
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct DME synthesis on CZZ/H-FER from variable CO2/CO syngas feeds†
Stefan Wild a,
Sabrina Polierera,
Thomas A. Zevacoa,
David Guseb,
Matthias Kindb,
Stephan Pitter*a,
Karla Herrera Delgado*a and
Jörg Sauera
a,
Sabrina Polierera,
Thomas A. Zevacoa,
David Guseb,
Matthias Kindb,
Stephan Pitter*a,
Karla Herrera Delgado*a and
Jörg Sauera
aIKFT – Institute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany. E-mail: karla.herrera@kit.edu; stephan.pitter@kit.edu
bTVT – Institute of Thermal Process Engineering, Karlsruhe Institute of Technology, Kaiserstraße 12, D-76131 Karlsruhe, Germany
First published on 12th January 2021
Abstract
Catalyst systems for the conversion of synthesis gas, which are tolerant to fluctuating CO/CO2 gas compositions, have great potential for process-technical applications, related to the expected changes in the supply of synthesis gas. Copper-based catalysts usually used in the synthesis of methanol play an important role in this context. We investigated the productivity characteristics for their application in direct dimethyl ether (DME) synthesis as a function of the CO2/COx ratio over the complete range from 0 to 1. For this purpose, we compared an industrial Cu/ZnO/Al2O3 methanol catalyst with a self-developed Cu/ZnO/ZrO2 catalyst prepared by a continuous coprecipitation approach. For DME synthesis, catalysts were combined with two commercial dehydration catalysts, H-FER 20 and γ-Al2O3, respectively. Using a standard testing procedure, we determined the productivity characteristics in a temperature range between 483 K and 523 K in a fixed bed reactor. The combination of Cu/ZnO/ZrO2 and H-FER 20 provided the highest DME productivity with up to 1017 gDME (kgCu h)−1 at 523 K, 50 bar and 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mlN (g h)−1 and achieved DME productivities higher than 689 gDME (kgCu h)−1 at all investigated CO2/COx ratios under the mentioned conditions. With the use of Cu/ZnO/ZrO2//H-FER 20 a promising operating range between CO2/COx 0.47 and 0.8 was found where CO as well as CO2 can be converted with high DME selectivity. First results on the long-term stability of the system Cu/ZnO/ZrO2//H-FER 20 showed an overall reduction of 27.0% over 545 h time on stream and 14.6% between 200 h and 545 h under variable feed conditions with a consistently high DME selectivity.
000 mlN (g h)−1 and achieved DME productivities higher than 689 gDME (kgCu h)−1 at all investigated CO2/COx ratios under the mentioned conditions. With the use of Cu/ZnO/ZrO2//H-FER 20 a promising operating range between CO2/COx 0.47 and 0.8 was found where CO as well as CO2 can be converted with high DME selectivity. First results on the long-term stability of the system Cu/ZnO/ZrO2//H-FER 20 showed an overall reduction of 27.0% over 545 h time on stream and 14.6% between 200 h and 545 h under variable feed conditions with a consistently high DME selectivity.
Introduction
Power-to-fuels concepts play a major role for the future integration of carbon neutral technologies within complex energy supply systems.1,2 Amongst potential non-fossil carbon resources for the production of synthetic hydrocarbons, carbon dioxide plays a dominant role. Once used in combination with sustainable, economically viable hydrogen production, CO2 would allow the production of carbon neutral fuels and industrial chemicals3 and, on the other hand, contribute to a mitigation of its environmental impact.4 In particular, the foreseeable dynamic character in power generation demands the development of robust processes that enable highly adaptive operation modes. A flexible production of chemical energy carriers from CO2-rich syngas, catalysed by efficient and long-term stable catalysts is hereby one of the most promising options. Besides other synthetic hydrocarbon-based energy carriers, dimethyl ether (DME) is a particularly interesting candidate due to its promising physical and chemical properties.5–7 It can be either directly used as diesel substitute8 or as intermediate for the production of a wide range of synthetic hydrocarbons.The DME synthesis is technically feasible in a one-step (i.e. reactions (R1) to (R4) in a single reactor)9–14 or two-step process (i.e. reactions (R1) to (R3) in one reactor, and reaction (R4) in a second reactor),15–18 typically using a Cu/ZnO-based catalyst (e.g. Cu/ZnO/Al2O3) for MeOH formation and a solid-acid catalyst such as γ-Al2O3, silica-modified alumina or zeolites for MeOH dehydration to DME. Compared to the industrially applied two-step process, the direct process allows higher COx conversion and a simplified reactor design resulting in reduced investment costs.9–13 In both processes, catalyst productivity strongly depends on the syngas composition, i.e. the ratios between H2, CO and CO2.19,20 Theoretical studies suggest that the synergistic effect of Cu and Zn containing domains in the MeOH forming catalyst is largely dependent on the feed composition.21 Also in situ investigations22,23 showed that changes in the catalytic activity of Cu/ZnO-based catalysts are caused by altered syngas composition leading to reversible changes of the catalyst morphology during MeOH formation from CO and CO2 hydrogenation.
CO hydrogenation to MeOH
| CO + 2H2 ⇌ CH3OH ΔH° 298 K = −90.4 kJ mol−1 | (R1) |
CO2 hydrogenation to MeOH
| CO2 + 3H2 ⇌ CH3OH + H2O ΔH° 298 K = −49.4 kJ mol−1 | (R2) |
Water-gas shift (WGS) and its reverse reaction (rWGS)
| CO + H2O ⇌ CO2 + H2 ΔH° 298 K = −41.0 kJ mol−1 | (R3) |
MeOH dehydration
| 2CH3OH ⇌ CH3OCH3 + H2O ΔH° 298 K = −23.5 kJ mol−1 | (R4) |
The use of CO2 as co-feed in the direct DME synthesis has been encouraged, however, this brings additional challenges predominantly associated with loss of catalyst activity,19,20,24,25 since additional water is formed through reaction (R2) and (R3). This challenge requires robust catalytic systems, particularly with higher water tolerance.24,26–28 Catalytic systems enabling both, CO and CO2 hydrogenation should therefore be equipped with a dehydration component with sufficient acidity for effective MeOH dehydration and concurrently, with appropriate hydrophobic surface characteristics to reduce the adsorption of water.29,30
Although Cu/ZnO/Al2O3 (CZA) catalysts are highly active and selective for MeOH synthesis from CO/H2, their activity towards CO2 hydrogenation is reduced.31–33 Amongst several alternative catalytic systems studied, it was proposed to improve CO2 conversion by using less hydrophilic promoters, such as ZrO2 instead of Al2O3.34–36 A large number of publications on the direct DME synthesis refer to the conversion of either CO or CO2 as the sole carbon source.25,37 However, the use of CO-pure syngas promotes coke formation,38 catalyst deactivation39,40 and CO2 formation, whereas CO2-pure syngas increases H2 requirement, water formation and lowers thermodynamic equilibrium.41 Consequently, a logical trade-off seems to be a syngas mixture involving CO and CO2. Although the issue of variable CO/CO2 feed compositions has been addressed in some previous studies, no truly satisfactory catalytic system has been thoroughly investigated for a wide variation range of CO/CO2 in combination with its long-term stability.19,42–45
Recently we showed that a novel continuous co-precipitation process leads to a Cu/ZnO/ZrO2 (CZZ) catalyst, which in combination with a ferrierite dehydration co-catalyst shows improved productivity for DME.46
The scope of our work is to investigate the tolerance of different catalytic systems, especially CZZ/FER, to variable changes in process parameters, particularly the influence of the volumetric CO2/COx inlet-ratio on DME productivity, with the aim of simultaneously maintaining productivity at a high level over a longer period of time. To understand the interplay of the MeOH forming catalyst with the MeOH dehydrating catalyst depending on the syngas feed composition, we compared two dehydration catalysts, γ-Al2O3, which is known to offer high DME productivity in CO-rich feeds while the formation of olefins is inhibited, due to its low acidity,47 and a FER-type zeolite with increased Brønsted acidity, having shown a reasonable water tolerance in the direct DME synthesis from CO2.48
Our hypothesis is that in this way it will be possible to determine what are the appropriate operating parameters under which reasonable DME production with a variable syngas composition takes place.
Experimental
Catalyst preparation
The CZZ catalyst was prepared by continuous co-precipitation method from metal nitrate solution and sodium bicarbonate at pH 7 using a micro jet mixer. The resulting solution was aged at 313 K for 2 h. The precipitate was filtered, dried at 383 K for 16 h and calcined at 623 K with 3 K min−1 for 4 h. The method was described in detail by Polierer et al.46A commercial CZA catalyst was used for comparison purposes. Commercial γ-Al2O3 (Alfa Aesar) or a ferrierite-type zeolite H-FER 20 (FER) (Zeolyst International) were used as dehydration catalysts. Before use, FER was calcined at 823 K for 4 h in air.
For activity tests all catalyst components were finely powdered, pressed and sieved into sieve fractions of 250–500 μm and then physically mixed with a mass ratio of 1:1 resulting in three catalytic systems: CZA/FER, CZZ/γ-Al2O3 and CZZ/FER. Since reactions (R1) to (R4) are exothermic, the catalysts were diluted with silicon carbide (SiC, Hausen Mineraliengroβhandel GmbH) with the same grain size in a mass ratio of 1:10 in order to minimize hot spot formation and therefore ensure largely isothermal operation.
Catalyst characterization
For a detailed characterization of the CZZ and the commercial CZA pre-catalysts we refer to our recent study.46 Selected properties of the MeOH pre-catalysts are shown in Table 1. Physico-chemical properties of the commercial acid dehydration catalysts are taken from Kim et al.49 and shown in Table 2.| Catalyst | Cu/wt% | Zn/wt% | Zr/wt% | Al/wt% | SBET/m2 g−1 | SCu/m2 g−1 | dCuO/nm calcined catalyst | dCuO/nm spent catalyst |
|---|---|---|---|---|---|---|---|---|
| CZZ | 61 | 31 | 8 | — | 125 | 27 | 4 | 10 |
| Com. CZA | 64 | 29 | — | 6 | 98 | 13 | 4 | 8 |
| Catalyst | NH3-TPD peak position/°C | Acid amount/mmol NH3 per gcat | ||||
|---|---|---|---|---|---|---|
| SBET/m2 g−1 | LT region | HT region | Total acidity | LT region | HT region | |
| γ-Al2O3 | 213 | 239 | 351 | 0.37 | 0.18 | 0.19 |
| FER | 390 | 208 | 383 | 0.70 | 0.31 | 0.39 |
Activity tests
Direct DME synthesis was performed in a stainless steel fixed bed reactor with an inner diameter of 12 mm and a length of 460 mm, filled with a physical mixture of 2 g admixed catalyst and 20 g SiC. The reactor was heated by four independent heating zones depicted in Fig. 1, to ensure an axial temperature difference within the catalyst bed of typically less than 2 °C. The gas supply was controlled using mass flow controllers (Bronkhorst Hi-Tec). Feed gases, carbon monoxide (CO, 99.97%), argon (Ar, 99.9999%), nitrogen (N2, 99.9999%), hydrogen (H2, 99.9999%) and a mixture carbon dioxide/nitrogen (CO2/N2, 50:50 ± 1.0 vol%) were provided by Air Liquid Germany GmbH. Product gas composition was analyzed by a gas chromatograph (Agilent G1530A), equipped with thermal conductivity (TCD) and flame ionization (FID) detectors connected to RT®-U-BOND and RT®-Molecular sieve 5A columns. Volumetric water concentration was determined with a FTIR CX4000 (Gasmet Technologies GmbH). Reduction of CZA and CZZ catalyst was performed at 1 bar with 5 vol% H2 diluted in Ar, while temperature was increased from 373 K to 473 K with a ramp of 20 K h−1, followed by further heating to a final reduction temperature of 513 K with 50/50 vol% H2/Ar at a rate of 12 K h−1. Reduction temperature was kept for another 5 h, before the reactor was purged with Ar and cooled to 493 K. Subsequently, the pressure was increased to 50 bar to perform direct DME synthesis. Feed gas compositions used are shown in Table 3. As CO2 hy`drogenation to MeOH (R2) requires stoichiometrically 1.5 equivalents more H2 than CO hydrogenation (R1), the H2 content was adjusted along different CO2/COx inlet-ratios according to (1).| yH2,in = 2.3 (yCO2,in + yCO,in) + yCO2,in | (1) |
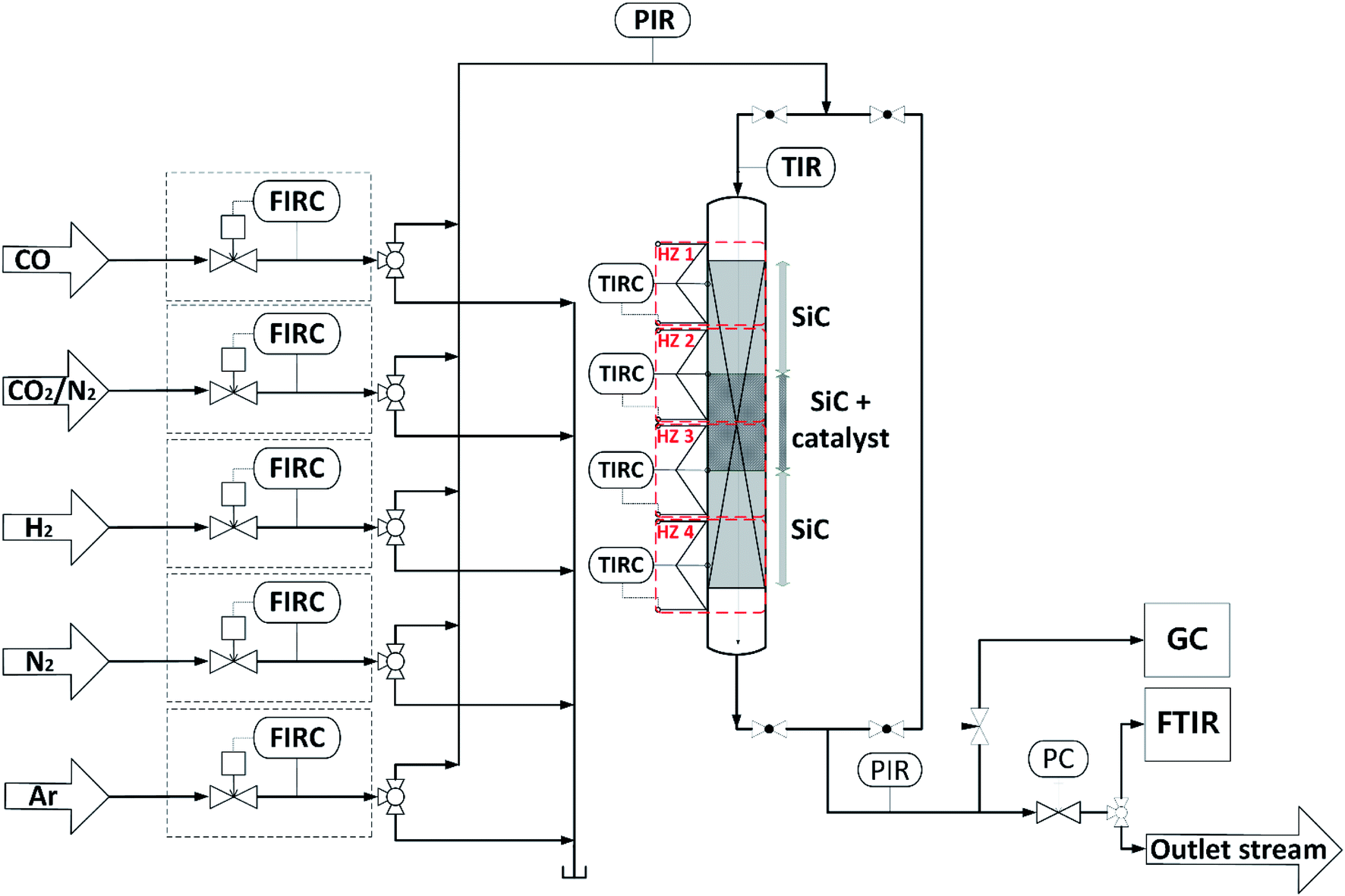 | ||
| Fig. 1 Schematic flowchart of the experimental setup used for the investigation of the direct DME synthesis. | ||
| CO2/COx | H2/vol% | CO/vol% | CO2/vol% | N2/vol% | Ar/vol% |
|---|---|---|---|---|---|
| 0.00 | 34.5 | 15.0 | 0.0 | 15.0 | 35.5 |
| 0.07 | 35.5 | 14.0 | 1.0 | 15.0 | 34.5 |
| 0.20 | 37.5 | 12.0 | 3.0 | 15.0 | 32.5 |
| 0.47 | 41.5 | 8.0 | 7.0 | 15.0 | 28.5 |
| 0.80 | 46.5 | 3.0 | 12.0 | 15.0 | 23.5 |
| 1.00 | 49.5 | 0.0 | 15.0 | 15.0 | 20.5 |
Each feed gas composition was investigated at five temperatures between 483 and 523 K and two gas-hourly space velocities (GHSV) of 18000 and 36000 mlN (g h)−1 with regard to the mass of Cu-based catalyst.
The general sequence for the process parameters variation is shown in Fig. S1†. After finishing the variation loops of CO2/COx values for each temperature, the reactor was purged with Ar for two hours, followed by setting a chosen reference point of 18000 mlN (g h)−1, 503 K and CO2/COx inlet-ratio of 0.8 at 50 bar. Repeated measurements at the reference point were performed to monitor catalyst stability.
Indexes of performance
In all experiments, the carbon balance presented a maximum deviation of ±3%, calculation were performed using eqn S1†. The performance indicators were calculated as follows:COx conversion:
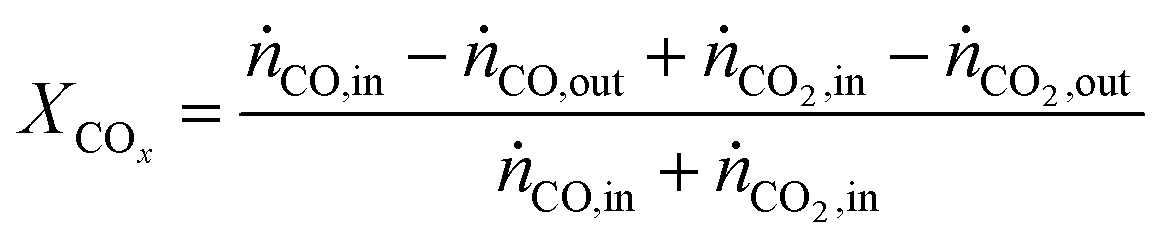 | (2) |
Cu-mass-specific DME/MeOH productivies:
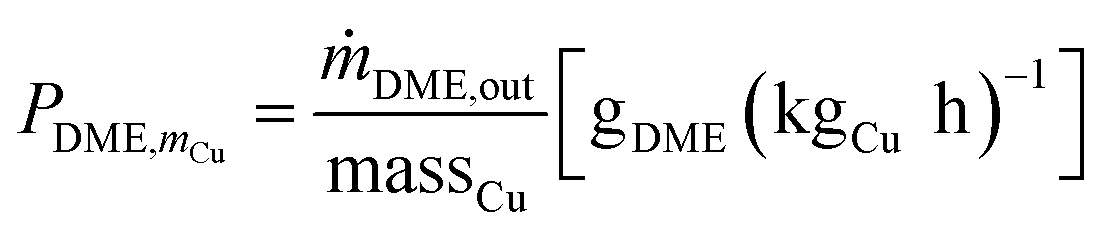 | (3) |
 | (4) |
In order to show the influence of the CO2/COx ratio on CO and CO2 hydrogenation, each in their role (i.e. reactant or product) on DME and MeOH formation three different cases were defined for the selectivity calculation.
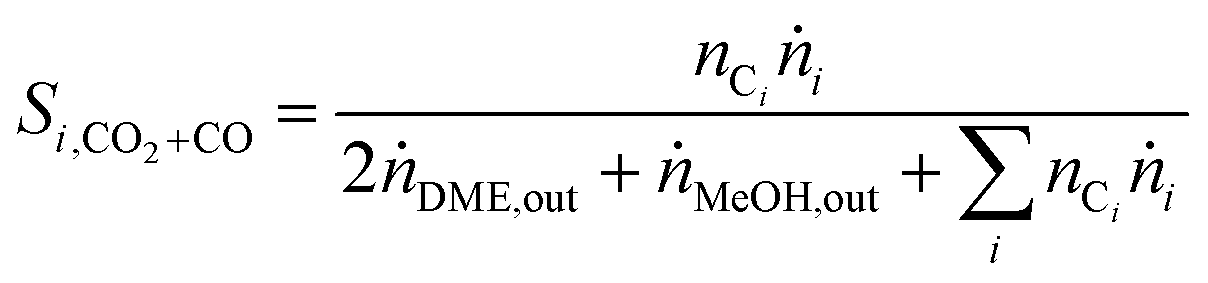 | (5) |
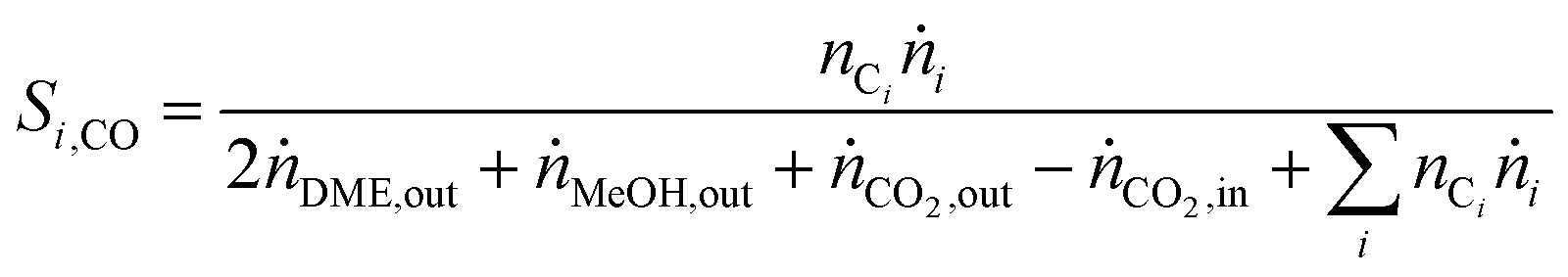 | (6) |
 | (7) |
Results and discussion
Comparison of γ-Al2O3 and FER as dehydration catalysts
In Fig. 2, CZZ/γ-Al2O3 is compared to CZZ/FER at different CO2/COx inlet-ratios with regard to COx conversion (Fig. 2a, bars), selectivities to MeOH and DME (Fig. 2a, lines) and, productivities of MeOH and DME (Fig. 2b). Regarding the COx conversion (Fig. 2a), a slight increase of CZZ/FER in comparison to CZZ/γ-Al2O3 is observable. As the MeOH catalyst is the same in both systems, this difference is attributed to the dehydration catalysts. Since the DME selectivity of CZZ/FER is higher than the one of CZZ/γ-Al2O3, there is an increased intermediate product (MeOH) removal with CZZ/FER. Consequently, there is an increase in MeOH production due to an equilibrium shift of the CO and CO2 hydrogenation ((R1) and (R2)), resulting in a slightly higher COx conversion. As the COx conversion is strongly kinetically controlled under the respective operating conditions (Fig. S2†) the enhancement due to equilibrium shift is only slightly pronounced. The CZZ/FER system reaches its highest DME selectivity of 92.1% at a CO2/COx inlet-ratio of 0.47, and even at 0.8 selectivity is still above 80%. CZZ/γ-Al2O3 shows a reduced DME selectivity up to 60% at CO2/COx inlet-ratios below 0.2, a further increase of the CO2 content leads to a strongly declining DME selectivity with a minimum of 4.8% at CO2/COx inlet-ratio of 1.00. Accordingly, CZZ/FER generally achieves higher DME productivity, with the difference to CZZ/γ-Al2O3 becoming more noticeable at higher CO2/COx inlet-ratios (Fig. 2b). Interestingly, CZZ/FER already enables a significantly improved DME productivity (67%) compared to CZZ/γ-Al2O3 at a relatively low CO2/COx inlet-ratio of 0.20, what can be attributed to the strong hydrophilic behaviour of γ-Al2O3 as reported in literature.12,19,33,50 On the other hand FER is marked by better water resistance, it has a higher acidity compared to γ-Al2O3 (see Table 2) and additionally well distributed acid sites with a suitable strength and a good resistance to coke formation in the presence of water,12,30,33,51 and therefore is superior for dehydration of MeOH formed at high CO2 content. The nearly constant COx conversion (Fig. 2a) as well as the improved DME selectivity of CZZ/FER compared to CZZ/γ-Al2O3 (Fig. 2b) lead to a superior DME productivity between 1017 gDME (kgCu h)−1 (CO2/CO = 0.20) and 689 gDME (kgCu h)−1 (CO2/COx = 1.00). Due to the high DME productivities at variable CO2/COx feed compositions, FER was chosen for further investigations. | ||
| Fig. 2 Influence of the CO2/COx inlet-ratio on direct DME synthesis with CZZ/FER (1:1 wt%) (black) and CZZ/γ-Al2O3 (1:1 wt%) (red) at 50 bar, 523 K and 36000 mlN (g h)−1. (a) COx conversion (right axis) and selectivities of MeOH and DME (left axis). (b) Productivities of MeOH and DME. | ||
Comparison of MeOH catalysts CZA and CZZ
We further studied the catalytic activity of the self-prepared CZZ and a commercial CZA catalyst as a benchmark, which is typically used for MeOH synthesis from CO-rich syngas, both in combination with FER. In Fig. 3 we compare CZA/FER (blue) and CZZ/FER (black) at different CO2/COx inlet-ratios. Fig. 3a displays COx conversion (bars) and selectivities to MeOH and DME (lines) at 523 K, while Fig. 3b represents the productivities of MeOH and DME at 503 and 523 K. CZZ/FER enables significantly elevated COx conversion for all investigated CO2/COx inlet-ratios compared to CZA/FER (Fig. 3a), resulting in correspondingly higher DME productivity values (Fig. 3b). We attribute the enhanced COx conversion to the properties of the continuously co-precipitated CZZ, i.e. its high Cu surface area (Table 1) and the presence of ZrO2, which is known to promote Cu dispersion11 and increase the activity of Cu-based catalysts in CO2 hydrogenation to MeOH and DME.11,35,46,52 It is interesting to note that although CZA has a relatively low copper surface area of 13 m2 g−1 CZA/FER offers high DME productivities: with pure H2/CO2 (according to CO2/COx = 1) at 523 K, the productivity is only 9% lower than using CZZ/FER (CZZ-SCu: 27 m2 g−1).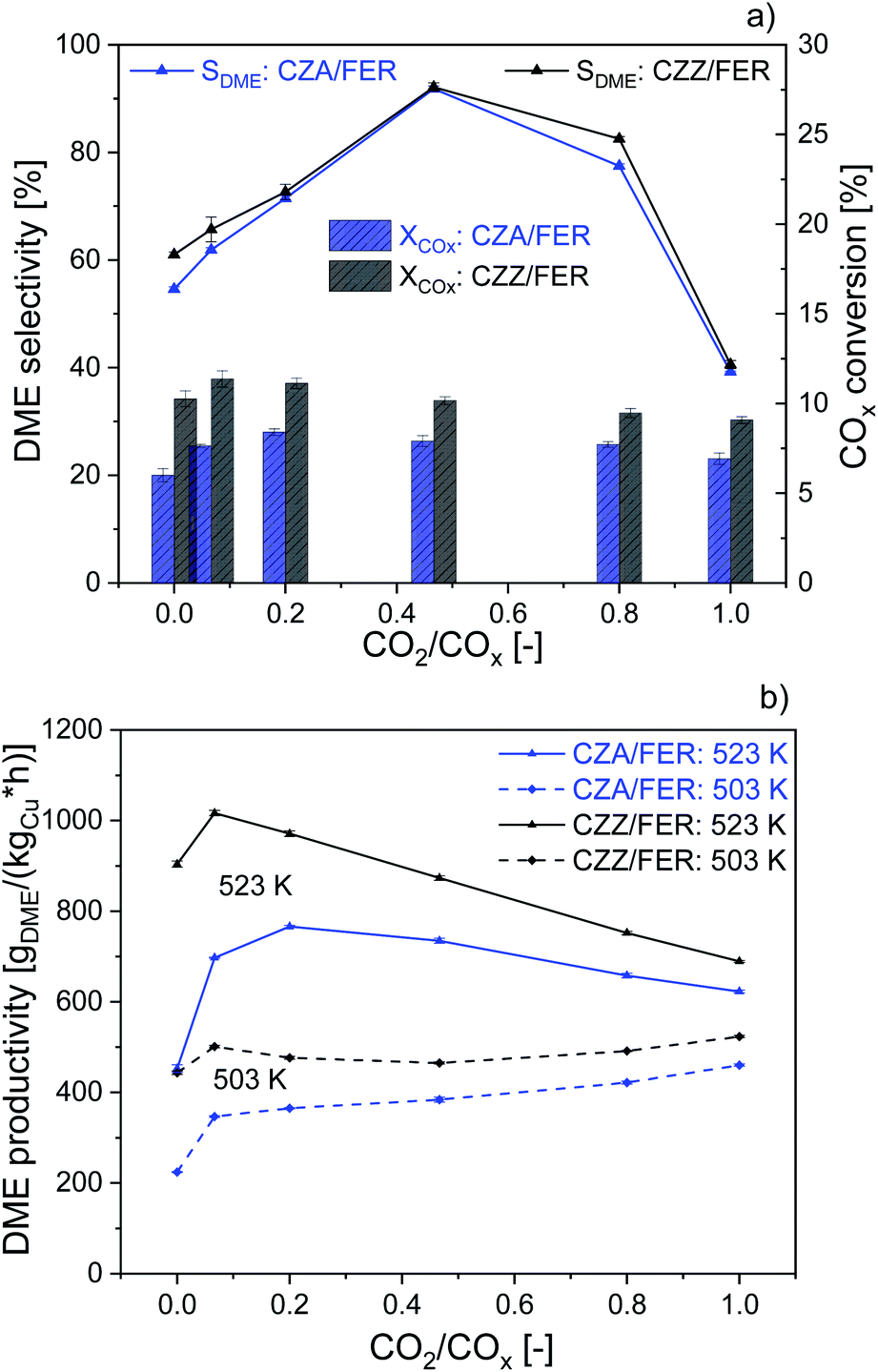 | ||
| Fig. 3 Influence of CO2/COx inlet-ratio on the DME synthesis with com. CZA catalyst (blue) and the CZZ catalyst (black) at 50 bar and GHSV: 36000 mlN (g h)−1, mixed with FER (1:1 wt%). DME selectivity and COx conversion ((a) 523 K). DME productivity ((b) 503 and 523 K). | ||
Similar observations were made by Kurtz et al.53 showing a pronounced linear dependence of MeOH activity and SCu using Cu/ZnO catalysts, whereby using a self-prepared CZA the MeOH activity increased non-linearly to copper surface area. Moreover, uncharacterized additional components of the commercial CZA could also influence its activity. Fig. 3a shows that this increased DME productivity in Fig. 3b is caused by higher COx conversion and DME selectivity. This observation is consistent with results presented in the literature.54–56 According to Behrens et al.57 and Studt et al.,21 CO2 hydrogenation (R2) is significantly faster than CO hydrogenation (R1) on a Cu/ZnO-based catalyst. Therefore, with additional CO2 in the feed, MeOH formation takes place more quickly at the beginning of the catalyst bed, whereas with a pure H2/CO feed, CO2 hydrogenation is only accelerated when part of the DME has already been produced and additional CO2 is generated via the WGS with the water formed in the process. By lowering the reactor temperature (503 K), the DME productivity of both catalyst systems changes only slightly over the entire CO2/COx feed range. With CZZ/FER DME productivity ranges between 433 and 523 gDME (kgCu h)−1. We consider this to be a combination of different effects: firstly, a reduced rate of endothermic rWGS (R3) results in less water being formed, which is able to inhibit the activity of the admixed catalyst,58 and secondly the positive effect on the thermodynamic equilibrium of COx conversion (R1), (R2) and MeOH dehydration (R4). Similar observations have been made by Sahibzada et al.59 using a CZA catalyst for MeOH synthesis, by increasing the CO2/COx inlet-ratio a continuously increasing MeOH productivity takes place as long as differential conditions prevail. The benefit of a slight increase of CO2 in feed (CO2/COx inlet-ratio from 0.00 to 0.07) leads to a maximum in DME productivity of 1017 gDME (kgCu h)−1 using CZZ/FER at 523 K. The DME productivity of CZZ/FER then gradually decreases to 689 gDME (kgCu h)−1 using CO2 as the sole carbon source, which we regard as an important argument for process operation with dynamically variable feed compositions. An increasing CO2 content changes the thermodynamic equilibrium,41 increases water formation and leads to a more oxidative atmosphere – resulting in a change of the Cu/Zn and Cu sites23,60 which negatively affects CO2 hydrogenation – leading to a performance levelling of the two catalyst systems in terms of COx conversion and DME productivity. Frusteri et al.12 investigated admixed catalyst systems of CZA and CZZ in combination with HZSM-5 under similar reaction conditions: at 533 K, 50 bar and a syngas mixture CO2/H2/N2 of 3/9/1 (cf. Table 3), the reported DME productivities were approx. 250 gDME (kgcat h)−1 with CZA/HZSM-5 and 190 gDME (kgcat h)−1 with CZZ/HZSM-5. In our experiments, at 523 K, 50 bar and with a CO2/COx inlet-ratio of 1.00, the CZZ/FER system achieves a DME productivity of 421 gDME (kgcat h)−1 (MeOH catalyst specific), demonstrating the particular suitability of continuously co-precipitated CZZ in combination with FER.
Influence of temperature and CO2/COx inlet-ratio on selectivity
Fig. 4 shows the influence of CO2/COx inlet-ratio and temperature on DME, MeOH, CO and CO2 selectivity using CZZ/FER. This diagram complements Fig. 3a, as it points out the influence of the feed composition on the selectivity of the four main carbon-containing species. At CO-rich feed compositions, CO2 is formed via the exothermic WGS (Case 2), resulting in a maximum CO2 selectivity of 43.1% (CO2/COx = 0.00) at the lowest measured temperature of 483 K. Increasing the amount of CO2 in the feed reduces the rate of WGS (R3), resulting in a decrease of CO2 selectivity, which in turn increases the selectivity to MeOH and DME. At a CO2/COx inlet-ratio of 0.47, both CO2 and CO are converted (Case 1), with increasing CO2 content the endothermic rWGS takes over and CO is formed (Case 3) with a maximum CO selectivity of 49.1% (CO2/COx = 1.00) at the highest measured temperature of 523 K. The impact of temperature on CO and CO2 selectivities, described before, leads to the respective differences in DME selectivity with changing temperature. The MeOH selectivity increases constantly from CO-rich feed compositions until a maximum at CO2/COx = 0.80 is reached. This can be attributed to the dehydration of MeOH to DME (R4), which can be negatively affected thermodynamically by higher water concentrations produced at higher CO2/COx inlet-ratios. Direct DME synthesis with feed gas compositions close to CO2/COx = 0.00 or 1.00 causes selectivity issues that might complicate an industrial process feasibility, as it would require an intensified CO/CO2 separation/recycling step.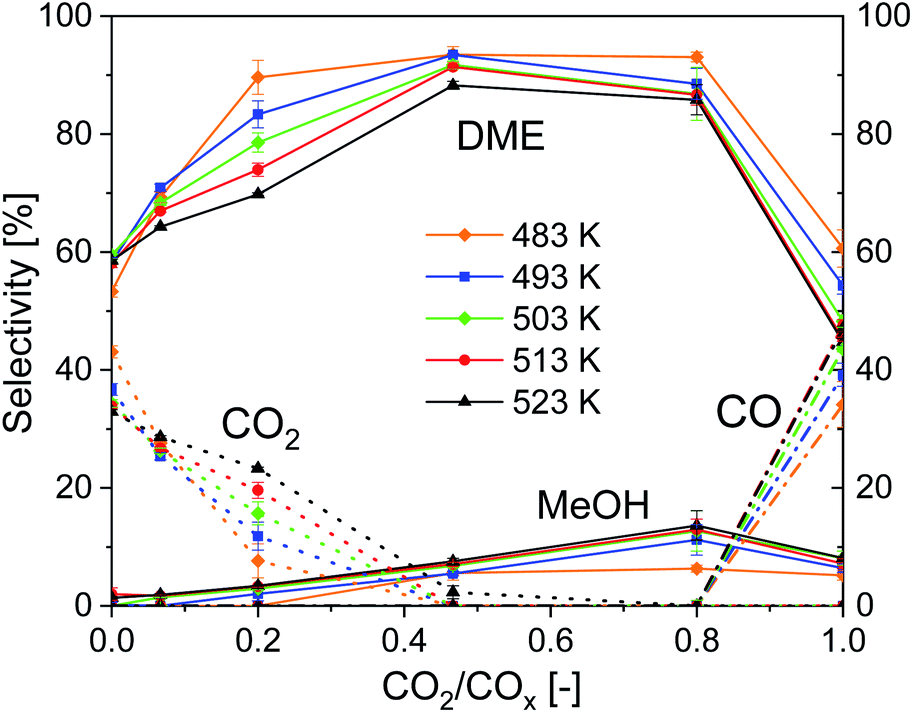 | ||
| Fig. 4 Influence of temperature and CO2/COx inlet-ratio on the selectivity, 18000 mlN (g h)−1, 50 bar, CZZ/FER 1:1 wt%. | ||
Working with CO2 as sole carbon source lowers the thermodynamic equilibrium of COx conversion41 and reduces the efficiency of hydrogen use, since water is produced in a higher ratio compared to the valuable products (i.e. MeOH and DME).
Given the high DME selectivity, an average CO2/COx inlet ratio (i.e. approximately between 0.4 and 0.8) is not only a reasonable operating range within which both CO and CO2 are converted to DME, but it also offers the option of achieving a high DME productivity with a dynamic variation of the CO2/CO ratio.
Catalyst stability
To assess the stability of the CZZ/FER catalyst, direct DME synthesis was operated over 550 h (Fig. 5). According to Fichtl et al.,61 the elevated water concentration formed in CO2-rich feed is the driving factor for irreversible deactivation effects. Therefore, and based on the above-mentioned arguments for a reasonable operating range, it seems appropriate to define a value of 0.80 for CO2/COx as reference point of the feed composition for this study. | ||
| Fig. 5 DME productivity between 126 and 545 h ToS at reference point: CO2/COx inlet-ratio = 0.8, 18000 mlN (g h)−1, 503 K, 50 bar, CZZ/FER 1:1 wt% with static reaction conditions in first 200 h ToS followed by various reaction conditions in temperature, CO2/COx inlet-ratio and GHSV. Dashed line fitted via OriginPro. | ||
For the period up to 200 h ToS, DME synthesis was performed under static reaction conditions, i.e. 503 K, 18000 mlN (g h)−1, CO2/COx = 0.80, 50 bar (reference point conditions). During this period, the activity of the catalyst in terms of DME productivity decreases to 326.6 gDME (kgCu h)−1 (85.5%, 175 h) of the initial DME productivity (100%, 0–20 h ToS). Subsequently, the process was subjected to feed variation as described in Table 3 with 10 K temperature steps from 483 K to 523 K at five different GHSV between 18000 and 42000 mlN (g h)−1 monitoring the recurring reference point after each variation cycle (Fig. S1†). DME productivity decreases to 283.6 gDME (kgCu h)−1 (74.2%) up to a ToS of 384 h and remains almost unchanged at 278.9 gDME (kgCu h)−1 (73.0%, 545 h) until the end of the observation period. DME selectivity was found to remain nearly constant, after a short run-in period of 5 h, with values in the range between 87.4 and 91.6%. This leads to the assumption that no relevant changes have taken place on the active sites of FER. Analogously, Frusteri et al.12 did not detect relevant coke formation working with CO2 as the sole carbon source. This can also be explained by the results of Sierra et al.38 who found that a slight increase in the water content in the gas phase reduces coke formation. Our results can confirm that relation: at CO-rich syngas concentrations and elevated temperatures, ethane was detected with CZZ/FER up to a maximum selectivity of 6.9% at 523 K, 50 bar, 36000 mlN (g h)−1 and a CO2/COx inlet-ratio of 0.00. Since a relatively low COx conversion range was achieved in the operating ranges considered, product concentrations were generally relatively low. Use of FER in a higher conversion range may result in increased formation of by-products such as methyl acetate, methane, ethane, and higher hydrocarbons. Hydrocarbon species were measured up to C4H10, concentrations below 0.01% by volume were not considered.
Our findings clearly demonstrate that the CZZ/FER catalyst is robust against fluctuations in the operating conditions after the initial operating phase and largely maintains its activity within the limits of the process parameter ranges investigated here.
Conclusions
In this study, the admixed catalyst systems CZZ/FER, CZZ/γ-Al2O3 and CZA/FER were investigated in the direct DME synthesis from variable CO2/COx feeds. Our findings underline that a superior catalytic activity and a higher water resistance of a commercial FER-type zeolite clearly overtakes those of γ-Al2O3 leading to a consistent DME productivity applying different CO2/COx inlet-ratios. The effectiveness of FER occurs not only at high CO2/COx inlet-ratio but already at a slight increase of the CO2/COx ratio.Combining a CZZ catalyst prepared by continuous precipitation method admixed with FER shows higher COx conversion and a significantly improved DME productivity for both, CO-rich feed (CO2/COx = 0.20, 1017 gDME (kgCu h)−1) and CO2-rich feed conditions (CO2/COx = 1.00, 689 gDME (kgCu h)−1) at 523 K, than the respective combination of a commercial CZA catalyst with FER. For CZZ/FER, we also found the option of adjusting DME productivity at 503 K largely independent of the CO2/COx ratio.
For CO2/COx inlet-ratios ranging between 0.47 and 0.80, temperatures between 483 K and 513 K and a GHSV of 18000 mlN (g h)−1, both CO2 and CO are converted – resulting in DME selectivities around 90%.
Detailed experiments with the CZZ/FER system performed under static and variable operating conditions showed that this catalytic system retains the major proportion of its initial DME productivity after 545 h time on stream. The over all deactivation in terms of DME productivity in the period from 0 to 545 h is 27.0%, and 14.6% during the period of variable feed conditions from 200 up to 545 h. The DME selectivity remains largely constant between 87.4% and 91.6% over the entire investigation duration. The extent to which aging phenomena due to sintering or coking play a role under process conditions is the subject of a planned investigation.
Our results prove the excellent suitability of CZZ/FER mixed catalyst systems for direct, flexible COx hydrogenation to DME under variable conditions. We believe that this type of catalyst system represents a promising option for use in sustainable power-to-fuel technologies that address both the use of hydrogen from renewable energy and the use of CO2 as a C1 raw material. For this reason, we are currently working intensively on modelling the process and optimising the composition of the catalyst bed and will report on this accordingly. Part of our work is furthermore to generate a sufficient data basis for a later planned kinetic modeling.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Helmholtz Association, Research Programme “Storage and Cross-linked Infrastructures”, Topic “Synthetic Hydrocarbons”. We kindly acknowledge the Chair of Industrial Chemistry at Ruhr-University Bochum for H2-TPR and N2O-RFC measurements, the support of TVT-KIT group, analytics and chemical laboratory workers from IKFT-KIT.References
- N. Dahmen, U. Arnold, N. Djordjevic, T. Henrich, T. Kolb, H. Leibold and J. Sauer, J. Supercrit. Fluids, 2015, 96, 124–132 CrossRef CAS.
- A. Varone and M. Ferrari, Renewable Sustainable Energy Rev., 2015, 45, 207–218 CrossRef.
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC.
- G. A. Olah, A. Goeppert and G. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS.
- B. Niethammer, S. Wodarz, M. Betz, P. Haltenort, D. Oestreich, K. Hackbarth, U. Arnold, T. Otto and J. Sauer, Chem. Ing. Tech., 2018, 90, 99–112 CrossRef CAS.
- N. Dahmen, J. Abeln, M. Eberhard, T. Kolb, H. Leibold, J. Sauer, D. Stapf and B. Zimmerlin, Appl. Catal., A, 2017, 6, e236 Search PubMed.
- J. Sun, G. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2014, 4, 3346–3356 CrossRef CAS.
- P. Geng, E. Cao, Q. Tan and L. Wei, Renewable Sustainable Energy Rev., 2017, 71, 523–534 CrossRef CAS.
- Y. Wang, H. Liu, H. Zhang and W. Ying, React. Kinet., Mech. Catal., 2016, 119, 585–594 CrossRef CAS.
- S. M. K. Aboul-Fotouh, L. I. Ali, M. A. Naghmash and N. A. K. Aboul-Gheit, J. Fuel Chem. Technol., 2017, 45, 581–588 CrossRef CAS.
- M. Sánchez-Contador, A. Ateka, P. Rodriguez-Vega, J. Bilbao and A. T. Aguayo, Ind. Eng. Chem. Res., 2018, 57, 1169–1178 CrossRef.
- F. Frusteri, M. Migliori, C. Cannilla, L. Frusteri, E. Catizzone, A. Aloise, G. Giordano and G. Bonura, J. CO2 Util., 2017, 18, 353–361 CrossRef CAS.
- R. Ahmad, D. Schrempp, S. Behrens, J. Sauer, M. Döring and U. Arnold, Fuel Process. Technol., 2014, 121, 38–46 CrossRef CAS.
- R. Ahmad, M. Hellinger, M. Buchholz, H. Sezen, L. Gharnati, C. Wöll, J. Sauer, M. Döring, J.-D. Grunwaldt and U. Arnold, Catal. Commun., 2014, 43, 52–56 CrossRef CAS.
- S. S. Akarmazyan, P. Panagiotopoulou, A. Kambolis, C. Papadopoulou and D. I. Kondarides, Appl. Catal., B, 2014, 145, 136–148 CrossRef CAS.
- X. Jianchao, M. Dongsen, Z. Bin, C. Qingling and T. Yi, Catal. Lett., 2004, 98, 235–240 CrossRef.
- D. Liu, C. Yao, J. Zhang, D. Fang and D. Chen, Fuel, 2011, 90, 1738–1742 CrossRef CAS.
- J. Fei, Z. Hou, B. Zhu, H. Lou and X. Zheng, Appl. Catal., A, 2006, 304, 49–54 CrossRef CAS.
- K. L. Ng, D. Chadwick and B. A. Toseland, Chem. Eng. Sci., 1999, 54, 3587–3592 CrossRef CAS.
- A. Ateka, M. Sánchez-Contador, J. Ereña, A. T. Aguayo and J. Bilbao, React. Kinet., Mech. Catal., 2018, 124, 401–418 CrossRef CAS.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- R. van den Berg, G. Prieto, G. Korpershoek, L. I. van der Wal, A. J. van Bunningen, S. Laegsgaard-Jorgensen, P. E. de Jongh and K. P. de Jong, Nat. Commun., 2016, 7, 13057 CrossRef CAS.
- J. D. Grunwaldt, A. M. Molenbroek, N. Y. Topsøe, H. Topsøe and B. S. Clausen, J. Catal., 2000, 194, 452–460 CrossRef CAS.
- J. Abu-Dahrieh, D. Rooney, A. Goguet and Y. Saih, Chem. Eng. J., 2012, 203, 201–211 CrossRef CAS.
- Z. Azizi, M. Rezaeimanesh, T. Tohidian and M. R. Rahimpour, Chem. Eng. Process., 2014, 82, 150–172 CrossRef CAS.
- A. García-Trenco and A. Martínez, Appl. Catal., A, 2012, 411–412, 170–179 CrossRef.
- D. Wang, Y. Han, Y. Tan and N. Tsubaki, Fuel Process. Technol., 2009, 90, 446–451 CrossRef CAS.
- N. Diban, A. M. Urtiaga, I. Ortiz, J. Ereña, J. Bilbao and A. T. Aguayo, Chem. Eng. J., 2013, 234, 140–148 CrossRef CAS.
- A. J. Jones and E. Iglesia, Angew. Chem., Int. Ed., 2014, 53, 12177–12181 CrossRef CAS.
- E. Catizzone, A. Aloise, M. Migliori and G. Giordano, J. Energy Chem., 2017, 26, 406–415 CrossRef.
- M. Liu, Y. Yi, L. Wang, H. Guo and A. Bogaerts, Catalysts, 2019, 9, 275 CrossRef.
- F. Arena, K. Barbera, G. Italiano, G. Bonura, L. Spadaro and F. Frusteri, J. Catal., 2007, 249, 185–194 CrossRef CAS.
- E. Catizzone, M. Migliori, A. Purita and G. Giordano, J. Energy Chem., 2019, 30, 162–169 CrossRef.
- S. Tada, A. Katagiri, K. Kiyota, T. Honma, H. Kamei, A. Nariyuki, S. Uchida and S. Satokawa, J. Phys. Chem. C, 2018, 122, 5430–5442 CrossRef CAS.
- E. Lam, K. Larmier, P. Wolf, S. Tada, O. V. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 10530–10535 CrossRef CAS.
- W. Wang, Z. Qu, L. Song and Q. Fu, J. Energy Chem., 2020, 40, 22–30 CrossRef.
- E. Catizzone, G. Bonura, M. Migliori, F. Frusteri and G. Giordano, Molecules, 2018, 23, 31 CrossRef.
- I. Sierra, J. Erena, A. T. Aguayo, J. M. Arandes, M. Olazar and J. Bilbao, Appl. Catal., B, 2011, 106, 167–173 CAS.
- J. T. Sun, I. S. Metcalfe and M. Sahibzada, Ind. Eng. Chem. Res., 1999, 38, 3868–3872 CrossRef CAS.
- J. Ereña, I. Sierra, A. T. Aguayo, A. Ateka, M. Olazar and J. Bilbao, Chem. Eng. J., 2011, 174, 660–667 CrossRef.
- W.-J. Shen, K.-W. Jun, H.-S. Choi and K.-W. Lee, Korean J. Chem. Eng., 2000, 17, 210–216 CrossRef CAS.
- A. Ateka, J. Ereña, P. Pérez-Uriarte, A. T. Aguayo and J. Bilbao, Int. J. Hydrogen Energy, 2017, 42, 27130–27138 CrossRef CAS.
- A. Bayat and T. Dogu, Ind. Eng. Chem. Res., 2016, 55, 11431–11439 CrossRef CAS.
- M.-H. Huang, H.-M. Lee, K.-C. Liang, C.-C. Tzeng and W.-H. Chen, Int. J. Hydrogen Energy, 2015, 40, 13583–13593 CrossRef CAS.
- A. Ateka, J. Ereña, M. Sánchez-Contador, P. Perez-Uriarte, J. Bilbao and A. Aguayo, Appl. Sci., 2018, 8, 677 CrossRef.
- S. Polierer, D. Guse, S. Wild, K. Herrera Delgado, T. N. Otto, T. A. Zevaco, M. Kind, J. Sauer, F. Studt and S. Pitter, Catalysts, 2020, 10, 816 CrossRef CAS.
- A. I. Osman, J. K. Abu-Dahrieh, D. W. Rooney, S. A. Halawy, M. A. Mohamed and A. Abdelkader, Appl. Catal., B, 2012, 127, 307–315 CrossRef CAS.
- G. Bonura, F. Frusteri, C. Cannilla, G. Drago Ferrante, A. Aloise, E. Catizzone, M. Migliori and G. Giordano, Catal. Today, 2016, 277, 48–54 CrossRef CAS.
- Y. T. Kim, K.-D. Jung and E. D. Park, Microporous Mesoporous Mater., 2010, 131, 28–36 CrossRef CAS.
- S. P. Naik, T. Ryu, V. Bui, J. D. Miller, N. B. Drinnan and W. Zmierczak, Chem. Eng. J., 2011, 167, 362–368 CrossRef CAS.
- P. S. Sai Prasad, J. W. Bae, S.-H. Kang, Y.-J. Lee and K.-W. Jun, Fuel Process. Technol., 2008, 89, 1281–1286 CrossRef.
- F. Arena, G. Mezzatesta, G. Zafarana, G. Trunfio, F. Frusteri and L. Spadaro, Catal. Today, 2013, 210, 39–46 CrossRef CAS.
- M. Kurtz, N. Bauer, C. Büscher, H. Wilmer, O. Hinrichsen, R. Becker, S. Rabe, K. Merz, M. Driess, R. A. Fischer and M. Muhler, Catal. Lett., 2004, 92, 49–52 CrossRef CAS.
- F. Dadgar, R. Myrstad, P. Pfeifer, A. Holmen and H. J. Venvik, Catal. Today, 2016, 270, 76–84 CrossRef CAS.
- K. G. Chanchlani, R. R. Hudgins and P. L. Silveston, J. Catal., 1992, 136, 59–75 CrossRef CAS.
- G. Liu, D. Willcox, M. Garland and H. H. Kung, J. Catal., 1984, 90, 139–146 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS.
- H. Ruland, H. Song, D. Laudenschleger, S. Stürmer, S. Schmidt, J. He, K. Kähler, M. Muhler and R. Schlögl, ChemCatChem, 2020, 12, 3216–3222 CrossRef CAS.
- M. Sahibzada, I. S. Metcalfe and D. Chadwick, J. Catal., 1998, 174, 111–118 CrossRef CAS.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS.
- M. B. Fichtl, D. Schlereth, N. Jacobsen, I. Kasatkin, J. Schumann, M. Behrens, R. Schlögl and O. Hinrichsen, Appl. Catal., A, 2015, 502, 262–270 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09754c |
| This journal is © The Royal Society of Chemistry 2021 |