
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid iodine capture from radioactive wastewater by green and low-cost biomass waste derived porous silicon–carbon composite
Guiyang Qua,
Ying Han *a,
Junjun Qiab,
Xinyue Xinga,
Minjie Houa,
Yang Sunc,
Xing Wang*a and
Guangwei Suna
*a,
Junjun Qiab,
Xinyue Xinga,
Minjie Houa,
Yang Sunc,
Xing Wang*a and
Guangwei Suna
aLiaoning Key Laboratory of Pulp and Papermaking Engineering, Dalian Polytechnic University, Dalian, 116034, China. E-mail: wangxing@dlpu.edu.cn; hanying@dlpu.edu.cn
bChina National Paper-Industry Investment Corp, Beijing, 100032, China
cDepartment of Chemistry, Faculty of Engineering, Gunma University, Kiryu, Gunma 376-8515, Japan
First published on 27th January 2021
Abstract
The effective and safe capture and storage of radioactive iodine (129I or 131I) are of significant importance during nuclear waste storage and nuclear energy generation. Herein, a porous silicon–carbon (pSi–C) composite derived from paper mill sludge (PMS) is synthesized and used for rapid iodine capture. The influences of the activator type, the impregnation ratio of the paper mill sludge to the activator, carbonization temperature, and carbonization time on the properties of the pSi–C composite are investigated. The pSi–C composite produced in the presence of ZnCl2 as the activator and at an impregnation ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, a carbonization temperature of 550 °C, and a carbonization time of 90 min has a surface area of 762.13 m2 g−1. The as-synthesized pSi–C composite exhibits promising iodine capture performance in terms of superior iodine adsorption capacity (qt) of around 250 mg g−1 and rapid equilibrium adsorption with in 15 min. The devised method is environmentally friendly and inexpensive and can easily be employed for the large-scale production of porous silicon-activated carbon composites with excellent iodine capture and storage from iodine-contaminated water.
:1, a carbonization temperature of 550 °C, and a carbonization time of 90 min has a surface area of 762.13 m2 g−1. The as-synthesized pSi–C composite exhibits promising iodine capture performance in terms of superior iodine adsorption capacity (qt) of around 250 mg g−1 and rapid equilibrium adsorption with in 15 min. The devised method is environmentally friendly and inexpensive and can easily be employed for the large-scale production of porous silicon-activated carbon composites with excellent iodine capture and storage from iodine-contaminated water.
1. Introduction
In response to the energy shortage and global warming, nuclear energy is increasingly accepted and used by many countries. However, along with the growing utilization of nuclear energy, nuclear waste pollution has become an inevitable issue.1,2 Among all the radioactive contaminants produced, public concern has been seriously expressed over radioactive iodine since the radioactive iodine isotopes with long half-lives, e.g. 129I or 131I, can accumulate in the ecological environment and damage human metabolic systems.3,4Even worse, the pollution caused by 129I with a long half-life of 15.7 million years will last for millions of years.5,6 Therefore, the rapid removal of radioactive iodine from nuclear waste is crucial to both the daily clean-up of nuclear waste and emergency treatment of nuclear spills. In nuclear waste, radioactive iodine exists as molecular iodine (I2), iodide (I−), hypoiodite ion (IO−), and iodate (IO3−).7,8 Generally, inorganic composite adsorbents such as silver-based zeolites9,10 and silver-impregnated materials11 and porous carbon12,13 are the early reported traditional sorbents that have been employed for capturing iodine. However, its low ability to adsorb iodine, low specific surface area and high cost of silver limited its practical application.
In the past few years, porous crystalline metal organic frameworks (MOFs),14,15 porous organic polymers (POPs),16,17 silicon composite materials,18–20 biomass-derived carbon materials,21–24 graphene oxide,25 Layered double hydroxide26 as new types of functional materials with the relatively high capability to capture iodine, have aroused substantial interest in many researchers. These porous materials have a better capacity for adsorbing iodine compared with the silver-based adsorbents. However, due to MOFs and POPs materials only play the role of enrichment and capture of iodine, and subsequent curing treatment is still needed, so their actual use is limited. Graphene oxide and layered double hydroxide are less dispersed in water. In contrast, water stability of silicon composite materials and biomass-derived carbon materials is relatively good. Therefore, regarding the advantages of silicon-containing substances and biomass-derived porous carbon materials, developing a method for the simple and inexpensive production of novel porous silicon-activated carbon (pSi–C) composites for rapid iodine capture is of great significance.
Paper mill sludge (PMS), which accounts for 3–5% of about 408 million tons of pulp and paper production every year around the world, contains a large number of inorganic materials, such as silicon and organic substances including cellulose, lignin, etc. Moreover, it is a high-quality raw material for preparing silicon–carbon composite materials. In this context, we devise an easy method for the synthesis of porous silicon-activated carbon composites from PMS and, for the very first time, deal with the challenge of obtaining a PMS-derived porous silicon-activated carbon composite to be utilized for rapid iodine capture.
2. Experimental procedure
2.1. Materials
In this study, the paper mill sludge (PMS) investigated was provided by a paper mill in Liaoning Province in China, raw materials were dried in an oven at 105 °C for 24 h and then the materials were crushed and sieved by 100 meshes stainless steel screen. Iodine (Aladdin) and potassium iodide (Aladdin) were used to make contaminated water. HCl, KOH, NaOH, MgCl2, and ZnCl2 were purchased from Alfa Aesar China (Tianjin, China) and used as received. All the other reagents were analytical grade and used as received.2.2. Porous silicon–carbon composite derived from biomass waste
Porous silicon–carbon (pSi–C) composite derived from biomass waste was prepared by chemical activation, using KOH, NaOH, MgCl2, or ZnCl2. The mixture of the PMS powder and chemical activator with the ratios of 1:0.5,1:1, 1:1.5, 1:2, or 1:2.5 was heated at carbonization temperature (400–600 °C) for carbonization time (30–150 min) under N2 gas flow of 40 mL min−1 at heating rate of 5 °C min−1 in a tube furnace. At the end of carbonization, the samples were allowed to cool down to room temperature. The products obtained were washed by 1 mol L−1 HCl and then filtered and rinsed by warm distilled water several times until reached to neutral pH to remove residual inorganic matter. Finally, the pSi–C composite was obtained after drying at 105 °C to constant weight.
2.3. Characterization
The pSi–C composite morphologies were determined by a JSM-7800F scanning electron microscope (SEM) (JEOL Ltd.; Tokyo, Japan), the chemical composition of the samples were analyzed using an Oxford Instruments X-Max20 energy dispersive X-ray spectrometer (EDS). SEM images and EDS data were typically collected at 15 kV, a working distance of 8.5 mm and a 60 μm aperture. The functional groups of the pSi–C composite were characterized by Fourier transform infrared spectrum (FTIR). The samples were dried at 105 °C, and then 1 mg samples were mixed with 100 mg KBr in a full pellet after grinding then method ranging from 4000 to 400 cm−1.The crystalline of the pSi–C composite was investigated by X-ray diffraction (XRD) with an XRD7000S (Shimadzu Corp; Kyoto, Japan) using a copper (Cu) Kα radiation at 25 °C source in the 10–70° 2θ range with a scanning step length of 5° min−1. Nitrogen adsorption–desorption isotherms at 77 K were measured over relative pressures range 0.01–0.995 using an Autosorb IQ sorptiometer from Quantachrome. Samples were degassed under vacuum at 100 °C for 8 h (heating rate = 5 °C min−1) under vacuum was performed to remove most of adsorbed impurities and humidity. The specific surface area was calculated by the Brunauer–Emmett–Teller (BET) method, and the pore size distribution, pore volumes, and the pore diameters were determined by BJH (Barrett–Joyner–Halenda) model applied to the desorption branch of the isotherm. The pore size distribution was calculated by the density functional theory (DFT) for samples based on the N2 isotherm adsorption data. Thermogravimetric Analysis (TGA) measurements were performed on a CDR-4P TGA under nitrogen atmosphere (flow rate = 60 mL min−1), a thermal treatment was started up to 600 °C at a heating rate of 10 °C min−1. Raman scattering spectra were collected between 100 and 500 cm−1 on a Jobin-Yvon T64000 by exciting samples with 785 nm laser and 0.05 mW power. The initial and residual iodine concentrations in solution were determined via UV-vis spectra by a Hitachi U-2900 Ultraviolet-visible absorption spectrometer (Tokyo, Japan) with a 1 cm quartz cuvette, and measured with UV-vis absorption spectrometer by monitoring the absorbance change at a wavelength of 287 nm.2.4. Iodine adsorption
In order to measure the iodine uptake capacity of the pSi–C composites in aqueous solution, a pSi–C sample (20 mg) was soaked in a KI3 aqueous solution (600 mg KI and 300 mg I2 in 3 mL of H2O). After adsorption, the sample was collected by filtration, washed with water until the filtrate become clear, and dried in open air to yield I2@pSi–C. The filtrate was collected and combined for bisulfite starch titration, according to Lin et al.27 The amount of iodine adsorbed for pSi–C composite samples were calculated by the following equation:
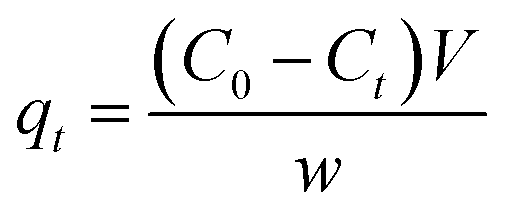 | (1) |
In order to monitor their iodine capture speed of the pSi–C composites in KI3 aqueous solution, a time-dependent UV-vis measurement was carried out on a UV-vis spectrophotometer. A pSi–C sample (3.0 mg) was added to a saturated KI3 aqueous solution (0.05 mol L−1) with stirring. The UV-vis spectrum of the solution was recorded every 2 min in the first 10 min.
3. Results and discussion
3.1. The component of paper mill sludge (PMS)
The component analysis of PMS was shown in Table 1. After dehydration and drying, the moisture content of PMS was 3.72%. The ash content of PMS was 52%, which were mainly due to the large amount of inorganic fillers and the sediment composition after wastewater treatment in the papermaking process. The content of organic components in the sludge was 41.78%. It can be known from the component analysis that the organic components are mainly composed of cellulose and lignin and contain a small amount of hemicellulose.| Moisture (%) | Ash (%) | Cellulose (%) | Hemicellulose (%) | Lignin (%) |
|---|---|---|---|---|
| 3.72 | 54.50 | 28.25 | 3.21 | 10.32 |
3.2. Effect of various activators on structure of pSi–C composite and its iodine capture
According to the related literature, several common activators,28–38 namely KOH, MgCl2, NaOH, and ZnCl2, are selected to carbonize PMS at a temperature of 550 °C and quality ratio (PMS/activator) of 1:1 for 120 min. The iodine adsorption capacity (qt) of the prepared composites is shown in Fig. 1a. The iodine capture capacity of the blank sample, the composite prepared without using any activator, was only 150 mg g−1, and the iodine capture capacity of the samples increases when the activators are added. Moreover, the pSi–C composite activated by ZnCl2 offers the best adsorption performance with an iodine capture capacity of 248.07 mg g−1.
 | ||
| Fig. 1 Effect of various activators on the structure of the pSi–C composite and its iodine capture: (a) iodine capture capacity; (b) FTIR spectra; (c) N2 adsorption–desorption isotherms; (d) PXRD patterns. | ||
The structures of the prepared composites are thoroughly analyzed to study their various adsorption characteristics. To this end, firstly, Fourier-transform infrared spectroscopy (FTIR) is utilized to identify the functional groups on the surface of the pSi–C composites synthesized using different activators, as delineated in Fig. 1b.
The adsorption peak at 3380 cm−1 is assigned to the H–O–H stretching vibration of the adsorbed water, and the strong peaks at 1185 and 789 cm−1 correspond to the asymmetric and symmetric stretching modes of SiO2 respectively. The aromatic skeletal vibrations observed at 1607 and 1430 cm−1 confirm the aromatic structure of the composites. It can be concluded from the FTIR analysis that the addition of the activator causes a certain change in the benzene ring of the pSi–C composites in such a way that the lignin may be degraded during the activation process.
Secondly, the nitrogen adsorption–desorption isotherms and the pore size distribution of the composites prepared by various activators are depicted in Fig. 1c, and all pore properties are shown in Table 2. The N2 adsorption–desorption isotherms of the as-prepared samples present characteristics of type IV (according to IUPAC classification) with hysteresis loops, indicating the presence of mesopores in the pSi–C composites. It can be seen in Fig. 1c that the hysteresis loops of the as-prepared samples are shaped, and the hysteresis occurring between the adsorption and desorption curves in a relative pressure range of 0.45–1.0 indicates the presence of mesopores as well as macropores in the composites. The N2 adsorption–desorption isotherms further imply that the pSi–C composite synthesized by ZnCl2 has a more porous structure, As can be seen from Table 2, as also proved by its high Brunauer–Emmett–Teller (BET) surface area of 518.60 m2 g−1, while the microporous structure is the most. Therefore, a higher surface area and a perfect porous structure are two important factors which can enhance the adsorption of iodine molecules onto the active sites of the composites. Thirdly, as can be seen in Fig. 1d, the powder X-ray diffraction (PXRD) patterns of the as-synthesized composites are almost similar. Moreover, the peak at 2θ angle of around 20° is attributed to the characteristic (002) plane of carbon, while the strong peak at 2θ angle of around 27° is ascribed to the quartz crystal, which is consistent with the SiO2 characterized in the FTIR spectra.
| Samples | SBETa (m2 g−1) | Vtotalb (cm3 g−1) | Vmicc (cm3 g−1) | Pore sized (nm) |
|---|---|---|---|---|
| a Specific surface area calculated by the BET method.b Total pore volume determined by N2 adsorption at a relative pressure of 0.99.c Micropore volume determined by DFT.d Mean pore diameter obtained from the desorption isotherm by the BJH method. | ||||
| PMS | 117.87 | 0.1376 | 0.02721 | 4.6698 |
| PMS-KOH | 180.39 | 0.2654 | 0.03657 | 5.8849 |
| PMS-NaOH | 222.13 | 0.4691 | 0.01562 | 8.4452 |
| PMS-MgCl2 | 239.59 | 0.5155 | 0.00785 | 8.6073 |
| PMS-ZnCl2 | 518.60 | 0.4665 | 0.10926 | 5.7021 |
3.3. Influence of synthesis conditions on iodine capture of composites
In addition to the activator, the main factors affecting the adsorption performance of the pSi–C composite include the quality ratio of the paper mill sludge powder to the activator, the carbonization temperature, and the carbonization time. The ratio is crucial to the development of the porous structure of the adsorbent materials. The mixtures of the PMS powder and the chemical activators at ratios of 1:0.5, 1:1, 1:1.5, 1:2, and 1:2.5 are heated at a temperature of 550 °C for 120 min in a tube furnace under a blanket of nitrogen gas. Fig. 2a shows the variation in the iodine capture capacity of the pSi–C composites with the different ratios of the PMS powder to the activators. When the ratio is less than 1:1, little ZnCl2 is participated in the formation of the carbon skeleton and poor development of the composites, so their BET surface area is very low, which leads to their relatively low iodine capture capacity. As long as the ratio is higher than 1:1, the maximum BET surface area of the samples is 519 m2 g−1, and they reach a maximum iodine capture capacity of 248.07 mg g−1. However, by increasing the amount of ZnCl2, the excess activator reacts with carbon owing to the low silica content of the solid residue, which may result in some larger pores because of the collapse of some micropores; hence, the BET surface area and the iodine adsorption capacity of the composites drop with the continual increment in the ratio. ZnCl2 is selected as the activator and mixed evenly with the PMS powder at ratio of 1:1, and the resultant mixture is carbonized at a carbonization temperature in the range of 400 to 600 °C for 120 min. According to the variation in the iodine adsorption capacity of the prepared composite with the carbonization temperature plotted in Fig. 2b, the iodine adsorption capacity increases from 183.20 to 248.07 mg g−1 below 550 °C, but it declines to 214.53 mg g−1 by further increasing the carbonization temperature to 600 °C. When the carbonization temperature is lower than 550 °C, a viscid tarry matter moves to the surface of the carbon matrix and forms oil particles, thereby blocking the pores of the composite and lowering its BET surface area and thus iodine adsorption capacity.
 | ||
| Fig. 2 Variation in the iodine adsorption capacity of the pSi–C composite with various factors: (a) the ratio of the PMS powder to ZnCl2, (b) carbonization temperature, (c) carbonization time. | ||
As the carbonization temperature gradually rises, the tar slowly volatilizes completely, which forms more pores and results in a larger specific surface area. However, when the carbonization temperature increases to 600 °C, the iodine adsorption capacity of the composite significantly falls because the micropores and mesopores will collapse and form larger pore structures.
At the same time, at high temperatures, the organic matter in the activated carbon may continue to decompose, and the functional groups controlling the adsorption process will change, thereby affecting the chemical adsorption of iodine.
In another series of experiments, the mixture of the PMS powder and ZnCl2 at ratio of 1:1 is carbonized at 550 °C for a period of 30 to 120 min. As can be seen in Fig. 2c, the iodine adsorption capacity of the pSi–C composite soars from 241.74 mg g−1 to a maximum of 248.85 mg g−1 as the carbonization time increases to 90 min but drops to 242.01 mg g−1 at longer times.
Thus, the activated carbon resulting from the precursor carbonized at a temperature of 550 °C for 90 min has a BET surface area of 762.13 m2 g−1, which leads to the composite with the highest iodine adsorption capacity is 248.85 mg g−1. In fact, in a shorter activation time frame, carbide materials such as the tarry matter cannot volatilize, which gives rise to a smaller BET surface area and thus lower iodine adsorption capacity.
On the other hand, a prolonged carbonization time may cause the over-carbonization of the carbon precursor, which results in the collapse of the pores and an increased proportion of the macropores, and consequently reduces the BET surface area and the iodine adsorption capacity of the corresponding composite.
As discussed above, we can conclude that a number of factors have a significant impact on the iodine adsorption capacity of the pSi–C composite. And the optimal experimental conditions can be achieved at an impregnation ratio of the PMS powder to ZnCl2 equal to 1:1, a carbonization temperature of 550 °C, a carbonization time of 90 min.
3.4. Adsorption kinetics
Fig. 3a presenting the influence of the contact time on the UV spectra of a saturated iodine aqueous solution in the presence of the pSi–C composite reveals that the iodine adsorption is a particularly rapid process. Indeed, after two minutes of adding the pSi–C composite to the iodine solution, the absorbance at 287 nm related to iodine is reduced from 1.22 to 0.25. | ||
| Fig. 3 Kinetics of the adsorption of iodine onto the pSi–C composite: (a) time-dependent UV spectra of a saturated iodine aqueous solution at various contact times; (b) time-dependent kinetics; (c) fitted pseudo first-order kinetic model; (d) fitted pseudo second-order kinetic model. | ||
Furthermore, the time-dependent iodine adsorption capacity presented in Fig. 3b confirms that the iodine adsorption of the pSi–C composite increases as the contact time extends. The adsorption of iodine onto the pSi–C composite reaches an equilibrium after 60 min, and the iodine adsorption capacity at equilibrium is equal to 252 ± 3 mg g−1. The variation in the iodine adsorption capacity with time (see Fig. 3b) implies that the pSi–C composite is highly effective in adsorbing iodine and can be utilized as a natural adsorbent for it. In addition, a pseudo first-order kinetic model (Fig. 3c) and a pseudo second-order kinetic model (Fig. 3d) expressed by eqn (2) and (3) respectively are fitted to the obtained experimental data:39
Pseudo first-order kinetics:
|
ln(qe − qt) = lnqe − k1t
| (2) |
Pseudo second-order kinetics:
 | (3) |
| Types of kinetics | qe (mg g−1) | k | R2 |
|---|---|---|---|
| First-order | 24.9706 | 0.04095 | 0.9688 |
| Second-order | 255.7545 | 0.005662 | 0.9999 |
The rate constants, the calculated qe values, and the linear regression coefficients (R2) are also presented in Table 2. Therefore, it can be concluded that the chemisorption plays a crucial role in the iodine adsorption process.
3.5. Adsorption isotherms
The isotherms of the adsorption of iodine onto the pSi–C composite are obtained at a temperature of 298, 308, and 318 K. In the same way, 20 mg of the adsorbent is added into 4 mL of iodine solution, and the concentration of the iodine solution and the concentration of the iodine solution is changed from 500 to 2100 mg L−1. According to Fig. 4a, at an iodine concentration of 2000 mg L−1, the iodine adsorption capacity of the pSi–C composite increases from 285.03 to 314.14 mg g−1 when the temperature rises from 298 to 318 K, indicating that the adsorption of iodine onto the pSi–C composite is an endothermic process. | ||
| Fig. 4 Isotherms of the adsorption of iodine onto the pSi–C composite: (a) experimental isotherms; (b) fitted Freundlich model; (c) fitted Langmuir model. | ||
Moreover, Langmuir and Freundlich adsorption isotherm models are used to analyze the isotherms of the adsorption of iodine onto the pSi–C composite, as shown in Fig. 4c and b, respectively.
The classical Langmuir40 and Freundlich models41 can be expressed by:
Langmuir model:
 | (4) |
Freundlich model:
 | (5) |
| Temperature (K) | Langmuir | Freundlich | ||||
|---|---|---|---|---|---|---|
| qm (mg g−1) | KL (L mg−1) | R2 | KF (mg g−1) | 1/n | R2 | |
| 298 | 300.30 | 0.02461 | 0.9917 | 55.0306 | 0.3095 | 0.8385 |
| 308 | 312.50 | 0.03473 | 0.9965 | 64.7388 | 0.2545 | 0.9366 |
| 318 | 328.95 | 0.04143 | 0.9983 | 64.6902 | 0.2771 | 0.9004 |
According to the results obtained, the R2 of Langmuir and Freundlich models range from 0.9917 to 0.9998 and from 0.87385 to 0.9778 respectively. Therefore, the adsorption of iodine onto the pSi–C composite can be better described by the Langmuir adsorption isotherm model. The maximum adsorption of iodine onto the pSi–C composite calculated by Langmuir model is 300.03, 312.50, and 328.95 mg g−1 at a temperature of 298, 308 K and 318 K respectively.
The adsorbent capacities of different adsorbents reported previously are compared with present work and given in Table 5. The maximum iodine adsorption capacity of pSi–C composite is higher than the previous works, such as Mg–Al (NO3) layered double hydroxide, and covalent organic polymers of JLUE-COP-3 and so on.
3.6. Morphology and structure of pSi–C composite and I2-adsorbed pSi–C composite
It can be inferred from Fig. 5a that the surface of the pSi–C composite is rough, which significantly increases its specific surface area, thereby leading to the higher adsorption of iodine onto it. After iodine adsorption, as shown in Fig. 5b, some pores of the activated carbon of the pSi–C composite are blocked, indicating that iodine is adsorbed onto the activated carbon.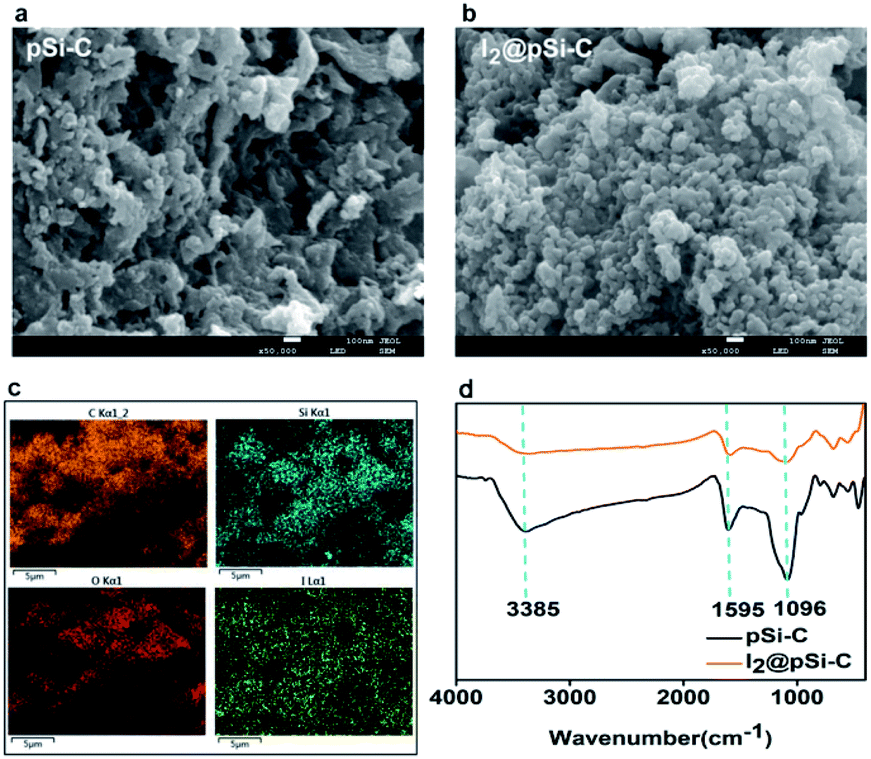 | ||
| Fig. 5 The SEM images of (a) the pSi–C composite and (b) the I2-adsorbed pSi–C composite; (c) EDS images of the I2@pSi–C composite; (d) FTIR spectra of the pSi–C composite and the I2@pSi–C composite. | ||
The energy dispersive spectroscopy (EDS) mapping is performed to reveal the distribution of each element, as presented by color treatment in Fig. 5c. The result shows the uniform distribution of iodine on the surface of the pSi–C composite. FTIR spectra are utilized to identify the functional groups on the surfaces of the pSi–C composite and the I2@pSi–C composite as delineated in Fig. 5d. There is a strong peak at around 3385 cm−1, which is ascribed to the respective vibration of the interlayer –OH groups and water. The peaks at 1607 and 1595 cm−1 are mainly the stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and CC bond in the benzene rings or aromatics available.
O bond and CC bond in the benzene rings or aromatics available.
The shoulder peaks appearing at 1096 cm−1 in the spectra of the pSi–C composite are also related to the stretching vibrations of Si–O–Si or Si–O–C. Moreover, the peaks at 789 and 470 cm−1 correspond to the symmetric stretching and symmetric bending modes of SiO2 respectively.
The intensity of the peak at 1595 cm−1 is deviation, so benzene rings or aromatics available may have a chemical reaction in the iodine adsorption process. SiO2 should only act as a skeleton, enhances the stability, and in the iodine adsorption process is only the physical adsorption.
As shown in Fig. 6a, the structure of the iodine inside the pores was revealed by Raman spectroscopy. The peak at 140 cm−1 is belong to the asymmetric stretching vibrations of I3−, the peak at 164 cm−1 is assigned to the I5− stretching vibration, which is form of I−[(I2)2],48,49 and the small peak at 176 cm−1 is characteristic of I2.50 The result is a further proof of most iodine had been converted polyiodide (I3−, I5−). As determined by the thermogravimetric analysis (TGA) (Fig. 6b). Since iodine is easy to sublimate, the calculated total weight loss rate is approximately the iodine adsorption amount. pSi–C composite shown weight loss of 10.02% from 25 to 600 °C, which was attributed to the decomposition of its original skeleton. The pSi–C composite after iodine capture, the I2@pSi–C composite, has a significant weight loss of 30.03% between 25 and 600 °C, so the iodine adsorption value is about 20%. This is different from the experimental data 255 mg g−1 because of the error caused by iodine sublimation when the I2@pSi–C composite was naturally dried after iodine adsorption.
 | ||
| Fig. 6 (a) Raman spectra of pSi–C composite and I2@pSi–C composite exhibiting the presence of iodine (b) TGA profiles of the pSi–C composite and the I2@pSi–C composite recorded in a nitrogen atmosphere. | ||
3.7. Adsorption mechanism
To investigate the mechanism of iodine adsorption, the composition of the paper mill sludge is analyzed. In the iodine adsorption process, the pSi–C composite plays a vital role not only in physical adsorption but also in chemical adsorption, which is consistent with the simulation of the adsorption dynamics. Lignin may play a key role in the process of iodine adsorption onto the pSi–C composite; thus, to prove this hypothesis, the following experiments are conducted. Lignin is extracted from the PMS three times using 2 mol L−1 NaOH to ensure that it is completely removed, then the obtained PMS is carbonized to produce a lignin-free pSi–C composite. As a normal sample, 20 wt% lignin is added to the lignin-free PMS sample, and the resultant product is carbonized to create a lignin-added pSi–C composite.The iodine adsorption capacity of the different samples is plotted in Fig. 7a. It is clear that the iodine adsorption capacity of the lignin-free pSi–C composite is only 80.72 mg g−1, which is reduced by 168.13 mg g−1 compared to the normal pSi–C composite with an iodine adsorption capacity of 248.85 mg g−1. However, the iodine adsorption capacity of the lignin-added pSi–C composite (273.22 mg g−1) is increased by 192.5 mg g−1 compared to that of the lignin-free pSi–C composite. According to the FTIR (Fig. 7b), the peak at 1600 cm−1 value of aromatic rings decreased significantly after the removal of lignin, and increased significantly after the addition of 20% lignin, and the types of other functional groups also increased. Therefore, from this comparison, we can deduce that the lignin component plays a significant role in the adsorption of iodine onto the pSi–C composite. This may be due to the benzene ring in the structure of lignin, which is an electron-rich group with a large π bond and is more prone to coordination reaction with iodine.51,52 It is also concluded that lignin plays a major role in paper mill sludge. Finally, the results are in agreement with the Raman spectroscopy analysis and FTIR analysis.
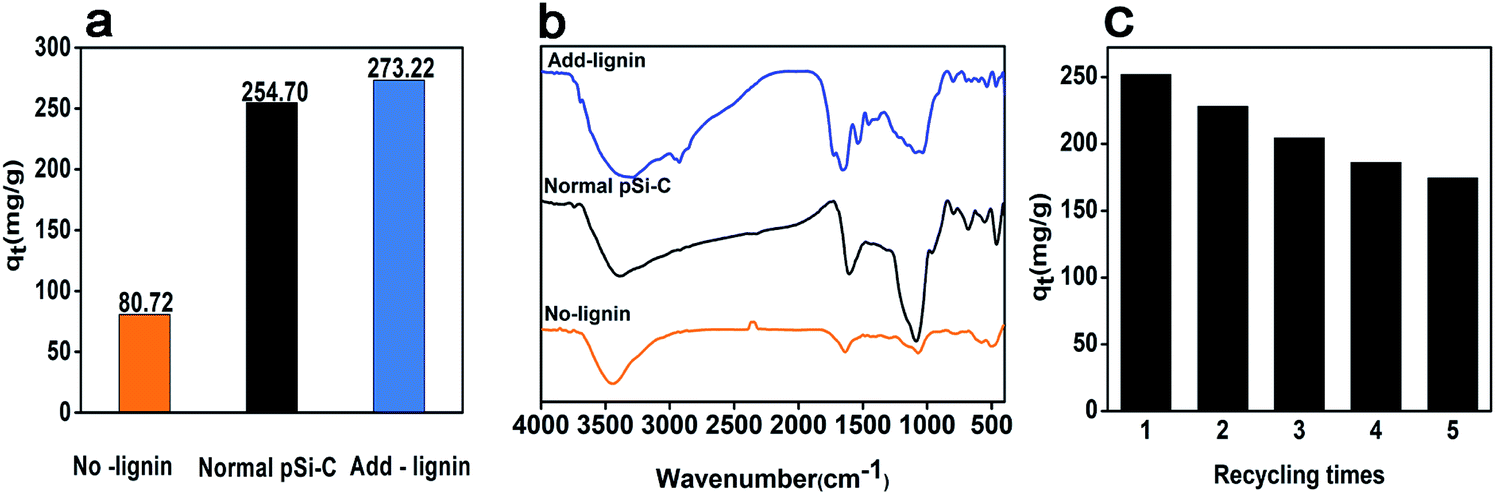 | ||
| Fig. 7 (a) Effect of lignin on the iodine adsorption of the pSi–C composite; (a) iodine adsorption value (b) FTIR spectra (c) repeated iodine adsorption experiments for pSi–C composite. | ||
The recyclability is also an important indicator for evaluating the performance of adsorbents. Due to the high solubility of iodine in ethanol solution, the I2@ pSi–C composite was cleaned by ethanol several times until the iodine was completely desorbed. After drying, adsorption was carried out. Fig. 7c showed that the iodine adsorption value was 174.45 mg g−1 after five recycles, which was maintained 69.6% of the initial capacity. It is demonstrated the pSi–C composite has good repeatable during treatment of iodine pollution.
4. Conclusions
A low-cost porous silicon-activated carbon composite is produced from paper mill sludge by chemical activation and utilized for rapid iodine capture. The pSi–C composite offers a high surface area up to 762.13 m2 g−1 and stable thermal stability. Further, according to the adsorption experiments and the fitted Langmuir adsorption isotherm model, the pSi–C composite can reach a maximum iodine adsorption capacity of 300.03 mg g−1. Also, the analysis of the structure of the pSi–C composite proves that the adsorption of iodine is mainly due to the coordination reaction between lignin and iodine. Finally, the synthesized pSi–C composite may serve as a cost-effective candidate for capturing and storing radioactive iodine.Conflicts of interest
The authors declare that they have no competing financial interests.Acknowledgements
This research was funded by Liaoning Education Department Project (No. J2020038), Liaoning University Innovation Talent Program in 2020 (No. 20200112) and the special financial grant from the China Postdoctoral Science Foundation (2020T130464).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS.
- G. Li, C. Yan, B. Cao, J. Jiang, W. Zhao, J. Wang and T. Mu, Green Chem., 2016, 18, 2522–2527 RSC.
- D. K. L. Harijan, V. Chandra, T. Yoon and K. S. Kim, J. Hazard. Mater., 2018, 344, 576–584 CrossRef CAS.
- X. L. Hou, C. L. Fogh, J. Kucera, K. G. Andersson, H. Dahlgaard and S. P. Nielsen, Sci. Total Environ., 2003, 308, 97–109 CrossRef CAS.
- H. Sun, A. Li, P. La, Z. Zhu, W. Liang and B. Yang, J. Mater. Sci., 2015, 50, 7326–7332 CrossRef CAS.
- S. Chen, Y. Qi, J. J. Cossa and S. I. Deocleciano Salomao Dos, Prog. Nucl. Energy, 2019, 117, 1–6 CrossRef.
- U. Von Gunten and Y. Bichsel, Water Res., 2000, 34, 3197–3203 CrossRef.
- T. Geng, W. Zhang, Z. Zhu and X. Kai, Microporous Mesoporous Mater., 2019, 273, 163–170 CrossRef CAS.
- D. Shetty, J. Raya, D. S. Han, Z. Asfari, J.-C. Olsen and A. Trabolsi, Chem. Mater., 2017, 29, 8968–8972 CrossRef CAS.
- Q. Cheng, W. Yang, Z. Li, Q. Zhu, T. Chu, D. He and C. Fang, J. Radioanal. Nucl. Chem., 2014, 303, 1883–1889 CrossRef.
- D. Li, D. I. Kaplan, K. A. Price, J. C. Seaman, K. Roberts, C. Xu, P. Lin, W. Xing, K. Schwehr and P. H. Santschi, J. Environ. Radioact., 2019, 208–209, 106017 CrossRef CAS.
- K. Xiao, H. Liu, Y. Li, G. Yang, Y. Wang and H. Yao, Chem. Eng. J., 2020, 382 Search PubMed.
- Q. M. Zhang, T. L. Zhai, Z. Wang, G. Cheng, H. Ma, Q. P. Zhang, Y. H. Zhao, B. Tan and C. Zhang, Adv. Mater. Interfaces, 2019, 6, 1–6 Search PubMed.
- D. F. Sava, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, P. S. Crozier, H. Zhao, P. J. Chupas and T. M. Nenoff, Chem. Mater., 2013, 25, 2591–2596 CrossRef CAS.
- D. Banerjee, X. Chen, S. S. Lobanov, A. M. Plonka, X. Chan, J. A. Daly, T. Kim, P. K. Thallapally and J. B. Parise, ACS Appl. Mater. Interfaces, 2018, 10, 10622–10626 CrossRef CAS.
- X. Qian, Z. Zhu, F. Ren, P. Mu, W. Liang, L. Chen and A. Li, ACS Appl. Mater. Interfaces, 2016, 8, 21063–21069 CrossRef CAS.
- S. Xiong, X. Tang, C. Pan, L. Li, J. Tang and G. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 27335–27342 CrossRef CAS.
- C. Li, Y. Wei, X. Wang and X. Yin, J. Taiwan Inst. Chem. Eng., 2018, 85, 193–200 CrossRef CAS.
- H. Li and Y. Li, Appl. Surf. Sci., 2019, 496, 1–9 Search PubMed.
- Z. Ye, L. Chen, C. Liu, S. Ning, X. Wang and Y. Wei, React. Funct. Polym., 2019, 135, 52–57 CrossRef CAS.
- H. Sun, B. Yang and A. Li, Chem. Eng. J., 2019, 372, 65–73 CrossRef CAS.
- F. Gao, C. Geng, N. Xiao, J. Qu and J. Qiu, Carbon, 2018, 139, 1085–1092 CrossRef CAS.
- A. Niksiar and B. Nasernejad, Biomass Bioenergy, 2017, 106, 43–50 CrossRef CAS.
- M. Schreiber, S. Vivekanandhan, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2014, 3, 33–41 CrossRef.
- S. Han, W. Um and W.-S. Kim, Dalton Trans., 2019, 48, 478–485 RSC.
- J. Kang, T. G. Levitskaia, S. Park, J. Kim, T. Varga and W. Um, Chem. Eng. J., 2020, 380 Search PubMed.
- Y. Lin, X. Jiang, S. T. Kim, S. B. Alahakoon, X. Hou, Z. Zhang, C. M. Thompson, R. A. Smaldone and C. Ke, J. Am. Chem. Soc., 2017, 139, 7172–7175 CrossRef CAS.
- Y. Liu, Z. Huo, Z. Song, C. Zhang, D. Ren, H. Zhong and F. Jin, J. Taiwan Inst. Chem. Eng., 2019, 96, 575–587 CrossRef CAS.
- W.-Q. Zhang, X. Sui, B. Yu, Y.-Q. Shen and H.-L. Cong, Integr. Ferroelectr., 2019, 199, 22–29 CrossRef CAS.
- Q. Liang, Y. Liu, M. Chen, L. Ma, B. Yang, L. Li and Q. Liu, Mater. Chem. Phys., 2020, 241, 122327 CrossRef CAS.
- Z. Xu, Z. Yuan, D. Zhang, W. Chen, Y. Huang, T. Zhang, D. Tian, H. Deng, Y. Zhou and Z. Sun, J. Cleaner Prod., 2018, 192, 453–461 CrossRef CAS.
- G. Zhong, S. Xu, J. Chao, X. Fu, W. Liao, Y. Xu, Z. Liu and Y. Cao, Ind. Eng. Chem. Res., 2020, 59, 21756–21767 CrossRef CAS.
- Y. Zhang, X. Song, Y. Xu, H. Shen, X. Kong and H. Xu, J. Cleaner Prod., 2019, 210, 366–375 CrossRef CAS.
- M. A. Islam, M. J. Ahmed, W. A. Khanday, M. Asif and B. H. Hameed, Ecotoxicol. Environ. Saf., 2017, 138, 279–285 CrossRef CAS.
- A. Arnelli, U. Putri, F. Cholis and Y. Astuti, J. Kim. Sains Apl., 2019, 22, 283–291 CrossRef.
- Ş. Ö. Teğin, Ö. Şahin, O. Baytar and M. S. Izgi, Int. J. Chem. Technol., 2020, 4, 130–137 Search PubMed.
- D. Hong, J. Zhou, C. Hu, Q. Zhou, J. Mao and Q. Qin, Fuel, 2019, 235, 326–335 CrossRef CAS.
- R. Ma, X. Qin, Z. Liu and Y. Fu, Materials, 2019, 12, 1377–1392 CrossRef CAS.
- Y. S. Ho, Water Res., 2006, 40, 119–125 CrossRef CAS.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- B. Liu, X. Ren, L. Chen, X. Ma, Q. Chen, Q. Sun, L. Zhang, P. Si and L. Ci, J. Hazard. Mater., 2019, 373, 705–715 CrossRef CAS.
- T. Zhang, X. Yue, L. Gao, F. Qiu, J. Xu, J. Rong and J. Pan, J. Cleaner Prod., 2017, 144, 220–227 CrossRef CAS.
- L. Kentjono, J. C. Liu, W. C. Chang and C. Irawan, Desalination, 2010, 262, 280–283 CrossRef CAS.
- H. Guan, D. Zou, H. Yu, M. Liu, Z. Liu, W. Sun, F. Xu and Y. Li, Front. Mater., 2019, 6, 1–12 CrossRef.
- T. Madrakian, A. Afkhami, M. A. Zolfigol, M. Ahmadi and N. Koukabi, Nano-Micro Lett., 2012, 4, 57–63 CrossRef CAS.
- B. Zheng, X. Liu, J. Hu, F. Wang, X. Hu, Y. Zhu, X. Lv, J. Du and D. Xiao, J. Hazard. Mater., 2019, 368, 81–89 CrossRef CAS.
- Q. Li, Q. Mao, C. Yang, S. Zhang, G. He, X. Zhang and W. Zhang, Int. J. Biol. Macromol., 2019, 141, 987–996 CrossRef CAS.
- X. Li, G. Chen, J. Ma and Q. Jia, Sep. Purif. Technol., 2019, 210, 995–1000 CrossRef CAS.
- E. Tanaka and N. Robertson, J. Mater. Chem. A, 2020, 8, 19991–19999 RSC.
- A. Hijazi, B. Azambre, G. Finqueneisel, F. Vibert and J. L. Blin, Microporous Mesoporous Mater., 2019, 288 Search PubMed.
- Z. Yan, Y. Yuan, Y. Tian, D. Zhang and G. Zhu, Angew. Chem., Int. Ed. Engl., 2015, 54, 12733–12737 CrossRef CAS.
- K. Su, W. Wang, B. Li and D. Yuan, ACS Sustainable Chem. Eng., 2018, 6, 17402–17409 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |