battery assembly
The as-prepared catalysts were evaluated by using a home-made Zn–air battery device (OMS-T4, Changsha Spring New Energy Technology Co., Ltd., China). Discharge–charge cycling tests were recorded on a LAND testing system (CT2001A, Wuhan, China) with eight channels. Carbon paper was cut into circular with a diameter of 20 mm to be directly acted as the air cathode. A certain amount of the catalyst ink containing the same amount of ruthenium dioxide was casted on the carbon paper with the mass loading of 1.5 ± 0.1 mg cm−2. A polished Zn plate with a diameter of 20 mm was used as anode. The electrolyte was prepared by adding 0.2 M ZnCl2 to 6.0 M KOH solution. The membrane adopted a Whatman glass microfiber filter with a diameter of 25 mm. The assembled battery maintained air contact during testing.Solid Zn–air battery assembly
All as-prepared catalysts casted on carbon paper directly explored as air cathode and the well-polished Zn plate was used as anode. The solid electrolyte was prepared as follows: PVA (3 g), KOH (3 g), and Zn(CH3COO)2 were dissolved in 24 mL deionized water, then the mixture heated at 90 °C under vigorous stirring until the solution become clear. Glass microfiber membrane was soaked in the as-prepared PVA–Zn–KOH electrolyte to obtain the solid electrolyte. Both carbon paper and polished Zn plate with same size were coated each side of the solid electrolyte membrane to fabricate the solid Zn–air battery.DFT calculations
Structural stability of graphitic N and pyridinic N: structural models of N species including graphene (G) and N-doped graphene with different ratios of pyridinic and graphitic N (Pr N:Gr N) were constructed to investigate the structure stability. For the seven Gr_N models (denoted as G_0PrN_6GrN, G_1PrN_5GrN, G_2PrN_4GrN, G_3PrN_3CN, G_4PrN_2GrN, G_5PrN_1GrN and G_0PrN_6GrN), the number of pyridinic atoms was set to increase from 0 to 6, meanwhile, the number of graphitic nitrogen atoms changed from 6 to 0. All the geometries were optimized by the B3LYP density functional with the 6-311+g(d) basis set using Gaussian 09 program. Different spin states were also considered to determine the most stable states which were chosen for the calculations of reaction pathways and diagrams analysis of free energy. Moreover, frequency calculations were performed for all the optimized modes to verify the stable structures and to obtain free energies. Zero-point energy and thermal corrections to the total energy were computed within the harmonic approximation using standard expressions of statistical mechanics at 298.15 K. The free energy diagram for the ORR pathway of each stable electrocatalyst model was performed according to the methods mentioned in our previous study.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Jiangsu Province Department of Education Higher Education surface project and Research Fund of Nantong University (03083030) and Large Instruments Open Foundation of Nantong University (KFJN2032) for financial support. The authors thank the Nantong University Analysis & Testing Center for structure and morphology characterization.Notes and references
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS
.
- M. Karimi, R. Borthakur, C. L. Dorsey, C.-H. Chen, S. Lajeune and F. P. Gabbai, J. Am. Chem. Soc., 2020, 142, 13651–13656 CrossRef CAS
- Y.-J. Wang, W. Long, L. Wang, R. Yuan, A. Ignaszak, B. Fang and D. P. Wilkinson, Energy Environ. Sci., 2018, 11, 258–275 RSC
- Y.-J. Wang, B. Fang, D. Zhang, A. Li, D. P. Wilkinson, A. Ignaszak and L. Zhang, Electrochem. Energy Rev., 2018, 1, 1–34 CrossRef CAS
- J. Zhang, Carbon, 2020, 167, 75–84 CrossRef
- S. Yu, W. Cai, L. Chen, L. Song and Y. Song, J. Energy Chem., 2021, 55, 533–548 CrossRef
- Q. Li, D. Xu, J. Guo, X. Ou and F. Yan, Carbon, 2017, 124, 599–610 CrossRef CAS
- T. Najam, S. S. Ahmad Shah, H. Ali, Z. Song, H. Sun, Z. Peng and X. Cai, Carbon, 2020, 164, 12–18 CrossRef CAS
- L. Yu, C. Yang, W. Zhang, W. Liu, H. Wang, J. Qi and L. Xu, J. Colloid Interface Sci., 2020, 575, 406–415 CrossRef CAS
- R. Zhao, Q. Li, Z. Chen, V. Jose, X. Jiang, G. Fu, J. M. Lee and S. Huang, Carbon, 2020, 164, 398–406 CrossRef CAS
- J. A. Behan, E. Mates-Torres, S. N. Stamatin, C. Domínguez, A. Iannaci, K. Fleischer, M. K. Hoque, T. S. Perova, M. García-Melchor and P. E. Colavita, Small, 2019, 15, 1502081 Search PubMed
- G. L. Chai, Z. Hou, D. J. Shu, T. Ikeda and K. Terakura, J. Am. Chem. Soc., 2014, 136, 13629–13640 CrossRef CAS
- J. P. McClure, J. D. Thornton, R. Jiang, D. Chu, J. J. Cuomo and P. S. Fedkiw, J. Electrochem. Soc., 2012, 159, F733–F742 CrossRef CAS
- J. Zhang, G. Zhang, S. Jin, Y. Zhou, Q. Ji, H. Lan, H. Liu and J. Qu, Carbon, 2020, 163, 154–161 CrossRef CAS
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS
- S. Kundu, T. C. Nagaiah, W. Xia, Y. Wang, S. V. Dommele, J. H. Bitter, M. Santa, G. Grundmeier, M. Bron, W. Schuhmann and M. Muhler, J. Phys. Chem. C, 2009, 113, 14302–14310 CrossRef CAS
- P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83–96 CrossRef CAS
- S. Wang, J. Qin, L. Zheng, D. Guo and M. Cao, Small, 2018, 5, 1801149 Search PubMed
- Y. Cai, Z. Zhang, Y. Ding, L. Hu, J. Wang, T. Chen and Y. Yao, Chin. Chem. Lett., 2020 DOI:10.1016/j.cclet.2020.10.036
- J. Liang, X. Du, C. Gibson, X. W. Du and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 17269–17272 CrossRef
- S. Zhanga, W. Xiab, Q. Yang, Y. V. Kaneti, X. Xub, S. M. Alshehri, T. Ahamad, M. S. A. Hossaine, J. Na, J. Tange and Y. Yamauchi, Chem. Eng. J., 2020, 396, 125154–125160 CrossRef
- H. Begum, M. S. Ahmed and Y.-B. Kim, Sci. Rep., 2020, 10, 12431–12440 CrossRef CAS
- E. Haque, A. Zavabeti, N. Uddin, Y. Wang, M. A. Rahim, N. Syed, K. Xu, A. Jannat, F. Haque, B. Y. Zhang, M. A. Shoaib, S. Shamsuddin, M. Nurunnabi, A. I. Minett, J. Z. Ou and A. T. Harris, Chem. Mater., 2020, 32, 1384–1392 CrossRef CAS
- W. Ding, Z. Wei, S. Chen, X. Qi, T. Yang, J. Hu, D. Wang, L.-J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 52, 1–6 CrossRef
- B. Zhang, C. Wang, D. Liu, Y. Liu, X. Yu and L. Wang, ACS Sustainable Chem. Eng., 2018, 6, 13807–13812 CrossRef CAS
- N. D. K. Tu, S. O. Park, J. Park, Y. Kim, S. K. Kwak and S. J. Kang, ACS Appl. Energy Mater., 2020, 3, 1602–1608 CrossRef
- Y. Xie, Y. Chen, L. Liu, P. Tao, M. Fan, N. Xu, X. Shen and C. Yan, Adv. Mater., 2017, 29, 1702268 CrossRef
- J. Wu, L. Ma, R. M. Yadav, Y. Yang, X. Zhang, R. Vajtai, J. Lou and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2015, 7, 14763–14769 CrossRef CAS
- L. Li, P. Dai, X. Gu, Y. Wang, L. Yan and X. Zhao, J. Mater. Chem. A, 2017, 5, 789–795 RSC
- J. Zhang, Y. Sun, J. Zhu, Z. Kou, P. Hu, L. Liu, S. Li, S. Mu and Y. Huang, Nano Energy, 2018, 52, 307–314 CrossRef CAS
- C. Zhao, S. Zhang, M. Han, X. Zhang, Y. Liu, W. Li, C. Chen, G. Wang, H. Zhang and H. Zhao, ACS Energy Lett., 2019, 4, 377–383 CrossRef CAS
- H. Han, Y. Noh, Y. Kim, W. S. Jung, S. Park and W. B. Kim, Nanoscale, 2019, 11, 2423–2433 RSC
- Q. Li, H. Zhu, Y. Tang, P. Zhu, H. Ma, C. Ge and F. Yan, Chem. Commun., 2019, 55, 12108–12111 RSC
- J. H. Dumont, U. Martinez, K. Artyushkova, G. M. Purdy, A. M. Dattelbaum, P. Zelenay, A. Mohite, P. Atanassov and G. Gupta, ACS Appl. Nano Mater., 2019, 2, 1675–1682 CrossRef CAS
- S. M. Lyth, Y. Nabae, S. Moriya, S. Kuroki, M. Kakimoto, J. Ozaki and S. Miyata, J. Phys. Chem. C, 2009, 113, 20148–20151 CrossRef CAS
- Z. Chen, D. Higgins and Z. W. Chen, Carbon, 2010, 48, 3057–3065 CrossRef CAS
- C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. Lett., 2010, 1, 2622–2627 CrossRef CAS
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS
- H. T. Larijani and M. Khorshidian, Appl. Surf. Sci., 2019, 492, 826–842 CrossRef CAS
- Q. Lv, W. Y. Si, J. J. He, L. Sun, C. F. Zhang, N. Wang, Z. Yang, X. D. Li, X. Wang, W. Q. Deng, Y. Z. Long, C. S. Huang and Y. L. Li, Nat. Commun., 2018, 9, 3376–3386 CrossRef
- C. S. Huang, Y. J. Li, N. Wang, Y. R. Xue, Z. C. Zuo, H. B. Liu and Y. L. Li, Chem. Rev., 2018, 118, 7744–7803 CrossRef CAS
- D. Liu, X. Zhang, Y.-J. Wang, S. Song, L. Cui, H. Fan, X. Qiao and B. Fang, Nanoscale, 2020, 12, 9524–9532 RSC
- Q. Li, J. Guo, H. Zhu and F. Yan, Small, 2019, 15, 1804874 CrossRef
- T. Qin, J. Zhao, R. Shi, C. Ge and Q. Li, Chem. Eng. J., 2020, 399, 125656–125668 CrossRef CAS
- L. Li, P. Dai, X. Gu, Y. Wang, L. Yan and X. Zhao, J. Mater. Chem. A, 2017, 5, 789–795 RSC
- H. Miao, S. Li, Z. Wang, S. Sun, M. Kuang, Z. Liu and J. Yuan, Int. J. Hydrogen Energy, 2017, 42, 28298–28308 CrossRef CAS
- S. Y. Wang, L. P. Zhang, Z. H. Xia, A. Roy, D. W. Chang, J. B. Baek and L. M. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS
- Y. C. Lin, P. Y. Teng, C. H. Yeh, M. Koshino, P. W. Chiu and K. Suenaga, Nano Lett., 2015, 15, 7408–7413 CrossRef CAS
- C. H. Chou, H. K. Lim, M. W. Chung, J. C. Park, H. Y. Shin, H. J. Kim and S. I. Woo, J. Am. Chem. Soc., 2014, 136, 9070–9077 CrossRef
- X. Zou, L. Wang and B. I. Yakobson, Nanoscale, 2018, 10, 1129–1134 RSC
- Z. Zhao, Z. K. Yuan, Z. S. Fang, J. H. Jian, J. Li, M. J. Yang, C. S. Mo, Y. Zhang, X. H. Hu, P. Li, S. Y. Wang, W. Hong, Z. K. Zheng, G. F. Ouyang, X. D. Chen and D. S. Yu, Adv. Sci., 2018, 5, 1800760 CrossRef
- J. W. Zhu, W. Q. Li, S. H. Li, J. Zhang, H. Zhou, C. T. Zhang, J. A. Zhang and S. C. Mu, Small, 2018, 14, 1800563 CrossRef
- Y. H. Qian, T. An, K. E. Birgersson, Z. L. Liu and D. Zhao, Small, 2018, 14, 1704169 CrossRef
- S. Chen, L. Zhao, J. Ma, Y. Wang, L. Dai and J. Zhang, Nano Energy, 2019, 60, 536–544 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10072b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |