
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient synthesis of 4-phenoxy-quinazoline, 2-phenoxy-quinoxaline, and 2-phenoxy-pyridine derivatives using aryne chemistry†
Mahesh K. Zilla‡
ab,
Sheena Mahajan‡b,
Rajni Khajuriac,
Vivek K. Guptad,
Kamal K. Kapoore and
Asif Ali*bf
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER), Berhampur, Odisha 760010, India. E-mail: mzilla@iiserbpr.ac.in
bCSIR-Indian Institute of Integrative Medicine, Canal Road, Jammu-180001, India. E-mail: sheena01mahajan@gmail.com
cDepartment of Chemistry, GDC for Women Kathua, Kathua-184101, India. E-mail: khajuriarajni36@gmail.com
dPost-Graduate Department of Physics & Electronics, University of Jammu, Jammu Tawi- 180006, India. E-mail: vivek.gupta2k9@gmail.com
eDepartment of Chemistry, University of Jammu, Jammu Tawi-180006, India. E-mail: kamalkka@gmail.com
fCSIR-Traditional Knowledge Digital Library (TKDL), 14-Satsang Vihar, Vigyan Suchna Bhawan, New Delhi-110067, India. E-mail: asifali@csir.res.in
First published on 15th January 2021
Abstract
Herein we report the mild and efficient synthesis of 4-phenoxyquinazoline, 2-phenoxyquinoxaline, and 2-phenoxypyridine derivatives from the starting materials viz. quinazolin-4(3H)-one, quinoxalin-2(1H)-one, and pyridin-2(1H)-one and aryne generated in situ from 2-(trimethylsilyl)phenyl trifluoromethanesulfonate and cesium fluoride. This synthetic methodology gives a new environmentally benign way for the preparation of several unnatural series of 4-phenoxyquinazoline, 2-phenoxyquinoxaline and 2-phenoxypyridine compounds with high yields and broad substrate scope.
Introduction
Aryne intermediates are one of the most important classes of organic species and are useful as substrates in various reactions for developing new synthetic methodologies,1 preparing heterocyclic molecules2 and in the total synthesis of natural products.3 Although aryne was generated from different starting materials, among all the reported methods, generation of aryne developed using the Kobayashi et al.4 method from trimethylsilyl phenyl triflate and cesium fluoride has been widely used for the last two decades because of its easy formation in situ and generation under normal conditions. Due to the highly electrophilic nature of arynes,5 these readily participated in reactions with the reactants and gave addition,6 insertion type products with good yields and atom economy.7 Because of their high reactivity in the reactions, arynes readily participate in reactions with substrates like styrenes without the use of any catalyst.8 After reading several reviews published by top authors on aryne chemistry,9 the main focus of the present work is the development of a mild, simple, eco-benign methodology using aryne chemistry with heterocyclic compounds.Literature reports reveals very few reactions of arynes with amides. Pintori et al.10 reported the insertion of benzene ring into the amide bond by using aryne chemistry. Later on, facile N-arylation of acetanilides with arynes was reported by Haber et al.11 Herein our method, we have got the result of aryl substituted iminoxy derivatives on amide group containing heterocyclic compounds (Scheme 1).
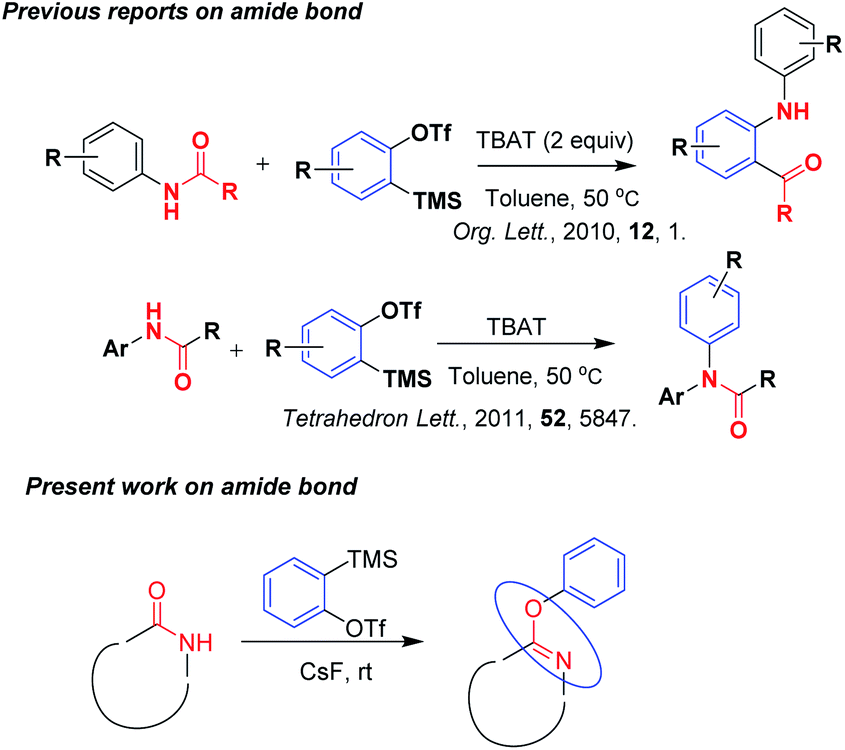 | ||
| Scheme 1 Reactions of arynes with amides. | ||
Heterocyclic compounds have unique importance in organic synthesis and in biological activity. Among these quinazolines, quinoxalines are the important class of building blocks found in a variety of pharmaceuticals. Quinazolines are the compounds showing different medicinal properties such as anti-convulsant,12 anti-bacterial,13 anti-diabetic,14 anti-carcinogen15 etc., whereas quinoxalines are having anti-biotic,16 anti-mycobacterial,17 anti-viral,18 anti-osteoclast,19 anti-convulsant20 properties. Some of the important biologically active quinazoline and quinoxaline derivatives are shown in Fig. 1.
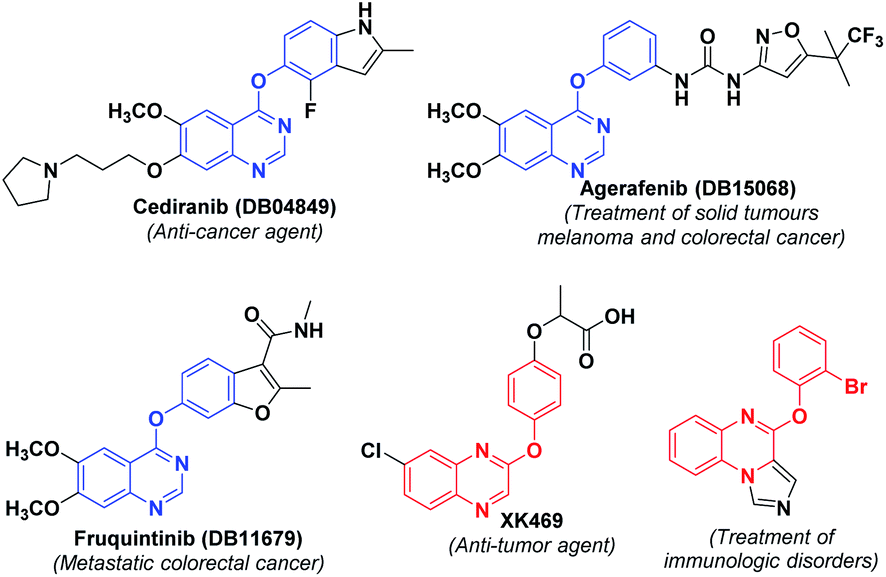 | ||
| Fig. 1 Representative biologically active quinazolines and quinoxalines. | ||
When we processed the work on developing mild and simple synthetic methodologies with aryne, we got an unfortunate result of aryl substituted iminoxy quinazoline of unnatural compound with an appropriate yield which would be useful for the preparation of a series of compounds in efficient manner. So the main objective of the present methodology was the diversification in preparation of unnatural heterocyclic compounds.
Results and discussion
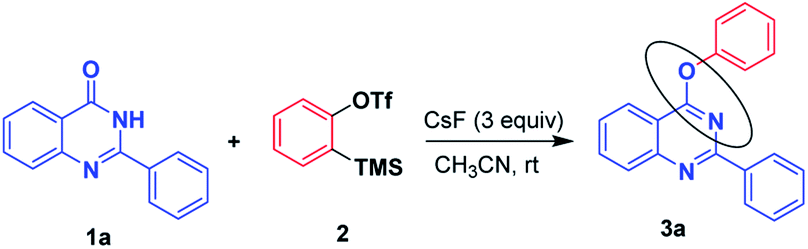
We commenced our investigation using 2-phenylquinazolin-4(3H)-one 1a and readily available 2-(trimethylsilyl)phenyl trifluoromethanesulfonate 2 (Table 1). When the reaction was performed with KF/18-crown-6 as the fluoride source for the generation of benzyne from 2-(trimethylsilyl)phenyltrifluoro methane sulfonate 2 at room temperature in acetonitrile, desired product 3a was obtained in a moderate yield of 45% (Table 1, entry 1). However by performing the same reaction with the increased amount of KF/18-crown-6 and KF/18-crown-6, minor effect in the product 3a yield i.e. 48% (Table 1, entry 2) was observed. Also by changing the solvent of the reaction as DME, poor yield of the desired product 3a (Table 1, entry 3) was obtained. Further, with different fluoride sources such as TBAF, TBAT the product 3a in moderate yields of 55% and 48% was obtained. Further, by employing CsF as a fluoride source, product 3a was obtained in high yield (72%). Later, by increasing the amount of CsF, to our delight, yield of the product 3a increased to 89%. Also by carrying out the reaction in different solvents such as DCM, toluene, DME less yields of product was obtained (Table 1, entry 8, 9, 10). Thus, 1 mmol of 2-phenyl quinazolin-4(3H)-one 1a, 1.2 mmol of 2-(trimethylsilyl) phenyl trifluoromethanesulfonate 2 in acetonitrile in presence of 3 mmol of CsF at room temperature were the optimized reaction conditions.
|
||||
|---|---|---|---|---|
| Entry | F− source | Equiv. | Solvent | Yieldb (%) |
| a Reaction conditions (unless otherwise stated): 1a (0.45 mmol, 1.0 equiv.), 2 (0.54 mmol, 1.2 equiv.), F− source, solvent (8 mL), at room temperature for 4 h.b Isolated yield.c Tetrabutylammonium fluoride.d Tetrabutylammonium difluorotriphenylsilicate. | ||||
| 1. | KF/18-crown-6 | 2.0 | CH3CN | 45 |
| 2. | KF/18-crown-6 | 3.0 | CH3CN | 48 |
| 3. | KF/18-crown-6 | 3.0 | DME | 22 |
| 4. | TBAFc | 2.0 | CH3CN | 55 |
| 5. | TBATd | 2.0 | CH3CN | 48 |
| 6. | CsF | 2.0 | CH3CN | 72 |
| 7. | CsF | 3.0 | CH3CN | 89 |
| 8. | CsF | 3.0 | DCM | 5 |
| 9. | CsF | 3.0 | Toluene | 5 |
| 10. | CsF | 3.0 | DME | 48 |
With the optimized reaction conditions in hand, we examined the substrate scope with different substituted 2-phenylquinazolin-4(3H)-one derivatives, and the results are depicted in Scheme 2. With electron donating groups (OCH3, SCH3) substituted on 2-phenylquinazolin-4(3H)-one, high yield of the desired product was obtained (Scheme 2: ex. OCH3, SCH3). Whereas with electron withdrawing groups, less yield of the desired product was obtained (Scheme 2: ex. CF3, OCF3).
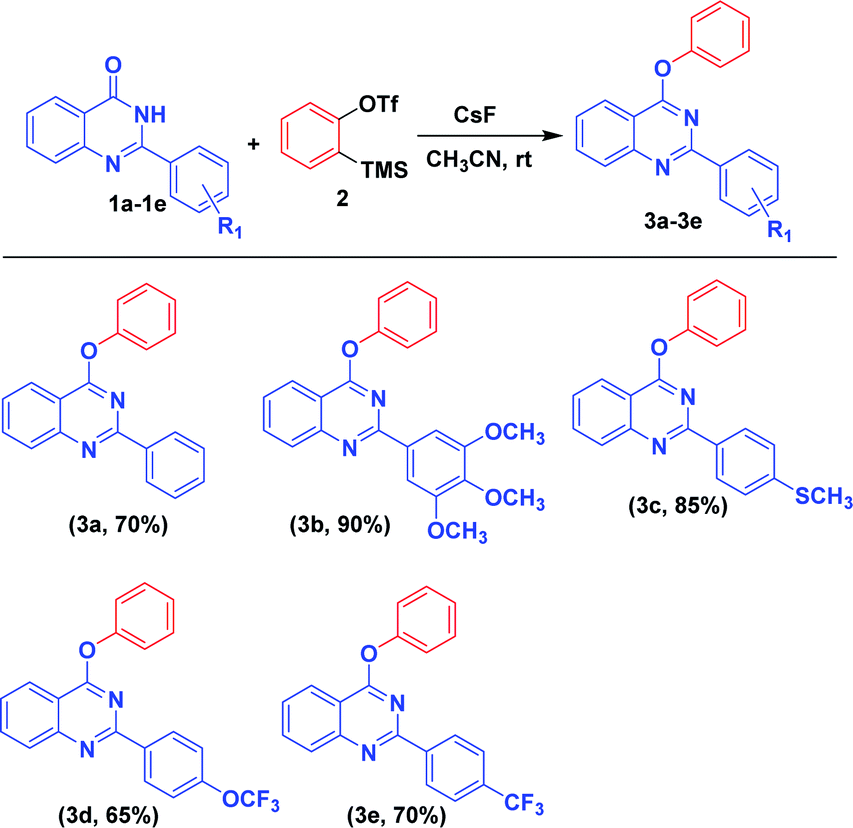 | ||
| Scheme 2 Substrate scope of different substituted 2-phenylquinazolin-4(3H)-one derivatives with benzynea. aReaction conditions (unless otherwise stated): 1a–e (0.45 mmol, 1.0 equiv.), 2 (0.54 mmol, 1.2 equiv.), CsF (1.35 mmol, 3 equiv.), acetonitrile solvent (8 mL), at room temperature for 4–6 h. | ||
Next, we extended to apply the same optimized reaction condition on quinoxalin-2(1H)-one derivatives which resulted in two different compounds viz. 2-phenoxyquinoxaline and 1-phenylquinoxalin-2(1H)-one in the ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. 2-Phenoxyquinoxaline was obtained as major product which was confirmed by the absence of carbonyl stretching peak in the IR spectrum indicated that it was arylated to iminoxy compound. Whereas the minor quantity compound, 1-phenylquinoxalin-2(1H)-one structure was confirmed by the IR spectrum having specific peak at 1663 cm−1 for the carbonyl group of amide indicating that no change in the carbonyl of the amide group confirming the N-arylated product. Further, we have enlarged the work for different substituted quinoxalin-2(1H)-one derivatives and the results are shown in Scheme 3.
:3. 2-Phenoxyquinoxaline was obtained as major product which was confirmed by the absence of carbonyl stretching peak in the IR spectrum indicated that it was arylated to iminoxy compound. Whereas the minor quantity compound, 1-phenylquinoxalin-2(1H)-one structure was confirmed by the IR spectrum having specific peak at 1663 cm−1 for the carbonyl group of amide indicating that no change in the carbonyl of the amide group confirming the N-arylated product. Further, we have enlarged the work for different substituted quinoxalin-2(1H)-one derivatives and the results are shown in Scheme 3.
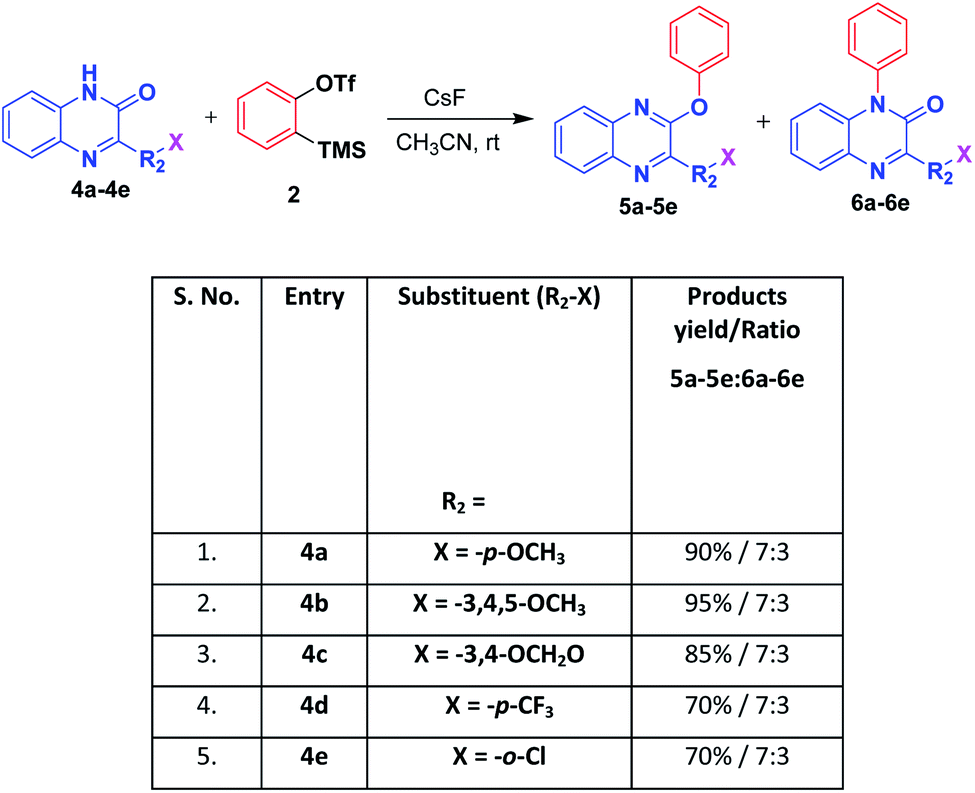 | ||
| Scheme 3 Application of the optimized reaction condition on different substituted (E)-3-styrylquinoxalin-2(1H)-one derivatives with benzynea. aReaction conditions (unless otherwise stated): 4a–e (0.28 mmol, 1.0 equiv.), 2 (0.33 mmol, 1.2 equiv.), CsF (0.84 mmol, 3 equiv.), acetonitrile solvent (8 mL), at room temperature for 4–6 h. b Isolated yield. | ||
Further, we have applied the optimized reaction on the pyridin-2(1H)-ones, predominantly resulting 2-phenoxy pyridine derivatives in high yield. Although the reaction of pyridones with aryne was reported by Masayuki Kuzuva et al. in 1984, the authors have generated aryne from the anthranilic acid with the use of hazardious chemical isopentyl nitrile in a mixture of solvents acetone and chloroform at a temperature of 70 °C with moderate yield. Where as in our method, we have generated aryne from 2-(trimethylsilyl)phenyl trifluoromethanesulfonate with CsF at room temperature resulting in a high yield of product in an efficient manner. Further, we have applied this reaction to enlarge the substrate scope, and the results are mentioned in Scheme 4. Structure of product 8e was confirmed by X-ray crystallographic studies. An ORTEP view of the compound 8e with atomic labeling is shown in Scheme 4.21 The geometry of the molecule was calculated using the WinGX,22 PARST23 and PLATON24 softwares. The crystallographic data is summarized in ESI.†
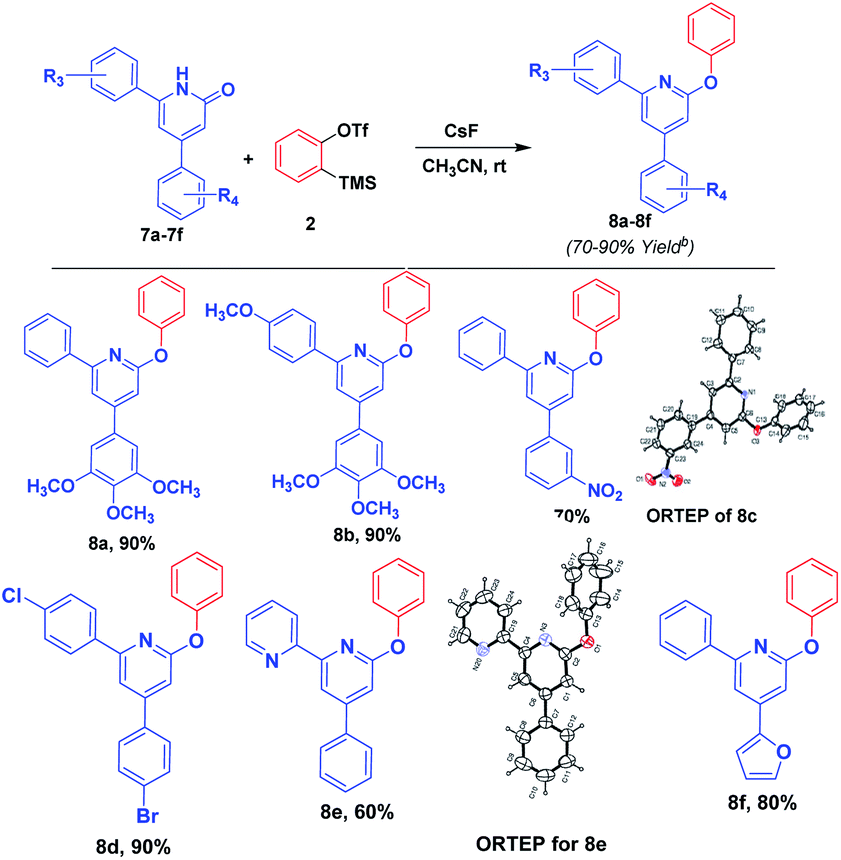 | ||
| Scheme 4 Application of the optimized reaction condition on different substituted pyridine-2(1H)-one derivatives with benzynea. aReaction conditions (unless otherwise stated): 7a–f (0.24 mmol, 1.0 equiv.), 2 (0.29 mmol, 1.2 equiv.), CsF (0.72 mmol, 3 equiv.), acetonitrile solvent (8 mL), at room temperature for 4–6 h. bIsolated yield. | ||
Experimental
Materials and methods
All the experiments were performed in an oven dried glass apparatus. All the commercially available reagents were purchased from Aldrich and were used without further purification. Melting points (°C) were measured in open glass capillaries using Perfit melting point apparatus and are uncorrected. The progress of reaction was monitored by thin layer chromatography (TLC) using silica gel pre-coated aluminium sheets (60 F254, Merck). Visualization of spots was effected by exposure to ultraviolet light (UV) at 365 nm and 254 nm, iodine vapours and 2% 2,4-dinitrophenylhydrazine in methanol containing few drops of H2SO4, dragendroff reagent and anisaldehyde reagent. Solvents used in purification were distilled before use. Recrystallization was achieved with ethanol. IR spectra (ν, cm−1) were recorded on Perkin-Elmer FTIR spectrophotometer using KBr discs. 1H and 13C NMR were recorded on Bruker AC-400 spectrometer operating at 400 MHz for 1H and 100 MHz for 13C with tetramethylsilane (TMS) as an internal standard. The chemical shifts are expressed in δ (ppm) downfield from TMS. All the 13C NMR spectra are proton decoupled. J values are given in Hertz (Hz). The abbreviations s, d, dd, t, q and m in 1H NMR spectra refer to singlet, doublet, doublet of doublet, triplet, quartet and multiplet respectively. Solvents were removed using Heidolph rotary evaporator. Electrospray ionization mass spectra MS (ESI) were recorded on Micro Mass VG-7070 H mass spectrometer at 70 eV. Elemental analysis was performed on Leco CHNS-932 analyzer.Crystal structure determination and refinement for 4-(3-nitrophe-nyl)-2-phenoxy-6-phenylpyridine (8c)
X-ray intensity data of 7234 reflections (of which 3530 unique) were collected on X'calibur CCD area-detector diffractometer equipped with graphite monochromated MoKα radiation (λ = 0.71073 Å). The crystal used for data collection was of dimensions 0.30 × 0.20 × 0.20 mm. The cell dimensions were determined by least-squares fit of angular settings of 1788 reflections in the θ range 3.93 to 27.57°. The intensities were measured by ω scan mode for θ ranges 3.53 to 26.00°. 2176 reflections were treated as observed (I > 2σ (I)). Data were corrected for Lorentz, polarisation and absorption factors. The structure was solved by direct methods using SHELXS97.25 Full-matrix least-squares refinement was carried out using SHELXL97.25 The final refinement cycles converged to an R = 0.0445 and wR (F2) = 0.0911 for the observed data. Residual electron densities ranged from −0.126 < Δρ < 0.158 eÅ−3. Atomic scattering factors were taken from International Tables for X-ray Crystallography (1992, vol. C, Tables 4.2.6.8 and 6.1.1.4). The crystallographic data are summarized in ESI.†Crystal structure determination and refinement for 6-phenoxy-4-phenyl-2,2′-bipyridine (8e)
X-ray intensity data of 5807 reflections (of which 3223 unique) were collected on X'calibur CCD area-detector diffractometer equipped with graphite monochromated MoKα radiation (λ = 0.71073 Å). The crystal used for data collection was of dimensions 0.30 × 0.20 × 0.20 mm. The cell dimensions were determined by least-squares fit of angular settings of 1471 reflections in the θ range 4.12 to 26.09°. The intensities were measured by ω scan mode for θ ranges 3.52 to 26.00°. 1919 reflections were treated as observed (I > 2σ(I)). Data were corrected for Lorentz, polarisation and absorption factors. The structure was solved by direct methods using SHELXS97.25 All non-hydrogen atoms of the molecule were located in the best E-map. Full-matrix least-squares refinement was carried out using SHELXL97.25 The final refinement cycles converged to an R = 0.0533 and wR (F2) = 0.1235 for the observed data. Residual electron densities ranged from −0.162 < Δρ < 0.172 eÅ−3. Atomic scattering factors were taken from International Tables for X-ray Crystallography (1992, vol. C, Tables 4.2.6.8 and 6.1.1.4). The crystallographic data are summarized in ESI.†General procedure for the preparation of 4-phenoxy-2-arylquinazoline (3a–e) with aryne
To a stirred solution of 2-phenylquinazolin-4(3H)-one (1a–e) (0.45 mmol, 1.0 equiv.) and trimethylsilyl phenyl triflate (2) (0.54 mmol, 1.2 equiv.) in acetonitrile as solvent, was added cesium fluoride (1.35 mmol, 3.0 equiv.) at room temperature. The mixture was stirred at room temperature for 4 h, after completing the reaction, reaction mixture was concentrated in vacuo. To the resulting residue, water was added, and the mixture was extracted with ethyl acetate three times. The combined organic phase was dried over sodium sulphate, and concentrated in vacuo. The crude product was purified by column chromatography (EtOAc in hexane) resulting the product.General procedure for the preparation of (E)-2-phenoxy-3-styrylquinoxaline (5a–e) with aryne
To a stirred solution of (E)-3-styrylquinoxalin-2(1H)-one (4a–e) (0.28 mmol, 1.0 equiv.) and trimethylsilyl phenyl triflate (2) (0.33 mmol, 1.2 equiv.) in acetonitrile as solvent, was added cesium fluoride (1.35 mmol, 3.0 equiv.) at room temperature. The mixture was stirred at room temperature for 4 h, after completing the reaction, reaction mixture was concentrated in vacuo. To the resulting residue, water was added, and the mixture was extracted with ethyl acetate three times. The combined organic phase was dried over sodium sulphate, and concentrated in vacuo. The crude product was purified by column chromatography (EtoAc in hexane), resulting the product.General procedure for the preparation of 2-phenoxypyridine derivatives (8a–f) with aryne
To a stirred solution of 4, 6-diphenylpyridin-2(1H)-one (7a–f) (0.24 mmol, 1.0 equiv.) and trimethylsilyl phenyl triflate (2) (0.29 mmol, 1.2 equiv.) in acetonitrile as solvent, was added cesium fluoride (0.72 mmol, 3.0 equiv.) at room temperature. The mixture was stirred at room temperature for 4 h, after completing the reaction, reaction mixture was concentrated in vacuo. To the resulting residue, water was added, and the mixture was extracted with ethyl acetate three times. The combined organic phase was dried over sodium sulphate, and concentrated in vacuo. The crude product was purified by column chromatography (EtoAc in hexane), resulting the product.Conclusions
In conclusion, we have developed a simple, mild, metal free, efficient, room temperature method for the preparation of 4-phenoxy-2-phenylquinazoline, (E)-2-phenoxy-3-styrylquinoxaline and 2-phenoxypyridine derivatives via an aryne intermediate. The present method enables very easy preparation of phenoxylated quinazoline, quinaxazoline and pyridine derivatives that will be useful for a feasible access to the unnatural product molecules and their biological screening.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MKZ thankful to the Council of Scientific and Industrial Research (CSIR) for the financial assistance. We are thankful to Deepika Singh for NMR spectral data analysis. One of the authors (Vivek) acknowledges the Department of Science & Technology for single crystal X-ray diffractometer as a National Facility under Project No. SR/S2/CMP-47/2003.Notes and references
- (a) T. Kaicharla, M. Thangaraj and A. T. Biju, Org. Lett., 2014, 16, 1728 CrossRef CAS; (b) C. Xie and Y. Zhang, Org. Lett., 2007, 9, 781 CrossRef CAS; (c) H. Y. Li, L. J. Xing, M. M. Lou, H. Wang, R. H. Liu and B. Wang, Org. Lett., 2015, 17, 1098 CrossRef CAS; (d) T. Truong, M. Mesgar, K. K. A. Le and O. Daugulis, J. Am. Chem. Soc., 2014, 136, 8568 CrossRef CAS; (e) K. Neog, D. Dutta, B. Das and P. Gogoi, Org. Lett., 2017, 19, 730 CrossRef CAS.
- (a) E. Yoshikawa and Y. Yamamoto, Angew. Chem., Int. Ed., 2000, 39, 173 CrossRef CAS; (b) H. Yoshida, H. Fukushima, J. Ohshita and A. Kunai, J. Am. Chem. Soc., 2006, 128, 11040 CrossRef CAS; (c) C. D. Gilmore, K. M. Allan and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 1558 CrossRef CAS.
- (a) K. M. Allan and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 17270 CrossRef CAS; (b) P. M. Tadross, S. C. Virgil and B. M. Stoltz, Org. Lett., 2010, 12, 1612 CrossRef CAS; (c) U. K. Tambar, D. C. Ebner and B. M. Stoltz, J. Am. Chem. Soc., 2006, 128, 11752 CrossRef CAS; (d) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 12, 1211 CrossRef.
- A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140 RSC.
- H. Yoshida, E. Shirakawa, Y. Honda and T. Hiyama, Angew. Chem., Int. Ed., 2002, 41, 3247 CrossRef CAS.
- (a) Y. L. Liu, Y. Liang, S. F. Pi and J. H. Li, J. Org. Chem., 2009, 74, 5691 CrossRef CAS; (b) H. Yoshida, M. Watanabe, J. Ohshita and A. Kunai, Tetrahedron Lett., 2005, 46, 6729 CrossRef CAS.
- S. S. Bhojgude, A. Bhunia, R. G. Gonnade and A. T. Biju, Org. Lett., 2014, 16, 676 CrossRef CAS.
- (a) T. Roy and A. T. Biju, Chem. Commun., 2018, 54, 2580 RSC; (b) T. Matsuzawa, S. Yoshida and T. Hosoya, Tetrahedron Lett., 2018, 59, 4197 CrossRef CAS; (c) M. Asamdi and K. H. Chikhalia, Asian J. Org. Chem., 2017, 6, 1331 CrossRef CAS; (d) K. Okuma, Heterocycles, 2012, 85, 515 CrossRef CAS; (e) C. Wu and F. Shi, Asian J. Org. Chem., 2013, 2, 116 CrossRef CAS; (f) A. G. Flores and R. Martin, Synthesis, 2013, 45, 563 CrossRef; (g) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS; (h) S. S. Bhojgude and A. T. Biju, Angew. Chem., Int. Ed., 2012, 51, 1520 CrossRef CAS; (i) A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140 RSC; (j) C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766 CrossRef CAS; (k) I. S. Kovalev, D. S. Kopchuk, G. V. Zyryanov, P. A. Slepukhin, V. L. Rusinov and N. O. Chupakhin, Chem. Heterocycl. Compd., 2012, 48, 536 CrossRef CAS; (l) D. Perez, D. Pena and E. Guitian, Eur. J. Org. Chem., 2013, 5981 CrossRef CAS; (m) D. Wu, H. Ge, S. H. Liu and J. Yin, RSC Adv., 2013, 3, 22727 RSC; (n) J. Shi and Y. Li, Chem. Soc. Rev., 2017, 46, 1707 RSC.
- D. G. Pintori and M. F. Greaney, Org. Lett., 2009, 12, 168 CrossRef.
- J. C. Haber, M. A. Lynch, S. L. Spring, A. D. Pechulis, J. Raker and Y. Wang, Tetrahedron Lett., 2011, 52, 5847 CrossRef CAS.
- J. F. Wolfe, T. L. Rathman, M. C. Sleevi, J. A. Campbell and T. D. Greenwood, J. Med. Chem., 1990, 33, 161 CrossRef CAS.
- (a) P. P. Kung, M. D. Casper, K. L. Cook, L. Wilson-Lingardo, L. M. Risen, T. A. Vickers, R. Ranken, L. B. Blyn, J. R. Wyatt and P. D. Cook, J. Med. Chem., 1999, 42, 4705 CrossRef CAS; (b) L. S. Reddy and B. E. Naik, Heterocycl. Lett., 2019, 9(4), 487 CAS.
- M. Saeedi, M. Mohammadi-Khanaposhtani, P. Pourrabia, N. Razzaghi, R. Ghadimi, S. Imanparast, M. A. Faramarzi, F. Bandarian, E. N. Esfahani, M. Safavi, M. Safavi, H. Rastegar, B. Larijani, M. Mahdavi and T. Akbarzadeh, Bioorg. Chem., 2019, 83, 161 CrossRef CAS.
- X.-W. Wei, J.-M. Yuan, W.-Y. Huang, N.-Y. Chen, X.-J. Li, C.-X. Pan, D.-L. Mo and G.-F. Su, Eur. J. Med. Chem., 2020, 186, 111851 CrossRef CAS.
- (a) Y. B. Kim, Y. H. Kim, J. Y. Park and S. K. Kim, Bioorg. Med. Chem. Lett., 2004, 14, 541 CrossRef CAS; (b) J. Peng, D. Kong, L. Liu, S. Song, H. Kuang and C. Xu, Anal. Methods, 2015, 7, 5204 RSC.
- (a) E. Moreno, S. Ancizu, S. P. Silanes, E. Torres, I. Aldana and A. Monge, Eur. J. Med. Chem., 2010, 45, 4418 CrossRef CAS; (b) L. E. Seitz, W. J. Suling and R. C. Reynolds, J. Med. Chem., 2002, 45, 5604 CrossRef CAS.
- (a) E. Tombary and E. Hawash, Med. Chem., 2014, 10, 521 CrossRef; (b) M. A. Henen, S. E. Bialy, F. E. Goda, M. N. A. Nasr and H. M. Eisa, Med. Chem. Res., 2012, 21, 2368 CrossRef CAS; (c) L. M. Wilhelmsson, N. Kingi and J. Bergman, J. Med. Chem., 2008, 51, 7744 CrossRef CAS.
- Y.-C. Zhang, Q. Shen, M.-W. Zhu, J. Wang, Y. Du, J. Wu and J.-X. Li, ChemistrySelect, 2020, 5(4), 1526 CrossRef CAS.
- (a) A. A. Elhelby, R. R. Ayyad and M. F. Zayed, Arzneim. Forsch., 2011, 61, 379 CrossRef CAS; (b) E. Elkaeed, A. Ghiaty, A. E. Morsy, K. E. Gamal and H. Sakr, Chem. Sci. Rev. Lett., 2014, 3, 1375 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR, other spectra of all the compounds and crystal data of 8c & 8e. CCDC 1451640 and 1451639. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09994e |
| ‡ Mahesh K. Zilla and Sheena Mahajan have equally contributed as a first author. |
| This journal is © The Royal Society of Chemistry 2021 |