
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControllable preparation of one-dimensional Li1.2Mn0.54Ni0.13Co0.13O2 cathode materials for high-performance lithium-ion batteries
Zhi Gaoa,
Jiayi Zhaoa,
Xiaoliang Pan *a,
Lijun Liub,
Shikun Xiea and
Huiling Yuana
*a,
Lijun Liub,
Shikun Xiea and
Huiling Yuana
aSchool of Mechanical Engineering, Jinggangshan University, Jian, 343009, China. E-mail: xiaoliang_pan@163.com
bSchool of Chemistry and Chemical Engineering, Jinggangshan University, Jian, 343009, China
First published on 26th January 2021
Abstract
Lithium-rich layered oxides are attractive candidates of high-energy-density cathode materials for high-performance lithium ion batteries because of their high specific capacity and low cost. Nevertheless, their unsatisfactory rate capability and poor cycling stability have strongly hindered commercial applications in lithium ion batteries, mainly due to the ineffectiveness of the complicated synthesis techniques to control their morphologies and sizes. In this work, the Li1.2Mn0.54Ni0.13Co0.13O2 cathode materials with a one-dimensional rod-like morphology were synthesized via a facile co-precipitation route followed by a post-calcination treatment. By reasonably adding NH3·H2O in the co-precipitation reaction, the sizes of the metal oxalate precursors could be rationally varied. The electrochemical measurements displayed that the Li1.2Mn0.54Ni0.13Co0.13O2 short rods delivered a high capacity of 286 mA h g−1 at 0.1C and excellent capacity retention of 85% after 100 cycles, which could be contributed to the improvement of the electrolyte contact, Li+ diffusion, and structural stability of the one-dimension porous structure.
1. Introduction
The energy and power densities of lithium-ion batteries strongly depend on the properties of cathode materials.1,2 Developing cathode materials with large reversible capacities, high rate capabilities and good cycling stabilities are of great importance for the improvements of electrochemical properties of the whole cells.3,4 Among cathode materials explored so far, lithium-rich layered oxides, xLi2MnO3 (1 − x)LiMO2 (0 < x < 1; M = Mn, Ni or/and Co), have been considered as some of the most promising cathode materials to meet the requirements for high energy densities of lithium-ion batteries owing to their high reversible capacity (>250 mA h g−1) and high working voltage (>3.5 V vs. Li/Li+), as well as low cost.5–7 However, there are still some serious problems preventing large scale commercial applications of lithium-rich layered oxides, such as the unsatisfactory rate capability, the poor cycling stability and the rapid decay of average voltage upon repeated cycling.8–10Recently, cathode materials with a one-dimensional morphology have attracted intensive research interest for their enhanced electrochemical performance due to the short diffusion path, the high specific surface area, and the ability to ease the strain caused by the repeated insertion/extraction of lithium ions.11–13 Thus, much work has been devoted on the syntheses of lithium-rich layered oxides with a typical one-dimensional morphology to improve their electrochemical performance. For examples, the bars were prepared by a co-precipitation method and the following calcination process, and the aspect ratios of the bars were controlled by tuning the ethanol–water volume ratio, and 0.5Li2MnO3·0.5LiNi1/3Co1/3Mn1/3O2 bars demonstrated the excellent capacity and rate capability;14 Li1.2Ni0.13Co0.13Mn0.54O2 with rod-like, olive-like and shuttle-like morphologies were fabricated by the co-precipitation route followed by subsequent annealing treatment process, and the morphologies of the products were tailored by controlling the volume ratio of ethylene glycol with the assistance of surfactant, and the olives achieved better electrochemical performance;15 Li1.2Ni0.15Co0.1Mn0.55O2 cross-linked nanorods and agglomerate microrods were synthesized via using carbon-decorated MnO2 nanowires and MnO2 nanorods as templates, and the cross-linked nanorods exhibited superior discharge capacity and cycling stability.16
From above-mentioned literatures, the manipulations of the one-dimensional morphology for lithium-rich layered oxides have been proved as an accessible approach in modulating their electrochemical properties. Meanwhile, it is clear that the further exploration to optimize the morphology and size for lithium-rich layered oxides is of tremendous significance in improvements of their electrochemical properties. To date, limited success has been achieved for a simply size-controlled synthesis of lithium-rich layered oxides with the one-dimensional morphology.
Herein, we report a simple strategy to fabricate the Li1.2Mn0.54Ni0.13Co0.13O2 rods with controllable sizes. The whole strategy mainly consists of two steps. For the first step, the metal oxalate precursors are synthesized by a co-precipitation method at room temperature. Importantly, the size of the precursors can be tuned by simply adding NH3·H2O in the co-precipitation reaction. For the second step, the metal oxalate precursors are calcined in air, obtaining the final Li1.2Mn0.54Ni0.13Co0.13O2 rods. The effects of the size on the electrochemical performance of the as-prepared cathode materials are systematically investigated. This work provides a simple strategy for the design and control of the one-dimensional morphology and size for lithium-rich layered oxides using in lithium-ion batteries.
2. Experimental
2.1. Materials syntheses
The Li1.2Mn0.54Ni0.13Co0.13O2 cathode materials with the one-dimensional morphology were fabricated by a facile co-precipitation method followed by a post-calcination treatment. In a typical synthesis, 8.4 mmol lithium acetate (LiCH3COO·2H2O), 3.6 mmol manganese acetate (Mn(CH3COO)2·4H2O), 0.87 mmol nickel acetate (Ni(CH3COO)2·4H2O) and 0.87 mmol cobalt acetate (Co(CH3COO)2·4H2O) were sufficiently dissolved in the mixture of 30 ml ethanol and 5 ml NH3·H2O at room temperature, while 19.08 mmol oxalate (H2C2O4·2H2O) was dissolved into another 10 ml ethanol. Then, the oxalic acid solution was rapidly poured into the mixed metal-acetate solution under stirring for 4 h to form the pink precipitate which was subsequently dried at 80 °C overnight to obtain the metal oxalate precursor. Finally, the metal oxalate precursor was calcined at 450 °C for 5 h, 750 °C for 10 h in air at a heating rate of 3.5 °C min−1 and then quenched naturally in the furnace to obtain the Li1.2Mn0.54Ni0.13Co0.13O2 short rods. For comparison, the Li1.2Mn0.54Ni0.13Co0.13O2 long rods were fabricated without NH3·H2O, while other conditions were kept unchanged.2.2. Materials characterizations
The X-ray diffraction (XRD) patterns were recorded by an X-ray powder diffractometer (Rigaku D/max-rA diffractometer, Cu Kα radiation, λ = 1.5406 Å). The XRD refinements were conducted by Rietveld method with FullProf_suite program. The chemical compositions of the samples were analyzed by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Agilent 730). The morphologies and structures of the samples were examined by a field-emission electron microscope (FE-SEM, FEI Quanta 200F), and a transmission electron microscope (JEOL, JEM-2100) with a high-resolution transmission electron microscopy. The specific surface areas of the samples were determined by the Brunauer–Emmett–Teller (BET) method (Micromeritics ASAP, 2020 M) with nitrogen as adsorption–desorption gas.2.3. Electrochemical measurements
The electrochemical measurements were carried out using CR2032-type coin cells on a Land-CT2001A battery test system (Jinnuo Wuhan Corp.). All the tests were performed at room temperature.The CR2032-type coin cells were assembled in an argon-filled glovebox using a metallic lithium foil as the anode, 1 M LiPF6 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (in volume) mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) as the electrolyze, and Celgard 2400 polypropylene as the separator. Cathodes were fabricated by spreading a mixture of 80 wt% active material, 10 wt% acetylene black and 10 wt% polyvinylidene fluoride (PVDF) on an Al foil. Galvanostatic charge/discharge tests were carried out at different rates between 2.0 and 4.8 V (1C = 250 mA g−1). Cyclic voltammetry (CV) measurements were recorded on a CHI660E electrochemical workstation (Chenhua Instruments Shanghai Inc.). Electrochemical impedance spectroscopy (EIS) tests were also recorded on the CHI660E electrochemical workstation, and an AC voltage signal of 5 mV was used over a frequency range from 0.01 to 100000 Hz.
:1 (in volume) mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) as the electrolyze, and Celgard 2400 polypropylene as the separator. Cathodes were fabricated by spreading a mixture of 80 wt% active material, 10 wt% acetylene black and 10 wt% polyvinylidene fluoride (PVDF) on an Al foil. Galvanostatic charge/discharge tests were carried out at different rates between 2.0 and 4.8 V (1C = 250 mA g−1). Cyclic voltammetry (CV) measurements were recorded on a CHI660E electrochemical workstation (Chenhua Instruments Shanghai Inc.). Electrochemical impedance spectroscopy (EIS) tests were also recorded on the CHI660E electrochemical workstation, and an AC voltage signal of 5 mV was used over a frequency range from 0.01 to 100000 Hz.
3. Results and discussion
3.1. Characterizations of the rods
Fig. 1 shows the XRD patterns of the metal oxalate precursors and the Li1.2Mn0.54Ni0.13Co0.13O2 materials as well as the nitrogen adsorption–desorption isotherms. In Fig. 1a, the main diffraction peaks of the metal oxalate precursors are indexed to Li2C2O4 (JCPDS no. 24-0646), MnC2O4·2H2O (JCPDS no. 25-0544), NiC2O4·2H2O (JCPDS no. 01-0299), and CoC2O4·2H2O (JCPDS no. 25-0250) phases. In Fig. 1b, all of the XRD diffraction peaks are well indexed to a hexagonal α-NaFeO2 structure with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m where the Na-sites are occupied by Li while the Fe-sites are occupied by Mn, Ni, Co, and Li.17,18 Notably, the sharp diffraction peaks imply the high crystallinity and no impurity peaks is detected, denoting the high purity of the final lithium-rich materials. Meanwhile, there are a few weak peaks detected around 20–25°, which are caused by the arrangement of Li+ and Mn4+ in the transition metal layers of Li2MnO3 phase with C2/m monoclinic symmetry.19 Generally, the intensity ratio of (003) and (104) peaks corresponds to the degree of the Li+/Ni2+ cation disordering in the lithium layers owing to their similar ion radii, and an over 1.2 of the ratio represents a negligible degree of the cation disordering.20 The values of 1.8 and 1.7 for the short and long rods illustrate a low level of the cation disordering in their crystal lattices, respectively. Besides, the clear divisions of (006)/(012) and (108)/(110) doublets indicate their well-defined layered structure.21,22
m where the Na-sites are occupied by Li while the Fe-sites are occupied by Mn, Ni, Co, and Li.17,18 Notably, the sharp diffraction peaks imply the high crystallinity and no impurity peaks is detected, denoting the high purity of the final lithium-rich materials. Meanwhile, there are a few weak peaks detected around 20–25°, which are caused by the arrangement of Li+ and Mn4+ in the transition metal layers of Li2MnO3 phase with C2/m monoclinic symmetry.19 Generally, the intensity ratio of (003) and (104) peaks corresponds to the degree of the Li+/Ni2+ cation disordering in the lithium layers owing to their similar ion radii, and an over 1.2 of the ratio represents a negligible degree of the cation disordering.20 The values of 1.8 and 1.7 for the short and long rods illustrate a low level of the cation disordering in their crystal lattices, respectively. Besides, the clear divisions of (006)/(012) and (108)/(110) doublets indicate their well-defined layered structure.21,22
 | ||
| Fig. 1 XRD patterns (a and b) of the metal oxalate precursors and the Li1.2Mn0.54Ni0.13Co0.13O2 rods, the Rietveld refinement results (c and d) and the nitrogen adsorption–desorption isotherms (e and f, the insets are the pore-size distribution plots). | ||
To obtain the details of the structural differences, the XRD patterns of the two samples are analyzed by Rietveld refinement on the basis of Rm structure, as shown in Fig. 1c and d. The observed intensities are shown by the crosses, while the calculated patterns are shown by the solid lines. Bragg peak positions of Rm (JCPDS no. 09-0063) and C2/m (JCPDS No. 84-1634) are shown by the vertical light and dark green bars, respectively. The corresponding lattice constants are tabulated in Table 1. According to the refinement results, there are slight differences in lattice constants for the two samples. Their size ratios of c/a are larger than that of idea cubic close stacking (the ratio is 4.899), confirming both of the samples have a complete layered structure. Additionally, there are little differences in peak positions of (003) and (104) for the two samples, and their high values of I(003)/I(104) peak ratios confirm that both the short and long rods have a great level of cation arrangement between Li+ and Ni2+.
| Sample | Lattice constants/Å | c/a | Peak positions/deg. | I(003)/I(104) | ||
|---|---|---|---|---|---|---|
| a | c | (003) | (104) | |||
| The short rods | 2.851 | 14.217 | 4.987 | 18.676 | 44.645 | 1.812 |
| The long rods | 2.850 | 14.195 | 4.981 | 18.632 | 44.526 | 1.746 |
Fig. 1e and f show the nitrogen adsorption–desorption isotherms of the short and long rods. It can be seen that both of the samples have a H1 type absorption-desorption behavior, indicating the existence of the porous structure in the rods. Specially, the BET specific surface area of the short rods (6.674 m2 g−1) is slight bigger than that of the long rods (6.068 m2 g−1). Additionally, the pore-size distribution plots show that both of the samples have a mesoporous structure.
The compositions of the short and long rods were measured by ICP. The data of Li/Mn/Ni/Co molar ratios are listed in Table 2. The results show that the compositions of the short and long rods are Li1.210Mn0.537Ni0.126Co0.127O2 and Li1.208Mn0.536Ni0.131Co0.125O2, respectively, which are approximately consistent with the Li1.2Mn0.54Ni0.13Co0.13O2 stoichiometry.
| Sample | Li/Mn/Ni/Co molar ratio |
|---|---|
| The short rods | 1.210/0.537/0.126/0.127 |
| The long rods | 1.208/0.536/0.131/0.125 |
The SEM images in Fig. 2a–c show the metal oxalate precursor prepared with NH3·H2O has a one-dimensional rod-like morphology. After the post-calcination treatment, the one-dimensional morphology of the metal oxalate precursor is inherited as shown in Fig. 2d. The low-magnification image in Fig. 2e displays that the lithium-rich material is composed of the short rods with about 2 μm length, around 300 nm thickness, and a good dispersed state. The high-magnification image in Fig. 2f presents that the short rods comprise many interconnected nano-sized particles, and possess a porous framework.
 | ||
| Fig. 2 SEM images of the metal oxalate precursor prepared via a co-precipitation method with the addition of NH3·H2O (a–c) and the short rods obtained by a post-calcination treatment in air (d–f). | ||
TEM characterization was carried out to obtain the structural insight of the short rods. The TEM images in Fig. 3a and b display that the nanoparticles link together to form the short rods with homogeneous structural integrity. The size of the nanoparticles is ∼90 nm as shown in Fig. 3c, agreeing well with the results of the SEM observations. TEM image in Fig. 3d presents the network constructed by the nanoparticles. The high-resolution TEM image performed at the edge of the nanoparticle (Fig. 3d) clearly shows the parallel lattice fringes in Fig. 3e. The lattice fringe spacing is ∼0.47 nm, corresponding to (003) plane of the rhombohedral LiMO2 phase (space group Rm) and (001) facet of the monoclinic Li2MnO3 phase (space group C2/m), indicating the high-quality and single-crystalline nature of the lithium-rich material.23 In Fig. 3f, the selected area electron diffraction (SAED) pattern is taken from the zone axis along [001]rhombohedral and [103]monoclinic direction, suggesting the coexistence of Rm and C2/m phases. The marked hexagon can be considered as the superposition of the Rm structure and the C2/m structure rotated 120° around [103] direction, which is consent well with the overall hexagonal symmetry of lithium-rich layered materials.24,25
 | ||
| Fig. 3 TEM images at different magnifications (a–d), HRTEM image of the dashed region in (d) (e) and SAED pattern taken from the zone axis along [001]rhombohedral and [103]monoclinic direction (f) of the short rods. | ||
The added NH3·H2O in the co-precipitation reaction is an important factor to fabricate the metal oxalate precursor. Here, the comparative experiment was conducted to clarify the role of NH3·H2O in the co-precipitation reaction. For the comparative experiment, all the synthetic conditions were kept the same except without NH3·H2O. The SEM images in Fig. 4a–c show that the as-synthesized precursor is composed of the long rods with the length of 4–10 μm and the thickness of ∼600 nm. After the calcination process, the morphology of the precursor is almost retained as shown in Fig. 4d. The magnified SEM images in Fig. 4e and f display that the long rods have a relatively broad particle size distribution.
 | ||
| Fig. 4 SEM images of the metal oxalate precursor prepared via a co-precipitation method without the addition of NH3·H2O (a–c) and the long rods obtained by a post-calcination treatment in air (d–f). | ||
Obviously, the results of the comparative experiment suggest that the addition of NH3·H2O in the co-precipitation reaction plays a significant role in size-control of the metal oxalate precursors. Furthermore, the rods with a short length can offer an efficient electrolyte contact, a short Li+ diffusion path and a stabilized matrix, which are expected to exhibit superior electrochemical performance.26,27
3.2. Size- and morphology-control mechanism of the rods
The above results reveal that the co-precipitation reaction with NH3·H2O is favorable for the formation of the short rods, while the long rods are obtained by the reaction without NH3·H2O. Consequently, the size of the metal oxalate precursors with the one-dimensional morphology can be mainly determined by simply adding NH3·H2O in the co-precipitation reaction. In general, the size of crystals can be related to their nucleation and growth kinetics mainly controlled by the factors such as the pH value, oversaturation and additives.14,28 For the synthesis of the short rods, the extra ammonium hydroxide is added into the solution. As a result, a high pH value of the reaction medium may attribute to a high nucleation rate of the oxalate crystals.29,30 On the other hand, ammonia and the dissolved Mn2+, Ni2+ as well as Co2+ ions can be available for the formation of [Mn–Ni–Co–(NH3)6]2+ complexes according to the following reaction (eqn (1)), which have been reported in previous works.26,31 These [Mn–Ni–Co–(NH3)6]2+ complexes in the solution can be behaved as the insoluble suspended matters, while other Mn2+, Ni2+ and Co2+ ions in the solution may offer a circumstance with a proper oversaturation degree for the formation of the oxalates (eqn (2)). The proper oversaturation degree may lead to an appropriate growth rate of the oxalates. With the precipitation of the oxalates and the evaporation of ammonia, more Mn2+, Ni2+ and Co2+ ions may be released from [Mn–Ni–Co–(NH3)6]2+ complexes to the solution (eqn (3)) to keep the appropriate growth rate until all the complexes and lithium ions are consumed. The above discussion suggests that the high nucleation rate and appropriate growth speed of the oxalates originated from the adding NH3·H2O may readily form the short rods under the co-precipitation condition. Conversely, the long rods may undergo a low nucleation rate as well as a fast growth rate in the co-precipitation condition.| Mn2+ + Ni2+ + Co2+ + 6NH3 → [Mn–Ni–Co–(NH3)6]2+ | (1) |
| 1.2Li+ + 0.54Mn2+ + 0.13Ni2+ + 0.13Co2+ + C2O42− + nH2O → Li1.2Mn0.54Ni0.13Co0.13C2O4·nH2O | (2) |
| [Mn–Ni–Co–(NH3)6]2+ → Mn2+ + Ni2+ + Co2+ + 6NH3 | (3) |
In addition, the two rods possess a one-dimensional morphology. Generally, the morphology of the precursors is highly dependent on the solvent of the reactions.26,32 In this work, ethanol is used as the solvent in the co-precipitation reaction. Correspondingly, hydroxyl group of ethanol may preferentially adsorb on certain crystallite faces of the metal oxalate precursors.14,33 Specifically, the growth of the capped faces with hydroxyl group can be impeded, while the uncapped faces may mainly incline in one direction. As a result, the uncapped faces freely grow out, readily resulting in the one-dimensional morphology.
On the basis of the above discussions, the strategy to control of the size and morphology of the metal oxalate precursors is schematically illustrated in Fig. 5.
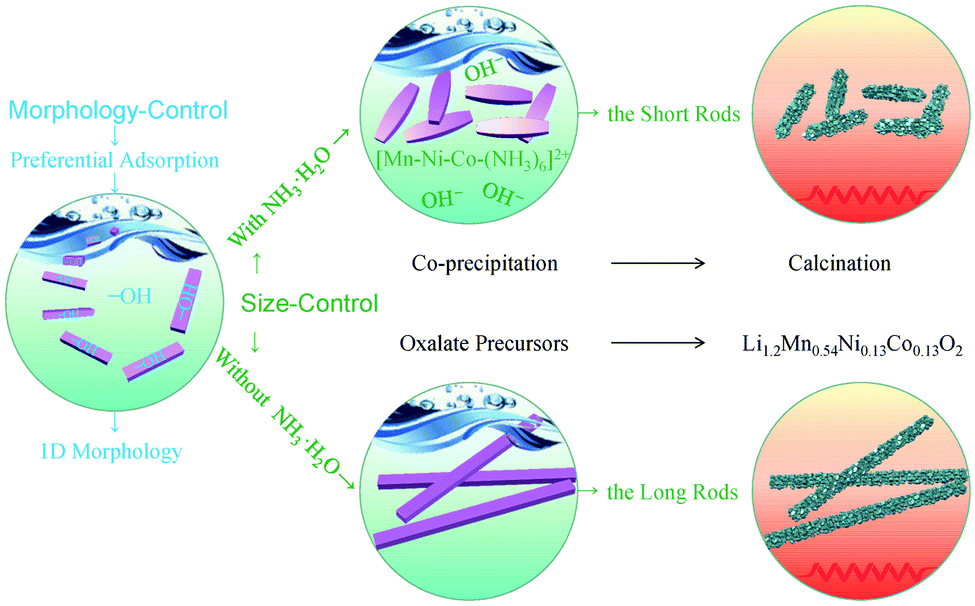 | ||
| Fig. 5 Schematic illustration of size- and morphology-control mechanism of the rods prepared by the ethanol mediated co-precipitation with/without NH3·H2O and the following calcination. | ||
3.3. Electrochemical properties of the rods
The charge/discharge curves of 1st, 2nd, 10th, 50th and 100th cycles between 2.0 and 4.8 V at 0.1C for the two rods are shown in Fig. 6a and b. There are two plateaus from the initial charge curves of the two rods. The plateau (below 4.5 V) corresponds to the deintercalation of lithium ions from the layered structure combined with the oxidation reactions of Ni2+ to Ni4+ as well as Co3+ to Co4+. The other plateau (above 4.5 V) relates to the oxygen extraction from the activation of Li2MnO3 phase.34,35 For the following charge processes, the slopes of the curves increase and there is no obvious steady plateau (above 4.5 V), indicating that the activation process of Li2MnO3 phase during the initial charge is irreversible.36 In detail, the short rods reach a discharge capacity of 286 mA h g−1 with a coulombic efficiency of 81% on the initial cycle, while the long rods deliver a discharge capacity of 280 mA h g−1 with a lower coulombic efficiency. In the subsequent cycles, high coulombic efficiencies over 95% can be achieved for both of the rods. | ||
| Fig. 6 Electrochemical performance of the short and long rods performed in the voltage range of 2.0–4.8 V vs. Li/Li+: the charge/discharge curves at 0.1C (a and b), CV plots at a scan rate of 0.1 mV s−1 (c), cycling properties at 0.1C (d), rate capability at various rates (e), Nyquist plots (the inset is the equivalent circuit) obtained before the cycle (f), the relationship between Z′ and ω−1/2 at low frequency region (g), and after 100 cycles (h) at 0.1C with the full discharging state. | ||
To further investigate the detail electrochemical behaviour during the cycles, cyclic voltammograms (CV) tests were conducted at a scan rate of 0.1 mV s−1 with a voltage range from 2.0 to 4.8 V. The CV plots of the two representative cycles for the two rods are shown in Fig. 6c. For the first cycle, both of the rods have two conspicuous oxidation peaks at about 4.0 V and 4.6 V. The former oxidation peak is associated to the deintercalation of lithium ions from the layered region, and the latter oxidation peak is related to oxygen loss from Li2MnO3 phase.37 There are two broad reduction peaks at about 3.6 V and 3.3 V for the two rods. Correspondingly, the reduction reactions of Ni4+ to Ni2+ and Co4+ to Co3+ occur at about 3.6 V, and the reduction reaction of Mn4+ to Mn3+ occurs at about 3.3 V.38 In the subsequent cycle, the oxidation peak of the two rods at 4.6 V disappears, and all the rest of peaks shift to lower voltages which correspond to the voltage decay of the discharge curves. In addition, a slow decay of voltage for the short rods can be observed by the comparison of the CV profiles, indicating that the short rods have a better reversibility.
Besides, the detailed contrasting cycle properties of the two rods at 0.1C are displayed in Fig. 6d. It can be obtained that the short and long rods maintain the discharge capacities of 243 and 213 mA h g−1 with the capacity retentions of 85 and 76% after 100 charge/discharge cycles, respectively. Obviously, the short rods show a better cycling stability.
Fig. 6e shows the discharge capacities of the two rods at high rates ranging from 0.5 to 10C. The discharge capacities of the short rods are 251 mA h g−1 at 0.5C, 227 mA h g−1 at 1C, 183 mA h g−1 at 5C and 152 mA h g−1 at 10C. When the rate reverts to 0.5C, the discharge capacity can still remain 248 mA h g−1. In contrast, the long rods show inferior rate capabilities, exhibiting the discharge capacities of 248, 220, 163 and 117 mA h g−1 at 0.5, 1, 5 and 10C, respectively. And only a discharge capacity of 241 mA h g−1 is recovered back at 0.5C. Higher discharge capacities at various rates suggest better reaction kinetics.39 These results indicate that the short rods exhibit the enhanced rate performance.
To further verify the improved reaction kinetics of the short rods, electrochemical impedance spectra (EIS) was performed before the cycle and after 100 cycles at 0.1C with the full discharging state. Nyquist plots of the two rods are shown in Fig. 6f–h. In Fig. 6f, the inset illustrates the corresponding equivalent circuit for fitting, where Rs, Rct, CPE and Zw correspond to the solution resistance, the charge transfer resistance, the capacitance and the Warburg resistance, respectively.40,41 Based on the equivalent circuit fitting, Rs and Rct of the short rods are 9.91 and 110.5 Ω, respectively, while Rs and Rct of the long rods are 10.53 and 240.2 Ω. According to the slopes (σ) of the short (186 Ω cm2 s−1/2) and long (221 Ω cm2 s−1/2) rods in Fig. 6g and the Warburg equations,33,35,37 Li+ diffusion coefficients of the short (1.73 × 10−15 cm2 s−1) and long (1.22 × 10−15 cm2 s−1) rods can be calculated. The enhanced diffusion kinetics of Li+ for the short rods could be contributed to their size advantages. Obviously, the short rods with a shorter length and smaller thickness can provide efficient electrolyte contact and fast Li+ diffusion channels, which has been proved by many literatures.11–14,42 Furthermore, the short rods exhibit a smaller change in the resistances after 100 cycles as shown in Fig. 6h, indicating that the short rods have a high structural stability during the cycle.
Based on the above electrochemical testing results, it is indicated that the short rods achieve the superior electrochemical performance in terms of the reversible capacity, rate capability as well as cycling stability in comparisons to the long rods. Particularly, the great dispersibility and homogeneous structural integration for the short rods which can be observed by the sem and tem images are advantageous for offering the appropriate contact area between the active material and the electrolyte, facilitating the efficient electrolyte infiltration into the internal regions of the rods, and alleviating the mechanical strain associated with the volume changes during the repeated insertion/extraction of lithium ions. Consequently, the size advantages of good dispersed state and homogeneous structural integrity for the short rods afford the overall achievements of their superior electrochemical properties.
4. Conclusions
In summary, the Li1.2Mn0.54Ni0.13Co0.13O2 rods with controllable sizes were fabricated by a facile co-precipitation method followed by a post-calcination treatment. It was demonstrated that the size of the metal oxalate precursors could be controlled by the adding NH3·H2O in the co-precipitation reaction. The electrochemical properties of the as-obtained Li1.2Mn0.54Ni0.13Co0.13O2 rods were influenced by their sizes. Particularly, the short rods exhibited a high capacity of 286 mA h g−1 at 0.1C and had a capacity retention of 85% after 100 cycles. The enhanced electrochemical performance of the short rods could be contributed to their size advantages, providing efficient electrolyte contact, fast Li+ diffusion channels and good structure stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant numbers 51762022), the Natural Science Foundation of Jiangxi Province (grant number 20171BAB206026 and 20192ACBL20029 and 20202BABL204031), and the Science & Technology Research Program of Jiangxi Provincial Education Department (grant number GJJ190539 and GJJ201013).Notes and references
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS.
- Y. Lu, Q. Zhang and J. Chen, Sci. China: Chem., 2019, 62, 533–548 CrossRef CAS.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS.
- W. Li, B. Song and A. Manthiram, Chem. Soc. Rev., 2017, 46, 3006–3059 RSC.
- S. H. Kang, Y. K. Sun and K. Amine, Electrochem. Solid-State Lett., 2003, 6, A183 CrossRef CAS.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- P. Rozier and J. M. Tarascon, J. Electrochem. Soc., 2015, 162, A2490–A2499 CrossRef CAS.
- S. Hy, H. Liu, M. Zhang, D. Qian, B. J. Hwang and Y. S. Meng, Energy Environ. Sci., 2016, 9, 1931–1954 RSC.
- H. Yu and H. Zhou, J. Phys. Chem. Lett., 2013, 4, 1268–1280 CrossRef CAS.
- K. Zhang, B. Li, Y. Zuo, J. Song, H. Shang, F. Ning and D. Xia, Electrochem. Energy Rev., 2019, 2, 606–623 CrossRef CAS.
- Q. Wei, F. Xiong, S. Tan, L. Huang, E. H. Lan, B. Dunn and L. Mai, Adv. Mater., 2017, 29, 1602300 CrossRef.
- B. Deng, Y. Chen, P. Wu, J. Han, Y. Li, H. Zheng, Q. Xie, L. Wang and D. L. Peng, J. Power Sources, 2019, 418, 122–129 CrossRef CAS.
- H. He, L. Zan, J. Liu and Y. Zhang, Electrochim. Acta, 2020, 333, 135558 CrossRef CAS.
- G. Ma, S. Li, W. Zhang, Z. Yang, S. Liu, X. Fan, F. Chen, Y. Tian, W. Zhang, S. Yang and M. Li, Angew. Chem., Int. Ed., 2016, 55, 3667–3671 CrossRef CAS.
- G. Wang, L. Yi, R. Yu, X. Wang, Y. Wang, Z. Liu, B. Wu, M. Liu, X. Zhang, X. Yang, X. Xiong and M. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 25358–25368 CrossRef CAS.
- S. Kong, Y. Gong, P. Liu, Y. Xiao, F. Qi and X. Z. Zhao, Ceram. Int., 2019, 45, 13011–13018 CrossRef CAS.
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey and S. A. Hackney, Electrochem. Commun., 2006, 8, 1531–1538 CrossRef CAS.
- K. A. Jarvis, Z. Deng, L. F. Allard, A. Manthiram and P. J. Ferreira, Chem. Mater., 2011, 23, 3614–3621 CrossRef CAS.
- C. S. Johnson, N. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, Chem. Mater., 2008, 20, 6095–6106 CrossRef CAS.
- J. M. Zheng, X. B. Wu and Y. Yang, Electrochim. Acta, 2011, 56, 3071–3078 CrossRef CAS.
- Y. Wu and A. Manthiram, J. Power Sources, 2008, 183, 749–754 CrossRef CAS.
- H. Z. Zhang, Q. Q. Qiao, G. R. Li, S. H. Ye and X. P. Gao, J. Mater. Chem., 2012, 22, 13104–13109 RSC.
- G. Z. Wei, X. Lu, F. S. Ke, L. Huang, J. T. Li, Z. X. Wang, Z. Y. Zhou and S. G. Sun, Adv. Mater., 2010, 22, 4364–4367 CrossRef CAS.
- H. Li, X. Wei, P. Yang, Y. Ren, S. Wang, Y. Xing and S. Zhang, Electrochim. Acta, 2018, 261, 86–95 CrossRef CAS.
- M. Vivekanantha, C. Senthil, T. Kesavan, T. Partheeban, M. Navaneethan, B. Senthilkumar, P. Barpanda and M. Sasidharan, Electrochim. Acta, 2019, 317, 398–407 CrossRef CAS.
- F. Fu, Y. Yao, H. Wang, G. L. Xu, K. Amine, S. G. Sun and M. Shao, Nano Energy, 2017, 35, 370–378 CrossRef CAS.
- D. Chen, Q. Yu, X. Xiang, M. Chen, Z. Chen, S. Song, L. Xiong, Y. Liao, L. Xing and W. Li, Electrochim. Acta, 2015, 154, 83–93 CrossRef CAS.
- X. Pan, Z. Gao, L. Liu, F. Xiao, F. Xiao, S. Xie and R. Yi, J. Alloys Compd., 2019, 783, 468–477 CrossRef CAS.
- N. Rapulenyane, E. Ferg and H. Luo, J. Alloys Compd., 2018, 762, 272–281 CrossRef CAS.
- L. Huang, L. Liu, H. Wu, Y. Wang, H. Liu and Y. Zhang, J. Alloys Compd., 2019, 775, 921–930 CrossRef CAS.
- X. Li, G. L. Xu, F. Fu, Z. Lin, Q. Wang, L. Huang, J. T. Li and S. G. Sun, Electrochim. Acta, 2013, 96, 134–140 CrossRef CAS.
- H. Li, Y. Ren, P. Yang, Z. Jian, W. Wang, Y. Xing and S. Zhang, Electrochim. Acta, 2019, 297, 406–416 CrossRef CAS.
- J. Zeng, Y. Cui, D. Qu, Q. Zhang, J. Wu, X. Zhu, Z. Li and X. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 26082–26090 CrossRef CAS.
- S. Hy, F. Felix, J. Rick, W. N. Su and B. J. Hwang, J. Am. Chem. Soc., 2014, 136, 999–1007 CrossRef CAS.
- Y. Ma, P. Liu, Q. Xie, G. Zhang, H. Zheng, Y. Cai, Z. Li, L. Wang, Z. Z. Zhu, L. Mai and D. L. Peng, Nano Energy, 2019, 59, 184–196 CrossRef CAS.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS.
- Y. Zhang, T. Zhu, L. Lin, M. Yuan, H. Li, G. Sun and S. Ma, J. Nanopart. Res., 2017, 19, 373 CrossRef.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Nat. Mater., 2013, 12, 827–835 CrossRef CAS.
- L. Zhang, N. Li, B. Wu, H. Xu, L. Wang, X. Q. Yang and F. Wu, Nano Lett., 2015, 15, 656–661 CrossRef CAS.
- D. Andre, M. Meiler, K. Steiner, C. Wimmer, T. Soczka-Guth and D. U. Sauer, J. Power Sources, 2011, 196, 5334–5341 CrossRef CAS.
- T. Liu, S. X. Zhao, L. L. Gou, X. Wu and C. W. Nan, Rare Met., 2019, 38, 189–198 CrossRef CAS.
- C. C. Xu, Y. Wang, L. Li, Y. J. Wang, L. F. Jiao and H. T. Yuan, Rare Met., 2019, 38, 29–34 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |