
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanocomposite membranes of polybenzimidazole and amine-functionalized carbon nanofibers for high temperature proton exchange membrane fuel cells
Li-Cheng Jheng†
a,
Afira Ainur Rosidah†b,
Steve Lien-Chung Hsu *b,
Ko-Shan Hoa,
Chun-Jern Pana and
Cheng-Wei Chengb
*b,
Ko-Shan Hoa,
Chun-Jern Pana and
Cheng-Wei Chengb
aDepartment of Chemical and Materials Engineering, National Kaohsiung University of Science and Technology, Kaohsiung, Taiwan, Republic of China
bDepartment of Materials Science and Engineering, National Cheng-Kung University, Tainan, Taiwan, Republic of China. E-mail: lchsu@mail.ncku.edu.tw; Fax: +86 6 2346290; Tel: +86 6 2757575 ext. 62904
First published on 8th March 2021
Abstract
Carbon nanofibers functionalized with aminobenzoyl groups (CNF–aminobenzoyl) were prepared via direct Friedel–Crafts acylation in polyphosphoric acid. The functionalization of CNFs was characterized using XPS, FTIR, TGA, and Raman analyses. Hexafluoroisopropylidene-containing polybenzimidazole (6FPBI) composite membranes containing pristine CNFs or CNF–aminobenzoyl were prepared using solvent-assisted dispersion and solvent-casting methods. In this work, the influence of the incorporation of functionalized CNFs on several physicochemical properties of the 6FPBI nanocomposite membranes, including their thermal stability, mechanical strength, and acid doping level, was studied. The results showed that CNF–aminobenzoyl provided better mechanical reinforcement for the nanocomposite membrane, compared to pristine CNF. The SEM observation confirmed the good compatibility between the CNF–aminobenzoyl fillers and the 6FPBI matrix. For the 0.3 wt% CNF–aminobenzoyl/6FPBI composite membrane, the tensile stress was increased by 12% to be 78.9 MPa (as compared to the 6FPBI membrane), the acid doping level was improved to 12.0, and the proton conductivity at 160 °C was measured above 0.2 S cm−1. Furthermore, the fuel cell performance of the membrane electrolyte assembly (MEA) for each nanocomposite membrane was evaluated. The maximum power density at 160 °C was found up to 461 mW cm−2 for the MEA based on the 0.3 wt% CNF–aminobenzoyl/6FPBI composite membrane.
1 Introduction
High-temperature proton exchange membrane fuel cells (HT-PEMFCs) operating in the temperature range between 100 °C and 200 °C under anhydrous conditions have been considered as a feasible alternative to the typical proton exchange membrane fuel cells based on Nafion® membranes running at relatively lower temperatures under a humidified atmosphere.1–4 The major advantages of HT-PEMFCs, such as the improved electrochemical kinetics, the higher tolerance to carbon monoxide contained in the hydrogen fuel, no need for humidification, and the simplified heat management, can ease the problems arising from the low-temperature operation and the use of a platinum catalyst.5–8Phosphoric acid-doped polybenzimidazole membranes, capable of conducting protons without humidification, are widely used as polymer electrolyte in HT-PEMFCs.8,9 The common type of polybenzimidazole (PBI) is poly(2,2′-m-(phenylene)-5,5′-benzimidazole) (mPBI), which possess superior thermal and chemical stabilities. However, mPBI exhibited insufficient solubility in most of organic solvents and poor processability during membrane fabrication due to its rigid polymer structure.10 Accordingly, the chemical structure of PBI was modified by introducing flexible spacers or bulky moieties into the polymer backbone to improve the solubility and processability. Some of solvent-soluble PBIs have been developed, including ether-containing PBI (OPBI),11 sulfone-containing PBI (SO2PBI),12 and hexafluoroisopropylidene-containing PBI (6FPBI).13–15 Among them, 6FPBI was found to exhibit better oxidative stability during the Fenton test than others.16
For phosphoric acid-doped PBI membranes, a higher acid content generally leads to a better proton conductivity. However, the presence of phosphoric acid will impair the mechanical strength of the membrane significantly due to the plasticization effect. To break the trade-off between proton conductivity and mechanical strength, incorporating fillers into acid-doped PBI membranes is an effective way. Because fillers incorporated in proton exchange membranes (PEMs) are possible to reduce the crystallinity, enhance the mechanical property, and improve the proton conductivity of the polymer simultaneously.3 Up to now, many different acid-doped PBI composite membranes prepared using a variety of fillers have been developed. The fillers can be categorized into several types, including inorganic oxides (e.g., SiO2, TiO2, ZrO2), heteropolyacids and their salts, ionic liquids, as well as carbon-based nanofillers.3,17,18 Among them, carbon-based nanofillers, such as carbon nanotube (CNT) and graphene oxide (GO), have received growing attention for HT-PEMFC applications in the past decade.19–29 These carbon-based nanofillers have several features superior to other types of fillers for composite membranes. For example, they can undergo modification processes more feasibly to introduce functional groups onto their surface.30 Also, it is not difficult to develop certain interactions (e.g., hydrogen bonding or π–π stacking interactions) between carbon-based nanofillers and a polymer matrix that can make them more compatible with each other.23,24,31,32 Additionally, the extraordinary mechanical properties and special dimensions of carbon-based nanofillers would allow them to provide better mechanical strength and fuel separation ability to the composite membrane.33
It is critical to choose proper surface modifications of carbon-based nanofillers for realizing their ideal properties or specific functions in a PEM.33 Many efforts have been made to functionalize carbon-based nanofillers to obtain more homogeneous filler dispersions, better interfacial interaction, higher acid doping, and improved proton conductivity in phosphoric acid doped nanocomposite membranes used for HT-PEMFCs. Examples of functionalized carbon-based nanofillers include sulfonated CNT,28 imidazole-functionalized CNT,28 PBI-functionalized CNT,20 Nafion functionalized CNT,20 phosphonated CNT,19 sulfonated GO,26 triazole modified GO,24 ionic liquid-functionalized GO,29 phosphonated GO,27 and isocyanate modified GO.22
Some studies reported amine-functionalized fillers could bring a beneficial effect on the proton conduction of PEMs.34–37 For example, T. Jana et al. prepared amine-functionalized silica nanoparticles for phosphoric acid doped PBI nanocomposite membranes.34,37 Their research findings revealed that a higher amine content on the silica nanoparticles resulted in higher holding capacity of phosphoric acid. The reason is that amine-functionalized silica may act as a base so that it could help the nanocomposite membrane dope more phosphoric acid molecules.
Carbon nanofiber (CNF) produced via catalyst chemical vapor deposition is known as vapor-grown carbon fiber (VGCF). Different from CNT, CNF opposes a cylindrical nanostructure with cup-stacked graphene layers and have relatively larger diameters ranging between 50 nm and 200 nm.38 The unique structure endows them with more reactive carbon edges that can be functionalized with specific functional groups to make it compatible with the polymer matrix.39 Besides, the lower production cost of CNTs is their major advantage over CNTs, which allows them to be used not only for research applications but also in the polymer industry. Baek et al. introduced an efficient way to covalently modify CNF via a simple reaction called direct Friedel–Crafts acylation.40 This method was suitable to modify CNT and graphene as well.41–43
To the best of our knowledge, there is almost no attempt so far to evaluate the application potential of nanocomposite membranes comprising PBI and CNF for HT-PEMFCs. Hence, we prepared two kinds of nanocomposite membranes of CNF/6FPBI and amine-functionalized CNF/6FPBI in this work. The amine-functionalized CNF was prepared by reacting CNF with aminobenzoic acid in polyphosphoric acid via Friedel–Crafts acylation, referred to the work done by S. Ahn and his coworkers.44 A hexafluoroisopropylidene containing PBI (6FPBI) was adopted as the polymer matrix for the nanocomposite membranes. The influence of incorporating amine-functionalized CNF on several physicochemical properties of the phosphoric acid doped nanocomposite membranes, including their thermal stability, mechanical strength, acid doping level, and proton conductivity, will be studied. Also, the fuel cell performances of the membrane electrolyte assembly (MEA) comprising the nanocomposite membranes will be evaluated in this work.
2. Experimental section
2.1 Materials
2,2-Bis(4-carboxyphenyl)hexafluoropropane (98%, from TCI), 3,3′-diaminobenzidine (99% from Alfa Aesar), phosphorus pentoxide (P2O5) (98%, from Showa), polyphosphoric acid (PPA) (≥83% as P2O5, from Sigma Aldrich), phosphoric acid (H3PO4) (85%, from Showa) and sodium hydroxide (NaOH) (98%, from Alfa Aesar) were used without further purification. N,N-dimethylacetamide (DMAc) (from Aldrich) and N,N-dimethylformamide (DMF) (99.8%, from Acros Organics) were used as received. 4-Aminobenzoic acid (ABA) (99%) were purchased from Alfa Aesar. In addition, carbon nanofibers were used as received from New Yongyu Applied Technology Materials Co., Ltd, Tainan, which were 110–150 nm in diameter and 7 μm in length.2.2 Synthesis of hexafluoroisopropylidene containing PBI (6FPBI)
6.066 g (0.015 mol) of 2,2-bis(4-carboxyphenyl)hexafluoropropane and 3.247 g (0.015 mol) of 3,3′-diaminobenzidine were added, followed by 17.5 g of P2O5 and 230 g of PPA into a 250 mL, three-necked flask equipped with a mechanical stirrer.13 At first, the mixture was slowly stirred using a mechanical stirrer and purged with a slow stream of nitrogen while heating from room temperature to 120 °C for 24 h. Then, the reaction temperature increased from 120 °C to 220 °C for 24 h and remain at 220 °C for another 24 h during the polymerization. After the polymerization, the mixture was poured into 1 L of deionized water. The resulting polymer was precipitated from the water and collected by filtration. To neutralize the polymer, it was washed with basic aqueous solution of NaOH and then washed thoroughly with deionized water five times. The 6FPBI polymer was obtained after drying in a vacuum oven at 100 °C for 24 h.2.3 Preparation of aminobenzoyl-functionalized carbon nanofibers (CNF–aminobenzoyl)
The procedures described below follow the synthesis method involving the Friedel–Crafts acylation reaction reported in the previous works.44 4-Aminobenzoic acid (0.5 g, 0.036 mol), CNFs (0.5 g), P2O5 (5 g), and PPA (20 g) was placed into a 250 mL, three-necked, round bottom flask equipped with a mechanical stirrer. The mixture was stirred under dry nitrogen at 100 °C for 4 h and at 130 °C for 48 h. At the end of the reaction, deionized water was added into the flask. After repeatedly washing with water until the pH reached 7, the resulting substance was isolated by filtration. Then, the product CNF–aminobenzoyl was obtained after drying in a vacuum oven for 24 h. Before use, CNF–aminobenzoyl was ground into fine powders using a mortar and pestle.2.4 Preparation of nanocomposite membranes and acid doping
A predetermined amount of the 6FPBI powder was dissolved in DMAc at 80 °C in a vial to make a 2 wt% solution. Meanwhile, a predetermined amount of CNF was dispersed in 3 mL of DMAc in another vial using a probe-type sonication (Q125 Sonicator) with an ice bath for 5 min. A small portion (about one-tenth) of the polymer solution was mixed with the CNF suspension at the beginning. Subsequently, a bath-type sonication for 30 min along with a probe-type sonication for 2 min was applied to the mixture. After that, the rest of the polymer solution was added to the mixture. A probe-type sonication for 2 min was applied to the mixture again for obtaining a homogeneous suspension. Finally, the resulting suspension was poured onto a petri dish. After most of the solvent was evaporated at 80 °C under reduced pressure for 12 h and the residual solvent was removed at 160 °C under vacuum for 12 h, a nanocomposite membrane was obtained. All the membranes used in this work was prepared with a thickness of 45 μm ± 2 μm.Acid-doped composite membranes were prepared by immersing the membranes in phosphoric acid at 80 °C for 3 h. Before measuring the doping levels, the membranes were dried in an air-flow oven at 110 °C for at least 12 h to remove the additional water within them and then stored at a temperature higher than 100 °C to avoid moisture absorption from the air. The doping level (χ), defined as the number of phosphoric acid per repeated unit of the polymer, was determined by weighing and calculated according to the following equation:
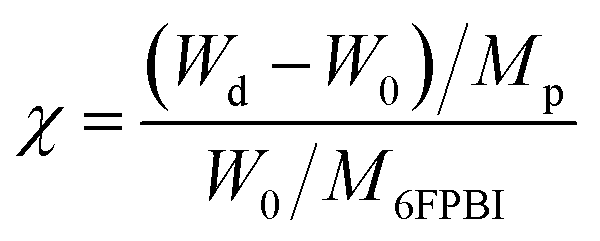
2.5 Characterizations and measurements
Fourier transform infrared spectroscopy (FTIR) analysis was performed on a JASCO FT/IR 4600 spectrometer with a KBr pellet containing a tiny amount of CNFs to identify the functional groups on CNFs. FTIR spectra in absorption mode were obtained by scanning 16 times in the wavenumber range from 400 cm−1 to 4000 cm−1 with a resolution of 2 cm−1. X-ray photoelectron spectroscopy (XPS) analysis was conducted on a ULVAC PHI5000 VersaProbe instrument to confirm the CNF functionalization. The XPS spectra were collected with a monochromatized Al Kα X-ray source generated by an electron beam (24.7 W). The XPSPEAK software version 4.1 was employed to deconvolute spectra into component peaks using a curve fitting method based on Gaussian Lorentzian sum functions. The background of the spectra was subtracted using the Shirley method. Thermogravimetric analysis (TGA) was carried out on a TA instrument model 2050 to examine the thermal stability and composition of materials. Each thermogram was recorded at a heating rate of 10 °C min−1 from 100 °C to 700 °C in a nitrogen flow (20 mL min−1). Scanning electron microscopy (SEM) was utilized to observe the morphology of the composite membranes. Before the observation, the cross-section of membrane was prepared by fracturing a membrane in liquid nitrogen and subsequently coating with gold. SEM micrographs were taken on a JEOL JSM-6700F high-resolution thermal field emission electron microscope with a voltage of 5.0 kV. The mechanical properties of composite membranes were analyzed using a Shimadzu AG-SI universal testing machine. The stress–strain curves were recorded at a 5 mm min−1 testing speed under ambient conditions. The samples were prepared with the dimensions of 30 mm in length, 4.5 mm in width, and approximately 30–40 μm in thickness. All the results are the average values for at least five samples. The proton conductivity of acid-doped membranes was measured using an Autolab PGSTAT 302N impedance analyzer at a frequency range of 102 to 105 Hz with an amplitude of 10 mV. The four-point-probe method was applied to measure the in-plane conductivity of the composite membranes. The impedance of the acid-doped composite membrane was determined from a Nyquist plot, the ohmic resistance of which was the approximate value of the real impedance-axis intercept. The proton conductivity of the acid-doped composite membrane was calculated according to the following equation:where σ, A, L, and RΩ are the proton conductivity (S cm−1), the cross-section area of the membrane (cm2), the length between the voltage measuring probes (0.425 cm), and the ohmic resistance (Ω), respectively. The dimensions of a membrane were 3 cm in length and 1 cm in width.
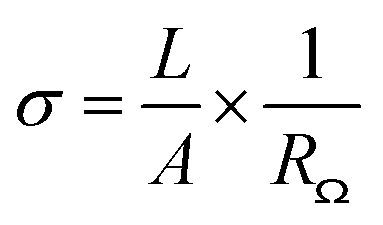
The oxidative stability of the membrane was evaluated using the Fenton test. The membrane was immersed in a 3 wt% hydrogen peroxide aqueous solution containing 4 ppm Fe2+ at 80 °C for 120 h. The remaining weight of the degraded membrane in the dry state was recorded every 24 h.
2.6 Fuel cell tests
The membrane electrode assemblies (MEAs) based on the acid-doped nanocomposite membranes of 6FPBI and CNFs were fabricated using the gas diffusion electrode (GDE) method. The catalyst ink consisting of carbon black supported platinum catalyst (20 wt% platinum on Vulcan XC-72, 0.16 g), 6FPBI ionomer (0.04 g), and DMAc solvent (9.8 g), was prepared by mixing and dispersing with a probe-type sonication for 3 min. A predetermined amount of the catalyst ink was coated on a microporous carbon paper (SGL, 24BC) by depositing consecutive drops. This procedure was conducted along with heating on a hot plate at 160 °C for evaporating the solvent. After that, a GDE with a platinum loading of 1.0 mg cm−2 was obtained. The ratio of carbon black supported platinum catalyst to 6FPBI was 4. The MEAs were fabricated by hot-pressing a piece of acid-doped nanocomposite membrane between two pieces of GDEs at 130 °C for 10 min. The active area of an MEA was 4 cm2.Fuel cell tests were carried out with single-cell hardware provided from HEPHAS Energy Co. During the operation, the fuel cell was fed with un-humidified hydrogen and oxygen at a constant flow rate of 100 mL min−1 under ambient pressure. Meanwhile, the I–V characteristics of fuel cells were recorded and controlled with a fuel cell testing system equipped with an electronic load unit controller and operation control software, which was provided by Tension Energy Inc.
3. Results and discussion
3.1 Characterization of CNF and CNF–aminobenzoyl
CNF–aminobenzoyl was prepared by the functionalization of CNF with aminobenzoic acid in polyphosphoric acid via Friedel–Craft acylation at temperatures between 100 °C and 130 °C, as shown in Scheme 1. The elements and the possible functional groups that belong to CNFs were analyzed using XPS. The full XPS spectra of pristine CNF (referred to the as-received CNF) and CNF–aminobenzoyl were provided in Fig. 1(a and b), respectively. Both spectra displayed two signals referring to C 1s and O 1s. Besides, CNF–aminobenzoyl exhibited an additional signal corresponding to N 1s as expected, which would come from aminobenzoyl groups. Fig. 1(c and d) show the narrow scan XPS spectra of C 1s in the range of binding energy from 280 eV to 294 eV for pristine CNF and CNF–aminobenzoyl, respectively. For pristine CNF, three sub-peaks at 284.6 eV, 286.3 eV, and 289 eV assigned to C–C, C–O–C, O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds can be identified by the peak deconvolution.45 We supposed that the oxygen-related bonds might originate from the defects at the edge of pristine CNF. On the other hand, the deconvoluted C 1s peak of the CNF–aminobenzoyl can be distinguished into four sub-peaks attributed to C–C bond at 284.6 eV, C–N bond at 285.9 eV, CO bond at 287.7 eV, as well as O–CO bond at 289 eV. The presence of C–N bond and CO bond would result from the aminobenzoyl groups.
O bonds can be identified by the peak deconvolution.45 We supposed that the oxygen-related bonds might originate from the defects at the edge of pristine CNF. On the other hand, the deconvoluted C 1s peak of the CNF–aminobenzoyl can be distinguished into four sub-peaks attributed to C–C bond at 284.6 eV, C–N bond at 285.9 eV, CO bond at 287.7 eV, as well as O–CO bond at 289 eV. The presence of C–N bond and CO bond would result from the aminobenzoyl groups.
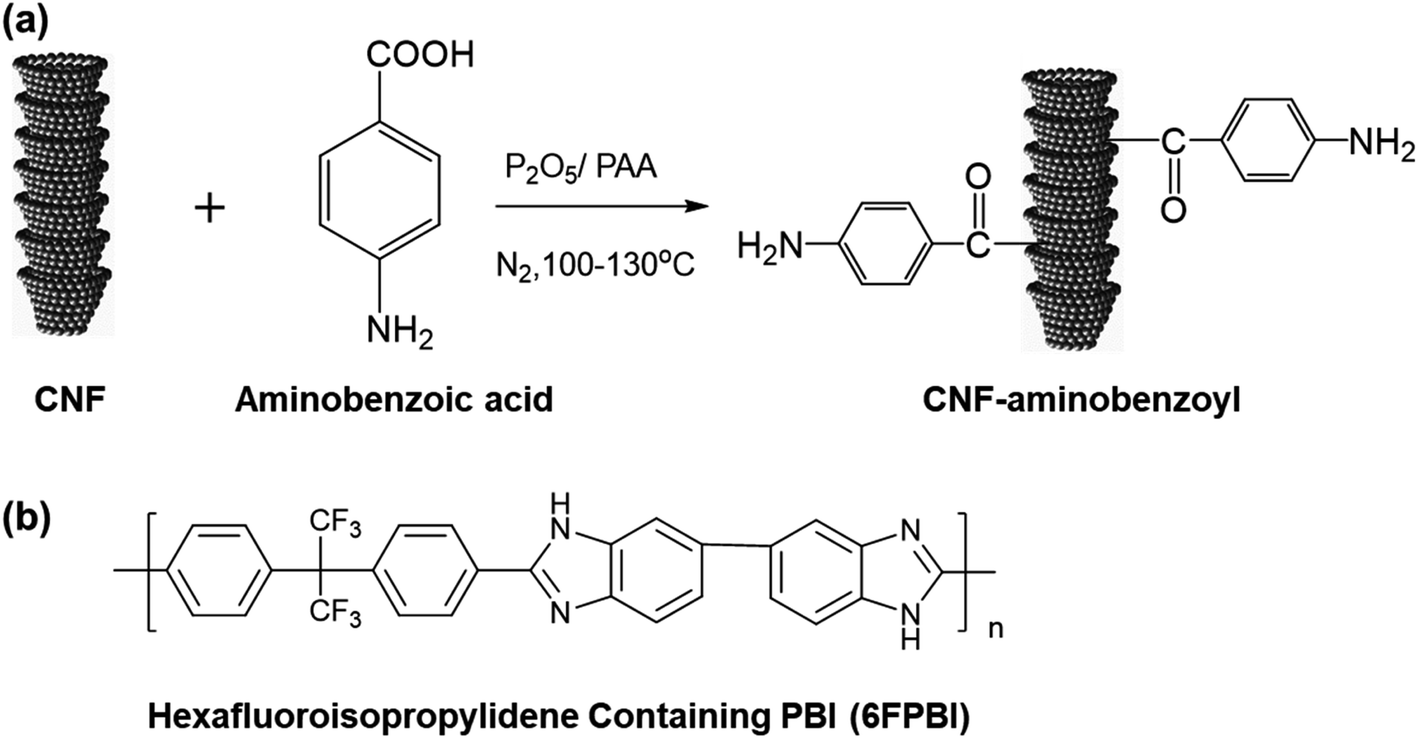 | ||
| Scheme 1 (a) Synthesis of CNF functionalized with aminobenzoyl groups (CNF–aminobenzoyl) via Friedel–Craft Acylation. (b) Chemical structure of hexafluoroisopropylidene containing PBI (referred as 6FPBI). | ||
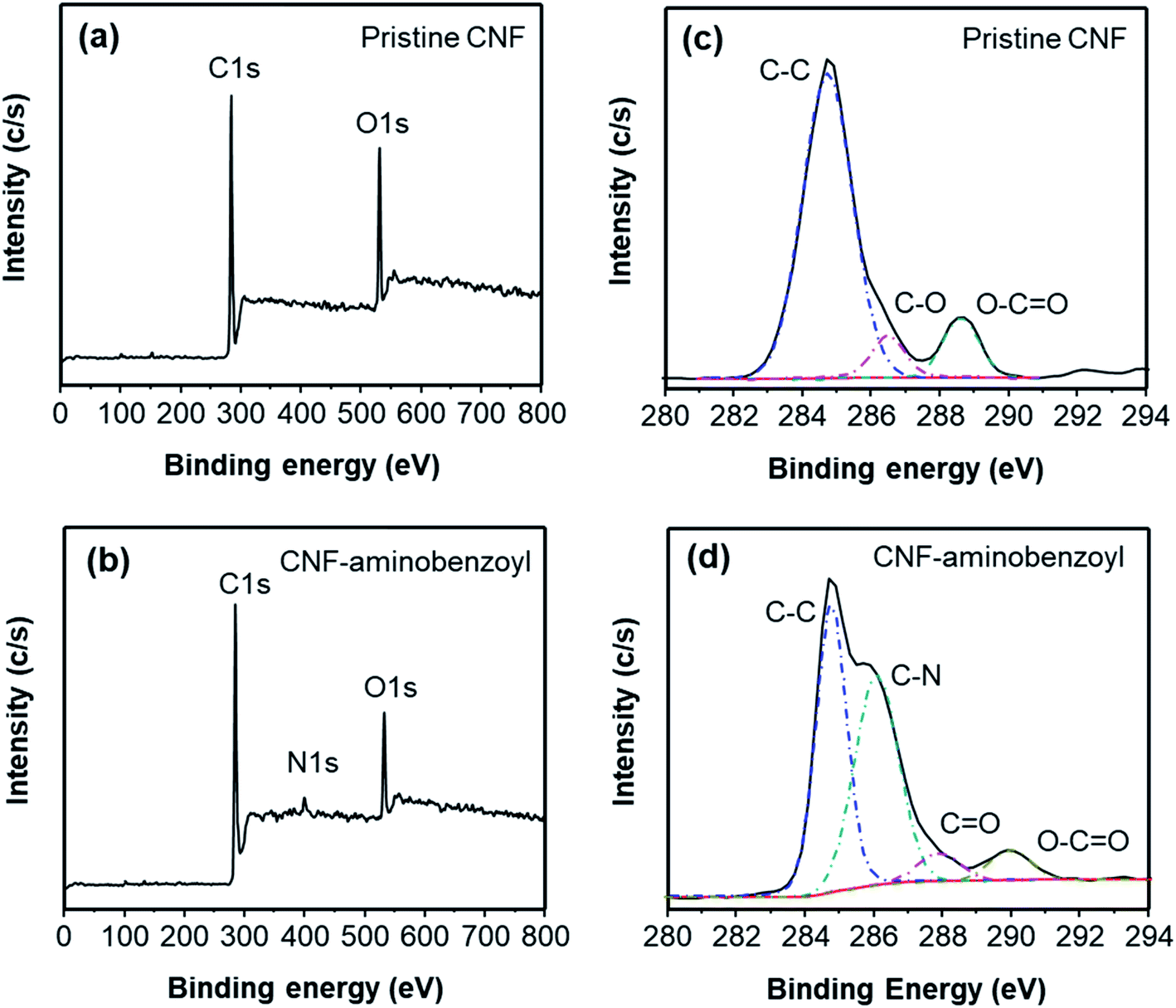 | ||
| Fig. 1 Full XPS spectra of (a) pristine CNF and (b) CNF–aminobenzoyl, as well as deconvoluted C 1s spectra of (c) pristine CNF and (d) CNF–aminobenzoyl. | ||
Fig. 2(a) shows the FTIR spectra of pristine CNF and CNF–aminobenzoyl. Both spectra exhibited a characteristic peak of CC bond at 1614 cm−1. In the spectrum of CNF–aminobenzoyl, two additional absorption peaks at 1710 cm−1 and 1518 cm−1 attributed to CO and N–H bonds were found.46 A joint analysis of XPS and FTIR confirmed the existence of aminobenzoyl groups on CNF–aminobenzoyl.
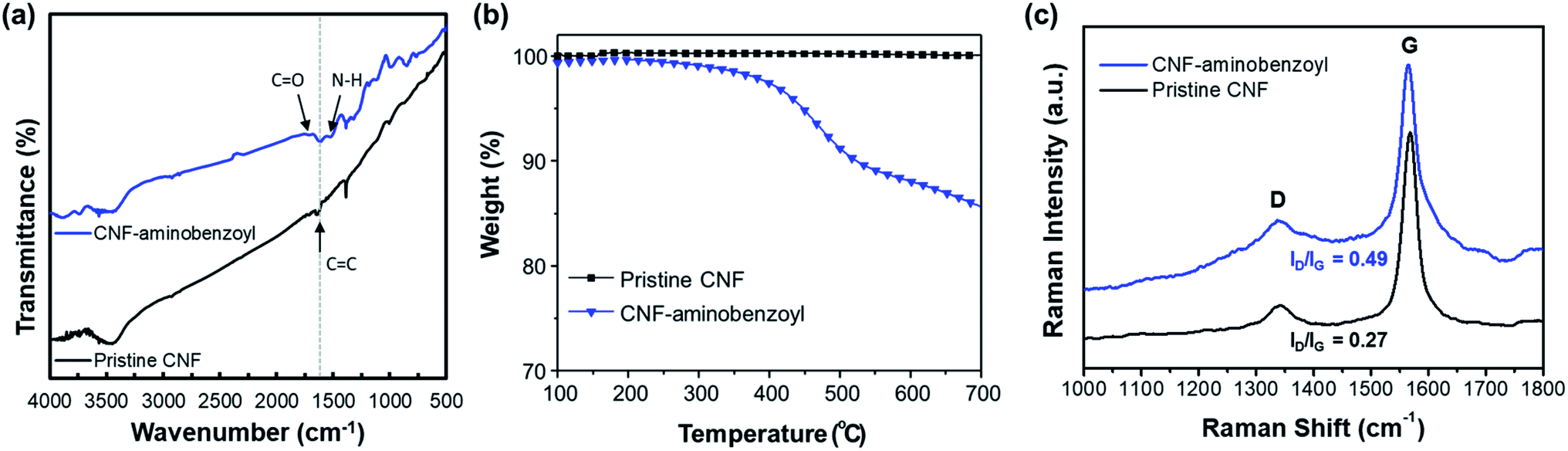 | ||
| Fig. 2 (a) FTIR spectra, (b) TGA thermograms, and (c) Raman spectra for pristine CNF and CNF–aminobenzoyl. | ||
The thermal stability of the pristine CNF and CNF–aminobenzoyl were evaluated using TGA under a nitrogen atmosphere. As shown in Fig. 2(b), the thermograms of pristine CNF exhibited almost no weight loss until 700 °C, indicating its excellent thermal stability. By comparison, the decomposition of CNF–aminobenzoyl began to take place from the temperature of approximately 400 °C. The weight loss of about 15% between 400 °C and 700 °C for CNF–aminobenzoyl would be attributed to the decomposition of attached groups.
Fig. 2(c) presents the Raman spectra of pristine CNF and CNF–aminobenzoyl. Similar to other carbon nanomaterials, both the pristine CNF and CNF–aminobenzoyl exhibited the two characteristic bands, D band at around 1350 cm−1 and G band at around 1560 cm−1.47 The D band is assigned to sp2-related defects and the structural disorder close to the edge of graphite structure. The presence of G band is due to the tangential C–C stretching mode.48 Therefore, comparing the intensity ratio of the D- and G-bands (ID/IG) allows us to evaluate the level of disorder or the quantity of defects within CNFs.47 The Raman spectra displayed that the ID/IG of CNF–aminobenzoyl was 0.49, higher than that of pristine CNF (ID/IG = 0.27). This result implied that the functionalization of CNF via the Friedel–Craft acylation likely created more defects within nanofibers.
3.2 Characterization of nanocomposite membranes
In this work, 6FPBI was adopted as the nanocomposite membranes' polymer matrix, and it was synthesized in polyphosphoric acid by condensation polymerization. The resulting 6FPBI exhibited a number average molecular weight of 237 k and an inherent viscosity of 2.1 dL g−1. Those were measured using a GPC analysis (calibrated with polystyrene standards) and a Ubbelohde tube viscometer in DMAc at 30 °C, respectively.The nanocomposite membranes with various CNF contents were prepared using solvent-assisted dispersion followed by solvent casting. During the solvent-assisted dispersion, we adopted a so-called priming method to avoid the CNF aggregations and obtain a homogeneous suspension49 The priming method was carried out by mixing the small part of polymer solution with the CNF suspension and subsequently sonicating the mixture. Then, the rest of the polymer solution was added into the mixture and the sonication was conducted again to obtain a good dispersion of CNF in the composite solution. It is believed that the stress at the interface between the fillers and the polymer matrix would be reduced after the priming process.50,51 Therefore, we can fabricate nanocomposite membranes with homogeneous filler distribution.
The morphology of the nanocomposite membranes was investigated using SEM. Fig. 3 presented the SEM micrographs of the pristine CNF/6FPBI nanocomposite membranes with various filler contents from the cross-section view. We found almost no aggregations of pristine CNFs within the 6FPBI matrix if the filler content was not higher than 0.3 wt%, as seen in Fig. 3(a and c). However, the filler agglomeration began to appear as soon as the loading of pristine CNF reached 0.5 wt%, as shown in Fig. 3(e). Meanwhile, it can be clearly seen in Fig. 3(b and d) that pristine CNFs were pulled out and floating on the fracture surface, indicating poor interfacial interaction between the pristine CNF and the 6FPBI matrix.39 Moreover, the poor adhesion between the filler and the polymer matrix caused some interfacial gaps, which can be found in Fig. 3(e and f).
 | ||
Fig. 3 SEM micrographs showing the fracture surface of pristine CNF/6FPBI nanocomposite membranes with (a and b) 0.1 wt%, (c and d) 0.3 wt%, and (e and f) 0.5 wt% loadings. (a, c and e) The images captured at low magnifications. (b, d and f) The enlarged images recorded at a high magnification of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. 000×. | ||
In Fig. 4, we observed that the CNF–aminobenzoyl/6FPBI nanocomposite membranes did not exhibit any filler agglomerations until the loading of CNF–aminobenzoyl was up to 0.5 wt%. This observation is similar with the finding of the CNF/6FPBI nanocomposite membranes. Despite that, we observed that some CNF–aminobenzoyl fillers were embedded in the matrix, and residual 6FPBI wrapped some of them. Also, it is not easy to distinguish the interfacial boundary between CNF–aminobenzoyl and 6FPBI. These results proved good interfacial interaction between CNF–aminobenzoyl and 6FPBI. Compared to pristine CNF, CNF–aminobenzoyl exhibited better compatibility with the 6FPBI matrix.
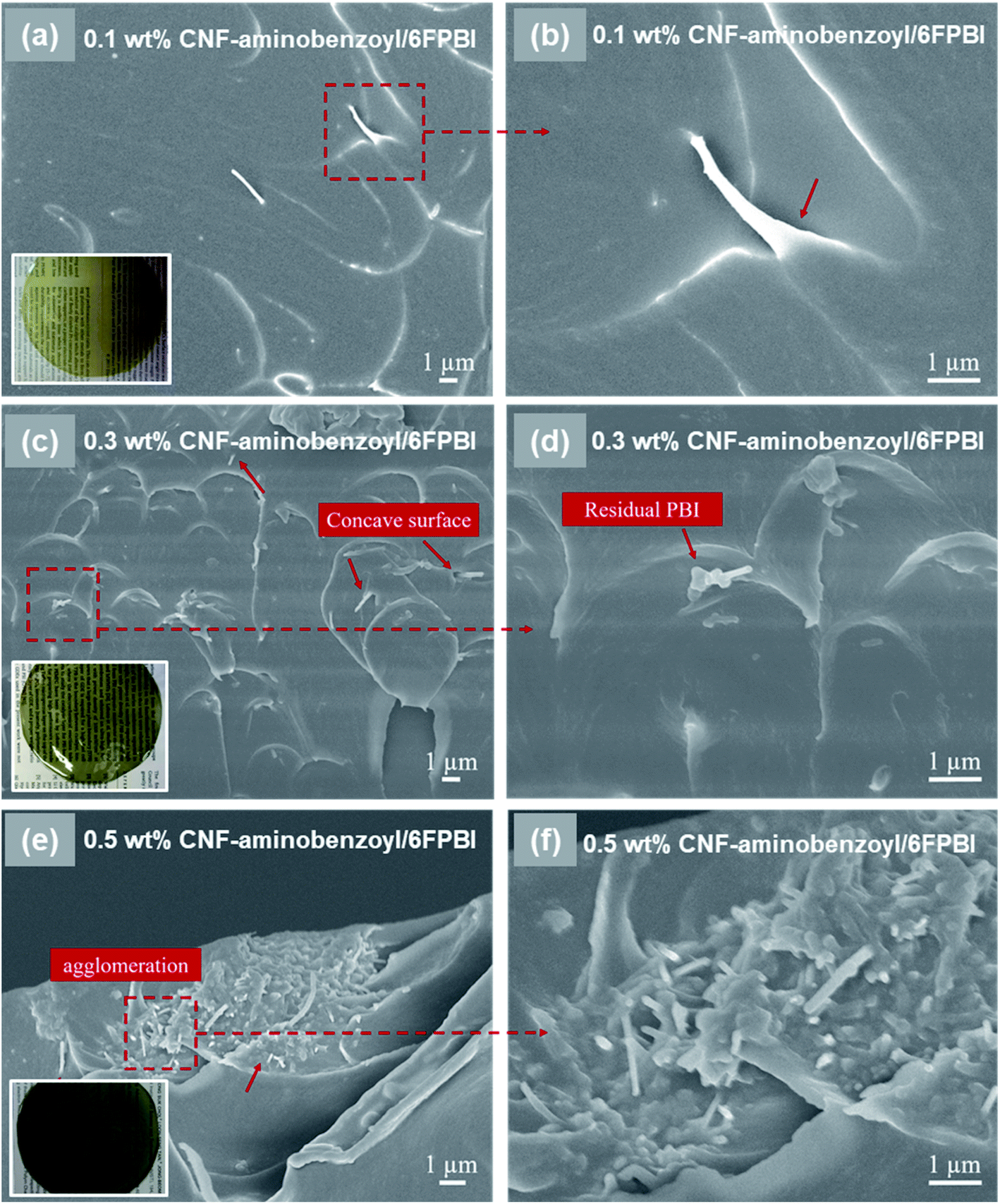 | ||
| Fig. 4 SEM micrographs showing the fracture surface of CNF–aminobenzoyl/6FPBI nanocomposite membranes with (a and b) 0.1 wt%, (c and d) 0.3 wt%, and (e and f) 0.5 wt% loadings. (a, c and e) The images captured at low magnifications. (b, d and f) The enlarged images recorded at a high magnification of 10000×. | ||
We conducted the XRD analyses to investigate the influences on the polymer structure once the nanofibers were incorporated into the polymer matrix. Fig. 5 shows the X-ray diffraction profiles of the 6FPBI, 0.3 wt% CNF/6FPBI, and 0.3 wt% CNF–aminobenzoyl/6FPBI nanocomposite membranes. We found that an additional tiny peak appeared at 26.6°, which corresponded to the CNF fillers.52 The broad band ranging between 10° and 30°, reflecting to the semicrystalline structure of the 6FPBI matrix, did not shift. Also, the shape of the diffraction profiles looks similar for the three membranes. However, we noticed a slight increase in the full width at half-maximum (FWHM) of the broad band after incorporating the nanofibers into the 6FPBI matrix. The FWHM value increased from 11.7° for 6FPBI to 14.3° for 0.3 wt% CNF/6FPBI and 13.2° for 0.3 wt% CNF–aminobenzoyl/6FPBI. An increase in FWHM without shifting the peak position revealed a reduction in crystallinity without changing the d-spacing for the nanocomposite membranes. The crystallinity reduction may be due to the fact that the nanofillers disturb the self-assembly of polymer chains though mutual interactions.53
 | ||
| Fig. 5 X-ray diffraction profiles of the 6FPBI, 0.3 wt% CNF/6FPBI, and 0.3 wt% CNF–aminobenzoyl/6FPBI membranes. | ||
To detect the change in thermal stability after the nanofibers were incorporated into the polymer matrix, we conducted TGA analyses for the nanocomposite membranes. As shown in Fig. 6, incorporating nanofibers into 6FPBI was found to produce an insignificant effect on the thermal stability of the nanocomposite membranes. All the thermograms of CNF/6FPBI and CNF–aminobenzoyl/6FPBI with various CNF contents looked almost identical. All of them exhibited a considerable weight loss in the temperature range between 500 °C and 600 °C, which resulted from the decomposition of the 6FPBI polymer backbone.54
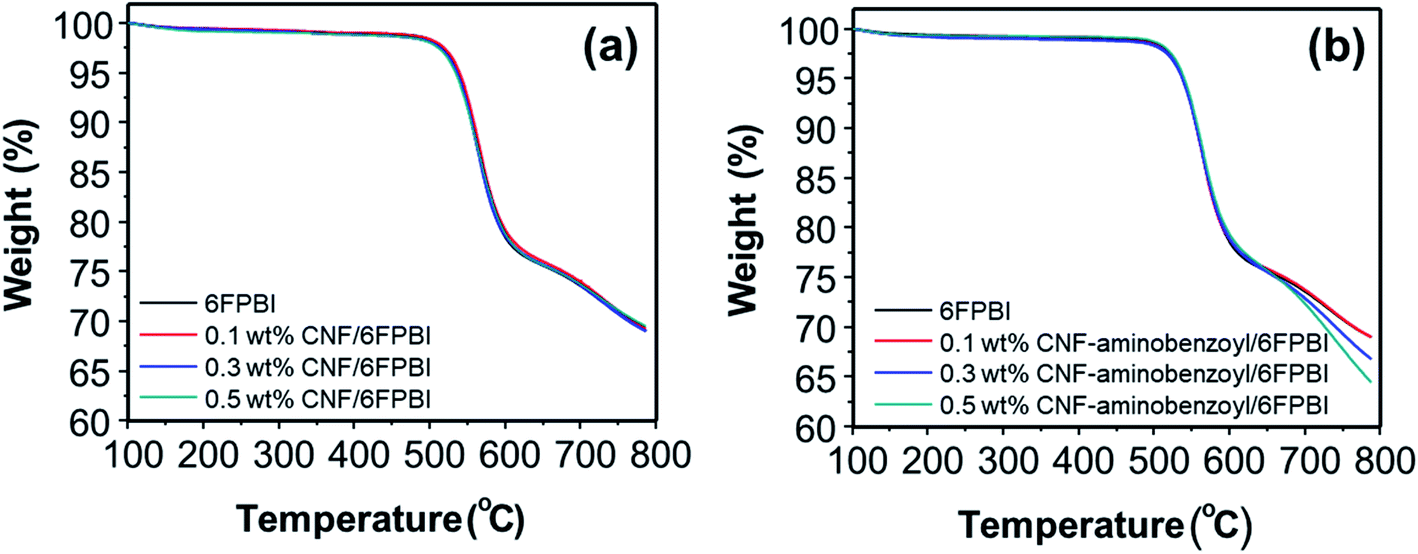 | ||
| Fig. 6 Thermograms of (a) CNF/6FPBI and (b) CNF–aminobenzoyl/6FPBI nanocomposite membranes with various filler contents under a nitrogen atmosphere. | ||
3.3 Mechanical properties and proton conductivity of nanocomposite membranes
We conducted a tensile test to examine the mechanical properties of nanocomposite membranes. Table 1 provides the tensile testing results for the 6FPBI and nanocomposite membranes without acid doping. It is found that all the nanocomposite membranes containing pristine CNF or CNF–aminobenzoyl had better tensile strength and strain at break compared to the 6FPBI membrane. This finding confirmed that the addition of CNF is capable of enhancing the mechanical properties of 6FPBI membranes.| Undoped membranes | Filler content (wt%) | Tensile properties | ||
|---|---|---|---|---|
| Elastic modulus (GPa) | Tensile strengtha (MPa) | Strain at break (%) | ||
| a Tensile strength is the maximum stress during the tensile deformation of the membrane before the break. | ||||
| 6FPBI | 0 | 0.96 ± 0.10 | 70.3 ± 1.8 | 21.8 ± 7.2 |
| CNF/6FPBI | 0.1 | 0.98 ± 0.15 | 71.4 ± 6.8 | 23.1 ± 3.1 |
| 0.3 | 1.09 ± 0.17 | 77.7 ± 1.4 | 27.3 ± 3.6 | |
| 0.5 | 0.91 ± 0.02 | 71.3 ± 0.8 | 25.9 ± 0.6 | |
| CNF–aminobenzoyl/6FPBI | 0.1 | 1.12 ± 0.13 | 72.6 ± 1.5 | 25.3 ± 8.5 |
| 0.3 | 1.21 ± 0.16 | 78.9 ± 2.0 | 28.1 ± 3.5 | |
| 0.5 | 0.85 ± 0.02 | 71.3 ± 0.7 | 26.1 ± 0.7 | |
However, the tensile performance was found dependent on the filler content. The optimum tensile performance was obtained when the filler content was 0.3 wt% for both the pristine CNF/6FPBI and CNF–aminobenzoyl/6FPBI nanocomposite membranes. When the filler content kept increasing to 0.5 wt%, the elastic modulus, tensile strength, and strain at break of the composite membrane went down instead. The decrease in tensile performance was supposed to result from filler agglomerations as observed previously in the SEM micrographs. Compared to pristine CNF, we found that CNF–aminobenzoyl enhanced the mechanical properties of the membrane more effectively. At the same filler loadings, the tensile performance of CNF–aminobenzoyl/6FPBI nanocomposite membranes was better than that of CNF/6FPBI. For example, the tensile strength values were measured to be 77.7 MPa and 78.9 MPa for 0.3 wt% CNF/6FPBI and 0.3 wt% CNF–aminobenzoyl/6FPBI nanocomposite membranes, respectively. That is because the better interfacial adhesion of 0.3 wt% CNF–aminobenzoyl/6FPBI can enhance stress transfer from the polymer matrix to the nanofillers, leading to better effectiveness in improving the mechanical properties of nanocomposite membranes.
The acid doping was carried out by immersing 6FPBI and nanocomposite membranes in phosphoric acid solution at 80 °C for 3 h. The amount of phosphoric acid absorbed by a 6FPBI membrane is referred to as the doping level (χ), which has a direct effect on the proton conduction ability of a membrane. The excess phosphoric acid, also called free acid, will be produced within 6FPBI when the doping level is higher than 2.2,55 It is known that the free acid plays a major role in assisting the proton transport via hopping mechanism, especially under anhydrous conditions.56 It presents in Fig. 7 that the doping level of the PBI pristine membrane was 10.9, and the presence of CNF or CNF–aminobenzoyl further improved the doping level to some extent. Both the highest doping levels achieved at the filler loading of 3 wt%, and they were measured to be 11.5 and 12.0 for the pristine CNF/6FPBI and CNF–aminobenzoyl/6FPBI nanocomposite membranes, respectively. Also, the doping level of the CNF–aminobenzoyl/6FPBI was higher than that of the pristine CNF/6FPBI. This result implied that the presence of the amine groups on the nanofillers would be helpful for high holding capacity of phosphoric acid in the PBI based nanocomposite membranes, was in line with the previous reports.34,37
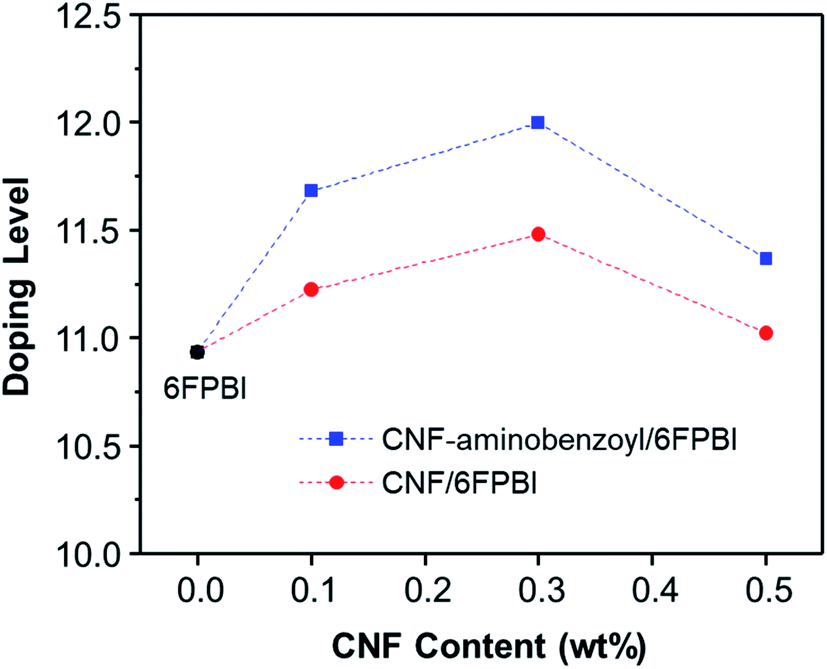 | ||
| Fig. 7 Doping level as a function of CNF content for the acid-doped nanocomposite membranes. | ||
It is noted that the doping level of the membrane varied with the filler content as shown in Fig. 7. When the filler content was 0.5 wt% higher than its optimum value (0.3 wt%), the doping level of the membrane turned to decline. In the work of Y. Devrim et al., they found similar result for the acid-doped graphene oxide/PBI nanocomposite membranes.25 Since both the trends of the doping level and the tensile performance varying with filler content were analogous, we presumed that the capacity of acid doping might be associated with the mechanical strength of the materials to a certain extent. Besides, the interactions of the hydrophilic oxygen-containing or amine groups on the surface of nanofibers with phosphoric acid via hydrogen bonding may have contributed to the increased doping level. However, the filler agglomerations may limit the total surface area of nanofibers within the membrane, leading to a reduced amount of phosphoric acid that can be attracted by the nanofibers. Accordingly, it can explain why the doping level decreased with increasing the filler content from 0.3 wt% to 0.5 wt%.
Table 2 shows the tensile test result of the acid-doped 6FPBI, 0.3 wt% CNF/6FPBI, and 0.3 wt% CNF–aminobenzoyl/6FPBI membranes. The elastic modulus and tensile strength for all the membranes decreased considerably after phosphoric acid doping. For example, the tensile strength of the 0.3 wt% CNF–aminobenzoyl/6FPBI membrane declined to 12.5 MPa from 78.9 MPa when it obtained a high doping level. This softening phenomenon is due to the plasticizing effect of phosphoric acid. Despite the occurrence of softening, the result confirmed that both the pristine CNF or amine-functionalized CNF nanofillers still enhanced the mechanical properties of the 6FPBI membrane in the presence of phosphoric acid. We noticed that all the acid-doped membranes swollen, and their thicknesses increased to be 95 μm ± 2 μm after phosphoric acid doping. The swelling ratios for all the membranes were similar (about 110%), which may be due to the small difference between their doping levels.
| Acid-doped membranesa | Filler content (wt%) | Tensile properties | ||
|---|---|---|---|---|
| Elastic modulus (GPa) | Tensile strengthb (MPa) | Strain at break (%) | ||
| a All the acid-doped nanocomposite membranes were prepared by immersing the membranes in phosphoric acid at 80 °C for 3 h before the tensile test.b Tensile strength is the maximum stress during the tensile deformation of the membrane before the break. | ||||
| 6FPBI | 0 | 0.23 ± 0.04 | 9.7 ± 0.9 | 86.1 ± 19.4 |
| CNF/6FPBI | 0.3 | 0.28 ± 0.07 | 10.2 ± 1.5 | 87.9 ± 15.5 |
| CNF–aminobenzoyl/6FPBI | 0.3 | 0.36 ± 0.11 | 12.5 ± 1.4 | 96.4 ± 13.1 |
The measurements of proton conductivities for the acid-doped membranes were conducted under anhydrous condition at temperatures ranging from 100 °C to 160 °C. The proton conductivity of a PBI based membrane is highly dependent on its acid doping level.57 Since that, we compared the temperature dependence of proton conductivity for the pristine CNF/6FPBI and CNF–aminobenzoyl/6FPBI nanocomposite membranes with the filler content of 0.3 wt% at their best doping levels. As shown in Fig. 8, the CNF–aminobenzoyl/6FPBI nanocomposite membrane having higher doping level (χ = 12.0) exhibited higher proton conductivity in the temperature range than others. The proton conductivity at 160 °C of the 6FPBI, 0.3 wt% CNF/6FPBI, and 0.3 wt% CNF–aminobenzoyl/6FPBI nanocomposite membranes was measured to be 0.14 S cm−1, 0.17 S cm−1, and 0.20 S cm−1, respectively. This result showed that the presence of nanofibers improved the proton conductivity of the acid-doped membranes, confirming the beneficial influence of doping level on the proton conductivity. However, the difference in the doping level between these membranes (Δχ < 1.1) was not large. This noticeable improvement in proton conductivity would be partially attributed to the hydrogen bonding networks between the hydrophilic oxygen-containing or amine groups on the nanofibers and phosphoric acid which could facilitate the hoping of protons through them. Similar explanations can be found in the previous studies.24,25,37 Furthermore, we supposed that the slight reduction in polymer crystallinity caused by the presence of nanofibers, which might reduce the hindrance of the phosphoric acid molecule or proton transport. This could be in part responsible for the increase in both the doping level and proton conductivity.
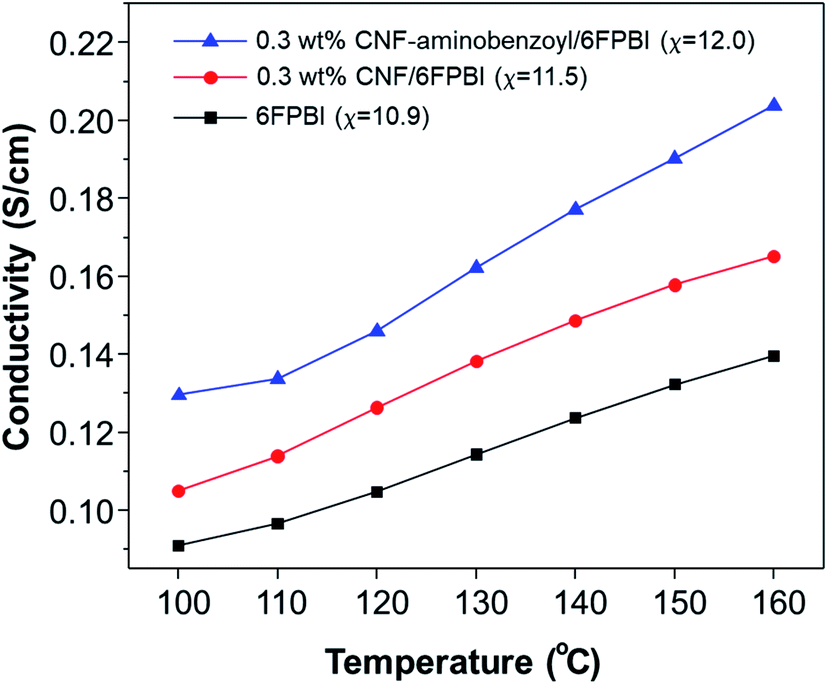 | ||
| Fig. 8 Temperature dependence of proton conductivity for acid-doped 6FPBI, 0.3 wt% CNF/6FPBI, and 0.3 wt% CNF–aminobenzoyl/6FPBI membranes. | ||
Table 3 summarizes the mechanical properties and proton conductivity for the PBI based nanocomposite and 6FPBI-related membranes in the recent studies for comparison.23,24,26,27,58–60 The doping level of the CNF–aminobenzoyl/6FPBI membrane is close to those of the PBI/MGO and PBI–clay membranes. These three membranes exhibited similar proton conductivities. However, the tensile strength of the acid-doped PBI–clay membranes was much less than those of the other membranes. This comparison implied that carbon-based nanofillers seem to enhance the membrane more effectively than inorganic clay fillers. On the other hand, the acid-doped 6FPBI membrane prepared in this work displayed higher proton conductivity at 160 °C compared to the previously reported value (the proton conductivity for F6PBI at χ = 8.8),59 which was attributed to its higher doping level (χ = 10.9). It was reported that the proton conductivity of phosphoric acid at a temperature between 150 °C and 170 °C ranged from 0.48 S cm−1 to about 0.6 S cm−1.61,62 For the proton conductivities listed in Table 3, most of them are higher than 0.1 S cm−1, and some of them even surpass the boundary of phosphoric acid's proton conductivity.
| Acid-doped membrane | Filler content (wt%) | Doping level | Tensile strength (MPa) | Strain at break (%) | Proton conductivity (S cm−1) | Reference |
|---|---|---|---|---|---|---|
| a The chemical structure of F6PBI is identical to 6FPBI used in the present work. | ||||||
| CNF–aminobenzyl/6FPBI | 0.3 | 12.0 | 12.5 | 96.4 | 0.20 (at 160 °C) | Present work |
| PBI/MGO | 1.2 | 12.2 | 12.6 | 150.1 | 0.13 (at 160 °C) | 24 |
| cPBI/RGO | 1.0 | 12.5 | 19.3 | — | 0.59 (at 170 °C) | 26 |
| PyPBI/PGO | 1.5 | 9.9 | 4.6 | 139.2 | 0.76 (at 140 °C) | 27 |
| PBI–CNT | 1.0 | 8.0 | 39 | ∼8 | 0.74 (at 180 °C) | 23 |
| PBI–clay | 15 | 12.0 | 0.9 | 53.1 | 0.12 (at 150 °C) | 58 |
| F6PBIa | 0 | 8.8 | 29.3 | 84.2 | 0.06 (at 160 °C) | 59 |
| 6FPBI | 0 | 10.9 | 9.7 | 86.1 | 0.14 (at 160 °C) | Present work |
| F6PBI–10% R3a | 0 | 14.0 | 9.7 | 20.7 | ∼0.65 (at 180 °C) | 60 |
3.4 Fuel cell performance
The key component of a proton exchange membrane fuel cell is the MEA, where the electrochemical reactions take place to generate electrical power. The fabrication of the MEAs in this work involved a GDE preparation process. Fig. 9 presented the results of fuel cell performance tested at 160 °C in an anhydrous condition for the MEAs of 6FPBI, 0.3 wt% CNF/6FPBI, and 0.3 wt% CNF–aminobenzoyl/6FPBI. Although, all the open-circuit voltages (OCVs) were found to be approximately 0.65 V for the three MEAs, pretty lower than the theoretical value. Their fuel cells still were able to produce high current densities exceeding 1800 mA cm−2 at a cell voltage of 0.2 V, as shown in Fig. 9(a). The low OCVs could be caused by either the fuel crossover problem or the internal current leakage of the fuel cell.63 Once the fuel crossover problem occurs, the polarization curve generally will shift down toward a lower cell voltage, and the activation loss in cell voltage will remain. On the contrary, the internal current leakage will offset the polarization curve in the left direction toward a lower current density instead, and the activation loss in cell voltage will diminish.64 In our case, the activation loss was found insignificant, implying that we may have the internal current leakage problem with the fuel cell system. However, the maximum power density of the MEA based on the acid-doped 6FPBI membrane was measured to be 410 mW cm−2, as shown in Fig. 9(b). This value is comparable to the previously reported values,14,65 indicating that the fuel cell performance of the MEA fabricated in this work was acceptable.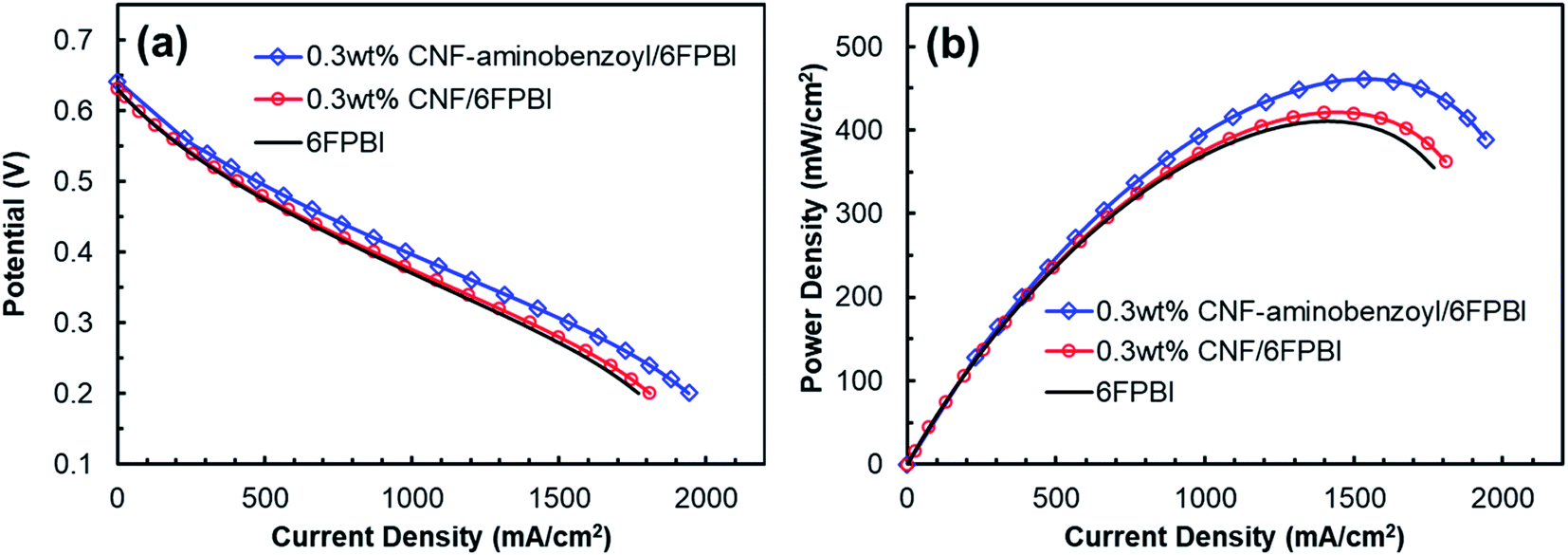 | ||
| Fig. 9 Plots of (a) potential as a function of current density and (b) power density as a function of current density for the MEAs of the acid-doped 6FPBI, 0.3 wt% CNF/6FPBI, and 0.3 wt% CNF–aminobenzoyl/6FPBI nanocomposite membranes tested in a fuel cell at 160 °C under the anhydrous condition. | ||
The intermediate region of the polarization curve, displaying a linear relation between the cell voltage and the current density, corresponds to the ohmic polarization resulting from the polymer electrolyte. As seen in Fig. 9(a), the polarization curve for the MEA of 0.3 wt% CNF–aminobenzoyl/6FPBI had a gentler slope during the ohmic polarization compared to the others, suggesting its ionic resistance was comparatively lower. This result is in agreement with the finding that the 0.3 wt% CNF–aminobenzoyl/6FPBI nanocomposite membrane exhibited relatively higher proton conductivity. It can be seen in Fig. 9(b) that the MEA of 0.3 wt% CNF–aminobenzoyl/6FPBI had relatively better fuel cell performance at 160 °C and achieved a maximum power density up to 461 mW cm−2.
3.5 Oxidative stability of nanocomposite membranes
A Fenton test was conducted to evaluate the oxidative stability of the membranes prepared in this work. As shown in Fig. 10, the remaining weights of all the membrane samples decreased as a function of time. The remaining weight of 6FPBI was found to be 71% at 120 h, which is similar to the value reported in our previous work.66 In addition, we found that both the 0.3 wt% CNF–aminobenzyl/6FPBI and 0.3 wt% CNF/6FPBI nanocomposite membranes exhibited less weight loss than the 6FPBI pristine membrane in presence of Fenton's agent (3% H2O2 containing 4 ppm Fe2+) during the 120 h test at 80 °C. This finding suggested that the incorporation of nanofillers like pristine CNF or amine-functionalized CNF into the 6FPBI matrix was capable of improving the membrane's chemical stability. The nanofillers were likely to shield the polar groups of PBI from the attack of free radical species.34 This result is similar to an earlier work of S. Mukhopadhyay and his coworkers using metal–organic framework (MOF) as nanofillers to enhance the oxidative stability the OPBI based nanocomposite membranes.67 Besides, CNF–aminobenzyl seemed enhance the membrane's oxidative stability more effectively than pristine CNF. The reason may be attributed to the better interaction between the CNF–aminobenzyl and the 6FPBI matrix.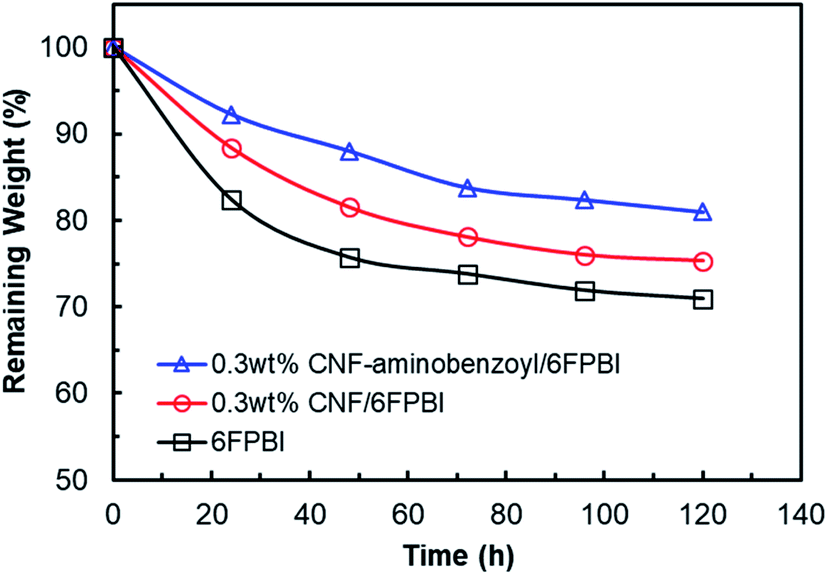 | ||
| Fig. 10 Oxidative stability of the 6FPBI, 0.3 wt% CNF/6FPBI, and 0.3 wt% CNF–aminobenzoyl/6FPBI membranes during the Fenton test (3% H2O2 containing 4 ppm Fe2+) for 120 h at 80 °C. | ||
In our previous study, the MEA based on the pristine 6FPBI membrane underwent a long-term fuel cell operation at a constant current of 200 mA cm−2 at 150 °C for 720 h with a small cell voltage decay of −0.294 mV h−1, indicating good durability of the 6FPBI membrane. On the other hand, the Fenton test is conducted to simulate the oxidative reaction by the attack of radical species (HO˙ and HOO˙) during fuel cell operation. Hence, the oxidative stability of the membrane can be used to estimate the membrane durability in oxidative environments as suggested by R. Gosalawit.68 Based on the Fenton test result above, we supposed that the 6FPBI nanocomposite membranes containing pristine CNF or amine-functionalized CNF would exhibit similar or even better durability for the fuel cell operation than the 6FPBI pristine membrane.
4. Conclusion
The amine-functionalized carbon nanofiber, namely CNF–aminobenzoyl, was successfully prepared by the Friedel–Craft acylation reaction of CNF with aminobenzoic acid, which was confirmed by XPS, FTIR, TGA, and Raman analyses. Two kinds of nanocomposite membranes of CNF/6FPBI and CNF–aminobenzoyl/6FPBI with various filler contents were fabricated using solvent-assisted dispersion followed by solvent casting. It was found that the incorporation of the nanofibers was capable of slightly decreasing the crystallinity, enhancing the mechanical strength, as well as increasing both the acid-doing level and the proton conductivity for these 6FPBI based nanocomposite membranes. As evidenced by the SEM observations, the 6FPBI matrix had better compatibility with CNF–aminobenzoyl than pristine CNF. As a result, CNF–aminobenzoyl enhanced the mechanical properties of the membrane more effectively than pristine CNF did at the same loading of filler. Furthermore, CNF–aminobenzoyl was found to obtain higher acid-doping level and proton conductivity of the membrane compared to pristine CNF, revealing that the presence of aminobenzoyl groups on the surface of nanofibers would be helpful for holding more phosphoric acid molecules in the membrane and facilitating the proton hopping. In the best case of present work, the 0.3 wt% CNF–aminobenzoyl/6FPBI nanocomposite membrane achieved the highest proton conductivity at 160 °C up to 0.2 S cm−1 and displayed the maximum power density of its MEA at 160 °C as high as 461 mW cm−2.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful for the financial support from the Ministry of Science and Technology (Taiwan, R.O.C.) through the project MOST 108-2218-E-992-306-MY2 and the project MOST 108-2221-E-006-119. Also, the authors acknowledge the use of XPS (ESCA, ULVAC PHI5000 VersaProbe) belonging to the Instrument Center of National Cheng Kung University and the operation service from Mr J. C. Li.References
- Q. Li, J. O. Jensen, R. F. Savinell and N. J. Bjerrum, Prog. Polym. Sci., 2009, 34, 449–477 CrossRef
.
- M. A. Haque, A. B. Sulong, K. S. Loh, E. H. Majlan, T. Husaini and R. E. Rosli, Int. J. Hydrogen Energy, 2017, 42, 9156–9179 CrossRef CAS
- C. Y. Wong, W. Y. Wong, K. Ramya, M. Khalid, K. S. Loh, W. R. W. Daud, K. L. Lim, R. Walvekar and A. A. H. Kadhum, Int. J. Hydrogen Energy, 2019, 44, 6116–6135 CrossRef CAS
- S. S. Araya, F. Zhou, V. Liso, S. L. Sahlin, J. R. Vang, S. Thomas, X. Gao, C. Jeppesen and S. K. Kær, Int. J. Hydrogen Energy, 2016, 41, 21310–21344 CrossRef CAS
- C. Yang, P. Costamagna, S. Srinivasan, J. Benziger and A. B. Bocarsly, J. Power Sources, 2001, 103, 1–9 CrossRef CAS
- Q. Li, R. He, J.-A. Gao, J. O. Jensen and N. J. Bjerrum, J. Electrochem. Soc., 2003, 150, A1599 CrossRef CAS
- Y. Shao, G. Yin, Z. Wang and Y. Gao, J. Power Sources, 2007, 167, 235–242 CrossRef CAS
- J. A. Asensio, E. M. Sánchez and P. Gómez-Romero, Chem. Soc. Rev., 2010, 39, 3210–3239 RSC
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS
- X. Li, X. Chen and B. C. Benicewicz, J. Power Sources, 2013, 243, 796–804 CrossRef CAS
- T.-H. Kim, S.-K. Kim, T.-W. Lim and J.-C. Lee, J. Membr. Sci., 2008, 323, 362–370 CrossRef CAS
- J. Yang, Q. Li, L. N. Cleemann, C. Xu, J. O. Jensen, C. Pan, N. J. Bjerrum and R. He, J. Mater. Chem., 2012, 22, 11185–11195 RSC
- S.-W. Chuang and S. L.-C. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4508–4513 CrossRef CAS
- G. Qian and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4064–4073 CrossRef CAS
- X. Li, G. Qian, X. Chen and B. C. Benicewicz, Fuel Cells, 2013, 13, 832–842 CAS
- Q. F. Li, H. C. Rudbeck, A. Chromik, J. O. Jensen, C. Pan, T. Steenberg, M. Calverley, N. J. Bjerrum and J. Kerres, J. Membr. Sci., 2010, 347, 260–270 CrossRef CAS
- J. J. Linares, L. C. Battirola and J. Lobato, in High Temperature Polymer Electrolyte Membrane Fuel Cells: Approaches, Status, and Perspectives, ed. Q. Li, D. Aili, H. A. Hjuler and J. O. Jensen, Springer International Publishing, Cham, 2016, pp. 275–295, DOI:10.1007/978-3-319-17082-4_13
- E. Quartarone, S. Angioni and P. Mustarelli, Materials, 2017, 10, 687 CrossRef
- R. Kannan, H. N. Kagalwala, H. D. Chaudhari, U. K. Kharul, S. Kurungot and V. K. Pillai, J. Mater. Chem., 2011, 21, 7223–7231 RSC
- Suryani, C.-M. Chang, Y.-L. Liu and Y. M. Lee, J. Mater. Chem., 2011, 21, 7480–7486 RSC
- J.-F. Wu, C.-F. Lo, L.-Y. Li, H.-Y. Li, C.-M. Chang, K.-S. Liao, C.-C. Hu, Y.-L. Liu and S. J. Lue, J. Power Sources, 2014, 246, 39–48 CrossRef CAS
- C. Xue, J. Zou, Z. Sun, F. Wang, K. Han and H. Zhu, Int. J. Hydrogen Energy, 2014, 39, 7931–7939 CrossRef CAS
- N. Guerrero Moreno, D. Gervasio, A. Godínez García and J. F. Pérez Robles, J. Power Sources, 2015, 300, 229–237 CrossRef CAS
- J. Yang, C. Liu, L. Gao, J. Wang, Y. Xu and R. He, RSC Adv., 2015, 5, 101049–101054 RSC
- N. Üregen, K. Pehlivanoğlu, Y. Özdemir and Y. Devrim, Int. J. Hydrogen Energy, 2017, 42, 2636–2647 CrossRef
- Y. Cai, Z. Yue, X. Teng and S. Xu, J. Electrochem. Soc., 2018, 165, F914–F920 CrossRef CAS
- E. Abouzari-Lotf, M. Zakeri, M. M. Nasef, M. Miyake, P. Mozarmnia, N. A. Bazilah, N. F. Emelin and A. Ahmad, J. Power Sources, 2019, 412, 238–245 CrossRef CAS
- L.-c. Jheng, C.-y. Huang and S. L.-c. Hsu, Int. J. Hydrogen Energy, 2013, 38, 1524–1534 CrossRef CAS
- E. Kowsari, A. Zare and V. Ansari, Int. J. Hydrogen Energy, 2015, 40, 13964–13978 CrossRef CAS
- V. D. Punetha, S. Rana, H. J. Yoo, A. Chaurasia, J. T. McLeskey, M. S. Ramasamy, N. G. Sahoo and J. W. Cho, Prog. Polym. Sci., 2017, 67, 1–47 CrossRef CAS
- M. W. Ahmad, B. Dey, G. Sarkhel, D. S. Bag and A. Choudhury, Mater. Chem. Phys., 2019, 223, 426–433 CrossRef CAS
- J.-Y. Kang, S.-M. Eo, I.-Y. Jeon, Y. S. Choi, L.-S. Tan and J.-B. Baek, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1067–1078 CrossRef CAS
- N. Shaari and S. K. Kamarudin, Int. J. Energy Res., 2019, 43, 2756–2794 CrossRef CAS
- S. Ghosh, S. Maity and T. Jana, J. Mater. Chem., 2011, 21, 14897–14906 RSC
- H. Wu, X. Shen, T. Xu, W. Hou and Z. Jiang, J. Power Sources, 2012, 213, 83–92 CrossRef CAS
- Y. Yin, T. Xu, G. He, Z. Jiang and H. Wu, J. Power Sources, 2015, 276, 271–278 CrossRef CAS
- B. Satheesh Kumar, B. Sana, D. Mathew, G. Unnikrishnan, T. Jana and K. S. Santhosh Kumar, Polymer, 2018, 145, 434–446 CrossRef CAS
- L. Feng, N. Xie and J. Zhong, Materials, 2014, 7, 3919–3945 CrossRef CAS
- M. H. Al-Saleh and U. Sundararaj, Composites, Part A, 2011, 42, 2126–2142 CrossRef
- J.-B. Baek, C. B. Lyons and L.-S. Tan, J. Mater. Chem., 2004, 14, 2052–2056 RSC
- H.-J. Lee, S.-W. Han, Y.-D. Kwon, L.-S. Tan and J.-B. Baek, Carbon, 2008, 46, 1850–1859 CrossRef
- I.-Y. Jeon, E.-K. Choi, S.-Y. Bae and J.-B. Baek, Nanoscale Res. Lett., 2010, 5, 1686 CrossRef PubMed
- C. K. Chua and M. Pumera, Chem.–Asian J., 2012, 7, 1009–1012 CrossRef CAS PubMed
- S.-N. Ahn, H.-J. Lee, B.-J. Kim, L.-S. Tan and J.-B. Baek, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7473–7482 CrossRef
- P. V. Lakshminarayanan, H. Toghiani and C. U. Pittman, Carbon, 2004, 42, 2433–2442 CrossRef
- D. A. Long, J. Raman Spectrosc., 2004, 35, 905 CrossRef
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS
- A. A. Mamedov, N. A. Kotov, M. Prato, D. M. Guldi, J. P. Wicksted and A. Hirsch, Nat. Mater., 2002, 1, 190–194 CrossRef CAS
- R. Lin, B. Villacorta Hernandez, L. Ge and Z. Zhu, J. Mater. Chem. A, 2018, 6, 293–312 RSC
- T. D. Kusworo, A. F. Ismail, A. Mustafa and T. Matsuura, Sep. Purif. Technol., 2008, 61, 249–257 CrossRef
- M. A. Aroon, A. F. Ismail, T. Matsuura and M. M. Montazer-Rahmati, Sep. Purif. Technol., 2010, 75, 229–242 CrossRef
- M. Endo, Y. A. Kim, T. Hayashi, T. Yanagisawa, H. Muramatsu, M. Ezaka, H. Terrones, M. Terrones and M. S. Dresselhaus, Carbon, 2003, 41, 1941–1947 CrossRef
- Y. Liu, W. Wu, P. Li, J. Lin, Z. Yang and J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 9964–9973 CrossRef PubMed
- S. L.-C. Hsu, Y.-C. Lin, T.-Y. Tasi, L.-C. Jheng and C.-H. Shen, J. Appl. Polym. Sci., 2013, 130, 4107–4112 CAS
- Q. Li, R. He, J. O. Jensen and N. J. Bjerrum, Fuel Cells, 2004, 4, 147–159 CrossRef
- R. He, Q. Li, G. Xiao and N. J. Bjerrum, J. Membr. Sci., 2003, 226, 169–184 CrossRef
- J. Yang, D. Aili, Q. Li, Y. Xu, P. Liu, Q. Che, J. O. Jensen, N. J. Bjerrum and R. He, Polym. Chem., 2013, 4, 4768–4775 RSC
- D. Plackett, A. Siu, Q. Li, C. Pan, J. O. Jensen, S. F. Nielsen, A. A. Permyakova and N. J. Bjerrum, J. Membr. Sci., 2011, 383, 78–87 CrossRef
- J. Yang, Q. Li, L. N. Cleemann, J. O. Jensen, C. Pan, N. J. Bjerrum and R. He, Adv. Energy Mater., 2013, 3, 622–630 CrossRef
- J. Yang, Y. Xu, P. Liu, L. Gao, Q. Che and R. He, Electrochim. Acta, 2015, 160, 281–287 CrossRef
- C. Korte, F. Conti, J. Wackerl and W. Lehnert, in High Temperature Polymer Electrolyte Membrane Fuel Cells, Springer International Publishing, 2016, ch. 8, pp. 169–194, DOI:10.1007/978-3-319-17082-4_8
- D.-T. Chin and H. H. Chang, J. Appl. Electrochem., 1989, 19, 95–99 CrossRef
- U. Chakraborty, Appl. Energy, 2016, 163, 60–62 CrossRef
- R. O'Hayre, S. -W. Cha, W. Colella and F. B. Prinz, in Fuel Cell Fundamentals, 2016, pp. 203–236, DOI:10.1002/9781119191766.ch6
- D. C. Villa, S. Angioni, S. D. Barco, P. Mustarelli and E. Quartarone, Adv. Energy Mater., 2014, 4, 1301949 CrossRef
- L.-C. Jheng, W. J.-Y. Chang, S. L.-C. Hsu and P.-Y. Cheng, J. Power Sources, 2016, 323, 57–66 CrossRef
- S. Mukhopadhyay, A. Das, T. Jana and S. K. Das, ACS Appl. Energy Mater., 2020, 3, 7964–7977 CrossRef CAS
- R. Gosalawit, S. Chirachanchai, S. Shishatskiy and S. P. Nunes, J. Membr. Sci., 2008, 323, 337–346 CrossRef CAS
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |