
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of bottom-up graphene oxide using citric acid and tannic acid, and its application as a filler for polypropylene nanocomposites†
Huiseob Shina,
Min-Young Lima,
Jinwoo Ohb,
Yonghoon Leec and
Jong-Chan Lee *a
*a
aSchool of Chemical and Biological Engineering, Institute of Chemical Processes, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul, 08826, Republic of Korea. E-mail: jongchan@snu.ac.kr
bPhoto-Electronic Hybrids Research Center, Korea Institute of Science and Technology (KIST), 5. Hwarang-ro 14-gil, Seongbuk-gu, Seoul, 02792, Republic of Korea
cChemical Pilot Bldg., S-OIL TS&D Center, 31 Magokjungang 8-ro 1-gil, Gangseo-gu, Seoul, 07793, Korea
First published on 17th February 2021
Abstract
The production of graphene oxide (GO) in large amounts for commercialization in the chemical industry has been limited because harsh and tedious process conditions are required. In this study, a novel carbon nanomaterial called ‘bottom-up graphene oxide (BGO)’ could be easily prepared for the first time by heat treatment of the mixture of citric acid (CA) and tannic acid (TA) with different weight ratios for the first time. BGO3 prepared using a 50/50 weight ratio of CA/TA was found to have an average lateral size of 250.0 nm and an average thickness of 7.2 nm, and it was further functionalized with cardanol to prepare cardanol functionalized BGO3 (CBGO3) to be used as a filler for the polypropylene (PP) nanocomposite, where cardanol was used to increase the compatibility between BGO3 and PP. The improved mechanical properties and thermal stability of PP nanocomposites containing CBGO3 could be ascribed to the intrinsic mechanical properties of the carbon nanomaterial and the increased compatibility by the attached cardanol on BGO3.
Introduction
Polypropylene (PP) is one of the most widely used thermoplastics because of its excellent mechanical properties, chemical stability, and easy processing conditions. Numerous studies have been conducted using various fillers to improve the mechanical properties, electric conductivity, and thermal stability of polyporpylene.1–6 Especially, when carbon nanomaterials such as carbon nanotube, graphene, and graphene derivatives were used as nanofillers of PP nanocomposites, physical properties were greatly improved even when a small amount of filler was added.7–14 The remarkable reinforcing efficiency of the carbon nanomaterials is due to the large specific surface area of the carbon nanomaterials, where a large interface is formed between the matrix and the filler.15–19Graphene oxide (GO) has a sheet-like structure where oxygen functional groups such as alcohol, carboxylic acid, epoxy, and ketone are on the basal plane or edge of a sheet composed of covalently bonded carbon atoms.20 GO has been used as a filler in polymer nanocomposites due to its excellent mechanical properties, large specific surface area, and abundant oxygen functional groups.21–23 However, the mass production of GO by Hummers' method has been limited due to the high production costs and legal regulations arising from the large amount of strong acids and oxidizing agents used in the oxidative exfoliation process of graphite. Especially, harsh reaction conditions and complicated purification processes are the obstacles for the commercialization of GO and its derivatives.24–26
Graphene quantum dot (GQD) is the small-size graphene with 100 nm in lateral size and less than 10 nm in thickness.27 There are two well-known methods to produce GQD: a top-down method to cut GO28 and a bottom-up method to use a precursor like citric acid (CA) as building block,29 where the bottom-up method has an advantage for the mass production because the precursor is just heated for the carbonization. These GQDs have been also used as the filler in the polymer nanocomposite application using epoxy,30–32 nitrile-butadiene rubber (NBR),33 poly(lactic acid) (PLA),34 and poly(vinyl alcohol) (PVA),35 etc. It is well known that many of the physical properties of GO are affected by the size of GO,36 ultimately affecting the physical properties of polymer nanocomposites containing GO as the filler.37 Therefore, if we can increase the lateral size of GQD that can be produced in larger quantity more easily, the improved reinforcing effect can be expected when the larger GQD is used as the filler.
In this study, a series of carbon nanomaterials were prepared using the bottom-up process by carbonizing the mixture of CA and tannic acid (TA), and they were found to have the average lateral size in the range of 50–5980 nm and the average thickness in the range of 1.4–175.1 nm. When a larger quantity of TA was used, the average lateral sizes of the products were found to be much larger than GQD prepared using only CA possibly because TA can increase the size in the bottom-up preparation process. We call the product as bottom-up GO (BGO) and it was further functionalized using cardanol, a major component of cashew nut-shell liquid (CNSL), to be used as a filler in the PP nanocomposite application. Cardanol was intentionally used for the functionalization of BGO because it is the natural product and the long alkyl chain in cardanol can increase the compatibility with PP.38–40 The detailed synthetic process for the preparation of BGO and the effect of the functionalized BGO on the property of PP nanocomposite is fully discussed in this paper.
Experimental
Materials
Polypropylene (PP) having melt index of 16 g/10 min was kindly supplied by S-Oil Corp. (Korea). Citric acid (CA), 4-dimethylaminopyridine (DMAP), and tannic acid (TA) were purchased from Alfa Aesar Korea. Graphite powders were purchased from BASF (Germany). N,N′-Dicyclohexylcarbodiimide (DCC), phosphorus pentoxide (P2O5), potassium permanganate (KMnO4), sodium nitrate (NaNO3) were purchased from Sigma-Aldrich Korea. Cardanol was received from Mercury Co., Ltd. (India). Ethanol, hydrogen peroxide (H2O2), petroleum ether, sulfuric acid (H2SO4), and tetrahydrofuran (THF) were purchased from Daejung Chemicals & Metals. All reagents and solvents were used as received.Preparation of bottom-up graphene oxide (BGO)
A series of BGOs were prepared using the mixture of CA and TA in different weight ratios such as 100/0, 75/25, 50/50, 25/75, and 0/100, where the total amount of the mixture as 10.0 g. The mixtures were put into a round bottom flask and were kept at 200 °C for 2 h in nitrogen (N2) atmosphere. Then, the obtained solids were filtered with an anode aluminium oxide (AAO) membrane filter with 0.02 μm pore size and washed with deionized water. BGOs from 100/0, 75/25, 50/50, 25/75 and 0/100 were obtained after drying in vacuum oven at 30 °C overnight in the yields of 14.2, 36.8, 42.0, 74.3 and 89.5%, respectively, with the average lateral size in the range of 50 nm to 5980 nm and the average thickness from 1.4 nm to 146.8 nm (Table 1). BGO3 prepared using a 50/50 weight ratio having the average lateral size of 250 nm and the average thickness of 7.2 nm was used for the functionalization and the preparation of PP nanocomposite in this study to study the effect of nanofiller in the nanocomposites.| Sample | CA/TAa | Average lateral size (nm) | Average thickness (nm) |
|---|---|---|---|
| a The weight ratio of CA/TA used for the preparation of BGO. | |||
| BGO1 | 100/0 | 50 ± 10 | 1.4 ± 0.3 |
| BGO2 | 75/25 | 100 ± 10 | 2.6 ± 0.3 |
| BGO3 | 50/50 | 250 ± 40 | 7.2 ± 1.2 |
| BGO4 | 25/75 | 1190 ± 250 | 175.1 ± 33.3 |
| BGO5 | 0/100 | 5980 ± 1530 | 146.8 ± 22.1 |
| CBGO3 | — | 270 ± 50 | 7.6 ± 0.2 |
| GO | — | 2040 ± 680 | 1.2 ± 0.2 |
| CGO | — | 1720 ± 340 | 2.8 ± 0.3 |
Preparation of cardanol functionalized BGO3 (CBGO3)
3.0 g of BGO3 and 3.0 g of cardanol were put in a round bottom flask and 30 mL of THF was added. After sonication for 30 min, 1.86 g of DCC and 0.12 g of DMAP were added to the mixture. The mixture was kept in 40 °C oil bath for 24 h under N2 atmosphere, and then THF was removed using rotary evaporator. Remaining solids were re-dispersed in 30 mL of THF by sonication and filtered with filter paper (Whatman grade 5) to remove N,N′-dicyclohexylurea (DCU). The eluent was poured into an excess amount of petroleum ether and filtered using a poly(tetrafluoroethylene) (PTFE) membrane filter with 0.2 μm pore size. The product was washed with chloroform several times. CBGO3 was obtained after drying in vacuum oven at 30 °C overnight with the yield of 32.0%.Preparation of graphene oxide (GO)
GO was prepared following the modified Hummers method. 1.0 g of graphite powders and 0.5 g of P2O5 were put in a vial and 6.0 mL of 98% H2SO4 was added to the mixture. The mixture was kept at 85 °C for 6 h. Then, the mixture was poured into 200 mL of deionized water and stirred overnight. The mixture was filtered through anode aluminium oxide (AAO) membrane filter with 0.2 μm pore size and washed with deionized water. The solid was dried in vacuum oven at 35 °C overnight. 1.0 g of the dried product and 0.5 g of NaNO3 were put into a round bottom flask in an ice bath and 23 mL of 98% H2SO4 was added to the mixture. The mixture was kept for 30 min without stirring. Then 3.0 g of KMnO4 was slowly added with stirring. The mixture was heated to 35 °C and stirred for 2 h. Then 140 mL of deionized water and 2.5 mL of 30% H2O2 were added. The mixture was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min and the supernatant decanted. Remaining solids were centrifuged at the same conditions, changing the solvent to deionized water, 10% HCl, deionized water (3 times), and ethanol. After the last centrifugation, the solids were filtered using an AAO membrane filter with 0.2 μm pore size, and the obtained product was dried overnight at 30 °C with the yield of 132.8%.
000 rpm for 30 min and the supernatant decanted. Remaining solids were centrifuged at the same conditions, changing the solvent to deionized water, 10% HCl, deionized water (3 times), and ethanol. After the last centrifugation, the solids were filtered using an AAO membrane filter with 0.2 μm pore size, and the obtained product was dried overnight at 30 °C with the yield of 132.8%.
Preparation of cardanol functionalized GO (CGO)
CGO was prepared by the previously reported method.41 0.3 g of GO was added to 150 mL of DMSO and sonicated for 30 min. 1.5 g of cardanol and 0.6 g of DMAP were added to the GO solution and the mixture was stirred at 100 °C for 3 days under N2 atmosphere. The product was obtained by filtration using an AAO membrane filter with 0.2 μm pore size, followed by washing with DMSO. The obtained product was dried overnight in a vacuum oven at 30 °C with the yield of 18.9%.Preparation of PP nanocomposites
A series of PP nanocomposites such as PP/BGO3, PP/CBGO3, PP/GO, and PP/CGO were prepared by mixing PP with BGO3, CBGO3, GO, and CGO, respectively. The contents of the fillers in PP nanocomposites were 0.01, 0.02, 0.05, and 0.1 wt%. The filler was first dispersed in 27.0 g of p-xylene by sonication for 30 min. Then, 3.0 g of PP was added to the dispersion, and then the mixture was placed in 140 °C oil bath for 30 min to dissolve PP granules. The polymer solution was cast on a glass Petri dish and dried using a vacuum oven at 80 °C overnight. Film type specimens (ASTM D638 type V) for the tensile test were prepared using a hot press (Carver Inc.) at 200 °C and a sample cutting machine (KM-130, Korinstech Inc.).Characterization
Fourier-transform infrared (FT-IR) spectra of BGOs and CBGO3 were collected with Tensor27 spectrometer (Bruker) at room temperature. Elemental analysis (EA) of BGOs and CBGO3 was performed with Trupec 4640 (Leco corp.). Transmission electron microscopy (TEM) images of the BGOs, CBGO3, GO, and CBGO were obtained with JEM-F200 (JEOL) with the accelerating voltage of 200 kV. The dispersion of each sample was dropped on the copper grid with carbon cloth (Ted Pella, Inc.). X-ray diffraction (XRD) spectra of BGO3 and CBGO3 were collected using SmartLab X-ray diffractometer (Rigaku) with Cu Kα radiation source and those of PP nanocomposites were obtained using D8 Discover (Bruker) with Cu Kα radiation source. X-ray photoelectron spectroscopy (XPS) spectra of BGO3 and CBGO3 were obtained with AXIS-His (Kratos Analytical) using Mg Kα (1254.0 eV) as the radiation source. Tapping-mode atomic force microscopy (AFM) measurements on the BGOs, CBGO3, GO and CGO were conducted using scanning probe microscopy MFP-3D Classic (Asylum Research-Oxford Instruments). Silicon cantilevers of the normal resonance frequency of 330 kHz (PPP-NCHR, Nanosensors) were used. Thermal gravimetric analysis (TGA) of BGO3, CBGO3, and PP nanocomposites was performed with Q-50 (TA Instruments) at a heating rate of 10 °C min−1 under N2 atmosphere. Differential scanning calorimetry (DSC) was conducted with Discovery DSC (TA Instrument). All samples were encapsulated in Tzero hermetic aluminium pans. All samples were first heated to 200 °C at 10 °C min−1, then cooled to −50 °C at cooling rate of 10 °C min−1 and heated again to 200 °C at the same heating rate in first heating under N2 atmosphere. The mechanical properties of PP nanocomposites were measured using a universal testing machine LS5 (LLOYD Instruments) when the samples were dried at 60 °C overnight before the test. The gauge length and cross head speed were 25.4 mm and 10 mm min−1 respectively. Scanning electron microscopy (SEM) was used to investigate the fractured surface of the PP nanocomposites with JSM-6701F (JEOL). TEM images of BGO3, CBGO3, GO, and CGO in PP nanocomposites were obtained using LIBRA 120 (Carl Zeiss) with the accelerating voltage of 120 kV. For TEM examination, the PP nanocomposite samples were prepared using ultramicrotomy. The samples were embedded in epoxy resin and subsequently sectioned at room temperature using EM UC7 (Leica). The thin sections were collected on the copper grids.Results and discussion
Synthesis and characterization of BGO
A series of BGOs named as BGO1, BGO2, BGO3, BGO4, and BGO5 were prepared by changing the weight ratios of citric acid (CA) to tannic acid (TA) from 100/0, 75/25, 50/50, 25/75, and 0/100, respectively (Fig. 1a). Graphene quantum dots (GQDs) with less than 100 nm in lateral size have been mostly prepared using citric acid as a precursor for the application in electrooptical devices,42,43 biomedical materials,44 and polymer nanocomposites.30–35 Though GQD showed reinforcing ability in the application for the polymer nanocomposite,30–35 we expected GQD with increased lateral size could be more effective considering the effect of the lateral size of graphene and graphene derivatives on the mechanical properties of polymer nanocomposites.32,36,37 In this study, we intentionally added TA in the synthesis to increase the lateral size of the product (Fig. 1a). The reaction temperature was decided to be 200 °C because the yield of the product was lower when the temperature was less than 200 °C and when the reaction was performed at higher than 200 °C, the oxygen content was not high enough for further modification. Also, the reaction time 2 h was found to be optimum considering the yield and the quality of the products.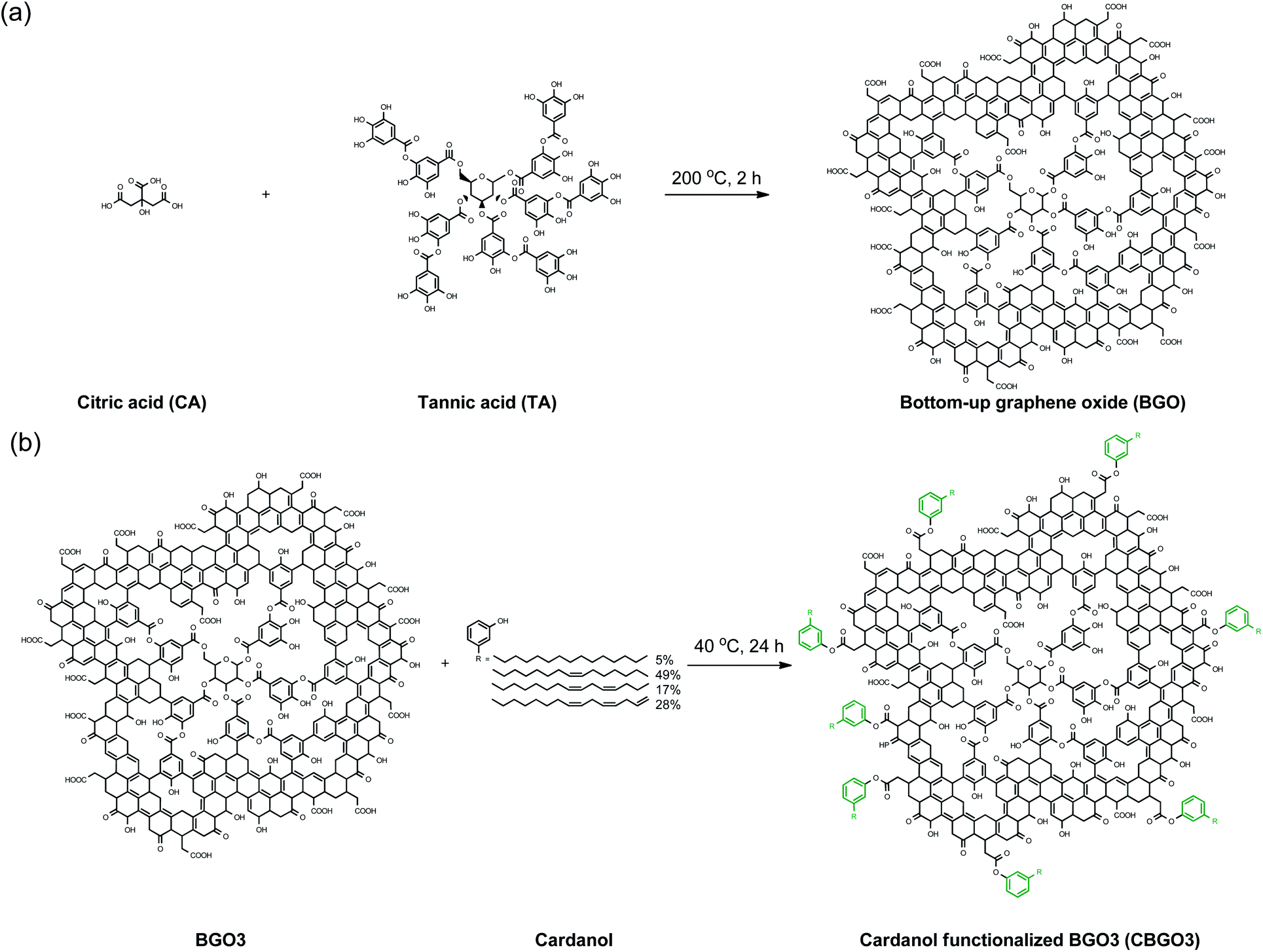 | ||
| Fig. 1 (a) Preparation of bottom-up graphene oxide (BGO) using citric acid (CA) and tannic acid (TA). (b) Functionalization of BGO3 using cardanol. | ||
When the weight ratio of CA/TA was changed from 100/0, 75/25, 50/50, 25/75, and 0/100, producing BGO1, BGO2, BGO3, BGO4, and BGO5, respectively, BGOs having the average lateral size in the range of 50 to 5980 nm and the average thickness in the rage of 1.4 to 175.1 nm were obtained (Table 1). BGO1 prepared using only citric acid was found to have a round shape with the average lateral size of 50 nm and the average thickness of 1.4 nm as shown in the TEM and AFM images in Fig. 2a that is close to the results of others in the preparation of GQDs.29,45–47 When the content of TA in the mixture for the preparation of BGOs is larger than 50 wt%, particles with irregular shapes and large distributions have been obtained as shown in Fig. 2d and e for BGO 4 and BGO5 from 25/75 and 0/100 mixtures of CA/TA. BGO3 prepared using 50/50 ratio of CA/TA shows mostly sheet-like shapes with the largest aspect ratio among the BGOs prepared in this study. The average lateral size and the average thickness values of BGO3 are 250 nm and 7.2 nm, respectively. Therefore, BGO3 itself and modified BGO3 were used as the nanofiller in the PP nanocomposite in this study because nanoplates having larger lateral size and/or surface area can have more interactions with polymer matrix48. FT-IR was used to follow the conversion of CA/TA mixture to BGOs. As the content of TA in the mixture increases, the peak intensities at around 1600 cm−1 from the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and around 1700 cm−1 from the ester CO bond increase, and the peak intensity at around 1750 cm−1 from the carboxylic acid CO bond decreases (Fig. S1†).49 Since TA has a large number of nucleophilic hydroxyl groups, the increase of TA content can increase the content of the connecting chemical bonds such as ester and double bonds. On the contrary, CA having three electrophilic carboxylic acid groups and one nucleophilic hydroxyl group has the limitation to grow the size. Therefore, BGO1 prepared only sing CA has the smallest average size, while BGO5 from only TA has the largest average size (Table 1).
C bond and around 1700 cm−1 from the ester CO bond increase, and the peak intensity at around 1750 cm−1 from the carboxylic acid CO bond decreases (Fig. S1†).49 Since TA has a large number of nucleophilic hydroxyl groups, the increase of TA content can increase the content of the connecting chemical bonds such as ester and double bonds. On the contrary, CA having three electrophilic carboxylic acid groups and one nucleophilic hydroxyl group has the limitation to grow the size. Therefore, BGO1 prepared only sing CA has the smallest average size, while BGO5 from only TA has the largest average size (Table 1).
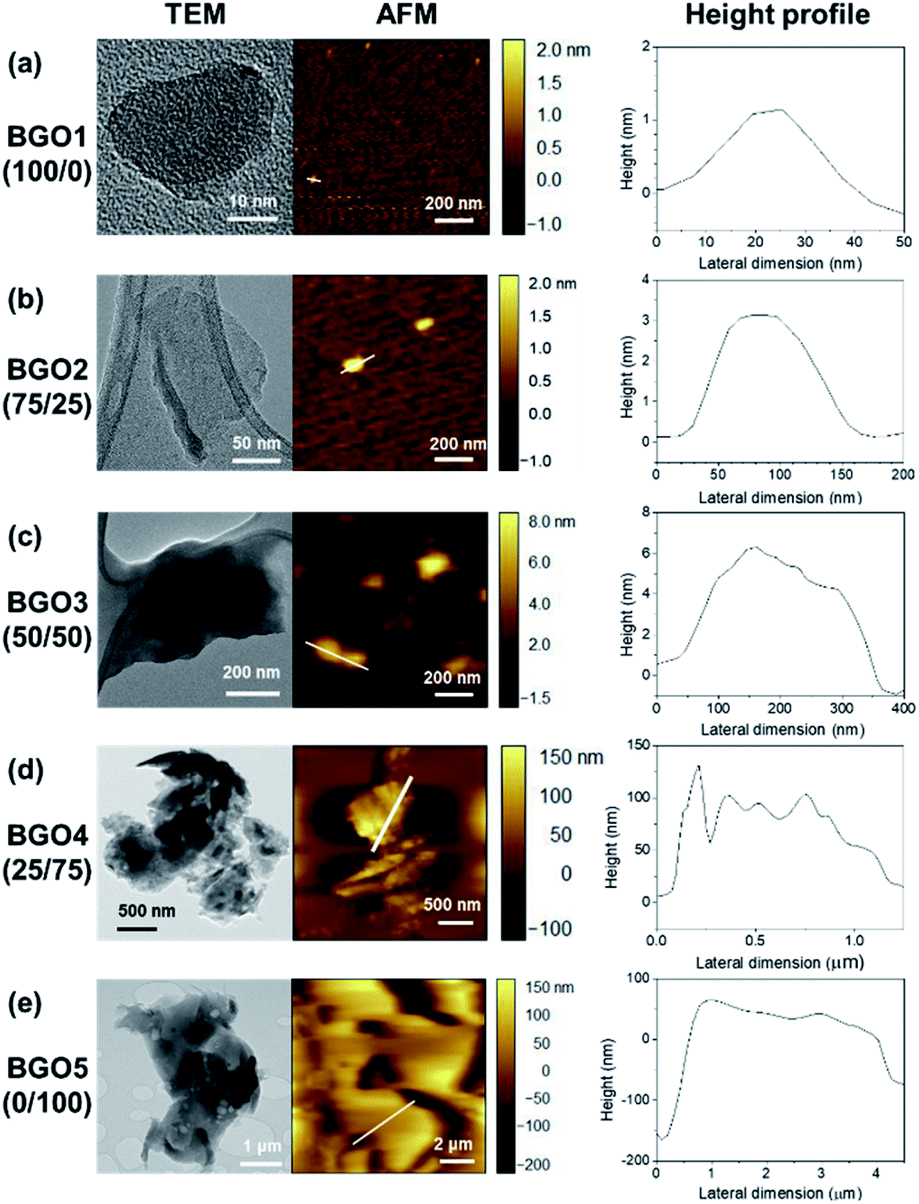 | ||
| Fig. 2 TEM images, AFM images, and height profiles of BGOs prepared using different weight ratio of CA/TA. The weight ratio of CA/TA is presented in the parenthesis. (a) BGO1, (b) BGO2, (c) BGO3, (d) BGO4 and (e) BGO5. | ||
Synthesis and characterization of cardanol functionalized BGO3 (CBGO3)
Cardanol was used to functionalize BGO3 to improve the compatibility of the filler with PP because cardanol has a long pentadecyl group (Fig. 1b).38,39 The functionalization of BGO3 with cardanol is possible because BGO3 has electrophilic COOH groups originated from CA in the edge part and cardanol has a nucleophilic OH group. The reaction between BGO3 and cardanol was performed using DCC and DMAP as the esterification catalysts in THF.50 The conversion from BGO3 to CBGO3 by cardanol could be confirmed by comparing the FT-IR spectrum of the reactants and that of the product (Fig. 3a). The intensity of CO bond peak from the carboxylic acid at 1757 cm−1 in BGO3 becomes much smaller in CBGO3,51,52 and new peaks at around 3000 cm−1 from attached cardanol appear.41
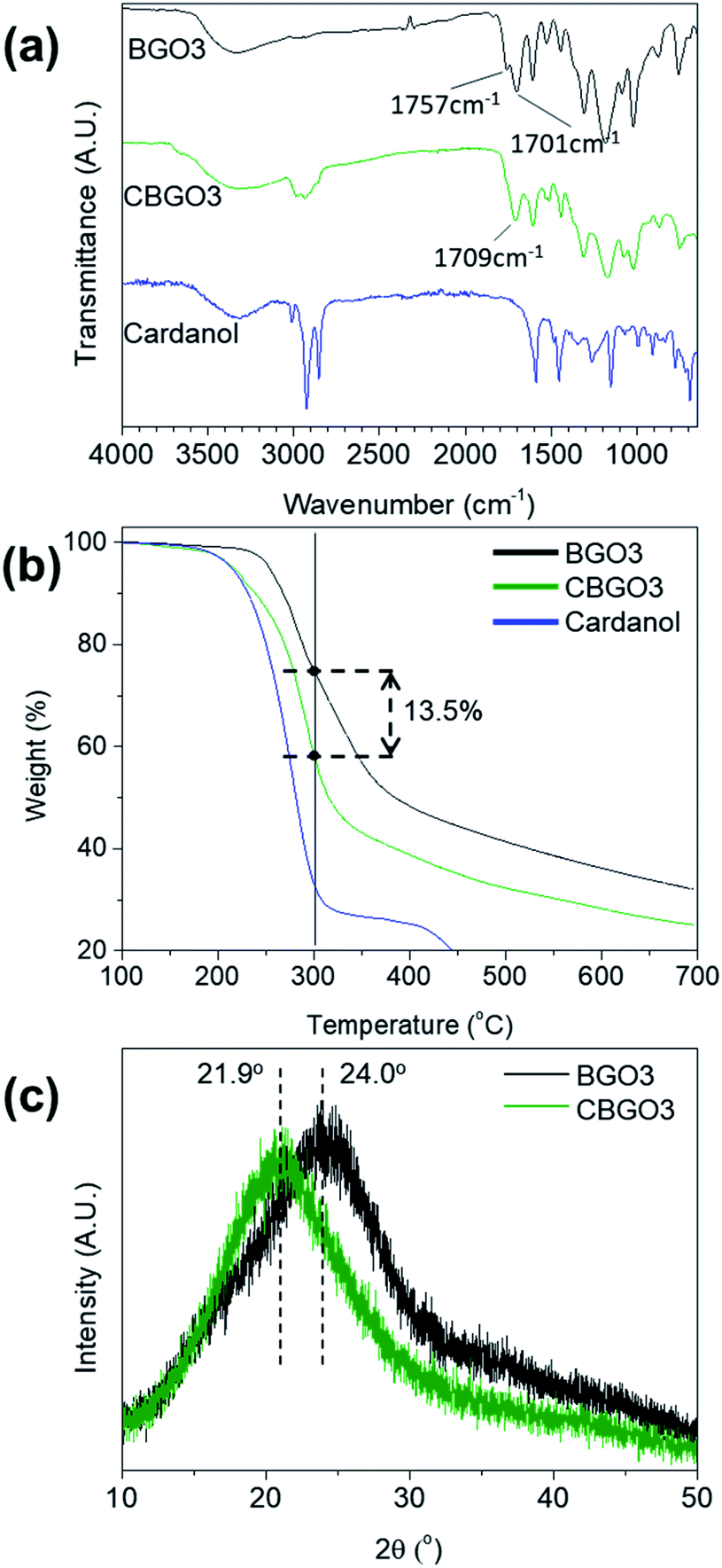 | ||
| Fig. 3 (a) FT-IR spectrum of BGO3, CBGO3, and cardanol. (b) TGA curves of BGO3, CBGO3, and cardanol. (c) XRD patterns of BGO3 and CBGO3. | ||
The conversion from BGO3 to CBGO3 was further confirmed by TGA and XRD (Fig. 3b and c). In the TGA curve of BGO3, the weight decrease can be divided into two stages, between 200 °C and 300 °C and above 300 °C. The thermal degradation between 200 °C and 300 °C can be ascribed to the thermal decomposition of oxygen functional groups19,53 and the thermal degradation above 300 °C can be ascribed to the pyrolysis of carbon structure.41 CBGO3 shows a smaller char yield than BGO3 due to the thermally unstable alkyl groups in CBGO3.54,55 Since the weight decrease at temperature under 300 °C are due to the decomposition of the oxygen functional groups in BGO319,53 and the decomposition of the alkyl groups in cardanol,55 the weight fraction of cardanol in CBGO3 can be determined as about 13.5 wt% as shown in Fig. 3b. In the XRD spectrum of BGO3 and CBGO (Fig. 3c), 2θ values of broad (002) peaks are 24.0° and 20.9°, respectively. The shift of the peak from 24.0° to 20.9° can be ascribed to the increase in d-spacing by the attachment of cardanol.56,57
In addition, elemental analysis (EA) and X-ray photoelectron spectroscopy (XPS) results confirm that cardanol is attached in CBGO3. In the EA results of BGO3 and CBGO3 (Table S1†), the carbon and hydrogen content of CBGO3 is 3.5 wt% and 0.9 wt% larger, respectively, and the oxygen content is 5 wt% smaller than those of BGO3. In the XPS C1s spectra of BGO3 and CBGO3 (Fig. S4b and c†), the C1s spectrum can be deconvoluted into three peaks: C–C/CC (284.7 eV), C–O (286.1 eV), and CO (288.9 eV).58–61 The relative peak intensity of C–C/CC of CBGO3 is larger than that of BGO3.
The morphology of CBGO3 nanoparticles was explored using TEM and AFM (Fig. 4c). The average lateral size and the average thickness of CBGO3 is found to be 270 nm and 7.6 nm, respectively (Table 1), which is slightly larger than those of BGO3. Comparing the TEM and AFM images of CBGO3 and those of BGO3 (Fig. 2 and 4c), the overall morphology of BGO3 is found to be maintained after the conversion from BGO3 to CBGO3. We tried to disperse BGO3 and CBGO3 in p-xylene, the good solvent for PP.62,63 BGO3 was not dispersed and precipitated in p-xylene, while CBGO3 was found to be more well-dispersed in p-xylene forming a more homogeneous dispersion state. Therefore, better miscibility of CBGO3 with PP than that of BGO3 is expected as reported by others.10,39,54
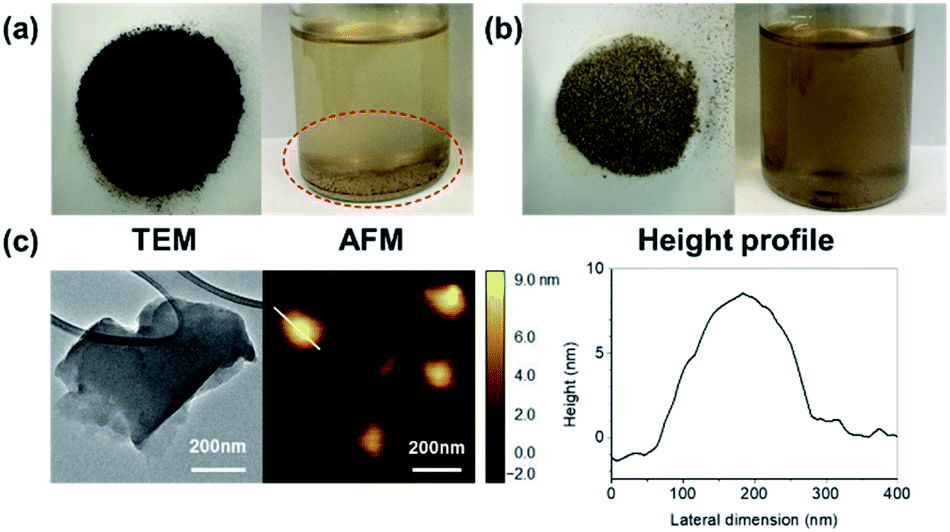 | ||
| Fig. 4 (a) Optical images of dried product and dispersion state in p-xylene (1 mg mL−1) of BGO3. (b) Optical images of dried product and dispersion state in p-xylene (1 mg mL−1) of CBGO3. (c) TEM image, AFM image, and height profile of CBGO3. | ||
Properties of PP nanocomposites
PP nanocomposites were prepared by mixing PP and CBGO3 with different contents from 0.01, 0.02, 0.05, to 0.1 wt% in p-xylene. The PP nanocomposite samples are called ‘PP/X-Y’, where X is the type of the filler such as GO, BGO3, CBGO3, and CGO and Y is the content of the filler in weight percent such as 0.01, 0.02, 0.05, and 0.1 and their mechanical properties are listed in Table S2.†The tensile strength values of PP/CBGO3 nanocomposites (Fig. 5a and Table 2) increase from 26.1 MPa (tensile strength of pristine PP) to 29.7, 31.4, 32.4, and 31.5 MPa for the nanocomposites containing 0.01, 0.02, 0.05, and 0.1 wt% of CBGO3, respectively. The maximum tensile strength value is obtained when the content of CBGO3 was 0.05 wt%. When the content is less than 0.05 wt% such as 0.01 and 0.02 wt%, the tensile strength value increases with the increase of CBGO3 content because further reinforcement effect can be achieved with the PP nanocomposite with larger CBGO3 content. When the content is larger than 0.05 wt% such as 0.1 wt%, the formation of the agglomerates that can act as mechanical defects or stress concentration points decreases the reinforcing efficiency,64,65 resulting in smaller tensile strength value for PP/CBGO3-0.1 than PP/CBGO3-0.05. Young's modulus values of PP/CBGO3 nanocomposites (Fig. 5b and Table 2) increase from 1853.4 MPa (Young's modulus of pristine PP) to 2132.3, 2260.6, 2392.5, and 2302.3 MPa for the nanocomposites containing 0.01, 0.02, 0.05, and 0.1 wt% of CBGO, respectively, and the trend is close to those of tensile strength behaviour. The elongation at break values of PP/CBGO3 nanocomposites (Fig. 5c and Table 2) are quite smaller than that of PP, and it decreases with the increase of CBGO3 content. This decrease in the PP nanocomposite system can be ascribed to the decrease of the chain mobility by the fillers as reported by others.8,66–68 Since their tensile strength and Young's modulus values of PP/CBGO3 nanocomposites were found to be maximum at PP/CBGO3-0.05, PP nanocomposites containing 0.05 wt% of BGO3, GO, and CGO were further prepared, and their mechanical properties were compared with PP/CBGO3-0.05, where BGO3 was prepared using CA/TA in weight ratio of 50/50 (Fig. 5 and Table 2).
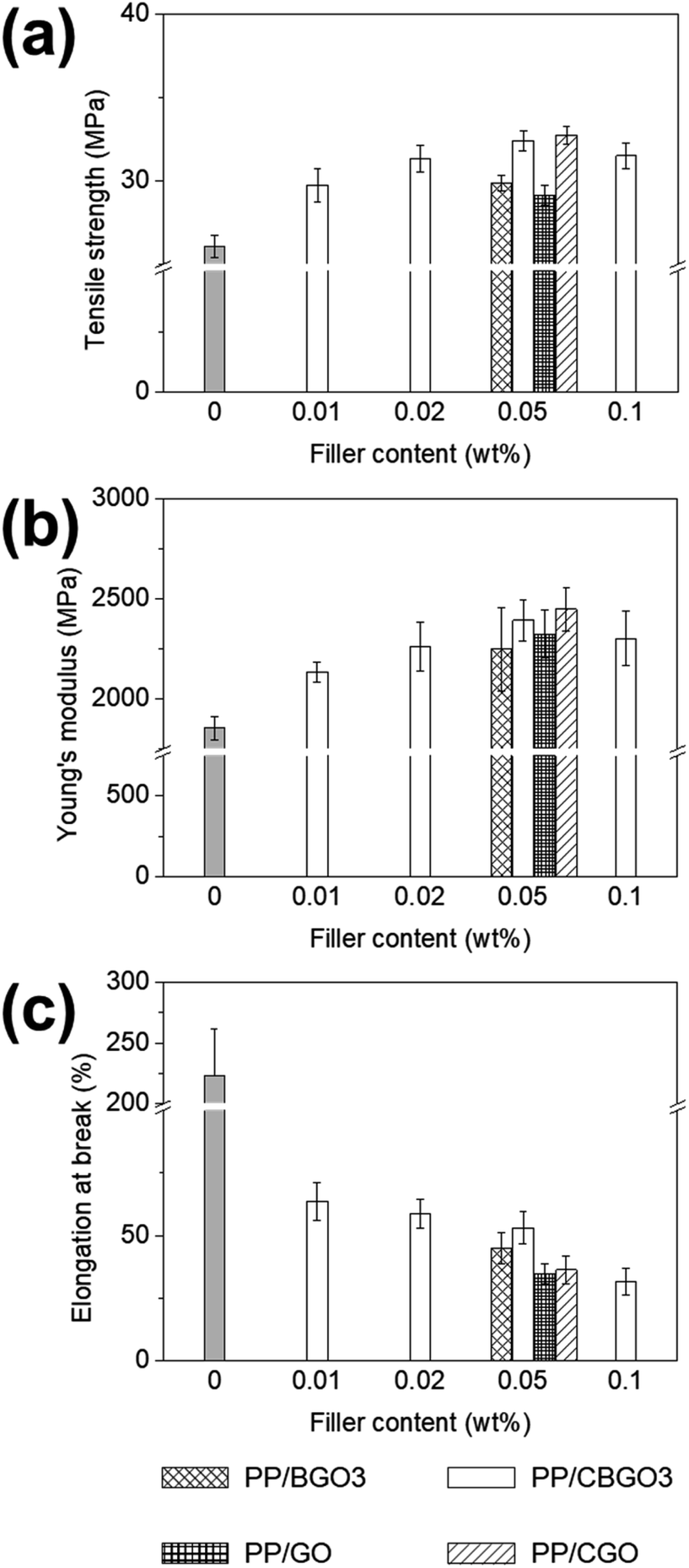 | ||
| Fig. 5 Tensile test results of PP/CBGO nanocomposites. (a) Tensile strength, (b) Young's modulus, and (c) elongation at break of PP/CBGO nanocomposites. | ||
| Sample | Tensile strength (MPa) | Young's modulus (MPa) | Elongation at break (%) | Td,5 (°C) |
|---|---|---|---|---|
| PP | 26.1 ± 0.3 | 1853.4 ± 57.1 | 222.7 ± 38.8 | 307.0 |
| PP/BGO3-0.05 | 29.9 ± 0.5 | 2247.5 ± 208.4 | 45.0 ± 6.3 | 396.4 |
| PP/CBGO3-0.05 | 32.4 ± 0.6 | 2392.5 ± 103.6 | 53.2 ± 6.4 | 411.2 |
| PP/GO-0.05 | 29.1 ± 0.6 | 2323.7 ± 119.4 | 34.7 ± 4.0 | 403.3 |
| PP/CGO-0.05 | 32.8 ± 0.5 | 2448.2 ± 109.7 | 36.3 ± 5.4 | 412.7 |
The order of the tensile strength value for PP nanocomposites containing 0.05 wt% of filler is PP/CGO > PP/CBGO3 > PP/BGO3 > PP/GO, and that of Young's modulus value is PP/CGO > PP/CBGO3 > PP/GO > PP/BGO3. The large tensile strength and Young's modulus values of PP/CGO and PP/CBGO3 nanocomposites than PP/GO and PP/BGO3 nanocomposites could be ascribed to the alkyl groups in the filler entangled with the polymer chains in PP that can facilitate the stress transfer from PP to filler69,70 and improve the dispersion state of the fillers in PP (Fig. 6a–d).48,71 The tensile strength and Young's modulus values of PP/CGO nanocomposite are slightly larger than those of PP/CBGO3 nanocomposite, possibly because CGO having the larger lateral size and aspect ratio of than those of CBGO3 as shown in the TEM and AFM images of CBGO3 and CGO (Fig. 4c and S3b†) can transfer the stress more efficiently.72 The elongation at break values of PP nanocomposites are smaller than that of pristine PP, and the order of the elongation at break values of PP nanocomposites containing 0.05 wt% filler is PP/CBGO3 > PP/BGO3 > PP/CGO > PP/GO. The PP nanocomposite containing CBGO3 has the largest elongation at break value among the PP nanocomposites due to the increased compatibility by the alkyl groups and the smaller size of the filler (Fig. 6e).54,73–75 Although CGO contains the alkyl groups to increase the compatibility, PP/BGO3 nanocomposites show larger elongation at break values than PP/CGO possibly because the size effect is more predominant than the compatibilization effect by the alkyl groups. For example, when two kinds of graphene with different lateral sizes were mixed with polyurethane (PU), the elongation at break value of PU nanocomposite with graphene having the average lateral size of 2.4 μm were 1.4-fold larger than those with graphene having the average lateral size of 8.3 μm.76
The thermal stability of PP nanocomposites containing 0.05 wt% of filler was investigated using TGA (Table 2 and S9†). The thermal degradation temperature values for 5 wt% loss (Td,5) of PP nanocomposites are higher than that of pristine PP (307.0 °C). The improved thermal stability of PP nanocomposites can be attributed to the radical scavenging effect, barrier effect, and tortuous path effect caused by the fillers.71,77–81 We also conducted XRD and DSC analyses to investigate the effect of the filler on the crystallinity and/or thermal transition behaviour (Fig. S9b–d†). The fillers generally decrease the crystallinity and the crystallization temperature, while there are not much differences in these properties between the PP nanocomposites because the content of filler is very small to affect the properties as reported by others.8,82,83
Although 0.05 wt% is not the optimum content to the maximum thermal stability and mechanical properties for PP/GO, PP/BGO3, and PP/CGO nanocomposites, we compared these properties of the nanocomposites containing 0.05 wt%. However, the main story that the filler containing alkyl chain (CBGO3 and CGO) can increase the mechanical strength and thermal stability and the nanocomposites with smaller filler size have the larger elongation at break value is still valid. Although CGO can also effectively increase the mechanical strength and the thermal stability as CBGO3, the maximum amount that can be produced in lab scale for CGO is less than a few grams due to the harsh and complicated preparation and purification conditions for GO, while the mass production such as a few hundred grams is possible for CBGO3 even in the lab scale because BGO3 can be prepared by just heating the mixture of CA and TA. Considering the facile preparation and purification condition of BGO, we believe that this study provides an alternative to prepare carbon nanomaterial to be used as a filler for polymer nanocomposites from natural resources for the first time and contributes to the development of polymer nanocomposites in the chemical industry.
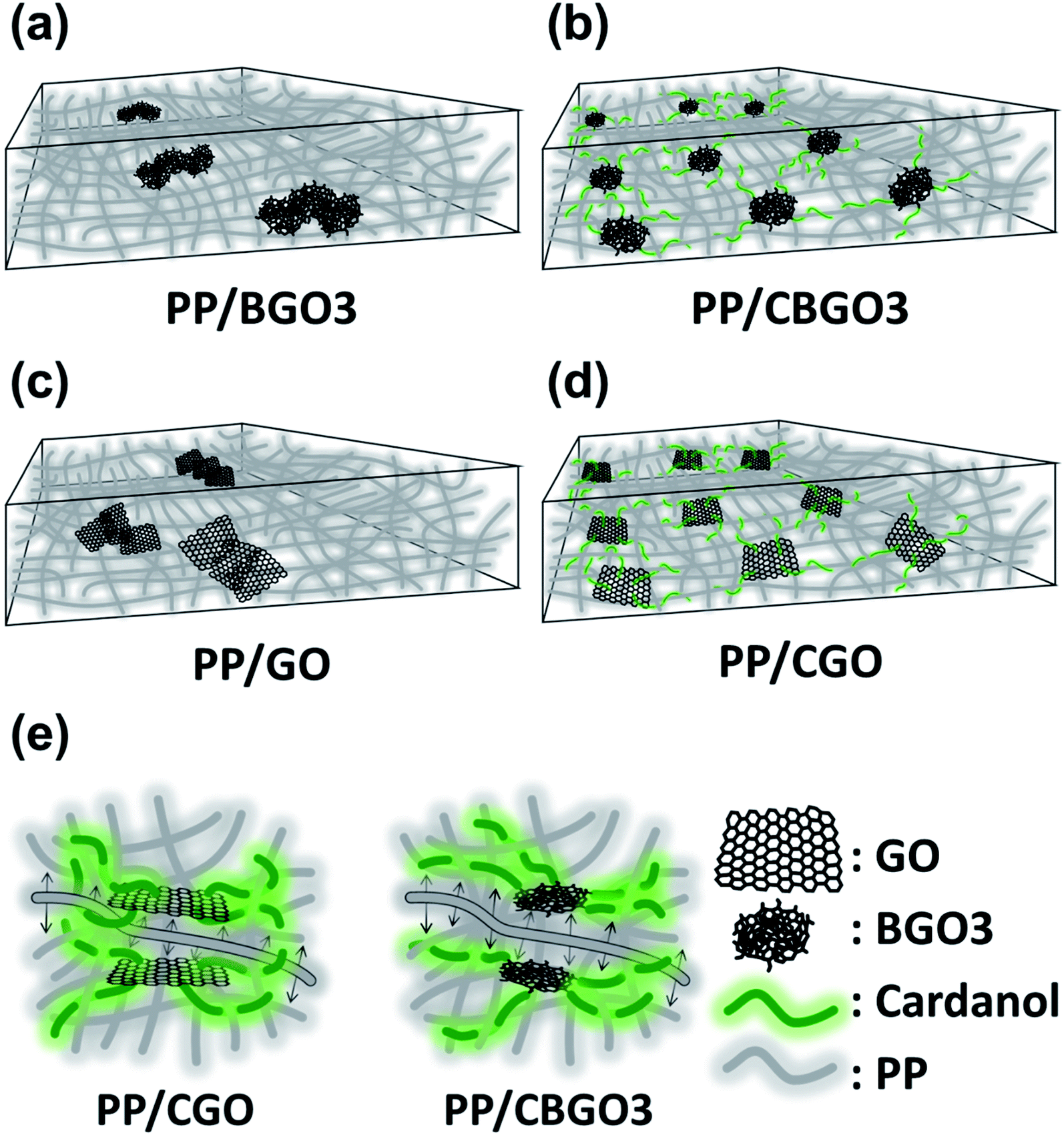 | ||
| Fig. 6 Graphical description of the dispersion state of fillers in PP nanocomposites. (a) PP/BGO3, (b) PP/CBGO3, (c) PP/GO and (d) PP/CGO nanocomposites. (e) Graphical description of the segmental motion of a model PP chain in PP/CGO and PP/CBGO3 nanocomposite. | ||
Conclusions
In this study, a novel carbon nanomaterial ‘bottom-up graphene oxide (BGO)’ was prepared by simply heating the mixture of the natural products such as citric acid (CA) and tannic acid (TA). When the weight ratio of CA/TA was 50/50, BGO3 having the largest aspect ratio between the lateral size of 250 nm and the thickness of 7.2 nm was obtained. This BGO3 was further functionalized with another natural product, cardanol, to be used as a filler in PP nanocomposite application because cardanol having alkyl group can increase the compatibility with PP. The optimum amount of the cardanol functionalized BGO3 called as CBGO3 to give the maximum mechanical strength was found to be 0.05 wt%. The effect of CBGO3 on the physical properties of the PP nanocomposites was studied by preparing other PP nanocomposites using BGO3, graphene oxide (GO), and cardanol functionalized graphene oxide (CGO). PP/CBGO and PP/CGO nanocomposites were found to have larger mechanical strength than PP/BGO3 and PP/GO nanocomposites because the alkyl chains in cardanol can increase the compatibility with PP matrix. Comparing PP/CBGO3 and PP/CGO nanocomposites, PP/CBGO3 nanocomposite was found to have larger elongation at break value (53.2%) than PP/CGO nanocomposite (36.3%) because the lateral size of CBGO3 is smaller than that of CGO. Therefore, BGO that can be prepared by the natural products, CA and TA, can be effectively used as a filler material by further functionalization with a natural product, cardanol, in improving the physical properties of PP nanocomposite. The mechanical properties and thermal stability of PP can be improved by utilizing natural resources as much as that can be achieved by the addition of GO.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT, MSIT) (No. NRF-2018R1A5A1024127 and NRF-2020R1A2C2008114).Notes and references
- P. Khare and N. Verma, Polym. Compos., 2017, 38, E359–E370 CrossRef CAS.
- L. Wang, M. Ando, M. Kubota, S. Ishihara, Y. Hikima, M. Ohshima, T. Sekiguchi, A. Sato and H. Yano, Composites, Part A, 2017, 98, 166–173 CrossRef CAS.
- A. Kaymakci, E. Birinci and N. Ayrilmis, Composites, Part B, 2019, 157, 43–46 CrossRef CAS.
- G. Cao, H. Lin, P. Kannan, C. Wang, Y. Zhong, Y. Huang and Z. Guo, Langmuir, 2018, 34, 14537–14545 CrossRef CAS.
- U. Gohs, L. Girdauskaite, L. Peitzsch, S. Rothe, C. Zschech, G. Heinrich and H. Rödel, Adv. Eng. Mater., 2016, 18, 409–416 CrossRef CAS.
- S. Kong, H. Seo, H. Shin, J.-H. Baik, J. Oh, Y.-O. Kim and J.-C. Lee, Polymer, 2019, 180, 121714 CrossRef CAS.
- Y. Huang, Y. Qin, Y. Zhou, H. Niu, Z.-Z. Yu and J.-Y. Dong, Chem. Mater., 2010, 22, 4096–4102 CrossRef CAS.
- B. Yuan, C. Bao, L. Song, N. Hong, K. M. Liew and Y. Hu, Chem. Eng. J., 2014, 237, 411–420 CrossRef CAS.
- F. Qiu, Y. Hao, X. Li, B. Wang and M. Wang, Composites, Part B, 2015, 71, 175–183 CrossRef CAS.
- R.-Y. Bao, J. Cao, Z.-Y. Liu, W. Yang, B.-H. Xie and M.-B. Yang, J. Mater. Chem. A, 2014, 2, 3190–3199 RSC.
- P. Song, Z. Cao, Y. Cai, L. Zhao, Z. Fang and S. Fu, Polymer, 2011, 52, 4001–4010 CrossRef CAS.
- F. You, D. Wang, X. Li, M. Liu, G.-H. Hu and Z.-M. Dang, RSC Adv., 2014, 4, 8799–8807 RSC.
- C.-Q. Li, J.-W. Zha, H.-Q. Long, S.-J. Wang, D.-L. Zhang and Z.-M. Dang, Compos. Sci. Technol., 2017, 153, 111–118 CrossRef CAS.
- J. Castillo, T. Lozano, R. Garcia, L. Morales-Zamudio, J. López-Barroso, P. G. Lafleur, S. Karami, S. Sanchez-Valdes, G. Martinez-Colunga, F. Rodriguez, C. Perez-Berumen, S. Flores and A. García, J. Appl. Polym. Sci., 2020, 137, 48258 CrossRef CAS.
- J. Ma, Q. Meng, A. Michelmore, N. Kawashima, Z. Izzuddin, C. Bengtsson and H.-C. Kuan, J. Mater. Chem. A, 2013, 1, 4255–4264 RSC.
- W.-H. Liao, S.-Y. Yang, J.-Y. Wang, H.-W. Tien, S.-T. Hsiao, Y.-S. Wang, S.-M. Li, C.-C. M. Ma and Y.-F. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 869–877 CrossRef CAS.
- R. Liu, S. Liang, X.-Z. Tang, D. Yan, X. Li and Z.-Z. Yu, J. Mater. Chem., 2012, 22, 14160–14167 RSC.
- L. Yang, S. L. Phua, C. L. Toh, L. Zhang, H. Ling, M. Chang, D. Zhou, Y. Dong and X. Lu, RSC Adv., 2013, 3, 6377–6385 RSC.
- M.-Y. Lim, H. J. Kim, S. J. Baek, K. Y. Kim, S.-S. Lee and J.-C. Lee, Carbon, 2014, 77, 366–378 CrossRef CAS.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS.
- K. H. Jung, H. J. Kim, M. H. Kim and J.-C. Lee, Macromol. Res., 2020, 28, 241–248 CrossRef CAS.
- Z. Tu, J. Wang, C. Yu, H. Xiao, T. Jiang, Y. Yang, D. Shi, Y.-W. Mai and R. K. Y. Li, Compos. Sci. Technol., 2016, 134, 49–56 CrossRef CAS.
- P. Pokharel, S. Choi and D. S. Lee, Composites, Part A, 2015, 69, 168–177 CrossRef CAS.
- H. Lee, J. Han, K. Kim, J. Kim, E. Kim, H. Shin and J.-C. Lee, J. Ind. Eng. Chem., 2019, 74, 223–232 CrossRef CAS.
- M.-Y. Lim, Y.-S. Choi, J. Kim, K. Kim, H. Shin, J.-J. Kim, D. M. Shin and J.-C. Lee, J. Membr. Sci., 2017, 521, 1–9 CrossRef CAS.
- M.-Y. Lim, H. Shin, D. M. Shin, S.-S. Lee and J.-C. Lee, Polymer, 2016, 84, 89–98 CrossRef CAS.
- P. Tian, L. Tang, K. S. Teng and S. P. Lau, Mater. Today Chem., 2018, 10, 221–258 CrossRef CAS.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS.
- Y. Dong, J. Shao, C. Chen, H. Li, R. Wang, Y. Chi, X. Lin and G. Chen, Carbon, 2012, 50, 4738–4743 CrossRef CAS.
- B. Karimi and B. Ramezanzadeh, J. Colloid Interface Sci., 2017, 493, 62–76 CrossRef CAS.
- N. Gobi, D. Vijayakumar, O. Keles and F. Erogbogbo, ACS Omega, 2017, 2, 4356–4362 CrossRef CAS.
- J. Kim, J. Kim, S. Song, S. Zhang, J. Cha, K. Kim, H. Yoon, Y. Jung, K.-W. Paik and S. Jeon, Carbon, 2017, 113, 379–386 CrossRef CAS.
- P. Sreenath, S. Singh, M. Satyanarayana, P. Das and K. D. Kumar, Polymer, 2017, 112, 189–200 CrossRef CAS.
- H. Xu, K. H. Adolfsson, L. Xie, S. Hassanzadeh, T. r. Pettersson and M. Hakkarainen, ACS Sustainable Chem. Eng., 2016, 4, 5618–5631 CrossRef CAS.
- P. Song, J. Dai, G. Chen, Y. Yu, Z. Fang, W. Lei, S. Fu, H. Wang and Z.-G. Chen, ACS Nano, 2018, 12, 9266–9278 CrossRef CAS.
- L. Dong, J. Yang, M. Chhowalla and K. P. Loh, Chem. Soc. Rev., 2017, 46, 7306–7316 RSC.
- J. Wang, X. Jin, C. Li, W. Wang, H. Wu and S. Guo, Chem. Eng. J., 2019, 370, 831–854 CrossRef CAS.
- S. H. Ryu and A. M. Shanmugharaj, Chem. Eng. J., 2014, 244, 552–560 CrossRef CAS.
- X. Yang, X. Wang, J. Yang, J. Li and L. Wan, Chem. Phys. Lett., 2013, 570, 125–131 CrossRef CAS.
- S. H. Ryu and A. M. Shanmugharaj, Mater. Chem. Phys., 2014, 146, 478–486 CrossRef CAS.
- M.-Y. Lim, Y.-S. Choi, H. Shin, K. Kim, D. M. Shin and J.-C. Lee, ACS Appl. Nano Mater., 2018, 1, 2600–2608 CrossRef CAS.
- Y. H. Kim, E. Y. Lee, H. H. Lee and T. S. Seo, ACS Appl. Mater. Interfaces, 2017, 9, 16375–16380 CrossRef CAS.
- X. Gong, Y. Liu, Z. Yang, S. Shuang, Z. Zhang and C. Dong, Anal. Chim. Acta, 2017, 968, 85–96 CrossRef CAS.
- R. Guo, S. Zhou, Y. Li, X. Li, L. Fan and N. H. Voelcker, ACS Appl. Mater. Interfaces, 2015, 7, 23958–23966 CrossRef CAS.
- C. Zhang, K. Wei, W. Zhang, Y. Bai, Y. Sun and J. Gu, ACS Appl. Mater. Interfaces, 2017, 9, 11082–11094 CrossRef CAS.
- Q. Fang, Y. Dong, Y. Chen, C.-H. Lu, Y. Chi, H.-H. Yang and T. Yu, Carbon, 2017, 118, 319–326 CrossRef CAS.
- S. Wang, Z.-G. Chen, I. Cole and Q. Li, Carbon, 2015, 82, 304–313 CrossRef CAS.
- J.-Z. Liang, Composites, Part B, 2019, 167, 241–249 CrossRef CAS.
- K. Ranoszek-Soliwoda, E. Tomaszewska, E. Socha, P. Krzyczmonik, A. Ignaczak, P. Orlowski, M. Krzyzowska, G. Celichowski and J. Grobelny, J. Nanopart. Res., 2017, 19, 273 CrossRef.
- H. Jiang and K. Zhu, Biomaterials, 2001, 22, 211–218 CrossRef CAS.
- M. Cano, U. Khan, T. Sainsbury, A. O'Neill, Z. Wang, I. T. McGovern, W. K. Maser, A. M. Benito and J. N. Coleman, Carbon, 2013, 52, 363–371 CrossRef CAS.
- J. Aguilera, V. Venegas, J. M. Oliva, M. J. Sayagués, M. de Miguel, J. A. Sánchez-Alcázar, M. Arévalo-Rodríguez and A. Zaderenko, RSC Adv., 2016, 6, 7279–7287 RSC.
- M.-Y. Lim, J. Oh, H. J. Kim, K. Y. Kim, S.-S. Lee and J.-C. Lee, Eur. Polym. J., 2015, 69, 156–167 CrossRef CAS.
- M. Y. Song, S. Y. Cho, N. R. Kim, S.-H. Jung, J.-K. Lee, Y. S. Yun and H.-J. Jin, Carbon, 2016, 108, 274–282 CrossRef CAS.
- W. Kiratitanavit, S. Ravichandran, Z. Xia, J. Kumar and R. Nagarajan, J. Renewable Mater., 2013, 1, 289–301 CrossRef.
- W.-S. Hung, C.-H. Tsou, M. De Guzman, Q.-F. An, Y.-L. Liu, Y.-M. Zhang, C.-C. Hu, K.-R. Lee and J.-Y. Lai, Chem. Mater., 2014, 26, 2983–2990 CrossRef CAS.
- W. Li, W. Wu and Z. Li, ACS Nano, 2018, 12, 9309–9317 CrossRef CAS.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S. J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- Z. Huang, Z. Li, L. Zheng, L. Zhou, Z. Chai, X. Wang and W. Shi, Chem. Eng. J., 2017, 328, 1066–1074 CrossRef CAS.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- B. Rodier, A. de Leon, C. Hemmingsen and E. Pentzer, ACS Macro Lett., 2017, 6, 1201–1206 CrossRef CAS.
- A. P. Bafana, X. Yan, X. Wei, M. Patel, Z. Guo, S. Wei and E. K. Wujcik, Composites, Part B, 2017, 109, 101–107 CrossRef CAS.
- J. Yang, Y. Huang, Y. Lv, P. Zhao, Q. Yang and G. Li, J. Mater. Chem. A, 2013, 1, 11184–11191 RSC.
- Y. Ren, H. Guo, Y. Liu, R. Lv, Y. Zhang, M. Maqbool and S. Bai, Compos. Sci. Technol., 2019, 183, 107787 CrossRef CAS.
- J. Phiri, L.-S. Johansson, P. Gane and T. Maloney, Composites, Part B, 2018, 147, 104–113 CrossRef CAS.
- D. Bagheriasl, P. J. Carreau, C. Dubois and B. Riedl, Compos. Sci. Technol., 2015, 117, 357–363 CrossRef CAS.
- L. Chen, S. C. Wong and S. Pisharath, J. Appl. Polym. Sci., 2003, 88, 3298–3305 CrossRef CAS.
- N. Hidayah, W.-W. Liu, C. Khe, C. Lai and N. Noriman, Mater. Today Commun., 2020, 22, 100775 CrossRef CAS.
- Y.-J. Wan, L.-X. Gong, L.-C. Tang, L.-B. Wu and J.-X. Jiang, Composites, Part A, 2014, 64, 79–89 CrossRef CAS.
- L.-X. Gong, Y.-B. Pei, Q.-Y. Han, L. Zhao, L.-B. Wu, J.-X. Jiang and L.-C. Tang, Compos. Sci. Technol., 2016, 134, 144–152 CrossRef CAS.
- J. Bian, Z. J. Wang, H. L. Lin, X. Zhou, W. Q. Xiao and X. W. Zhao, Composites, Part A, 2017, 97, 120–127 CrossRef CAS.
- S. Chatterjee, F. Nafezarefi, N. H. Tai, L. Schlagenhauf, F. A. Nüesch and B. T. T. Chu, Carbon, 2012, 50, 5380–5386 CrossRef CAS.
- G. C. Onuegbu and I. O. Igwe, Mater. Sci. Appl., 2011, 2, 810 Search PubMed.
- H. Ismail, H. D. Rozman, R. M. Jaffri and Z. A. M. Ishak, Eur. Polym. J., 1997, 33, 1627–1632 CrossRef CAS.
- Y. Li, R. Umer, Y. A. Samad, L. Zheng and K. Liao, Carbon, 2013, 55, 321–327 CrossRef CAS.
- J. T. Choi, D. H. Kim, K. S. Ryu, H. I. Lee, H. M. Jeong, C. M. Shin, J. H. Kim and B. K. Kim, Macromol. Res., 2011, 19, 809–814 CrossRef CAS.
- J. Yang, Y. Huang, Y. Lv, S. Li, Q. Yang and G. Li, Carbon, 2015, 89, 340–349 CrossRef CAS.
- W. Hu, J. Zhan, X. Wang, N. Hong, B. Wang, L. Song, A. A. Stec, T. R. Hull, J. Wang and Y. Hu, Ind. Eng. Chem. Res., 2014, 53, 3073–3083 CrossRef CAS.
- F. Fang, S. Ran, Z. Fang, P. Song and H. Wang, Composites, Part B, 2019, 165, 406–416 CrossRef CAS.
- S. G. Prolongo, A. Jiménez-Suárez, R. Moriche and A. Ureña, Eur. Polym. J., 2014, 53, 292–301 CrossRef CAS.
- J. Liang, J. Wang, G. C. Tsui and C. Tang, J. Thermoplast. Compos. Mater., 2018, 31, 246–264 CrossRef CAS.
- M. Floros, L. Hojabri, E. Abraham, J. Jose, S. Thomas, L. Pothan, A. L. Leao and S. Narine, Polym. Degrad. Stab., 2012, 97, 1970–1978 CrossRef CAS.
- J. Morawiec, A. Pawlak, M. Slouf, A. Galeski, E. Piorkowska and N. Krasnikowa, Eur. Polym. J., 2005, 41, 1115–1122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09856f |
| This journal is © The Royal Society of Chemistry 2021 |