DOI:
10.1039/D0RA09712H
(Paper)
RSC Adv., 2021,
11, 8107-8116
An ingenious method for the determination of the relative and absolute configurations of compounds containing aryl-glycerol fragments by 1H NMR spectroscopy†
Received
16th November 2020
, Accepted 11th February 2021
First published on 19th February 2021
Abstract
A concise method was established to determine the relative and absolute configurations of aryl-glycerols that depend on the chemical shift differences (Δδ) of the diastereotopic methylene protons (H-3) by 1H NMR spectroscopy. When using DMSO-d6 as the preferred solvent, the threo configuration corresponded to a larger ΔδH3a–H3b value (>0.15 ppm), whereas the erythro configuration (<0.07 ppm) corresponded to a smaller value. Furthermore, the absolute configurations were determined with the aid of a simple acylation reaction through camphanoyl chloride. In the threo enantiomers, the Δδ value of the 1R,2R configuration was <0.15 ppm, and that of the 1S,2S configuration was >0.20 ppm. In the erythro enantiomers, the Δδ value of 1R,2S was >0.09 ppm, and that of 1S,2R was <0.05 ppm. Remarkably, this empirical rule is invalid in CDCl3. In addition, this method was also verified by a quantum 1H NMR calculation.
Introduction
Chiral alcohols, such as chiral secondary alcohols, prim,sec-diols, sec,sec-diols and 1,2,3-prim,sec,sec-triols, are ubiquitous in natural products and synthetic products. The assignment of their relative and absolute configurations is always a challenging task. Because of the difficulty of growing crystals of these compounds and the lack of distinct Cotton effects in their electronic circular dichroism (ECD) spectra, NMR spectroscopy is suitable for this purpose and is readily available compared with other methods. For instance, the absolute configurations of chiral secondary alcohols were determined using Mosher's method by 1H NMR spectroscopy;1 the relative configurations of some sec,sec-diols were assigned using a JBCA (J-based configuration analysis) method2 or by comparison of their 13C-Δδ behaviors in a chiral bidentate NMR solvent.3 We have also developed two approaches to (1) discriminate the threo and erythro configurations of vicinal diol in polyacetylene glycosides by the 3JHH value using acetic acid-d4/D2O as the solvent;4 and (2) determine the absolute configurations of 2-hydroxy phenylethanoid glycosides (prim,sec-diol derivatives) by the chemical shift differences (Δδ) of the diastereotopic methylene protons.5
Aryl-glycerols (AGs), a kind of 1,2,3-prim,sec,sec-triols, are frequent in natural products such as phenylpropanoids and lignans6–15 and are important intermediates in many chemical syntheses.16–21 Due to the conformational flexibility of the triol chain in AGs, determining the relative and absolute configurations is significantly challenging. Recently, Mosher's method was developed to assign the absolute configuration of AGs by comparing the 1H NMR data of the tris-(R)- and the tris-(S)-MPA ester derivatives with those of the AGs.22,23 However, the inability to prepare tris-MPA ester derivatives for trace compounds and the complex rules governing the comparison of the ΔδRS(H-1) and |ΔδRSH(2) − ΔδRSH(1)| of AGs, to some extent, have limited the application of this method. For some naturally occurring AGs, the regular ΔδC1–C2 parameters have been used to differentiate only the threo and erythro relative configurations.9,15 However, we found that this rule is not applicable to AGs without substituent groups on the benzene ring. Therefore, a new reliable method to determine the relative and absolute configurations of AGs is a worthwhile task.
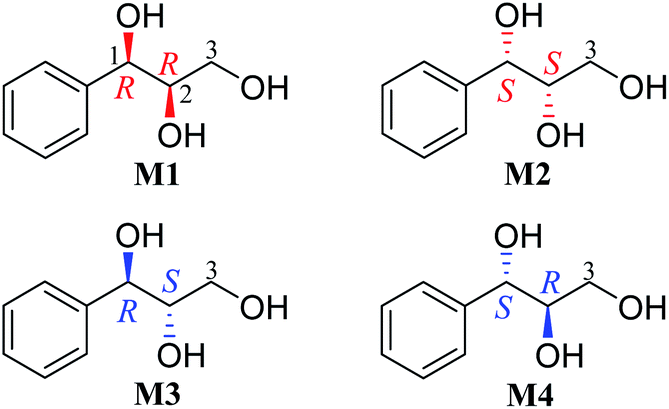
Results and discussion
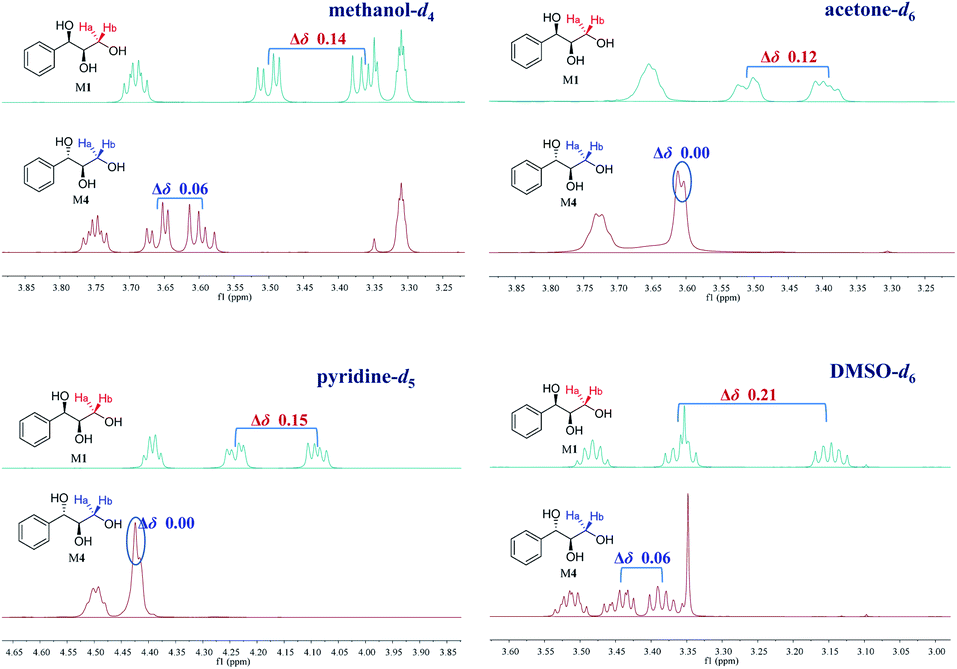
Our recent study found that the threo and erythro configurations of H-7 and H-8 in 8,4′-oxyneolignans can be discriminated by the values of ΔδH9a–H9b of methylene protons in 1H NMR spectroscopy.24 When we determined the configurations of a pair of threo syringylglycerol enantiomers (1 and 2) obtained from the Arnebia euchroma, a careful comparison of the 1H NMR spectroscopic data of 1 in methanol-d4 with the corresponding data in erythro syringylglycerol in the literature showed that there were significant differences.9 The value of ΔδH3a–H3b of methylene protons in 1 is 0.13 ppm, while the value is only 0.07 ppm in erythro syringylglycerol. Surprisingly, this result is similar to the rule in 8,4′-oxyneolignans. For AGs without large steric hindrance groups, can the ΔδH3a–H3b values of the methylene groups reflect their relative stereochemistry? To explore and summarize this phenomenon, two pairs of 1-phenylpropane-1,2,3-triol enantiomers were prepared (M1–M4) through the hydrolysis of (2R,3R)-3-phenyl-2-oxiranemethanol and (2S,3S)-3-phenyl-2-oxiranemethanol (Fig. 1). The 1H NMR spectra of compounds M1 and M4 in methanol-d4 were recorded. Obviously, the ΔδH3a–H3b (0.14 ppm) value of methylene protons in the threo configuration was remarkably larger than the corresponding value (0.06 ppm) in the erythro configuration (Fig. 3). Considering that deuterated solvents have a major impact on the chemical shifts of methylene protons, the 1H NMR spectra of compounds M1 and M4 were recorded in other deuterated solvents, acetone-d6, pyridine-d5, DMSO-d6, and CDCl3, as shown in Fig. 3. Similarly, in the threo configurations, the ΔδH3a–H3b values of methylene protons were 0.12 in acetone-d6, 0.15 in pyridine-d5, and 0.21 in DMSO-d6. In the erythro configurations, the ΔδH3a–H3b values of methylene protons were 0.00 in acetone-d6, 0.00 in pyridine-d5, and 0.06 in DMSO-d6. Remarkably, there was no obvious difference in CDCl3 (threo: 0.09 ppm; erythro: 0.07 ppm). In addition, the chemical shift values of methylene protons in the erythro configuration were obviously larger than those in the threo configuration.
 |
| | Fig. 1 Structures of compounds M1–M4. | |
 |
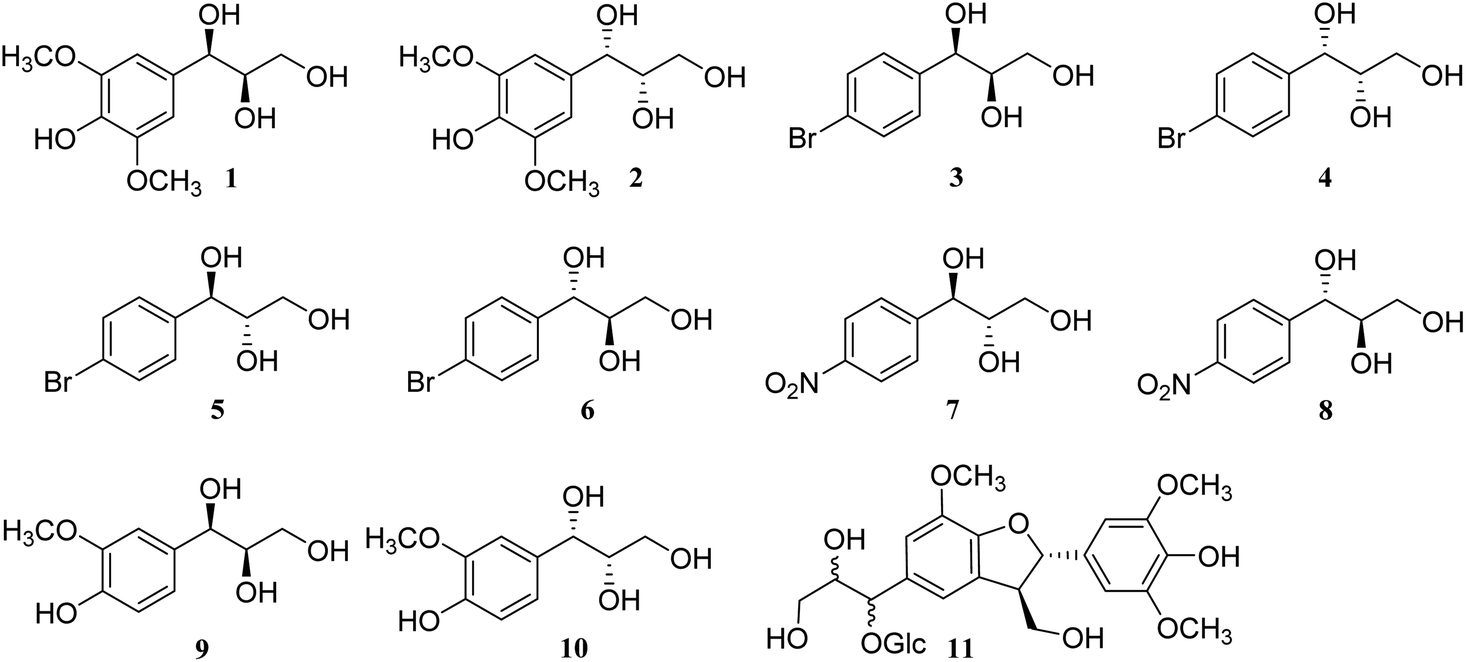
| | Fig. 2 Structures of compounds 1–11. | |
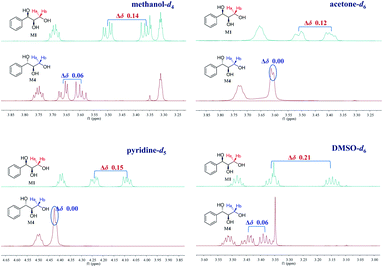 |
| | Fig. 3 Influence of different deuterated solvents on the ΔδH3a–H3b values of M1 and M4 (1H NMR 500 MHz). | |
To further confirm the accuracy and reliability of the method and investigate the influence of different substitution groups on the benzene ring, 1-(4-bromophenyl)propane-1,2,3-triol (3–6) and 1-(4-nitrophenyl)propane-1,2,3-triol (7 and 8) were synthesized through the Sharpless asymmetric epoxidation and hydrolysis reaction (Fig. 2).25 As expected, larger ΔδH3a–H3b values of methylene protons appeared in the threo configurations (3–4), and smaller ΔδH3a–H3b values of methylene protons appeared in the erythro configurations (5–8). Additionally, three natural products, threo-1-C-guaiacylglycerol (9 and 10), obtained from bamboo juice, and compound 11, obtained from Cortex Lycii, were also in accordance with the general trends in methanol-d4 and DMSO-d6 (Fig. 2). Thus, the relative configuration of AGs can be determined from the ΔδH3a–H3b values of methylene protons of the 1H NMR spectra in methanol-d4, acetone-d6, pyridine-d5, and DMSO-d6; DMSO-d6 is the preferred solvent (Table 1).
Table 1 ΔδH3a–H3b values (ppm) of the AGs 1–11 in different solvents (1H NMR 500 MHz)
| No. |
δH3a |
δH3b |
ΔδH3a–H3b |
Solvent |
Relative conf. |
| 1 & 2 |
3.50 |
3.37 |
0.13 |
Methanol-d4 |
Threo |
| 3.49 |
3.40 |
0.09 |
Acetone-d6 |
| 3 & 4 |
3.55 |
3.39 |
0.16 |
Methanol-d4 |
Threo |
| 3.56 |
3.42 |
0.14 |
Acetone-d6 |
| 4.27 |
4.09 |
0.18 |
Pyridine-d5 |
| 3.39 |
3.15 |
0.24 |
DMSO-d6 |
| 5 & 6 |
3.64 |
3.59 |
0.05 |
Methanol-d4 |
Erythro |
| 3.61 |
3.61 |
0.00 |
Acetone-d6 |
| 4.39 |
4.39 |
0.00 |
Pyridine-d5 |
| 3.42 |
3.37 |
0.05 |
DMSO-d6 |
| 7 & 8 |
3.68 |
3.65 |
0.03 |
Methanol-d4 |
Erythro |
| 3.67 |
3.63 |
0.04 |
Acetone-d6 |
| 3.42 |
3.42 |
0.00 |
DMSO-d6 |
| 9 & 10 |
3.47 |
3.34 |
0.13 |
Methanol-d4 |
Threo |
| 3.31 |
3.14 |
0.17 |
DMSO-d6 |
| 11 |
3.52 |
3.38 |
0.14 |
Methanol-d4 |
Threo |
| 3.35 |
3.18 |
0.17 |
DMSO-d6 |
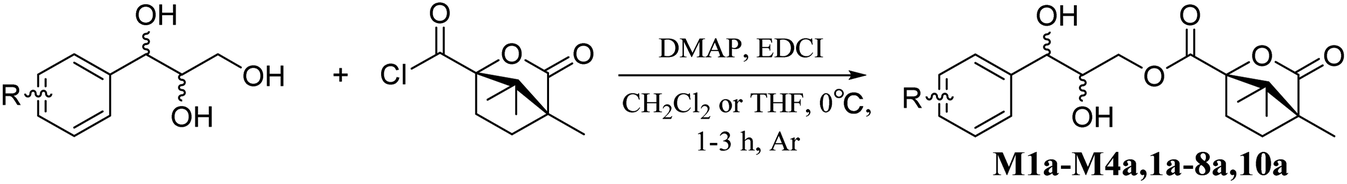
Then, a more challenging task is how to determine the absolute configurations of a pair of enantiomers. Considering the conformational flexibility of the triol chain in AGs, the introduction of a chiral auxiliary agent bearing a larger steric hindrance group may be an efficient approach. Camphanoyl chloride, a acylating agent containing two special stereogenic centers, is frequently utilized to determine enantiomeric purity in organic synthesis and its derivatives was also used to assign absolute configurations of chiral alcohols by NMR.26,27 It made molecules more rigid due to its steric hindrance in conformational studies. These features inspired us to thoroughly investigate the relationship between the chemical shift differences of methylene protons and the absolute configurations of mono-camphanoyl AGs.
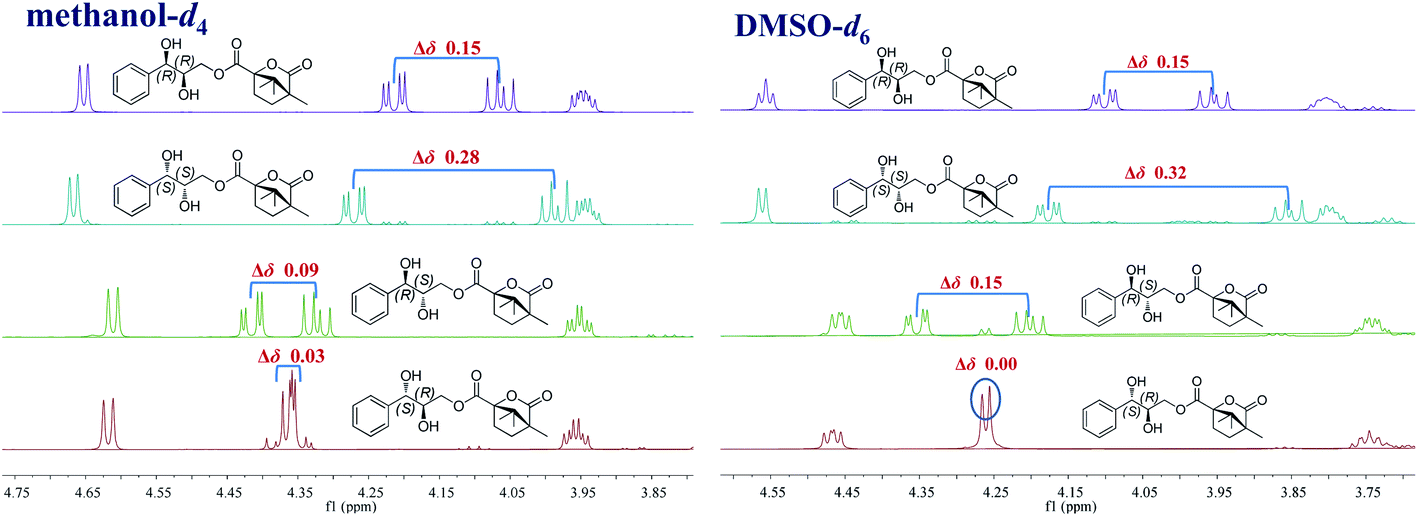
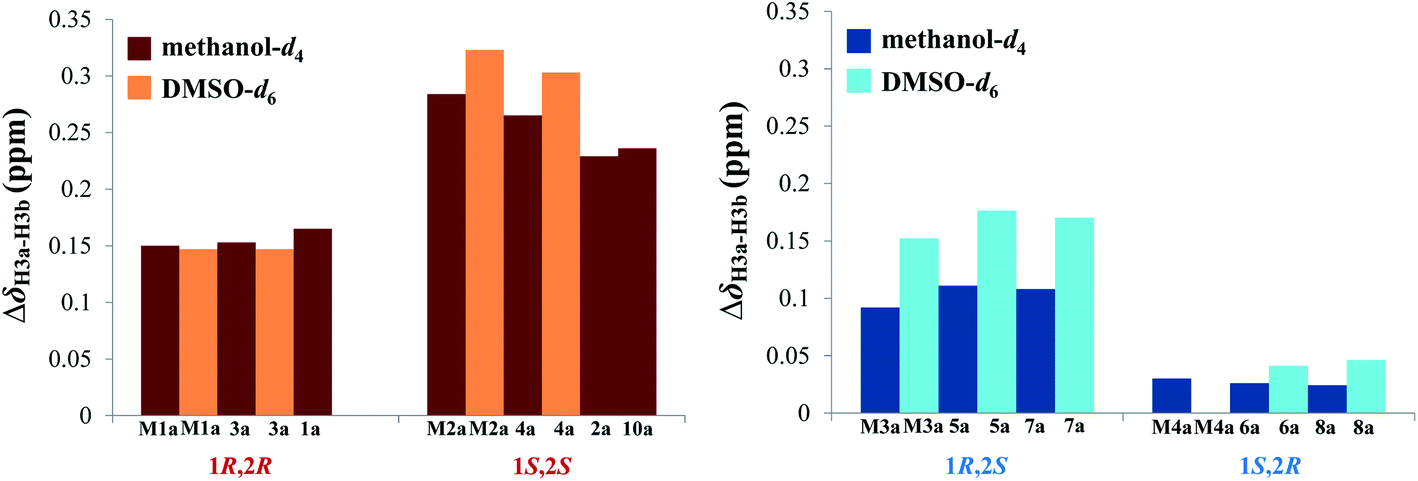
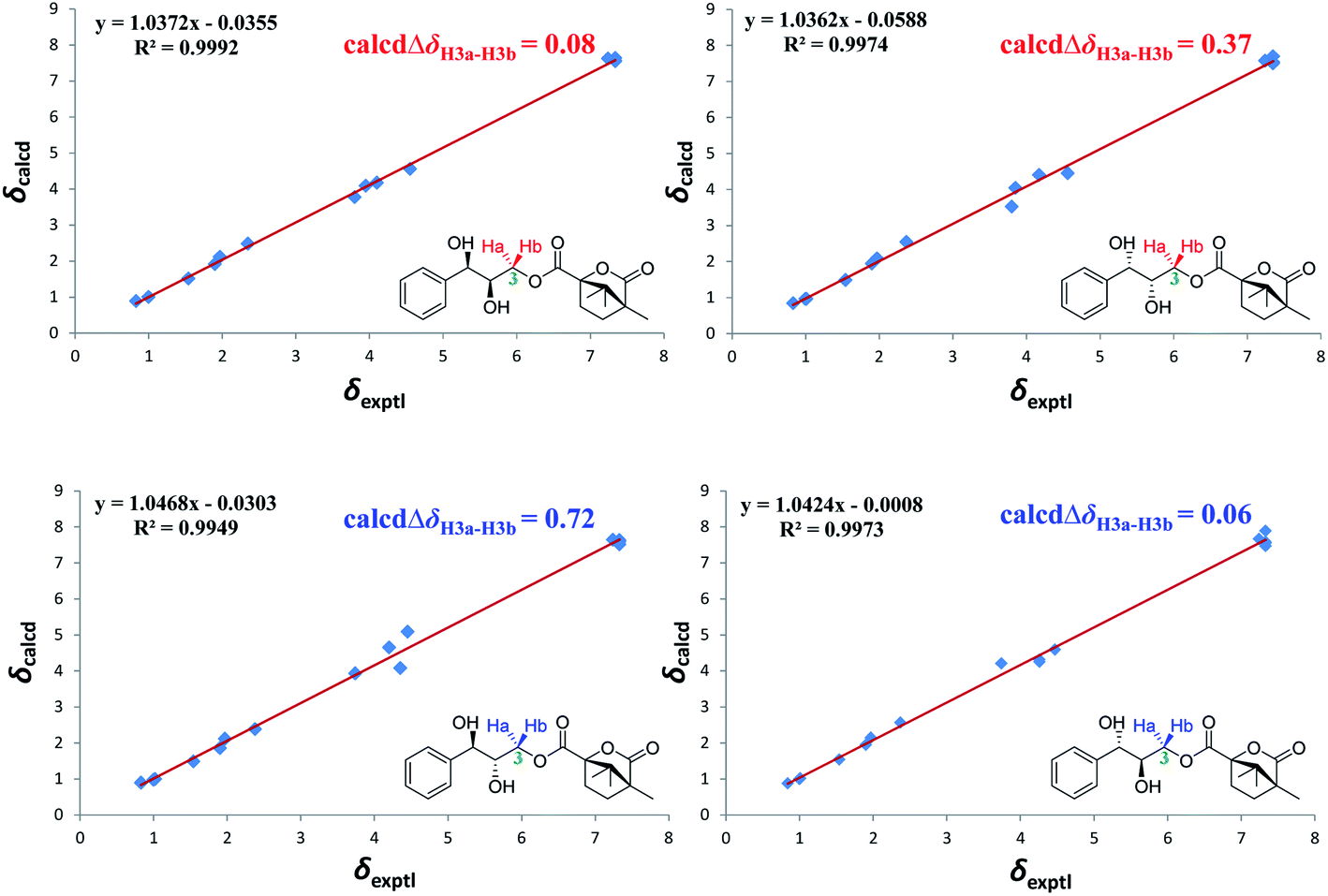
Accordingly, acylation reactions (Scheme 1) were carried out using (−)-1S,4R-camphanoyl chloride and two pairs of 1-phenyl glycerol enantiomers M1–M4 as raw materials, and four products (M1a–M4a) were successfully obtained. Their 1H NMR spectra were measured in several deuterated solvents, including methanol-d4, DMSO-d6, acetone-d6, pyridine-d5, and CDCl3. To our delight, in methanol-d4 and DMSO-d6, the ΔδH3a–H3b values of methylene protons showed obvious differences in the threo and erythro configurations. In the threo enantiomers, the ΔδH3a–H3b values of methylene protons in M1a (1R,2R) were 0.15 and 0.15, respectively, while the ΔδH3a–H3b values in M2a (1S,2S) were 0.28 and 0.32, respectively (Fig. 4). In the erythro system, the ΔδH3a–H3b values of methylene protons in M3a (1R,2S) were 0.09 and 0.15, respectively, while the ΔδH3a–H3b values in M4a (1S,2R) were 0.03 and 0.00, respectively. The chemical shift differences were high enough to discriminate the absolute configuration, especially when using DMSO-d6 as the solvent. However, in the other three deuterated solvents, acetone-d6, CDCl3, and pyridine-d5, the ΔδH3a–H3b values were irregular. These results suggested that the anisotropic effect of camphanoyl could be efficiently and selectively space oriented toward the methylene protons of mono-camphanoyl AGs in methanol-d4 and DMSO-d6. To further verify the reliability of the method, derivatives 1a–8a, and 10a were also successfully synthesized. As expected, the rule still applies in methanol-d4 and DMSO-d6 (Fig. 5). Furthermore, the theoretical 1H NMR calculation of M1a–M4a was carried out by GIAO NMR calculation method in DMSO (the detailed procedure see ESI†).28 The calculated results were better match with the experimental data (R2 ≥ 0.995) (Fig. 6). The calculated ΔδH3a–H3b values were M1a: 0.08, M2a: 0.37, M3a: 0.72, and M4a: 0.06, respectively. This result verified our method was also reliable by quantum chemical calculation. These results confirmed that camphanoyl chloride is a suitable pure chiral derivatizing agent to assign the absolute configuration of AGs.
 |
| | Scheme 1 The acylation reaction. | |
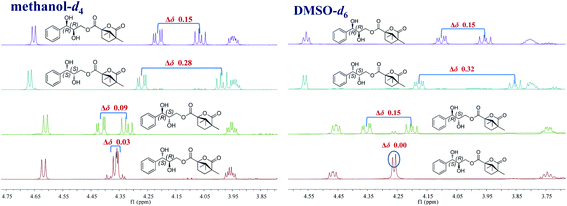 |
| | Fig. 4 Influence of different deuterated solvents on the ΔδH3a–H3b values of M1a–M4a (1H NMR 500 MHz). | |
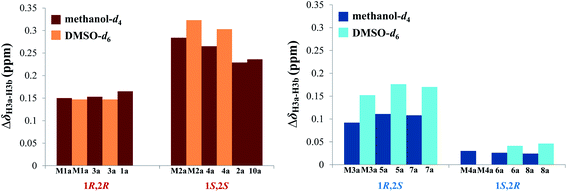 |
| | Fig. 5 ΔδH3a–H3b values (ppm) of the acylation products. | |
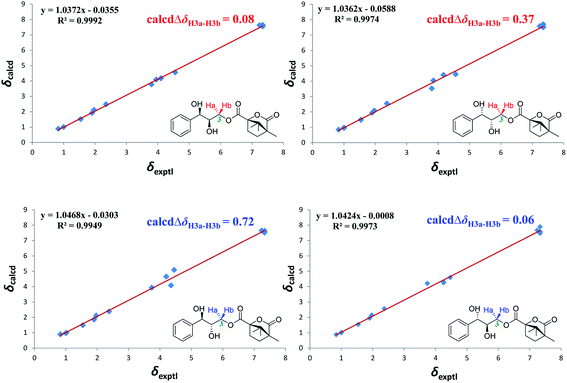 |
| | Fig. 6 Linear correlation plots of predicted versus experimental 1H NMR chemical shifts. | |
Experimental section
General experimental procedures
The optical rotations were recorded with a RUDOLPH automatic V polarimeter (RUDOLPH, Hackettstown, NJ, USA). The NMR spectra were recorded with a Bruker 500 MHz (Bruker-Biospin, Billerica, MA, USA). HRESIMS reports were obtained from an Agilent 6520 HPLC-Q-TOF (Agilent Technologies, Waldbronn, Germany) and Q Exactive Focus LCMSMS (Thermo Scientific, MA, USA). Preparative HPLC separations were performed using a Shimadzu LC-10AT with an ODS-A column (250 mm × 10 mm, 5 μm; YMC Corp., Kyoto, Japan). An Agilent 1200 series system with an Apollo C 18 column (250 mm × 4.6 mm, 5 μm; Alltech Corp., KY, USA) was used for HPLC-DAD analysis. All reactions were magnetically stirred with a DF-101S magnetic stirrer (Hengfengchangwei Technology Co. Ltd, Beijing, China). Commercially available reagents were used without further purification unless otherwise stated. All reactions were monitored by TLC with silica gel pre-coated GF254 plates and HPLC-DAD.
Structure characterization and synthesis procedures
(2S,3S)-3-Phenyl-2-oxiranemethanol and (2R,3R)-3-phenyl-2-oxiranemethanol. The asymmetric epoxidation was carried out according to the Sharpless asymmetric Epoxidation. Colorless oil; the specific rotations were [α]20D −47 (c 0.1 methanol) and [α]20D +70 (c 0.1 methanol), respectively; 1H-NMR (500 MHz, methanol-d4): δ 7.27–7.35 (5H, one mono-substituted benzene ring system), 3.87 (1H, dd, J = 12.5, 3.0 Hz), 3.83 (1H, d, J = 2.0 Hz), 3.67 (1H, dd, J = 12.5, 5.5 Hz), 3.15 (1H, m); 13C-NMR (125 MHz, CDCl3): δ 136.8, 128.7, 128.7, 128.5, 125.9, 125.9, 62.5, 61.3, 55.7; HRESIMS m/z 151.0754 [M + H]+ (calcd for C9H11O2 151.0753).A solution of (2R,3R)-3-phenyl-2-oxiranemethanol (100 mg, 0.6 mmol) and acetic acid (0.5 ml) in acetonitrile was stirred by rotary evaporator at 55 °C until the solution was dry. Through the analysis of HPLC, the (2R,3R)-3-phenyl-2-oxiranemethanol was completely hydrolyzed. The residue was separated by preparative HPLC (MeOH/H2O, 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75, v\v, HOAc, 0.1%) to afford M1 (45 mg) and M4 (50 mg). The hydrolysis procedure of (2S,3S)-3-phenyl-2-oxiranemethanol was same as the above. The residue was separated by preparative HPLC (MeOH/H2O, 25:75, v\v, HOAc, 0.1%) to afford M2 and M3. Compounds M1–M4 are known compounds and they were identified by comparing their spectral data with those of the known compounds.22
:75, v\v, HOAc, 0.1%) to afford M1 (45 mg) and M4 (50 mg). The hydrolysis procedure of (2S,3S)-3-phenyl-2-oxiranemethanol was same as the above. The residue was separated by preparative HPLC (MeOH/H2O, 25:75, v\v, HOAc, 0.1%) to afford M2 and M3. Compounds M1–M4 are known compounds and they were identified by comparing their spectral data with those of the known compounds.22
(1R,2R)-1-Phenylpropane-1,2,3-triol and (1S,2S)-1-phenylpropane-1,2,3-triol (M1 and M2). Colorless oil; the specific rotations were [α]20D −39 (c 0.1 methanol) and [α]20D +33 (c 0.1 methanol), respectively. 1H-NMR (500 MHz, methanol-d4): δ 7.25–7.40 (5H, one mono-substituted benzene ring system), 4.63 (1H, d, J = 5.5 Hz), 3.69 (1H, m), 3.50 (1H, dd, J = 11.5, 4.0 Hz), 3.36 (1H, dd, J = 11.5, 6.0 Hz). 1H-NMR (500 MHz, acetone-d6): δ 7.23–7.41 (5H, one mono-substituted benzene ring system), 4.68 (1H, d, J = 5.5 Hz), 3.65 (1H, m), 3.51 (1H, dd, J = 11.0, 3.5 Hz), 3.39 (1H, dd, J = 11.5, 5.5 Hz). 1H-NMR (500 MHz, pyridine-d5): δ 7.84 (2H, d, J = 7.5 Hz), 7.40 (2H, t, J = 7.5 Hz), 7.30 (1H, t, J = 7.5 Hz), 5.39 (1H, d, J = 5.5 Hz), 4.39 (1H, m), 4.24 (1H, dd, J = 11.0, 4.5 Hz), 4.09 (1H, dd, J = 11.0, 6.0 Hz). 1H-NMR (500 MHz, DMSO-d6): δ 7.19–7.34 (5H, one mono-substituted benzene ring system), 4.53 (1H, t, J = 5.0 Hz), 3.48 (1H, m), 3.36 (1H, m), 3.15 (1H, m). 1H-NMR (500 MHz, CDCl3): δ 7.28–7.33 (5H, one mono-substituted benzene ring system), 4.66 (1H, d, J = 5.5 Hz), 3.76 (1H, brs), 3.54 (1H, brd, J = 11.0 Hz), 3.46 (1H, brd, J = 11.0 Hz). 13C-NMR (125 MHz, CDCl3): δ 140.6, 128.6, 128.6, 128.2, 126.8, 126.8, 76.2, 74.8, 63.2; HRESIMS refer to compounds M3 and M4.
(1R,2S)-1-Phenylpropane-1,2,3-triol and (1S,2R)-1-phenylpropane-1,2,3-triol (M3 and M4). Colorless oil; the specific rotations were [α]20D −47 (c 0.1 methanol) and [α]20D +27 (c 0.1 methanol), respectively. 1H-NMR (500 MHz, methanol-d4): δ 7.24–7.41 (5H, one mono-substituted benzene ring system), 4.61 (1H, d, J = 6.5 Hz), 3.75 (1H, m), 3.66 (1H, dd, J = 11.5, 3.5 Hz), 3.60 (1H, dd, J = 11.5, 6.5 Hz); 1H-NMR (500 MHz, acetone-d6): δ 7.21–7.41 (5H, one mono-substituted benzene ring system), 4.67 (1H, d, J = 4.0 Hz), 3.73 (1H, m), 3.61 (2H, overlap). 1H-NMR (500 MHz, pyridine-d5): δ 7.88 (2H, d, J = 8.0 Hz), 7.40 (2H, t, J = 8.0 Hz), 7.30 (1H, t, J = 8.0 Hz), 5.35 (1H, d, J = 6.0 Hz), 4.50 (1H, m), 4.42 (2H, overlap). 1H-NMR (500 MHz, DMSO-d6): δ 7.19–7.34 (5H, one mono-substituted benzene ring system), 4.42 (1H, overlap), 3.51 (1H, m), 3.44 (1H, m), 3.38 (1H, m). 1H-NMR (500 MHz, CDCl3): δ 7.30–7.39 (5H, one mono-substituted benzene ring system), 4.89 (1H, d, J = 5.0 Hz), 3.85 (1H, m), 3.74 (1H, dd, J = 11.0, 5.5 Hz), 3.67 (1H, dd, J = 11.0, 3.5 Hz). 13C-NMR (125 MHz, CDCl3): δ 140.4, 128.8, 128.8, 128.3, 126.3, 126.3, 76.1, 74.6, 63.1; HRESIMS m/z 191.0679 [M + Na]+ (calcd for C9H12O3Na 191.0679).
(2R,3R)-2,3-Dihydroxy-3-phenylpropyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (M1a). Compound M1 (10 mg, 0.06 mmol) solution in dry CH2Cl2 or THF was added to a solution of camphanic acid chloride (13 mg, 0.06 mmol), EDCI (11.4 mg, 0.06 mmol) and DMAP (3.6 mg, 0.03 mmol) at 0 °C with the protection of argon gas. The reaction was stirred at 0 °C for 1 h and was detected by TLC. Then, the reaction was quenched by dropwise addition of H2O. The suspension was extracted with CH2Cl2. Removal of the solvent under reduced pressure and purification of the residue by preparative HPLC, eluting with 40% acetonitrile/H2O, gave the M1a 8.7 mg. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 7.27–7.42 (5H, one mono-substituted benzene ring system), 4.65 (1H, d, J = 5.5 Hz), 4.21 (1H, dd, J = 11.5, 3.5 Hz), 4.06 (1H, dd, J = 11.5, 7.0 Hz), 3.95 (1H, m), 2.47 (1H, m), 2.03 (1H, m), 1.96 (1H, m), 1.62 (1H, m), 1.09 (3H, s), 1.09 (3H, s), 0.94 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 7.23–7.38 (5H, one mono-substituted benzene ring system), 4.56 (1H, t, J = 5.0 Hz), 4.10 (1H, dd, J = 11.0, 3.5 Hz), 3.95 (1H, dd, J = 11.0, 7.5 Hz), 3.80 (1H, m), 2.35 (1H, m), 1.97 (1H, m), 1.90 (1H, m), 1.54 (1H, m), 1.01 (3H, s), 1.00 (3H, s), 0.83 (3H, s). 1H-NMR (500 MHz, acetone-d6): δ 7.26–7.45 (5H, one mono-substituted benzene ring system), 4.73 (1H, t, J = 5.0 Hz), 4.24 (1H, dd, J = 11.5, 3.5 Hz), 4.11 (1H, dd, J = 11.5, 7.0 Hz), 3.98 (1H, m), 2.46 (1H, m), 2.01 (1H, m), 1.95 (1H, m), 1.60 (1H, m), 1.09 (3H, s), 1.05 (3H, s), 0.92 (3H, s). 1H-NMR (500 MHz, pyridine-d5): δ 7.80 (2H, d, J = 7.5 Hz), 7.42 (2H, d, J = 7.5 Hz), 7.32 (1H, t, J = 7.5 Hz), 5.22 (1H, d, J = 5.0 Hz), 4.75 (1H, dd, J = 11.0, 7.5 Hz), 4.70 (1H, dd, J = 11.0, 4.0 Hz), 4.52 (1H, m), 2.47 (1H, m), 1.99 (1H, m), 1.82 (1H, m), 1.57 (1H, m), 1.06 (3H, s), 1.05 (3H, s), 1.02 (3H, s). 1H-NMR (500 MHz, CDCl3): δ 7.30–7.37 (5H, one mono-substituted benzene ring system), 4.68 (1H, d, J = 7.0 Hz), 4.25 (1H, dd, J = 11.5, 3.5 Hz), 4.11 (1H, dd, J = 11.5, 6.5 Hz), 3.98 (1H, m), 2.41 (1H, m), 2.02 (1H, m), 1.92 (1H, m), 1.69 (1H, m), 1.11 (3H, s), 1.05 (3H, s), 0.96 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.7, 142.9, 129.3, 129.3, 128.8, 127.9, 127.9, 92.9, 75.6, 74.5, 67.6, 56.0, 55.4, 31.5, 29.9, 17.1, 17.0, 9.9; HRESIMS m/z 371.1465 [M + Na]+ (calcd for C19H24O6Na 371.1458).
(2S,3S)-2,3-Dihydroxy-3-phenylpropyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (M2a). The synthesis procedure of M2a was same with the M1a. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 7.27–7.41 (5H, one mono-substituted benzene ring system), 4.67 (1H, d, J = 6.0 Hz), 4.27 (1H, dd, J = 11.0, 3.5 Hz), 3.99 (1H, dd, J = 11.0, 6.5 Hz), 3.94 (1H, m), 2.49 (1H, m), 2.04 (1H, m), 1.97 (1H, m), 1.63 (1H, m), 1.11 (3H, s), 1.09 (3H, s), 0.94 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 7.23–7.37 (5H, one mono-substituted benzene ring system), 4.56 (1H, d, J = 5.0 Hz), 4.18 (1H, dd, J = 11.0, 3.5 Hz), 3.85 (1H, dd, J = 11.0, 7.0 Hz), 3.80 (1H, m), 2.38 (1H, m), 1.97 (1H, m), 1.90 (1H, m), 1.54 (1H, m), 1.01 (3H, s), 1.00 (3H, s), 0.83 (3H, s). 1H-NMR (500 MHz, acetone-d6): δ 7.25–7.45 (5H, one mono-substituted benzene ring system), 4.73 (1H, d, J = 5.5 Hz), 4.29 (1H, dd, J = 11.0, 4.0 Hz), 4.03 (1H, dd, J = 11.0, 6.5 Hz), 3.97 (1H, m), 2.49 (1H, m), 2.02 (1H, m), 1.96 (1H, m), 1.60 (1H, m), 1.10 (3H, s), 1.05 (3H, s), 0.92 (3H, s). 1H-NMR (500 MHz, pyridine-d5): δ 7.80 (2H, d, J = 8.0 Hz), 7.42 (2H, d, J = 8.0 Hz), 7.32 (1H, t, J = 8.0 Hz), 5.24 (1H, d, J = 5.0 Hz), 4.80 (1H, dd, J = 11.0, 3.0 Hz), 4.62 (1H, dd, J = 11.0, 7.0 Hz), 4.53 (1H, m), 2.50 (1H, m), 2.01 (1H, m), 1.84 (1H, m), 1.58 (1H, m), 1.06 (3H, s), 1.06 (3H, s), 1.02 (3H, s). 1H-NMR (500 MHz, CDCl3): δ 7.30–7.38 (5H, one mono-substituted benzene ring system), 4.68 (1H, d, J = 6.5 Hz), 4.25 (1H, dd, J = 11.5, 3.5 Hz), 4.08 (1H, dd, J = 11.5, 6.5 Hz), 3.98 (1H, m), 2.42 (1H, m), 2.02 (1H, m), 1.92 (1H, m), 1.68 (1H, m), 1.11 (3H, s), 1.06 (3H, s), 0.97 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.8, 142.8, 129.3, 129.3, 128.8, 127.9, 127.9, 92.9, 75.6, 74.5, 67.6, 56.0, 55.4, 31.5, 30.0, 17.1, 17.0, 9.9; HRESIMS refer to compound M1a.
(2S,3R)-2,3-Dihydroxy-3-phenylpropyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (M3a). The synthesis procedure of M3a was same with the M1a. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 7.26–7.42 (5H, one mono-substituted benzene ring system), 4.61 (1H, d, J = 6.5 Hz), 4.42 (1H, dd, J = 11.5, 3.0 Hz), 4.32 (1H, dd, J = 11.5, 7.0 Hz), 3.95 (1H, m), 2.49 (1H, m), 2.04 (1H, m), 1.97 (1H, m), 1.62 (1H, m), 1.10 (3H, s), 1.09 (3H, s), 0.94 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 7.23–7.37 (5H, one mono-substituted benzene ring system), 4.46 (1H, dd, J = 6.5, 4.5 Hz), 4.35 (1H, dd, J = 11.5, 3.0 Hz), 4.20 (1H, dd, J = 11.5, 7.0 Hz), 3.74 (1H, m), 2.39 (1H, m), 1.98 (1H, m), 1.90 (1H, m), 1.55 (1H, m), 1.03 (3H, s), 1.00 (3H, s), 0.83 (3H, s). 1H-NMR (500 MHz, acetone-d6): δ 7.24–7.44 (5H, one mono-substituted benzene ring system), 4.72 (1H, dd, J = 6.5, 4.5 Hz), 4.39 (1H, dd, J = 11.5, 3.0 Hz), 4.33 (1H, dd, J = 11.5, 7.0 Hz), 4.01 (1H, m), 2.48 (1H, m), 2.00 (1H, m), 1.94 (1H, m), 1.59 (1H, m), 1.09 (3H, s), 1.05 (3H, s), 0.92 (3H, s). 1H-NMR (500 MHz, pyridine-d5): δ 7.82 (2H, d, J = 7.5 Hz), 7.41 (2H, d, J = 7.5 Hz), 7.31 (1H, t, J = 7.5 Hz), 5.22 (1H, d, J = 6.5 Hz), 5.06 (1H, dd, J = 11.5, 3.0 Hz), 4.98 (1H, dd, J = 11.5, 7.0 Hz), 4.55 (1H, m), 2.49 (1H, m), 1.99 (1H, m), 1.82 (1H, m), 1.57 (1H, m), 1.07 (3H, s), 1.06 (3H, s), 1.05 (3H, s). 1H-NMR (500 MHz, CDCl3): δ 7.29–7.38 (5H, one mono-substituted benzene ring system), 4.83 (1H, d, J = 5.5 Hz), 4.34 (1H, dd, J = 11.5, 7.5 Hz), 4.25 (1H, dd, J = 11.5, 3.0 Hz), 4.09 (1H, m), 2.40 (1H, m), 2.00 (1H, m), 1.92 (1H, m), 1.67 (1H, m), 1.10 (3H, s), 1.04 (3H, s), 0.95 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.9, 143.2, 129.2, 129.2, 128.6, 128.1, 128.1, 93.0, 75.8, 74.1, 67.8, 56.0, 55.4, 31.5, 30.0, 17.1, 17.0, 9.9; HRESIMS refer to compound M1a.
(2R,3S)-2,3-Dihydroxy-3-phenylpropyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (M4a). The synthesis procedure of M4a was same with the M1a. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 7.26–7.42 (5H, one mono-substituted benzene ring system), 4.62 (1H, d, J = 7.0 Hz), 4.38 (1H, dd, J = 11.5, 6.5 Hz), 4.35 (1H, dd, J = 11.5, 3.5 Hz), 3.96 (1H, m), 2.49 (1H, m), 2.03 (1H, m), 1.97 (1H, m), 1.62 (1H, m), 1.10 (3H, s), 1.09 (3H, s), 0.94 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 7.22–7.36 (5H, one mono-substituted benzene ring system), 4.47 (1H, dd, J = 6.5, 4.5 Hz), 4.26 (2H, overlap), 3.75 (1H, m), 2.38 (1H, m), 1.97 (1H, m), 1.90 (1H, m), 1.54 (1H, m), 1.01 (3H, s), 1.00 (3H, s), 0.84 (3H, s). 1H-NMR (500 MHz, acetone-d6): δ 7.24–7.44 (5H, one mono-substituted benzene ring system), 4.72 (1H, dd, J = 6.0, 4.5 Hz), 4.38 (1H, dd, J = 11.5, 7.0 Hz), 4.33 (1H, dd, J = 11.5, 3.5 Hz), 4.01 (1H, m), 2.47 (1H, m), 2.00 (1H, m), 1.94 (1H, m), 1.59 (1H, m), 1.09 (3H, s), 1.05 (3H, s), 0.92 (3H, s). 1H-NMR (500 MHz, pyridine-d5): δ 7.82 (2H, d, J = 7.5 Hz), 7.41 (2H, d, J = 7.5 Hz), 7.31 (1H, t, J = 7.5 Hz), 5.22 (1H, d, J = 7.0 Hz), 5.06 (1H, dd, J = 11.5, 7.0 Hz), 4.98 (1H, dd, J = 11.5, 3.0 Hz), 4.55 (1H, m), 2.50 (1H, m), 1.99 (1H, m), 1.83 (1H, m), 1.57 (1H, m), 1.07 (3H, s), 1.06 (3H, s), 1.04 (3H, s). 1H-NMR (500 MHz, CDCl3): δ 7.30–7.38 (5H, one mono-substituted benzene ring system), 4.84 (1H, d, J = 5.5 Hz), 4.38 (1H, dd, J = 11.5, 7.5 Hz), 4.24 (1H, dd, J = 11.5, 3.0 Hz), 4.08 (1H, m), 2.41 (1H, m), 2.01 (1H, m), 1.92 (1H, m), 1.68 (1H, m), 1.11 (3H, s), 1.04 (3H, s), 0.95 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.9, 143.2, 129.2, 129.2, 128.6, 128.1, 128.1, 93.0, 75.8, 74.2, 67.8, 56.0, 55.4, 31.5, 30.0, 17.1, 17.0, 9.9; HRESIMS refer to compound M1a.
(1R,2R)-1-(4-Hydroxy-3,5-dimethoxyphenyl)propane-1,2,3-triol and (1S,2S)-1-(4-hydroxy-3,5-dimethoxyphenyl)propane-1,2,3-triol (1 and 2). Colorless oil; the specific rotations were [α]20D −100 (c 0.1 methanol) and [α]20D +70 (c 0.1 methanol), respectively. 1H-NMR (500 MHz, methanol-d4): δ 6.68 (2H, brs), 4.53 (1H, d, J = 6.0 Hz), 3.67 (1H, m), 3.50 (1H, dd, J = 11.5, 4.0 Hz), 3.37 (1H, dd, J = 11.5, 6.5 Hz). 1H-NMR (500 MHz, acetone-d6): δ 6.69 (2H, brs), 4.56 (1H, d, J = 6.0 Hz), 3.62 (1H, m), 3.49 (1H, dd, J = 11.0, 4.0 Hz), 3.40 (1H, dd, J = 11.0, 6.0 Hz). 13C-NMR (125 MHz, methanol-d4): δ 149.1, 149.1, 135.9, 134.1, 105.0, 105.0, 77.6, 75.6, 64.2, 56.7, 56.7; HRESIMS m/z 267.0839 [M + Na]+ (calcd for C11H16O6Na 267.0837). Compounds 1 and 2 are known compounds and they were identified by comparing their spectral data with those of the known compounds.8
4-((1R,2R)-1,2-Dihydroxy-3-(((1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carbonyl)oxy)propyl)-2,6-dimethoxyphenyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (1a). Compound 1 (6 mg, 0.02 mmol) solution in dry CH2Cl2 or THF was added to a solution of camphanic acid chloride (9 mg, 0.04 mmol), EDCI (7.6 mg, 0.04 mmol) and DMAP (2.4 mg, 0.02 mmol) at 0 °C with the protection of argon gas. The reaction was stirred at 0 °C for 1 h and was detected by TLC. Then, the reaction was quenched by dropwise addition of H2O. The suspension was extracted with CH2Cl2. Removal of the solvent under reduced pressure and purification of the residue by preparative HPLC, eluting with 40% acetonitrile/H2O, gave the 1a 2.9 mg. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 6.84 (2H, brs), 4.68 (1H, d, J = 5.0 Hz), 4.33 (1H, dd, J = 11.5, 4.0 Hz), 4.16 (1H, dd, J = 11.5, 6.5 Hz), 3.97 (1H, m), 2.67 (1H, m), 2.48 (1H, m), 2.12 (2H, overlap), 2.04 (1H, m), 1.97 (1H, m), 1.70 (1H, m), 1.63 (1H, m), 1.22 (3H, s), 1.14 (3H, s), 1.10 (3H, s), 1.09 (3H, s), 1.08 (3H, s), 0.97 (3H, s); HRESIMS m/z 605.2593 [M + H]+ (calcd for C31H41O12 605.2593).
4-((1S,2S)-1,2-Dihydroxy-3-(((1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carbonyl)oxy)propyl)-2,6-dimethoxyphenyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (2a). Compound 2 (4.4 mg, 0.018 mmol) solution in dry CH2Cl2 or THF was added to a solution of camphanic acid chloride (7.8 mg, 0.036 mmol), EDCI (6.8 mg, 0.036 mmol) and DMAP (2.2 mg, 0.018 mmol) at 0 °C with the protection of argon gas. The reaction was stirred at 0 °C for 1 h and was detected by TLC. Then, the reaction was quenched by dropwise addition of H2O. The suspension was extracted with CH2Cl2. Removal of the solvent under reduced pressure and purification of the residue by preparative HPLC, eluting with 55% methanol/H2O, gave the 2a 2.0 mg. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 6.82 (2H, brs), 4.69 (1H, d, J = 5.0 Hz), 4.36 (1H, dd, J = 11.5, 4.0 Hz), 4.13 (1H, dd, J = 11.5, 6.5 Hz), 3.97 (1H, m), 2.67 (1H, m), 2.49 (1H, m), 2.12 (2H, overlap), 2.02 (2H, overlap), 1.70 (1H, m), 1.63 (1H, m), 1.22 (3H, s), 1.14 (3H, s), 1.11 (3H, s), 1.09 (3H, s), 1.08 (3H, s), 0.94 (3H, s); HRESIMS m/z 605.2593 [M + H]+ (calcd for C31H41O12 605.2600).
((2R,3R)-3-(4-Bromophenyl)oxiran-2-yl)methanol and ((2S,3S)-3-(4-bromophenyl)oxiran-2-yl)methanol. The asymmetric epoxidation was also carried out according to the Sharpless asymmetric Epoxidation. Colorless oil; the specific rotations were [α]20D +45 (c 0.1 methanol) and [α]20D −25 (c 0.1 methanol), respectively. 1H-NMR (500 MHz, methanol-d4): δ 7.49 (2H, d, J = 8.5 Hz), 7.22 (2H, d, J = 8.5 Hz), 3.85 (1H, dd, J = 12.5, 3.0 Hz), 3.83 (1H, d, J = 1.5 Hz), 3.67 (1H, dd, J = 12.5, 4.5 Hz), 3.13 (1H, m); 13C-NMR (125 MHz, methanol-d4): δ 138.2, 132.6, 132.6, 128.7, 128.7, 122.8, 63.9, 62.6, 56.2; HRESIMS m/z 228.9859 [M + H]+ (calcd for C9H10O2Br 228.9858).A solution of ((2R,3R)-3-(4-bromophenyl)oxiran-2-yl)methanol (65 mg, 0.3 mmol) and acetic acid (0.5 ml) in acetonitrile was stirred by rotary evaporator at 55 °C until the solution was dry. Through the analysis of HPLC, the ((2R,3R)-3-(4-bromophenyl)oxiran-2-yl)methanol was completely hydrolyzed. The residue was separated by preparative HPLC (MeOH/H2O, 45:55, v\v) to afford 3 (27 mg) and 6 (38 mg). The hydrolysis procedure of ((2S,3S)-3-(4-bromophenyl)oxiran-2-yl)methanol was same as the above. The residue was separated by preparative HPLC (MeOH/H2O, 45:55, v\v) to afford 4 and 5. Compounds 3–6 are known compounds and their absolute configurations were identified by comparing the optical rotations of M1–M4.
(1R,2R)-1-(4-Bromophenyl)propane-1,2,3-triol and (1S,2S)-1-(4-bromophenyl)propane-1,2,3-triol (3 and 4). Colorless oil; the specific rotations were [α]20D −20 (c 0.1 methanol) and [α]20D +16 (c 0.1 methanol), respectively. 1H-NMR (500 MHz, methanol-d4): δ 7.48 (2H, d, J = 8.5 Hz), 7.33 (2H, d, J = 8.5 Hz), 4.65 (1H, d, J = 5.0 Hz), 3.65 (1H, m), 3.55 (1H, dd, J = 11.5, 4.5 Hz), 3.39 (1H, dd, J = 11.5, 6.5 Hz). 1H-NMR (500 MHz, acetone-d6): δ 7.49 (2H, d, J = 8.5 Hz), 7.37 (2H, d, J = 8.5 Hz), 4.72 (1H, t, J = 4.5 Hz), 3.64 (1H, m), 3.56 (1H, m), 3.42 (1H, m). 1H-NMR (500 MHz, pyridine-d5): δ 7.71 (2H, d, J = 8.5 Hz), 7.55 (2H, d, J = 8.5 Hz), 5.38 (1H, d, J = 5.0 Hz), 4.35 (1H, m), 4.28 (1H, dd, J = 11.0, 4.5 Hz), 4.09 (1H, dd, J = 11.0, 6.0 Hz). 1H-NMR (500 MHz, DMSO-d6): δ 7.48 (2H, d, J = 8.5 Hz), 7.29 (2H, d, J = 8.5 Hz), 4.55 (1H, t, J = 5.0 Hz), 3.46 (1H, m), 3.39 (1H, m), 3.15 (1H, m). 13C-NMR (125 MHz, methanol-d4): δ 143.0, 132.2, 132.2, 129.8, 129.8, 122.0, 77.1, 74.4, 64.1; HRESIMS m/z 268.9784 [M + Na]+ (calcd for C9H11O3BrNa 268.9778).
(1R,2S)-1-(4-Bromophenyl)propane-1,2,3-triol and (1S,2R)-1-(4-bromophenyl)propane-1,2,3-triol (5 and 6). Colorless oil; the specific rotations were [α]20D −8 (c 0.1 methanol) and [α]20D +12 (c 0.1 methanol), respectively. 1H-NMR (500 MHz, methanol-d4): δ 7.48 (2H, d, J = 8.5 Hz), 7.33 (2H, d, J = 8.5 Hz), 4.58 (1H, d, J = 6.5 Hz), 3.70 (1H, m), 3.64 (1H, dd, J = 11.5, 4.0 Hz), 3.59 (1H, dd, J = 11.5, 6.5 Hz). 1H-NMR (500 MHz, acetone-d6): δ 7.48 (2H, d, J = 8.5 Hz), 7.37 (2H, d, J = 8.5 Hz), 4.65 (1H, d, J = 6.5 Hz), 3.69 (1H, m), 3.62 (2H, overlap). 1H-NMR (500 MHz, pyridine-d5): δ 7.74 (2H, d, J = 8.5 Hz), 7.55 (2H, d, J = 8.5 Hz), 5.30 (1H, dd, J = 5.5, 3.0 Hz), 4.42 (1H, m), 4.39 (2H, overlap). 1H-NMR (500 MHz, DMSO-d6): δ 7.47 (2H, d, J = 8.5 Hz), 7.28 (2H, d, J = 8.5 Hz), 4.41 (1H, t, J = 5.0 Hz), 3.47 (1H, m), 3.42 (1H, m), 3.37 (1H, m). 13C-NMR (125 MHz, methanol-d4): δ 142.9, 132.0, 132.0, 130.3, 130.3, 122.0, 76.5, 75.3, 64.3; HRESIMS refer to compounds 3 and 4.
(2R,3R)-3-(4-Bromophenyl)-2,3-dihydroxypropyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (3a). The synthesis procedure of 3a was same with the M1a. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 7.50 (2H, d, J = 8.5 Hz), 7.35 (2H, d, J = 8.5 Hz), 4.66 (1H, d, J = 5.0 Hz), 4.26 (1H, dd, J = 11.5, 4.0 Hz), 4.11 (1H, dd, J = 11.5, 7.0 Hz), 3.92 (1H, m), 2.46 (1H, m), 2.03 (1H, m), 1.97 (1H, m), 1.63 (1H, m), 1.09 (6H, overlap), 0.94 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 7.51 (2H, d, J = 8.5 Hz), 7.31 (2H, d, J = 8.5 Hz), 4.56 (1H, d, J = 3.5 Hz), 4.12 (1H, dd, J = 11.0, 3.5 Hz), 3.97 (1H, dd, J = 11.0, 8.0 Hz), 3.79 (1H, m), 2.35 (1H, m), 1.97 (1H, m), 1.90 (1H, m), 1.54 (1H, m), 1.00 (6H, overlap), 0.82 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.7, 142.4, 132.3, 132.3, 129.9, 129.9, 122.3, 92.9, 74.7, 74.2, 67.6, 56.1, 55.4, 31.5, 29.9, 17.1, 17.0, 9.9; HRESIMS m/z 449.0570 [M + Na]+ (calcd for C19H23O6BrNa 449.0562).
(2S,3S)-3-(4-Bromophenyl)-2,3-dihydroxypropyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (4a). The synthesis procedure of 4a was same with the M1a. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 7.50 (2H, d, J = 8.5 Hz), 7.34 (2H, d, J = 8.5 Hz), 4.67 (1H, d, J = 5.0 Hz), 4.31 (1H, dd, J = 11.5, 4.0 Hz), 4.04 (1H, dd, J = 11.5, 7.0 Hz), 3.91 (1H, m), 2.48 (1H, m), 2.04 (1H, m), 1.98 (1H, m), 1.63 (1H, m), 1.10 (3H, s), 1.09 (3H, s), 0.93 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 7.51 (2H, d, J = 8.0 Hz), 7.31 (2H, d, J = 8.0 Hz), 4.56 (1H, d, J = 3.5 Hz), 4.19 (1H, dd, J = 11.5, 3.5 Hz), 3.88 (1H, dd, J = 11.5, 7.0 Hz), 3.79 (1H, m), 2.36 (1H, m), 1.97 (1H, m), 1.90 (1H, m), 1.54 (1H, m), 1.00 (6H, overlap), 0.82 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.7, 142.4, 132.3, 132.3, 129.9, 129.9, 122.3, 92.9, 74.7, 74.1, 67.5, 56.0, 55.4, 31.5, 30.0, 17.1, 17.0, 9.9; HRESIMS refer to compounds 3a.
(2S,3R)-3-(4-Bromophenyl)-2,3-dihydroxypropyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (5a). The synthesis procedure of 5a was same with the M1a. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 7.50 (2H, d, J = 8.5 Hz), 7.34 (2H, d, J = 8.5 Hz), 4.57 (1H, d, J = 7.0 Hz), 4.42 (1H, dd, J = 11.5, 3.5 Hz), 4.31 (1H, dd, J = 11.5, 7.0 Hz), 3.89 (1H, m), 2.48 (1H, m), 2.03 (1H, m), 1.98 (1H, m), 1.63 (1H, m), 1.10 (3H, s), 1.09 (3H, s), 0.94 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 7.51 (2H, d, J = 8.5 Hz), 7.30 (2H, d, J = 8.5 Hz), 4.42 (1H, d, J = 6.0 Hz), 4.36 (1H, dd, J = 11.0, 3.0 Hz), 4.18 (1H, dd, J = 11.0, 6.5 Hz), 3.69 (1H, m), 2.38 (1H, m), 1.98 (1H, m), 1.91 (1H, m), 1.55 (1H, m), 1.02 (3H, s), 1.00 (3H, s), 0.83 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.9, 142.7, 132.2, 132.2, 130.2, 130.2, 122.3, 93.0, 75.0, 74.0, 67.7, 56.0, 55.4, 31.5, 30.0, 17.1, 17.0, 9.9; HRESIMS refer to compounds 3a.
(2R,3S)-3-(4-Bromophenyl)-2,3-dihydroxypropyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (6a). The synthesis procedure of 6a was same with the M1a. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 7.50 (2H, d, J = 8.5 Hz), 7.34 (2H, d, J = 8.5 Hz), 4.57 (1H, d, J = 6.5 Hz), 4.37 (1H, dd, J = 11.5, 4.0 Hz), 4.35 (1H, dd, J = 11.5, 6.0 Hz), 3.90 (1H, m), 2.48 (1H, m), 2.03 (1H, m), 1.97 (1H, m), 1.62 (1H, m), 1.10 (3H, s), 1.09 (3H, s), 0.94 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 7.51 (2H, d, J = 8.5 Hz), 7.30 (2H, d, J = 8.5 Hz), 4.43 (1H, d, J = 6.5 Hz), 4.28 (1H, dd, J = 11.5, 3.5 Hz), 4.23 (1H, dd, J = 11.5, 6.5 Hz), 3.69 (1H, m), 2.37 (1H, m), 1.97 (1H, m), 1.91 (1H, m), 1.54 (1H, m), 1.00 (6H, overlap), 0.84 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.9, 142.7, 132.2, 132.2, 130.1, 130.1, 122.3, 93.0, 75.1, 74.0, 67.7, 56.0, 55.4, 31.5, 30.0, 17.1, 17.0, 9.9; HRESIMS refer to compounds 3a.
((2R,3R)-3-(4-Nitrophenyl)oxiran-2-yl)methanol and ((2S,3S)-3-(4-nitrophenyl)oxiran-2-yl) methanol. The asymmetric epoxidation was also carried out according to the Sharpless asymmetric Epoxidation. Colorless oil; the specific rotations were [α]20D +44 (c 0.1 methanol) and [α]20D −46 (c 0.1 methanol), respectively. 1H-NMR (500 MHz, methanol-d4): δ 8.21 (2H, d, J = 9.0 Hz), 7.53 (2H, d, J = 9.0 Hz), 4.00 (1H, d, J = 2.0 Hz), 3.89 (1H, dd, J = 13.0, 3.0 Hz), 3.71 (1H, dd, J = 13.0, 4.5 Hz), 3.17 (1H, m); 13C-NMR (125 MHz, methanol-d4): δ 149.1, 146.6, 127.7, 127.7, 124.6, 124.6, 64.5, 62.3, 55.7; HRESIMS m/z 196.0604 [M + H]+ (calcd for C9H10O4N 196.0608).
(1R,2S)-1-(4-Nitrophenyl)propane-1,2,3-triol and (1S,2R)-1-(4-nitrophenyl)propane-1,2,3-triol (7 and 8). A solution of ((2R,3R)-3-(4-nitrophenyl)oxiran-2-yl)methanol (80 mg, 0.25 mmol) (which was dissolved by THF) and acetic acid (0.5 ml) in water was stirred at 80 °C until the solution was dry. Through the analysis of HPLC, the ((2R,3R)-3-(4-nitrophenyl)oxiran-2-yl)methanol was completely hydrolyzed. The residue was separated by chiral column on preparative HPLC (n-hexane/isopropanol, 80:20, v\v) to afford 7 (50 mg) and 8 (30 mg). The hydrolysis of ((2S,3S)-3-(4-nitrophenyl)oxiran-2-yl)methanol was also produce compounds 7 and 8. Compounds 7 and 8 are known compounds and they were identified by comparing their spectral data with those of the known compounds.29 Colorless oil; the specific rotations were [α]20D −14 (c 0.3 acetone) and [α]20D +15 (c 0.3 acetone), respectively. 1H-NMR (500 MHz, methanol-d4): δ 8.23 (2H, d, J = 8.5 Hz), 7.66 (2H, d, J = 8.5 Hz), 4.74 (1H, d, J = 7.0 Hz), 3.74 (1H, m), 3.68 (1H, dd, J = 11.5, 4.5 Hz), 3.65 (1H, dd, J = 11.5, 6.0 Hz). 1H-NMR (500 MHz, DMSO-d6): δ 8.17 (2H, d, J = 8.5 Hz), 7.60 (2H, d, J = 8.5 Hz), 4.72 (1H, d, J = 5.5 Hz), 3.52 (1H, m), 3.42 (2H, overlap). 1H-NMR (500 MHz, acetone-d6): δ 8.19 (2H, d, J = 8.5 Hz), 7.70 (2H, d, J = 8.5 Hz), 4.82 (1H, t, J = 6.0 Hz), 3.75 (1H, m), 3.67 (1H, dd, J = 11.0, 5.0 Hz), 3.63 (1H, dd, J = 11.0, 5.0 Hz). 13C-NMR (125 MHz, DMSO-d6): δ 151.8, 146.4, 128.5, 128.5, 122.6, 122.6, 75.2, 72.9, 62.9; found 258.0619. HRESIMS m/z 258.0619 [M + HCOO]+ (calcd for C10H12O7N 258.0620).
(2S,3R)-2,3-Dihydroxy-3-(4-nitrophenyl)propyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (7a). The synthesis procedure of 7a was same with the M1a. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 8.23 (2H, d, J = 8.5 Hz), 7.66 (2H, d, J = 8.5 Hz), 4.72 (1H, d, J = 7.0 Hz), 4.46 (1H, dd, J = 11.5, 3.5 Hz), 4.35 (1H, dd, J = 11.5, 6.5 Hz), 3.91 (1H, m), 2.50 (1H, m), 2.03 (1H, m), 1.98 (1H, m), 1.63 (1H, m), 1.11 (3H, s), 1.09 (3H, s), 0.94 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 8.20 (2H, d, J = 9.0 Hz), 7.62 (2H, d, J = 9.0 Hz), 4.58 (1H, dd, J = 6.5, 3.5 Hz), 4.39 (1H, dd, J = 11.5, 3.0 Hz), 4.22 (1H, dd, J = 11.5, 6.5 Hz), 3.73 (1H, m), 2.39 (1H, m), 1.98 (1H, m), 1.91 (1H, m), 1.55 (1H, m), 1.03 (3H, s), 1.00 (3H, s), 0.83 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.9, 151.4, 148.8, 129.3, 129.3, 124.1, 124.1, 92.9, 74.8, 74.0, 67.6, 56.0, 55.4, 31.5, 29.9, 17.0, 17.0, 9.9; HRESIMS m/z 394.1496 [M + H]+ (calcd for C19H24O8N 394.1494).
(2R,3S)-2,3-Dihydroxy-3-(4-nitrophenyl)propyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (8a). The synthesis procedure of 8a was same with the M1a. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 8.23 (2H, d, J = 8.5 Hz), 7.66 (2H, d, J = 8.5 Hz), 4.71 (1H, d, J = 7.5 Hz), 4.42 (1H, dd, J = 11.5, 4.0 Hz), 4.39 (1H, dd, J = 11.5, 5.5 Hz), 3.92 (1H, m), 2.50 (1H, m), 2.03 (1H, m), 1.97 (1H, m), 1.62 (1H, m), 1.10 (3H, s), 1.09 (3H, s), 0.95 (3H, s). 1H-NMR (500 MHz, DMSO-d6): δ 8.20 (2H, d, J = 9.0 Hz), 7.63 (2H, d, J = 9.0 Hz), 4.59 (1H, d, J = 7.0 Hz), 4.31 (1H, dd, J = 11.5, 3.5 Hz), 4.26 (1H, dd, J = 11.5, 6.5 Hz), 3.74 (1H, m), 2.38 (1H, m), 1.97 (1H, m), 1.91 (1H, m), 1.54 (1H, m), 1.01 (3H, s), 1.00 (3H, s), 0.84 (3H, s). 13C-NMR (125 MHz, methanol-d4): δ 180.3, 168.9, 151.4, 148.8, 129.2, 129.2, 124.2, 124.2, 92.9, 74.9, 74.1, 67.6, 56.0, 55.4, 31.5, 30.0, 17.0, 17.0, 9.9; HRESIMS refer to compound 7a.
(1R,2R)-1-(4-Hydroxy-3-methoxyphenyl)propane-1,2,3-triol and (1S,2S)-1-(4-hydroxy-3-methoxyphenyl)propane-1,2,3-triol (9 and 10). Colorless oil; the specific rotations were [α]20D −19 (c 0.1 methanol) and [α]20D +23 (c 0.1 methanol), respectively. 1H-NMR (500 MHz, methanol-d4): δ 6.99 (1H, d, J = 1.5 Hz), 6.80 (1H, dd, J = 8.5, 1.5 Hz), 6.76 (1H, d, J = 8.5 Hz), 4.52 (1H, d, J = 6.5 Hz), 3.86 (3H, s), 3.66 (1H, m), 3.47 (1H, dd, J = 11.0, 4.0 Hz), 3.34 (1H, dd, J = 11.0, 6.5 Hz). 1H-NMR (500 MHz, DMSO-d6): δ 6.89 (1H, brs), 6.69 (2H, overlap), 4.38 (1H, d, J = 5.0 Hz), 3.74 (3H, s), 3.44 (1H, m), 3.31 (1H, dd, J = 11.0, 4.0 Hz), 3.14 (1H, dd, J = 11.0, 6.0 Hz). 13C-NMR (125 MHz, DMSO-d6): δ 147.0, 145.3, 134.3, 119.1, 114.7, 111.0, 75.9, 72.9, 62.6, 55.6; HRESIMS m/z 237.0733 [M + Na]+ (calcd for C10H14O5Na 237.0719). Compounds 9 and 10 are known compounds and they were identified by comparing their spectral data with those of the known compounds.7
4-((1S,2S)-1,2-Dihydroxy-3-(((1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carbonyl)oxy)propyl)-2-methoxyphenyl(1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (10a). Compound 10 (3 mg, 0.014 mmol) solution in dry CH2Cl2 or THF was added to a solution of camphanic acid chloride (6 mg, 0.028 mmol), EDCI (5.3 mg, 0.028 mmol) and DMAP (1.7 mg, 0.014 mmol) at 0 °C with the protection of argon gas. The reaction was stirred at 0 °C for 1 h and was detected by TLC. Then, the reaction was quenched by dropwise addition of H2O. The suspension was extracted with CH2Cl2. Removal of the solvent under reduced pressure and purification of the residue by preparative HPLC, eluting with 55% methanol/H2O, gave the 10a 1.0 mg. Colorless oil; 1H-NMR (500 MHz, methanol-d4): δ 7.23 (1H, d, J = 1.5 Hz), 7.08 (1H, d, J = 8.0 Hz), 7.02 (1H, dd, J = 8.0, 1.5 Hz), 4.71 (1H, d, J = 5.0 Hz), 4.34 (1H, dd, J = 11.5, 4.0 Hz), 4.11 (1H, dd, J = 11.5, 6.5 Hz), 3.96 (1H, m), 3.86 (3H, s), 2.65 (1H, m), 2.49 (1H, m), 2.16 (2H, overlap), 2.02 (2H, overlap), 1.70 (1H, m), 1.63 (1H, m), 1.21 (3H, s), 1.14 (3H, s), 1.11 (3H, s), 1.09 (3H, s), 1.09 (3H, s), 0.94 (3H, s); HRESIMS m/z 575.2487 [M + H]+ (calcd for C30H39O11 575.2490).
Compound 11. White powder; 1H-NMR (500 MHz, DMSO-d6): δ 6.88 (2H, overlap), 6.64 (2H, brs), 5.41 (2H, overlap), 4.56 (1H, d, J = 6.0 Hz), 4.40 (1H, d, J = 7.5 Hz), 3.77 (3H, s), 3.73 (6H, s), 3.68 (2H, m), 3.64 (1H, m), 3.59 (1H, m), 3.46 (1H, m), 3.38 (2H, overlap), 3.17 (2H, overlap), 3.06 (3H, overlap). 13C-NMR (125 MHz, methanol-d4): δ 147.9, 147.9, 146.6, 142.9, 135.3, 133.5, 131.3, 128.7, 115.7, 112.2, 104.2, 103.7, 103.7, 87.3, 82.0, 76.9, 76.5, 75.2, 74.2, 70.0, 62.7, 61.9, 61.0, 56.0, 56.0, 55.7, 53.1; HRESIMS m/z 607.2026 [M + Na]+ (calcd for C27H36O14Na 607.2000).
Conclusions
For natural AGs, there was previously no efficient method to determine their absolute configurations. Our present study presents a convenient and quick method for determining the relative and absolute configurations that only depends on the chemical shift difference (ΔδH3a–H3b) of the diastereotopic methylene protons (H-3) in 1H NMR spectroscopy. In particular, the method is a good way to determine the limited amounts of natural AGs; such limited amounts prevent the use of other strategies. Remarkably, the empirical rule is invalid in CDCl3. In addition, the introduction of an economic camphanoyl group with larger steric hindrance in this method may provide a reference for solving the stereochemistry problems of other flexible conformer compounds.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was funded by the National Natural Science Foundation of China (No. 81973194) and the Drug Innovation Major Project (No. 2018ZX09711001-008). We appreciate Ms. Y. H. Wang (Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College) for testing the NMR spectra.
References
- X. F. Hou, S. Yao, A. Mándi, T. Kurtán, C. P. Tang, C. Q. Ke, X. Q. Li and Y. Ye, Bicunningines A and B, two new dimeric diterpenes from Cunninghamia lanceolate, Org. Lett., 2012, 14, 460 CrossRef CAS
 .
. - N. Matsumori, D. Kaneno, M. Murata, H. Nakamura and K. Tachibana, Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. a method of configuration analysis for natural products, J. Org. Chem., 1999, 64, 866 CrossRef CAS .
- S. Higashibayashi and Y. Kishi, Assignment of the relative and absolute configurations of acyclic secondary 1,2-diols, Tetrahedron, 2004, 60, 11977 CrossRef CAS .
- K. Xu, P. F. Yang, Y. N. Yang, Z. M. Feng, J. S. Jiang and P. C. Zhang, Direct assignment of the threo and erythro configurations in polyacetylene glycosides by 1H NMR spectroscopy, Org. Lett., 2017, 19, 686 CrossRef CAS .
- S. Y. Shao, F. Zhang, Y. N. Yang, Z. M. Feng, J. S. Jiang and P. C. Zhang, An approach for determining the absolute configuration of C-2 in 2-oxygenated phenylethanoid glycosides by 1H NMR spectroscopy, Org. Lett., 2016, 18, 4084 CrossRef CAS .
- T. Deyama, T. Ikawa, S. Kitagawa and S. Nishibe, The constituents of Eucommia ulmoides OLIV. V. isolation of dihydroxydehydrodiconiferyl alcohol isomers and phenolic compounds, Chem. Pharm. Bull., 1987, 35, 1785 CrossRef CAS .
- T. Ishikawa, E. Fujimatu and J. Kitajima, Water-soluble constituents of anise: new glucosides of anethole glycol and its related compounds, Chem. Pharm. Bull., 2002, 50, 1460 CrossRef CAS .
- H. Matsuura, H. Miyazaki, C. Asakawa, M. Amano, T. Yoshihara and J. Mizutani, Isolation of α-glusosidase inhibitors from hyssop (Hyssopus officinalis), Phytochemistry, 2004, 65, 91 CrossRef CAS .
- S. Lin, S. J. Wang, M. T. Liu, M. L. Gan, S. Li, Y. C. Yang, Y. H. Wang, W. Y. He and J. G. Shi, Glycosides from the stem bark of Fraxinus sieboldiana, J. Nat. Prod., 2007, 70, 817 CrossRef CAS .
- M. L. Gan, Y. L. Zhang, S. Lin, M. T. Liu, W. X. Song, J. C. Zi, Y. C. Yang, X. N. Fan, J. G. Shi, J. F. Hu, J. D. Sun and N. H. Chen, Glycosides from the root of Iodes cirrhosa, J. Nat. Prod., 2008, 71, 647 CrossRef CAS .
- X. Y. Chai, H. Y. Ren, Z. R. Xu, C. C. Bai, F. R. Zhou, S. K. Ling, X. P. Pu, F. F. Li and P. F. Tu, Investigation of two Flacourtiaceae plants: Bennettiodendron leprosipes and Flacourtia ramontchi, Planta Med., 2009, 75, 1246 CrossRef CAS .
- L. Wang, F. Li, C. Y. Yang, A. A. Khan, X. Liu and M. K. Wang, Neolignans, lignans and glycoside from the fruits of Melia toosendan, Fitoterapia, 2014, 99, 92 CrossRef CAS .
- Y. L. Li, Y. X. Gao, H. Z. Jin, L. Shan, W. L. Chang, X. W. Yang, H. W. Zeng, N. Wang, A. Steinmetz and W. D. Zhang, Chemical constituents of Abies fabri, Phytochemistry, 2015, 117, 135 CrossRef CAS .
- X. X. Huang, M. Bai, L. Zhou, L. L. Lou, Q. B. Liu, Y. Zhang, L. Z. Li and S. J. Song, Food byproducts as a new and cheap source of bioactive compounds: lignans with antioxidant and anti-inflammatory properties from Crataegus pinnatifida seeds, J. Agric. Food Chem., 2015, 63, 7252 CrossRef CAS .
- F. H. Li, J. Zhang, M. B. Lin, X. M. Su, C. K. Li, H. Q. Wang, B. M. Li, R. Y. Chen and J. Kang, Anti-inflammatory terpenes from Schefflera rubriflora C. J. Tseng & G. Hoo with their TNF-α and IL-6 inhibitory activities, Phytochemistry, 2019, 163, 23 CrossRef CAS .
- P. Chakraborty, S. Jana, S. Saha and S. C. Roy, Titanocene (III) chloride mediated formal synthesis of magnofargesin and 7’-epimagnofargesin, Tetrahedron Lett., 2012, 53, 6584 CrossRef CAS .
- K. Hernández, T. Parella, J. Joglar, J. Bujons, M. Pohl and P. Clapés, Role of the bridge in photoinduced electron transfer in porphyrin–fullerene dyads, Chem.–Eur. J., 2015, 21, 1 CrossRef .
- A. R. Patel, X. G. Hu, A. Lawer, M. I. Ahmed, C. Au, R. Jwad, J. Trinh, C. Gonzalez, E. Hannah, M. M. Bhadbhade and L. Hunter, Scalable, stereoselective syntheses of α,β-difluoro-γ-amino acids, Tetrahedron, 2016, 72, 3305 CrossRef CAS .
- D. M. M. Barrett, A. R. Neal, C. Hand, J. R. D. Montgomery, I. Panovic, O. S. Ojo, C. S. Lancefield, D. B. Cordes, A. M. Z. Slawin, T. Lebl and N. J. Westwood, The synthesis and analysis of lignin-bound Hibbert ketone structures in technical lignins, Org. Biomol. Chem., 2016, 14, 10023 RSC .
- D. M. M. Barrett, J. R. D. Montgomery, C. S. Lancefield, D. B. Cordes, A. M. Z. Slawin, T. Lebl, R. Carr and N. J. Westwood, Use of bisulfite processing to generate high-β-O-4 content water-soluble lignosulfonates, ACS Sustainable Chem. Eng., 2017, 5, 1831 CrossRef .
- L. Monsigny, E. Feghali, J. C. Berthet and T. Cantat, Efficient reductive depolymerization of hardwood and softwood lignins with Brookhart's iridium (iii) catalyst and hydrosilanes, Green Chem., 2018, 20, 1981 RSC .
- E. Lallana, F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, The 1H NMR method for the determination of the absolute configuration of 1,2,3-prim,sec,sec-triols, Org. Lett., 2006, 8, 4449 CrossRef CAS .
- F. Freire, E. Lallana, E. Quiñoá and R. Riguera, The Stereochemistry of 1,2,3-triols revealed by 1H NMR spectroscopy: principles and applications, Chem.–Eur. J., 2009, 15, 11963 CrossRef CAS .
- Y. N. Yang, B. Han, P. F. Yang, Z. M. Feng, J. S. Jiang and P. C. Zhang, A concise approach for determining the relative configuration of H-7 and H-8 in 8,4’-oxyneolignans by 1H NMR spectroscopy, Org. Chem. Front., 2019, 6, 886 RSC .
- Y. Gao, R. M. Hanson, J. M. Klunder, S. Y. Ko, H. Masamune and K. B. Sharpless, Catalytic asymmetric epoxidation and kinetic resolution: modified procedures including in situ derivatization, J. Am. Chem. Soc., 1987, 109, 5765 CrossRef CAS .
- C. S. Philbin and S. J. Schwartz, Resolution of diastereomeric flavonoid (1S)-(−)-camphanic acid esters via reversed-phase HPLC, Phytochemistry, 2007, 68, 1206 CrossRef CAS .
- C. Meier, W. H. G. Laux and J. W. Bats, Asymmetric synthesis of chiral, nonracemic dialkyl α-hydroxyarylmethyl and α-, β- and γ-hydroxyalkylphosphonates from keto phosphonates, Liebigs Ann. Chem., 1995, 1995, 1963 CrossRef .
- L. Pisano, L. Degennaro, M. Carraro, U. Azzena, F. Fanelli, P. Mastrorilli and R. Luisi, Computational NMR as useful tool for predicting structure and stereochemistry of four-membered sulfur heterocycles, Eur. J. Org. Chem., 2016, 2016, 3252 CrossRef CAS .
- X. Hao, T. Nguyen, D. B. Kearns, C. C. Arpin, R. Fall and T. Sammakia, New inhibitors of colony spreading in Bacillus subtilis and Bacillus anthracis, Bioorg. Med. Chem. Lett., 2011, 21, 5583 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: 1D NMR, 2D NMR, and HRMS spectra. See DOI: 10.1039/d0ra09712h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Jian-Shuang Jiang and
Pei-Cheng Zhang
,
Jian-Shuang Jiang and
Pei-Cheng Zhang