
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiol-yne click reaction: an interesting way to derive thiol-provided catechols†
Fabiana Nador *a,
Juan Mancebo-Aracila,
Duham Zanottoa,
Daniel Ruiz-Molinab and
Gabriel Radivoya
*a,
Juan Mancebo-Aracila,
Duham Zanottoa,
Daniel Ruiz-Molinab and
Gabriel Radivoya
aInstituto de Química del Sur (INQUISUR-CONICET), Universidad Nacional del Sur, Av. Alem 1253, 8000 Bahía Blanca, Argentina. E-mail: fabiana.nador@uns.edu.ar
bInsitut Català de Nanociència i Nanotecnologia (ICN2), Edifici ICN2, UAB Campus, Bellaterra, 08193 Barcelona, Spain
First published on 7th January 2021
Abstract
The hydrothiolation of activated alkynes is presented as an attractive and powerful way to functionalize thiols bearing catechols. The reaction was promoted by a heterogeneous catalyst composed of copper nanoparticles supported on TiO2 (CuNPs/TiO2) in 1,2-dichloroethane (1,2-DCE) under heating at 80 °C. The catalyst could be recovered and reused in three consecutive cycles, showing a slight decrease in its catalytic activity. Thiol derivatives bearing catechol moieties, obtained through a versatile Michael addition, were reacted with different activated alkynes, such as methyl propiolate, propiolic acid, propiolamide or 2-ethynylpyridine. The reaction was shown to be regio- and stereoselective towards anti-Markovnikov Z-vinyl sulfide in most cases studied. Finally, some catechol derivatives obtained were tested as ligands in the preparation of coordination polymer nanoparticles (CNPs), by taking the advantage of their different coordination sites with metals such as iron and cobalt.
Introduction
Vinyl sulfides are of great interest because they can be used as versatile building blocks in organic synthesis1 and as intermediates for the synthesis of biologically active compounds2 and new materials,3 among others.The thiol-yne click (TYC) reaction, also known as alkyne hydrothiolation, is one of the simplest and most atom-economical approach to produce alkenyl sulfides from thiols and alkynes.4,5 The TYC reaction commonly occurs in the presence of free radicals,6 strong acids,7 bases8 or transition metals9 and, as shown in Scheme 1, in principle can lead to one of the regio- and stereoisomeric vinyl sulfides: A (branched) through a Markovnikov orientation, B (E linear) and C (Z linear), or produce mixtures of them through an anti-Markovnikov orientation (Scheme 1).10,11
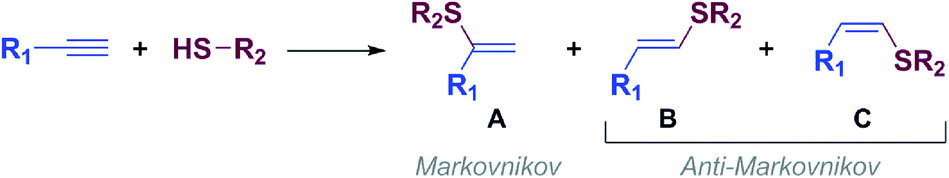 | ||
| Scheme 1 Schematic representation of alkyne hydrothiolation. | ||
Regardless of the method used to promote the reaction, it is important to bear in mind that both alkyne and thiol groups are inherently nucleophilic, hence the activation of one or both groups is necessary for the construction of vinyl sulfides.5 In this sense, the use of activated alkynes, meaning alkynes bearing an adjacent electron-withdrawing group (such as carboxylic acid, ester or amide group), resulted in a reduction of the electronic density on the alkyne, thus enhancing the nucleophilic attack of the thiol. The reaction of thiols, especially aliphatic ones, with propiolic acid esters is very slow in the absence of catalysts, but is remarkably accelerated by bases12 due to the increased concentration of thiolate ions in an alkaline solution.13 Because of its click characteristics, which include a fast, quantitative and selective reaction under mild conditions, the hydrothiolation of activated alkynes has been used in many interesting applications, such as polymer syntheses14 or post-polymerization modifications,15,16 the design of covalent adaptable networks (CANs)17 and the chemoselective modification of amino acids side chains in unprotected peptides.13,18
On the other hand, in recent years, an increasing number of works have focused on the synthesis of new bioinspired catechol-based molecules, which have shown to be useful in the fabrication of novel functional materials,19 including adhesives,20 capsules,21 coatings,22 hydrogels23 and in the formation of coordination polymer nanoparticles (CNPs).24 However, despite the extraordinary characteristics of catechol-derived structures, their preparation is challenging mainly due to the vulnerability of catechol to oxidation. As a result, only a few synthetic methodologies have been described and even among these, relatively lengthy synthetic routes, comprising protection/deprotection of the catechol ring, harsh reaction conditions and overall poor atom economy have been reported.25 Accordingly, some members of our group recently reported a systematic and straightforward methodology for the synthesis of functional catechols with thiol pendant groups, obtained by oxidation of the simplest catechol (pyrocatechol) to the corresponding o-quinone, followed by the conjugated nucleophilic addition of different dithiol molecules.26,27
Although there are several examples of methodologies with catechol derivatives where the thiol–ene reaction has been exploited, the thiol-yne reaction in the presence of catechol moiety has been less explored. To the best of our knowledge, only two very interesting examples related to this issue have been published. A functional copolymer polyPDP, containing blocks with alkyne and catechol pendant groups respectively, easily reacted with 4-arm thiol PEG via a TYC reaction under UV exposure, to produce self-adhesive hydrogels,28 while another study described the reduction of graphene oxide to graphene by alkynyl-terminated dopamine, and post-functionalization with thiols by photochemical click reaction between the alkyne and thiols.29 As described in both articles, the TYC reaction was activated by UV irradiation, but resulted in a double addition of thiol molecules on account of the fact that radicals react faster with the vinyl sulfide intermediate than with the starting alkyne. Thus, no vinyl sulfide could be obtained through the TYC reaction in the presence of catechols.
On this basis, we attempted to apply TYC reaction to the modification of catechol structures bearing a thiol functionality as end-group. As already mentioned, in most cases the addition of a base to promote the hydrothiolation reaction of activated alkynes, is mandatory. However, the catechol moiety is not compatible with even moderately basic media, due to its oxidation to o-quinone by atmospheric oxygen or another mild oxidant. An alternative is to carry out the hydrothiolation of alkynes substituted with electron-withdrawing groups and promoted by transition metals, thus taking advantage of the beneficial effect of an increase in the nucleophilicity of the thiol caused by the presence of metal species. In this context, copper-based catalytic systems are of particular interest since the activation of both the alkyne and the thiol is thought to be the key step in this reaction.5
Here we report our results on the thiol-yne click reaction between activated alkynes, substituted with electron-withdrawing groups, and different thiol derivatives bearing catechol pendant moieties. The reaction was regio- and stereoselective towards the anti-Markovnikov Z-vinyl sulfide in the vast majority of cases studied. The heterogeneous and low cost CuNPs/TiO2 catalyst also showed a high activity and good recyclability. Additionally, some of the vinyl sulfides obtained were used in the preparation of CNPs, taking advantage of their chelating properties.
Results and discussion
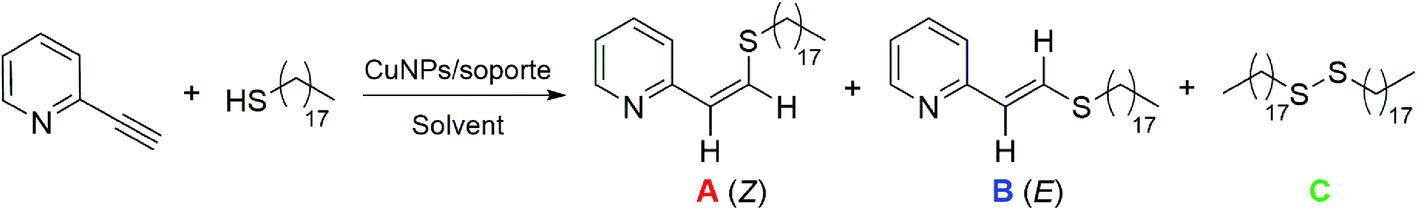
The catalysts were prepared by the addition of the support to a suspension of freshly prepared copper nanoparticles (CuNPs). The CuNPs were generated by the fast reduction of anhydrous copper(II) chloride, using lithium sand and a catalytic amount of DTBB (4,4′-di-tert-butylbiphenyl, 10 mol%) as the reducing system, in THF at room temperature (see Scheme S1†). The catalysts were ready for use as prepared, after filtration and drying, and without any pre-treatment. Different inorganic materials, such as activated carbon, nanosized silica coated maghemite (MagSilica®), TiO2, zeolite Y (ZY), MgO, Al-MCM-41, ZnO and CeO2, were tested as supports for the CuNPs.As listed in Table 1, 2-ethynylpyridine and 1-octadecanethiol were used as model substrates in order to test the activity of different catalysts in the TYC reaction. The reaction of 2-ethynylpyridine (0.1 mmol) with 1-octadecanethiol (0.1 mmol) was conducted in the presence of different CuNPs/support catalysts (10 mg) in THF at 25 °C for 20 h. The most promising systems are shown in Table 1, entries 1–3. In these cases, the regioselectivity was excellent, providing only the anti-Markovnikov adduct, but the stereoselectivity was moderate, giving an average Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E isomers ratio of near 35:65. The observed conversion was low for both the CuNPs/TiO2- and CuNPs/ZnO-catalyzed TYC reactions (entries 2 and 3), with 40% of 1-octadecanethiol remaining unreacted. In contrast, the reaction promoted by the CuNPs/C catalyst gave a full conversion to disulfide C as the main product, together with vinyl sulfides A and B in a lower proportion (entry 1). For the reaction catalyzed by CuNPs/TiO2, an increase in the catalyst loading significantly improved the conversion to products A and B, but with a marked drop in the stereoselectivity (entry 4). In addition, a rise in temperature to 66 °C led to a 77% conversion to the desired products, leaving no unreacted thiol (entry 5). However, the very good conversion towards A and B was accompanied by an increase in the amount of disulfide formed, even working under a nitrogen atmosphere.
:E isomers ratio of near 35:65. The observed conversion was low for both the CuNPs/TiO2- and CuNPs/ZnO-catalyzed TYC reactions (entries 2 and 3), with 40% of 1-octadecanethiol remaining unreacted. In contrast, the reaction promoted by the CuNPs/C catalyst gave a full conversion to disulfide C as the main product, together with vinyl sulfides A and B in a lower proportion (entry 1). For the reaction catalyzed by CuNPs/TiO2, an increase in the catalyst loading significantly improved the conversion to products A and B, but with a marked drop in the stereoselectivity (entry 4). In addition, a rise in temperature to 66 °C led to a 77% conversion to the desired products, leaving no unreacted thiol (entry 5). However, the very good conversion towards A and B was accompanied by an increase in the amount of disulfide formed, even working under a nitrogen atmosphere.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (Cu mg) | Solvent | Temp. (°C) | Conv. A + Bb (%) | Conv. Cb (%) | Selectivity Z:E |
| a Conditions: alkyne (0.1 mmol), thiol (0.1 mmol), catalyst, solvent (2 mL) and a reaction time of 20 h.b Conversion determined by 1H-NMR analysis of the mixture.c Reaction carried out under a N2 atmosphere.d Conversion could be not calculated due to the presence of many by-products. | ||||||
| 1 | CuNPs/C (10) | THF | 25 | 23 | 77 | 40:60 |
| 2 | CuNPs/TiO2 (10) | THF | 25 | 44 | 15 | 32:68 |
| 3 | CuNPs/ZnO (10) | THF | 25 | 31 | 30 | 31:69 |
| 4 | CuNPs/TiO2 (30) | THF | 25 | 65 | 14 | 43:57 |
| 5c | CuNPs/TiO2 (30) | THF | 66 | 77 | 23 | 49:51 |
| 6 | CuNPs/ZnO (15) | THF | 66 | 44 | 10 | 70:30 |
| 7 | CuCI2 (100) | THF | 66 | — | 100 | — |
| 8 | CuNPs/TiO2 (30) | Dioxane | 90 | d | — | 37:63 |
| 9 | CuNPs/TiO2 (30) | DMSO | 70 | — | 100 | — |
| 10 | CuNPs/TiO2 (30) | DCM | 40 | 75 | 22 | 85:15 |
| 11 | CuNPs/TiO2 (30) | DCE | 80 | 82 | 18 | 86:14 |
| 12 | TiO2 (0) | DCM | 40 | — | 60 | — |
A similar behaviour was observed with the CuNPs/ZnO catalyst, as the conversion also improved at a higher reaction temperature and catalyst loading, but in this case the stereoselectivity markedly favoured the Z-vinyl sulfide (entry 6). The use of CuCl2 as the catalyst gave a complete conversion to disulfide C (entry 7). With regard to the reaction solvent, previous reports have shown that the use of THF in the presence of Cu(I) and O2 favours the formation of the disulfide product.30 In view of this, we tested other solvents, such as dioxane, DMSO, DCM and 1,2-DCE. As shown in Table 1 (entries 8–11), when the reaction was carried out in DCM, almost the same conversion as in THF was observed but with a notably improved stereoselectivity to Z-vinyl sulfide (Z/E ratio = 85/15) (entry 10). Moreover, the use of 1,2-DCE as the solvent and the possibility that higher temperatures could be reached, substantially improved the conversion to the desired product and reduced the amount of disulfide formed (entry 11). The use of 1,2-DCE in the hydrothiolation reaction has been previously reported by other authors as leading to excellent results.31,32
Finally, no conversion to products A and B was observed in the presence of the support alone (entry 12). In view of these experimental observations, the best conditions found were those corresponding to entry 11 in Table 1.
The copper-on-titanium oxide catalyst was characterized by different techniques. The copper content in the catalysts, ca. 6.4–7.0 wt%, was determined by inductively coupled plasma optical emission spectroscopy (ICP-MS). Analysis by TEM revealed the presence of small spherical aggregates with an average size of 30 ± 15 nm, homogeneously distributed over seed-like particles of the support with a length between of 100–300 nm (see Fig. S1 and S2†). Energy-dispersive X-ray (EDX) analysis on various regions showed the presence of copper on the small spherical aggregates located on the surface, whereas titanium was detected mainly in the seed particles (Fig. S3†). Electron energy loss spectroscopy (EELS) was performed on those small spherical aggregates, confirming the presence of copper.
The heterogeneous nature of the CuNPs/TiO2 catalyst led us to test its recyclability. Thus, the hydrothiolation of propiolamide with 1-octadecanothiol was carried out and the catalyst could be recovered by filtration, washed with DCM and reused at least three times without a significant loss of its regio- or stereoselectivity. As shown in Fig. 1, the conversion value showed a drop of about 10% in the activity of the first cycle, but remained constant along the last two cycles.
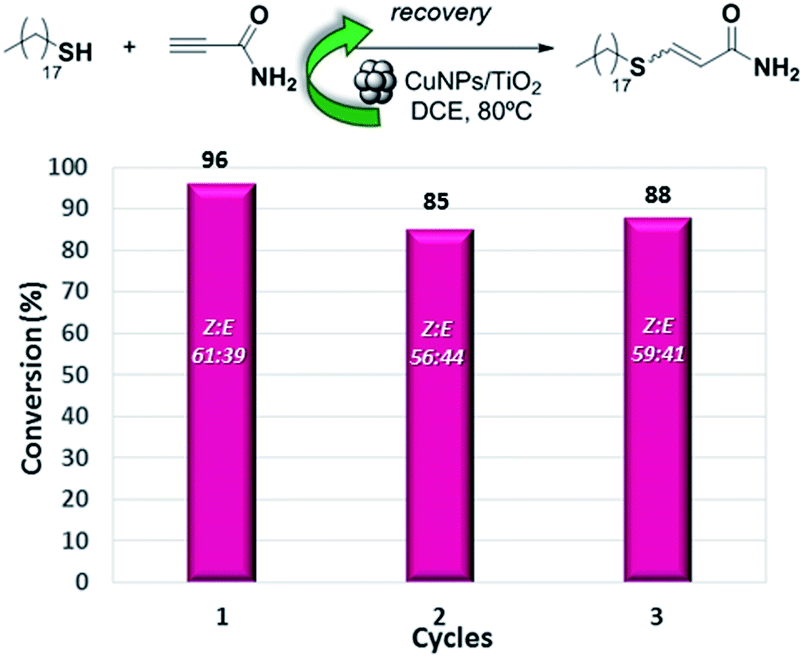 | ||
| Fig. 1 Recycling of the CuNPs/TiO2 catalyst in the synthesis of 3-(heptadecylthio)acrylamide. | ||
With the optimized conditions in hand, the scope of the TYC reaction was studied for a series of alkynes (2), mainly terminal ones, with several thiols bearing catechol pendant groups. The starting catechols were synthesized according to the procedure previously reported by our group, which was based on the attachment of dithiolated chains to pyrocatechol in two stages (see Scheme 2a).26 Here, pyrocatechol is first oxidized to o-benzoquinone with NaIO4 in an aqueous solution. Then, the solution is extracted with DCM, and the dry organic extracts are added over a solution of a dithiol and an excess of trifluoracetic acid (TFA) in DCM. At this point, the thiol produces a nucleophilic attack on the quinoid ring, giving a keto–enol intermediate that spontaneously tautomerizes into the substituted catecholic form, yielding the Michael adduct 1.
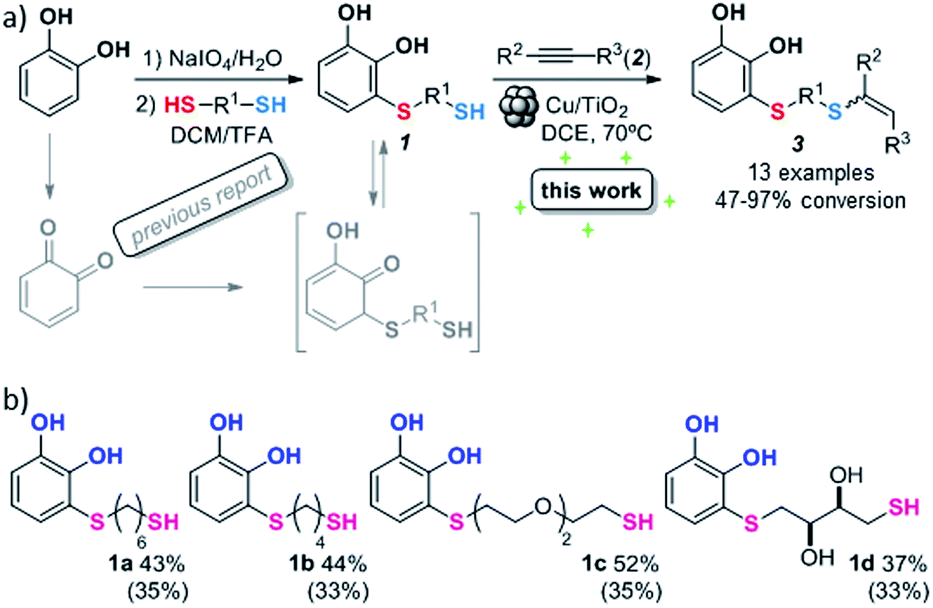 | ||
| Scheme 2 (a) Thiol conjugate addition and hydrothiolation of alkynes. (b) Synthesized Michael adducts with their respective conversion values. Isolated product yields are indicated in brackets. | ||
Applying this previously reported procedure, four Michael adducts were obtained with conversions in the range of 37–52% (Scheme 2b). Although the conversion values to the Michael adduct may seem to be rather low, it should be noted that the oxidation step from pyrocatechol to quinone occurs at around 75% conversion, thus leaving at least 25% of the starting dithiol unreacted. Considering this, and referencing the values of mono-adduct obtained for that initial 75% of quinone, the conversion values would be around 52–75%.
We then studied the conjugation of dithiols molecules bearing an apolar alkyl chain (1,6-hexanedithiol and 1,4-butanedithiol, 1a and 1b, respectively), a polar moiety (2,2-(ethylenedioxy)diethanethiol, 1c), and pendant hydroxyl groups (DL-dithiothreitol, 1d). The carbon chain between both thiol groups should function as a spacer in the catechol derivative, providing a degree of flexibility, as well as enough spatial separation when needed. Moreover, changes in the polarity of the spacer could modify the solubility of these adducts in aqueous or polar media. Additionally, the procedure for obtaining the Michael adduct 1 could be successfully scaled up to a 9 mmol scale, keeping the same conversion value as that of the reaction carried out at the 1 mmol scale.
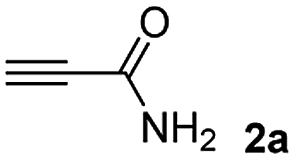
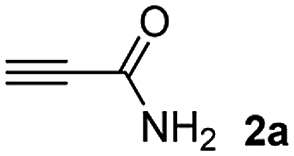
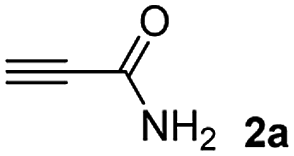
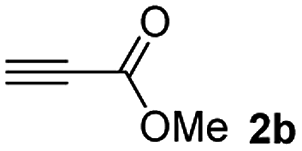
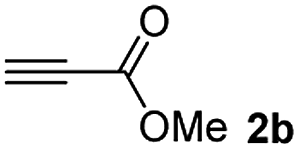
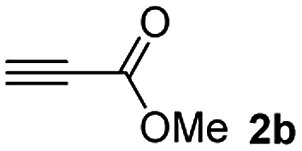
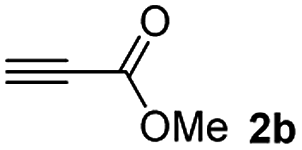
The results of the TYC reaction between the prepared Michael adducts 1a–d and different activated alkynes with adjacent electron-withdrawing groups are presented in Table 2. The starting alkynes were readily synthesized from methyl propiolate and acetylenedicarboxylic acid, respectively (see Experimental section). Under the optimized conditions shown in Table 1, the alkyne (0.4 mmol) and the thiol (0.4 mmol) were then reacted upon heating in 1,2-DCE at 80 °C in the presence of the CuNPs/TiO2 catalyst (120 mg, 30.2 mol%). Fortunately, the hydrothiolation of propiolamide (2a) with 1a produced an excellent conversion to 3aa with a very good stereoselectivity (Z/E ratio = 83/17) after 41 h (Table 2, entry 1). Similar promising results were obtained with the Michael adducts 1b and 1c (entries 2 and 3). When other activated alkynes, such as methyl propiolate 2b, were tested, the products of the TYC reaction followed a similar trend as that observed for 2a, that is, good to excellent conversions into the anti-Markovnikov vinyl sulfide and very good Z stereoselectivity (entries 4–7).
| Entry | Michael adduct | Alkyne | Time (h) | Product | % Conv.b,c | Selectivity Z:Ed |
|---|---|---|---|---|---|---|
| a Conditions: alkyne (0.4 mmol), Michael adduct (0.4 mmol), catalyst (120 mg, 30.2 mol%), DCE (120 mg, 30.2 mol%), DCE (4 ml) and heating at 80 °C.b Conversion determined by 1H-NMR analysis of the crude mixture.c The yields of products are shown in brackets.d The selectivities Z:E of isolated products are shown in brackets are shown in brackets.e Conditions: alkyne (0.4 mmol), Michael adduct (0.5 mmol), catalyst (120 mg, 30.2 mol%), DCE (4 mL) and heating at −80 °C. |
||||||
| 1 | 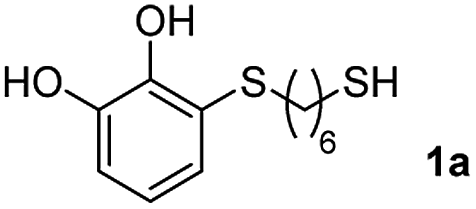 |
 |
41 | 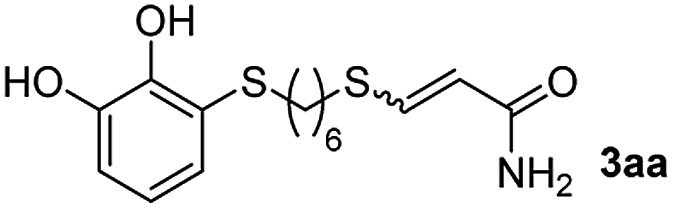 |
86 (40) | 83:17 (83:17) |
| 2 | 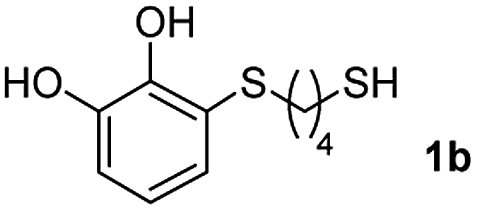 |
 |
22 | 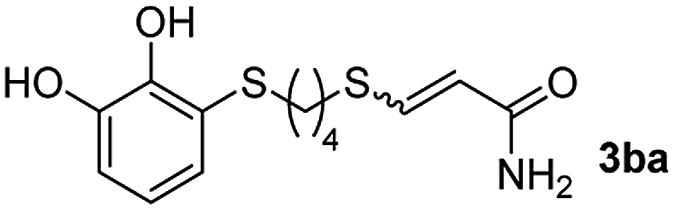 |
87 (53) | 86:14 (87:13) |
| 3 | 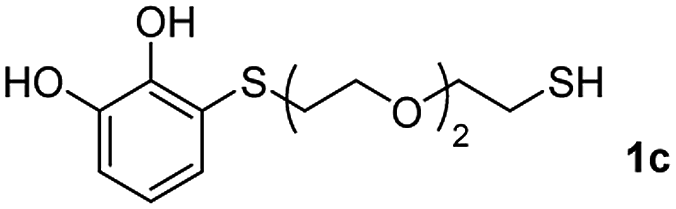 |
 |
24 | 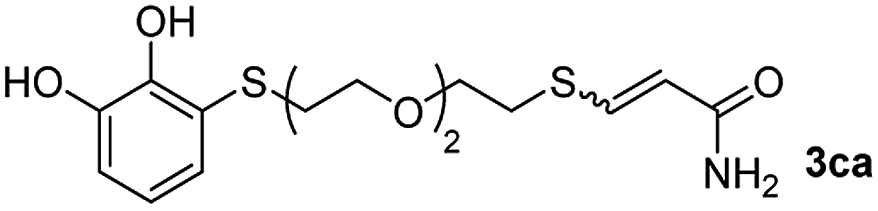 |
97 (69) | 85:15 (99:1) |
| 4 | 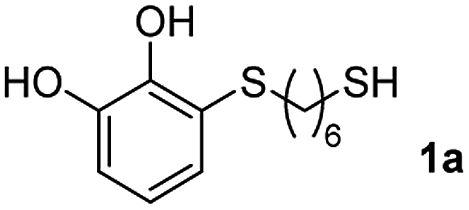 |
 |
48 | 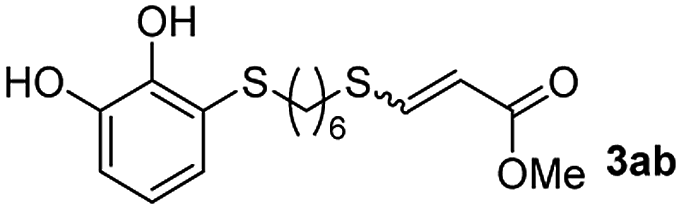 |
78 (39) | 82:18 (81:19) |
| 5 | 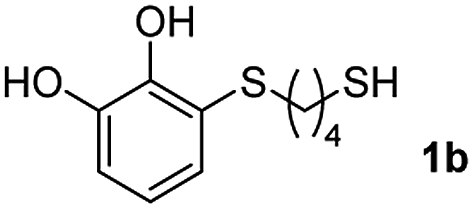 |
 |
48 | 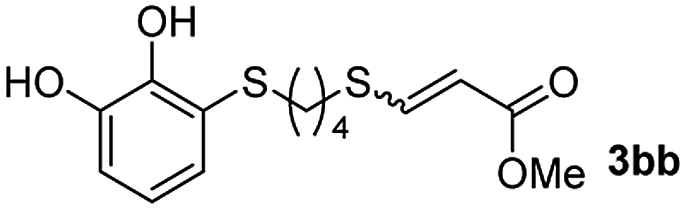 |
90 (46) | 86:14 (86:14) |
| 6 | 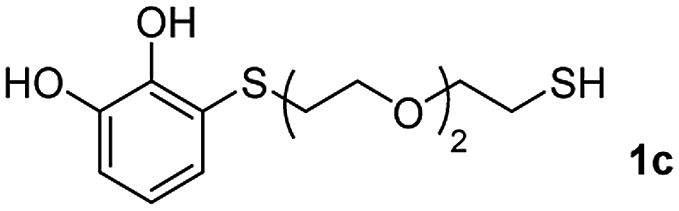 |
 |
48 | 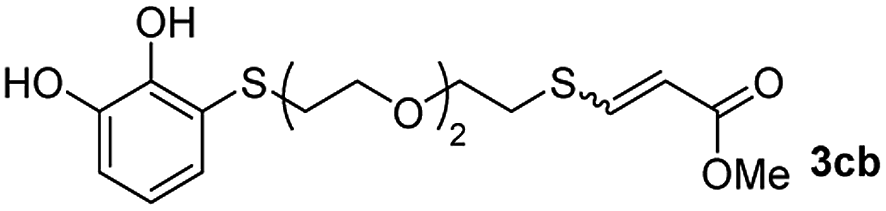 |
78 (36) | 79:21 (80:20) |
| 7 | 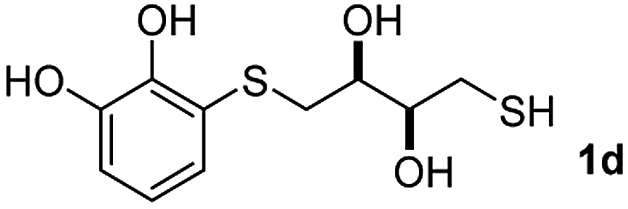 |
 |
72 | 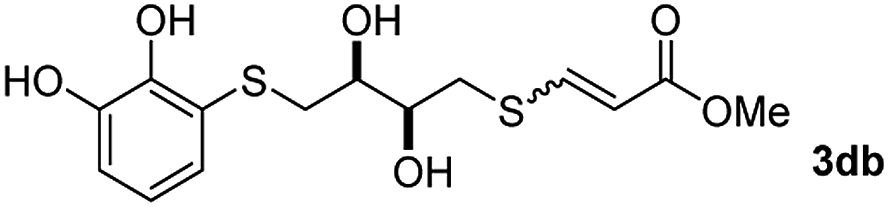 |
45 (23) | 90:10 (90:10) |
| 8 | 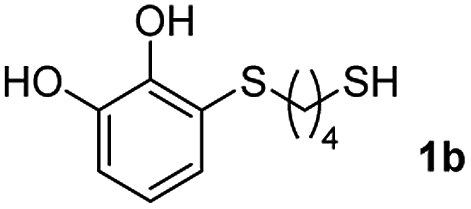 |
 |
24 | 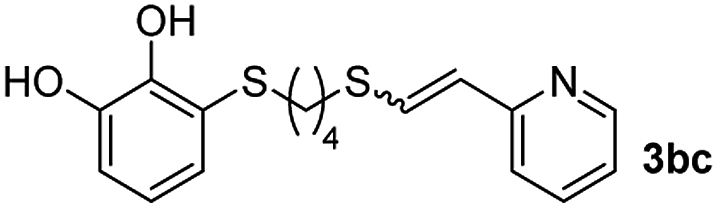 |
67 (50) | 81:19 (77:23) |
| 9 | 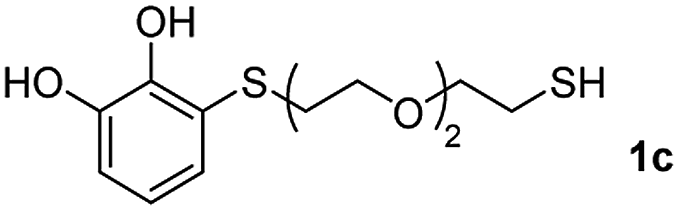 |
 |
24 | 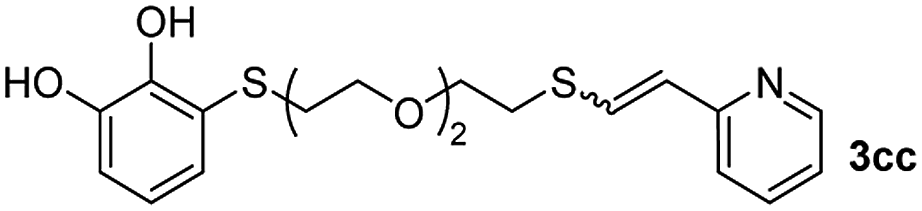 |
69 (37) | 58:44 (56:44) |
| 10e | 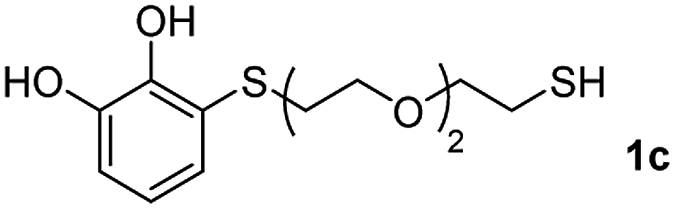 |
 |
24 | 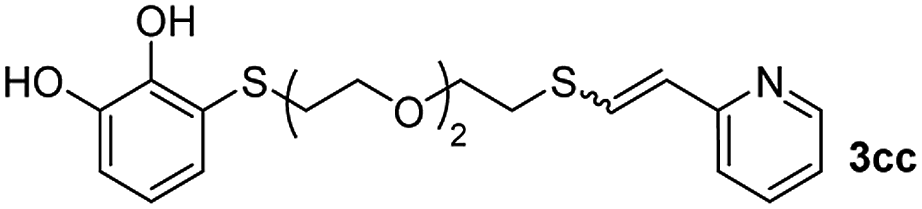 |
60 | 52:48 |
| 11 |  |
 |
46 | 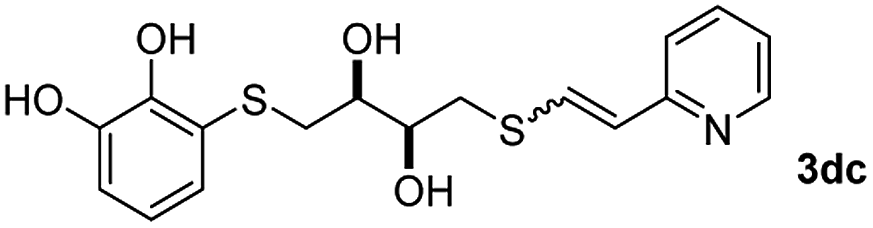 |
53 (21) | 64:36 (67:33) |
| 12 | 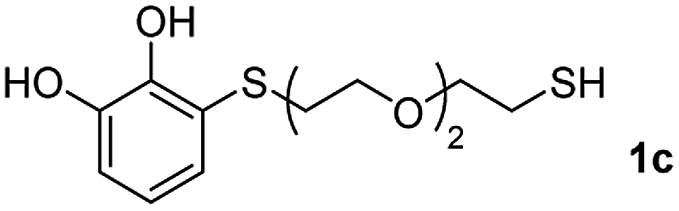 |
 |
96 | 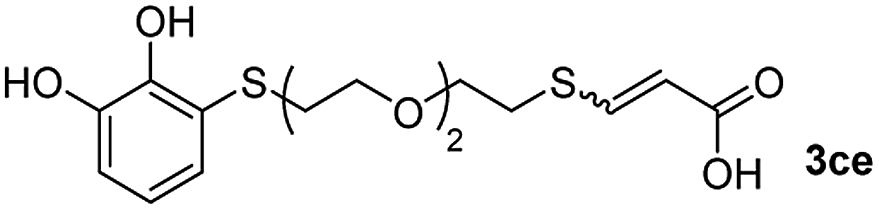 |
47 (14) | 99:1 (99:1) |
| 13 | 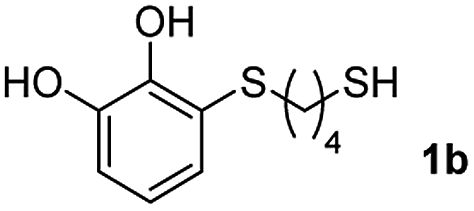 |
 |
24 | 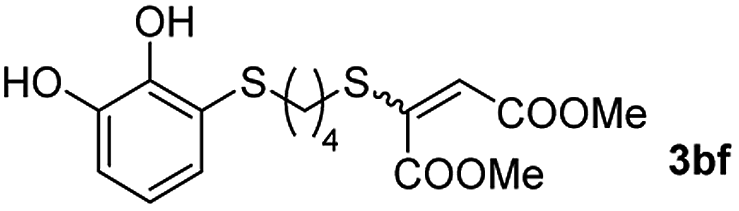 |
92 (31) | 52:48 (55:45) |
| 14 | 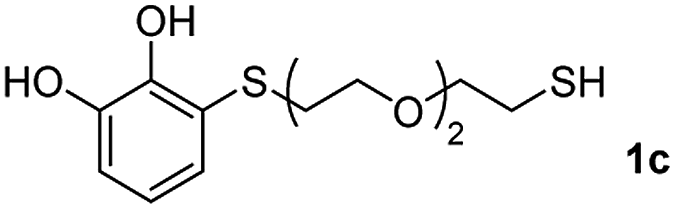 |
 |
24 | 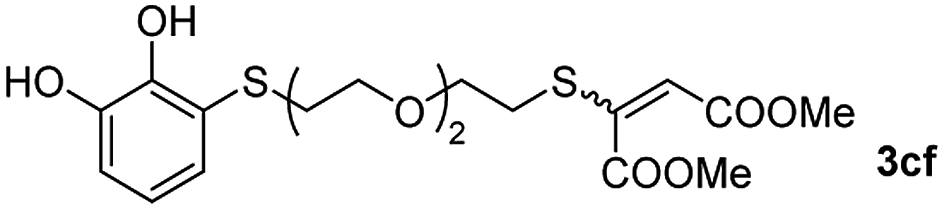 |
90 (63) | 40:60 (39:61) |
Interestingly, the corresponding s-cis and s-trans rotamers were detected for both products in entries 3 and 6. It is known that these types of conformers are usually observed in acyclic α,β-unsaturated esters, amides or carboxylic acids.33 In our case, both the cis isomer and s-cis rotamer were the major products, with a 1:1 ratio between them.
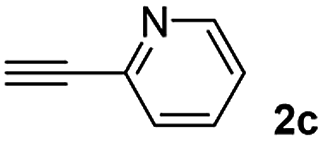
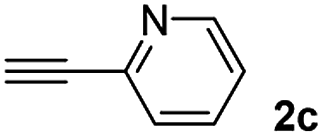
When 2-ethinyl pyridine (2c) was used as the starting alkyne, the conversions were significantly lower than those observed with propiolamide (2a) and methyl propiolate (2b). In all cases, it was observed that much of the starting thiol was quickly oxidized to the corresponding disulfide, with a considerable amount of 2c remaining unreacted (entries 8–11). The use of an excess of the thiol (1.25 equivalents) produced practically no change in conversion to 3cc (entry 10, footnote c).
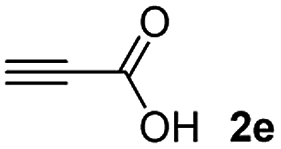
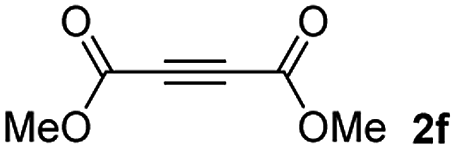
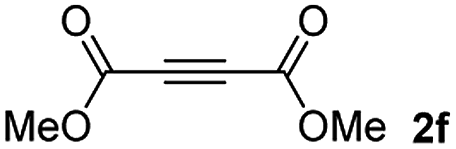
Among the terminal alkynes tested, the lowest conversion was obtained with propiolic acid (entry 12, 47%), even after 96 h of reaction. However, in this case, the stereoselectivity to the Z-isomer was excellent with a Z:E ratio of 99:1. On the other hand, the internal alkyne dimethyl acetylenedicarboxylate 2f gave the best conversion values when reacted with the Michael adducts 1b and 1c (entries 13–14). It is well known that the higher reactivity of 2f is due to the low electron density on the triple carbon–carbon bond which is connected to two carbonyl groups, and following this trend, when we tried to carry out the reaction using acetylenedicarboxylic acid, the conversion to the hydrothiolation product was relatively good. However, a considerable amount of decarboxylated product was observed.
With regard to the reaction time, it is important to note that all the thiols used (Michael adducts) were aliphatic and had different carbon-chain lengths. As has already been reported,10,12a,31 in many cases, aliphatic thiols require both longer reaction times and higher temperatures, to achieve satisfactory conversions compared to aromatic ones.
While the conversions observed in Table 2 ranged from good to excellent, a significant decrease in the isolated product yields was observed when the crude reaction products were purified. This fact was mainly attributed to catechol's numerous interactions,34 such as non-covalent forces and chemical bonding, with both the TiO2 and silica present in the catalyst and the stationary phase in the chromatographic column, respectively. A strategy that worked very effectively in some cases was to wash the crude reaction mixture with different mixtures of solvents (see Experimental section). For example, the products 3ca and 3bc could be recovered with only a loss of about 25% compared to the conversion values determined by NMR. On the other hand, it is important to note that practically in all cases, the relationship between the Z and E isomers was maintained even after column chromatography purification (see Table 2).
Although the exact mechanism involved is difficult to ascertain at this stage, based on our results and previous reports by other groups in the same area,5,35 a plausible mechanistic pathway could be proposed that proceeds via an external nucleophilic anti-Markovnikov attack from activated thiol species to the corresponding activated alkyne. In this context, it is worth mentioning that in our CuNPs/TiO2 catalyst, the Lewis acid character of the transition metal (Cu) and the acid–base character of the support (TiO2) could lead to activation of the alkyne, thiol or both.
On the one hand, the catalyst could be increasing the nucleophilicity of the thiol by deprononation of the SH group by the Lewis basic sites present at the “non-innocent” support (TiO2). This necessity for free basic sites on the catalyst surface to activate the thiol could also serve as an explanation for the low reactivity observed when propiolic acid was used as a starting alkyne (see entry 12, Table 2). Alternatively, the activation of the S–H bond by the very active CuNPs should not be disregarded. At this point, it should be noted that the TiO2 support could be playing another important role, that is, as a fixer of the catechol moiety to the surface of the catalyst by coordination between the Lewis acid sites on titanium and one or both of the oxygen atoms of the hydroxyl groups at the catechol aromatic ring.34 On the other hand, based on our experience working with CuNPs as catalysts for various alkyne transformations,36 one could assume that the activation of the carbon–carbon triple bond on the alkyne could proceed via π-coordination with copper. Another classical mode of activation for terminal alkynes, that cannot be disregarded involves the formation of metal–vinylidene species.35b If this were the case, the thiol would attack the electrophilic α-carbon of the cumulene to selectively provide the anti-Markovnikov products. Work in this area remains ongoing and we are actively carrying out some additional experiments and DFT calculations in order to disclose the precise mechanism for this CuNPs/TiO2-catalyzed thiol-yne reaction.
Application of compound 3 as a ligand in the synthesis of CNPs
As a preliminary test and taking into account the experiences of our group in the preparation of CNPs for different purposes, such as contrast agents,37 carriers in HIV/AIDS therapy38 or platforms for drug delivery,39 we decided to carry out the preparation of CNPs with some of the synthesized ligands and different metallic salts, such as FeCl3·6H2O and CoCl2·6H2O. The synthesis of CNPs is very straightforward and consisted of mixing the organic ligand in EtOH under magnetic stirring, and adding, drop by drop, the metallic salt dissolved in water, over the alcoholic phase. Following this methodology, several of the ligands synthesized were tested with Fe(III), showing the formation of an iron–catecholate complex in all cases as the solution instantaneously turned to blue-violet, followed by the formation of a precipitate. Then, the solution was centrifuged and washed several times with water and EtOH, and finally analyzed by SEM and EDX (Fig. 2). Among the cases evaluated, only some of them presented characteristics of a regularly structured material. For example, Fig. 2a and b show the SEM micrographs of the nanoparticles generated by the combination of ligands 3ca and 3ab with the FeCl3·6H2O salt, respectively. The images revealed the formation of oval-shaped nanoparticles, arranged in chains and clusters, with average diameters between 100–300 nm. Moreover, EDX analysis evidenced the presence of S, C, O and Fe elements in the entire analyzed surface.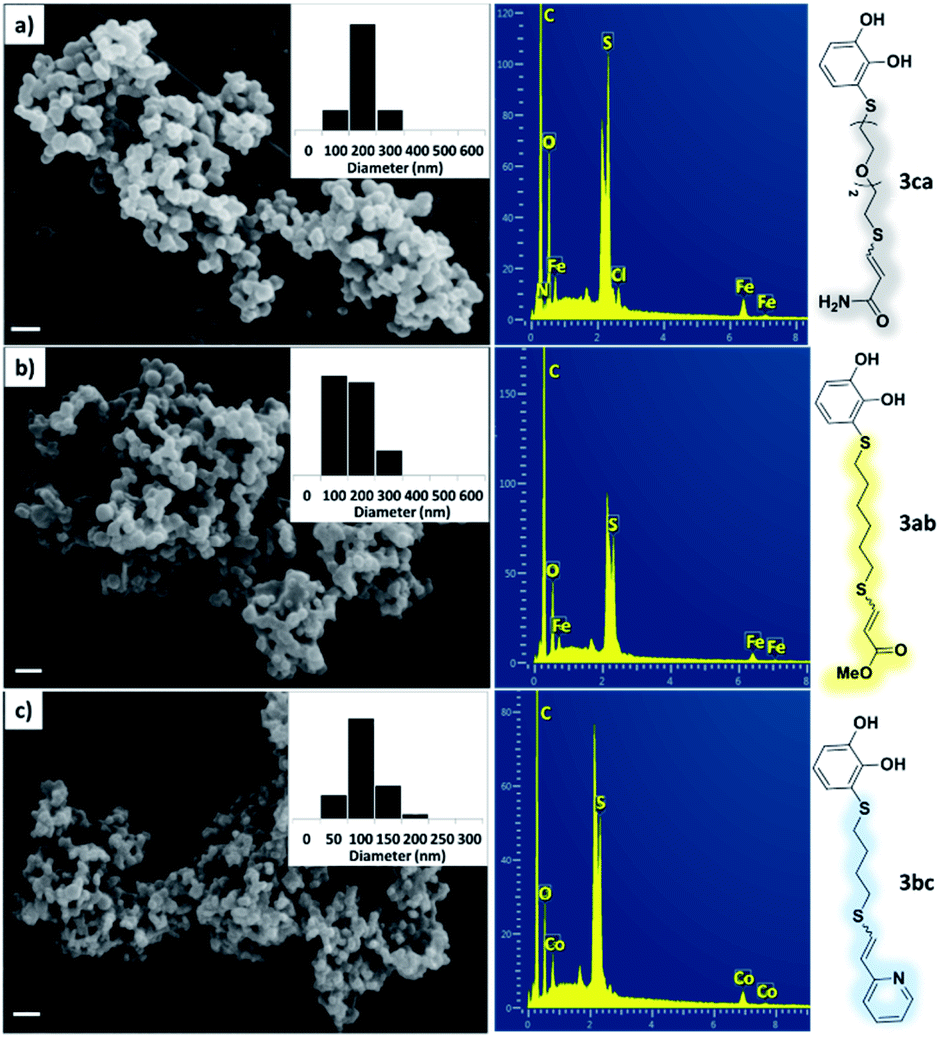 | ||
| Fig. 2 SEM micrographs, histograms and EDX spectra for CNPs prepared by mixing: (a) 3ca ligand with Fe(III), (b) 3ab ligand with Fe(III) and (c) 3bc ligand with Co(II). Scale bars are 500 nm. | ||
Fig. 2c shows the results obtained by analysis of the precipitate formed by mixing compound 3bc, with a pyridinic ring, and a Co(II) salt. Thus, the SEM micrograph revealed the formation of oval-shaped nanoparticles grouped in chains and clusters, with an average size of 100 ± 50 nm. Meanwhile, EDX analysis showed the presence of S, C, O and Co elements on the entire analyzed surface. This result was particularly interesting since the traditional methodology in our group for preparing CNPs has been one whereby the metallic salt is mixed with two types of ligands: corresponding to a catechol derivative and to a ditopic N,N′-ligand (1,4-bis(imidazole-1-ylmethyl)benzene (bix) or 4,4′-bipyridyl). In this way, it is estimated that the general structure achieved is that represented in Fig. S4.†40 However, more recently, it has been determined that, depending on the metallic salt used, there could be a higher proportion of coordinated catechol than bix, or quite the reverse, affecting both the structure of the polymer obtained as well as the correct elucidation of its repeating unit. The synthetic approach presented here, would allow us to have in the same ligand either the catechol group and the pyridine ring, which are both able to coordinate with the metal. It is important to mention that those ligands 3 without a pyridine ring in their structure were not able to form a precipitate with Co(II). Moreover, those ligands, such as 3cc bearing a pyridinic ring but with a longer spacer chain, were less prone to forming a precipitate, producing a slight turbidity, even after 24 h.
Conclusions
In summary, we have reported an interesting approach for the synthesis of vinyl sulfides bearing catechol pendant groups, via a thiol-yne click reaction, promoted by a heterogeneous catalyst based on CuNPs supported on TiO2, in 1,2-DCE and under heating at 80 °C. The results showed that the alkynes activated with electron-withdrawing groups adjacent to the triple bond were the most efficient in obtaining the desired products. Under these conditions, the reaction was regio- and stereoselective towards the formation of anti-Markovnikov Z-vinyl sulfides in the vast majority of cases, with good to excellent conversions (47–97%). The heterogeneous and low cost CuNPs/TiO2 catalyst also showed a high activity and good recyclability. While there are numerous reports in which the alkyne hydrothiolation is carried out by employing activated alkynes, this reaction remains poorly explored and even rarer in the presence of a catechol moiety.Regarding the mechanism of the reaction, although it is difficult to ascertain at this stage, we believe that, based on the experimental evidence, there may be a nucleophilic attack through activated thiol species, mainly by deprotonation of the SH group by the basic sites of TiO2. This necessity of free basic sites on the catalyst surface to activate the thiol could also serve as an explanation for the low reactivity observed when propiolic acid was tested as a starting alkyne. On the other hand, we assume that the activation of the carbon–carbon triple bond on the alkyne could proceed via π-coordination with copper.
Finally, as a preliminary test to demonstrate the potential of these types of new compounds as ligands, we carried out the preparation of CNPs by using selected vinyl sulfides and metallic salts, such as Fe(III) and Co(II). Analysis of the resulting CNPs by SEM and TEM demonstrated the formation of oval-shaped nanoparticles with average diameters between 100–300 nm with some of the ligands tested. This approach, in which several coordinating groups can coexist in the same ligand, would allow improvements in monitoring and characterizing these CNPs.
Overall, through this methodology, new products with potential application in the field of materials chemistry were obtained.
Experimental
General
All moisture-sensitive reactions were carried out under a nitrogen atmosphere. Anhydrous tetrahydrofuran was freshly distilled from sodium/benzophenone ketyl. All the starting materials were of the best available grade (Aldrich, Merck, Alfa Aesar) and were used without further purification. Nitric acid 69% and hydrochloric acid 30% used for analysis of the metal traces were purchased from Scharlab SL. The standard solution of Cu was obtained from Perkin Elmer. Commercially available copper(II) chloride dihydrate was dehydrated upon heating in an oven (150 °C, 45 min) prior to use for the preparation of the CuNPs. Column chromatography was performed with Merck silica gel 60 (0.040–0.063 μm, 240–400 mesh) and hexane/EtOAc as the eluent. The reactions were monitored by thin-layer chromatography on silica gel plates (60F-254) visualized under UV light and/or using FeCl3 in water for staining. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker ARX-300 spectrometer using CDCl3 or CD3OD as solvents. Nitric acid 69% and metal traces and chlorhydric acid 30% used for analysis of the metal traces by ICP-MS were purchased from Scharlab SL. The standard solution of Cu was obtained from Perkin Elmer.For CuNPs/TiO2 characterization, TEM and STEM images, together with EDX and EELS spectra, were obtained with an FEI Tecnai G2 F20 instrument coupled to an EDAX detector and a Quantum SE 963 Gatan Imaging Filter (GIF). The samples were prepared by casting a drop of the corresponding sample dispersion on a holey carbon copper grid, and then by evaporating off the solvent at room temperature. The copper content in the supported catalyst was determined by using an ICP-MS NexION 300X instrument from Perkin Elmer. Before its use, all glass material was washed with HNO3 40% for 72 h and rinsed with MilliQ water. Plastic material was washed with HNO3 0.5% and rinsed with MilliQ water before use. The standard solution of 1000 ppm Cu in 5% HNO3 was diluted up to 1 ppm and used to prepare the calibration curves (20, 40, 80, 120 and 240 ppb Cu). Digestion of the samples was carried out in 3 mL of concentrated ultrapure HNO3/HCl (1:1) in a Milestone ETHOS EASY microwave digester. The microwave system was set at 1800 W with a temperature ramp of 25 min up to 230 °C, then being kept for 15 min more at this temperature. Digested samples were diluted with HNO3 0.5% to a final dilution of 1/100, 1/400 and 1/1000 for later analysis. Elemental analysis was carried out using a Flash EA 2000 CHNS, Thermo Fisher Scientific system.
For CNPs characterization, SEM images were performed in a LEO EVO 40XVP system operated at 10 kV coupled to an EDX detector Oxford X-Max 50. Samples were prepared by drop casting of the corresponding dispersion on an aluminium tape followed by evaporation of the solvent under room conditions, then metallized with gold in a sputter coater.
Preparation of the CuNPs/TiO2 catalyst
Anhydrous copper(II) chloride (404 mg, 3 mmol) was added to a suspension of lithium (63 mg, 9 mmol) and 4,4′-di-tert-butylbiphenyl (DTBB, 80 mg, 0.3 mmol) in THF (10 mL) at room temperature under a nitrogen atmosphere. The reaction mixture, which was initially dark blue, rapidly changed to black, indicating that the suspension of copper nanoparticles was formed. This suspension was diluted with THF (10 mL) followed by the addition of the TiO2 (2.4 g). The resulting mixture was stirred for 1 h at room temperature, quenched with water and then filtered. The solid was successively washed with EtOH (15 mL) and dried under vacuum.Preparation of Michael adducts 1
For the preparation of 1a–d, 2 mmol of the corresponding dithiol was dissolved in 3 mL of CH2Cl2 in a Schlenk flask under nitrogen. For more polar dithiols, 3 mL of a 1:2 mixture MeOH:CH2Cl2 was used. To this solution, 460 μL of trifluoroacetic acid (TFA, 2 mmol) was added with stirring. In a separate flask, a solution of 468 mg of NaIO4 (2.2 mmol) in 80 mL of H2O was prepared and cooled in an ice bath. Next, 220 mg of pyrocatechol (2 mmol) dissolved in 1 mL of Et2O was added to the aqueous solution and left stirring vigorously for 15 min. The orange-reddish o-benzoquinone was extracted with 4 × 15 mL of CH2Cl2, and the organic phase was dried over anhydrous Na2SO4, filtered and immediately added to the dithiol solution. The reaction mixture was stirred in the dark at room temperature under nitrogen for 6 h. After this, the solvent and the TFA were evaporated under reduced pressure and the crude was purified by flash column chromatography (hexane–EtOAc) to give the corresponding Michael adducts. Compounds 1a–d were characterized by comparison of their physical and spectroscopic data with those described in the literature (see ESI S3†).26
Preparation of the alkynes
General procedure for the synthesis of vinyl sulfide, 3, catalyzed by CuNPs/TiO2
The Michael adduct 1 (0.4 mmol) and alkyne 2 (0.4 mmol) were added to a reaction tube containing CuNPs/TiO2 (120 mg, 30.2 mol% Cu) in 1,2-DCE (4 mL) under air. The reaction mixture was warmed to 80 °C and monitored by TLC until total conversion of the starting material. The catalyst was removed by filtration and washing with hexane–AcOEt. Finally, the filtrate was concentrated under vacuum and the product was purified by column chromatography to give the corresponding vinyl sulfide, 3. Full data for all the compounds are provided below (see ESI S3†).Authors' contributions
FN carried out the funding acquisition, conceptualization, investigation, methodology and writing. JMA participated in the conceptualization, investigation, methodology and writing. DZ participated in the methodology. DRM participated in the investigation. GR carried out the funding acquisition, investigation and writing. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ANPCyT (Project PICT 2016-0385), SGCyT-UNS (Project PGI 24/Q106) and CONICET (Project PIP 11220130100498CO). We thank Belén Ballesteros and Marcos Rosado from Electron Microscopy Unit of Institut Català de Nanociència i Nanotecnologia (ICN2) in Barcelona, Spain; and Maria Julia Yañez from Electron Microscopy Unit in Centro Científico Tecnológico – CONICET, Bahía Blanca, Argentina for their measurements and assistance. We thank Miguel Ángel Moreno-Villaécija for carrying out the ICP/MS analysis.Notes and references
- (a) J. Doroszuk, M. Musiejuk, Ł. Ponikiewski and D. Witt, Eur. J. Org. Chem., 2018, 6333–6337 CrossRef CAS; (b) A. Riesco-Domínguez, J. van de Wiel, T. A. Hamlin, B. van Beek, S. D. Lindell, D. Blanco-Ania, F. M. Bickelhaupt and F. P. J. T. Rutjes, J. Org. Chem., 2018, 83, 1779–1789 CrossRef.
- (a) M. S. Kabir, M. Lorenz, M. L. Van Linn, O. A. Namjoshi, S. Ara and J. M. Cook, J. Org. Chem., 2010, 75, 3626–3643 CrossRef CAS; (b) Q. Li, Ti. Dong, X. Liu and X. Lei, J. Am. Chem. Soc., 2013, 135, 4996–4999 CrossRef CAS.
- (a) A. B. Lowe, C. E. Hoyleb and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC; (b) M. M. Bassaco, M. Monçalves, F. Rinaldi, T. S. Kaufman and C. C. Silveira, J. Photochem. Photobiol., A, 2014, 290, 1–10 CrossRef CAS.
- K. Choudhuri, M. Pramanik, A. Mandal and P. Mal, Asian J. Org. Chem., 2018, 7, 1849–1855 CrossRef CAS.
- R. Castarlenas, A. Di Giuseppe, J. J. Pérez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2013, 52, 211–222 CrossRef CAS.
- (a) M. Lo Conte, S. Pacifico, A. Chambery, A. Marra and A. Dondoni, J. Org. Chem., 2010, 75, 4644–4647 CrossRef CAS; (b) L. Benati, L. Capella, P. C. Montevecchi and P. Spagnolo, J. Chem. Soc., Perkin Trans. 1, 1995, 1035–1038 RSC.
- (a) S. Kanagasabapathy, A. Sudalai and B. C. Benicewicz, Tetrahedron Lett., 2001, 42, 3791–3794 CrossRef CAS; (b) C. G. Screttas and M. Micha-Screttas, J. Org. Chem., 1979, 44, 713–719 CrossRef CAS.
- (a) A. Kondoh, K. Takami, H. Yorimitsu and K. Oshima, J. Org. Chem., 2005, 70, 6468–6473 CrossRef CAS; (b) Z.-L. Wang, R.-Y. Tang, P.-S. Luo, C.-L. Deng, P. Zhong and J.-H. Li, Tetrahedron, 2008, 64, 10670–10675 CrossRef CAS; (c) N. Zhao, C. Lin, L. Wen and Z. Li, Tetrahedron, 2019, 75, 3432–3440 CrossRef CAS.
- (a) S. N. Riduan, J. Y. Ying and Y. Zhang, Org. Lett., 2012, 14, 1780–1783 CrossRef CAS; (b) H. Ma, X. Ren, X. Zhou, C. Ma, Y. He and G. Huang, Tetrahedron Lett., 2015, 56, 6022–6029 CrossRef CAS.
- Y. Yang and R. M. Rioux, Chem. Commun., 2011, 47, 6557–6559 RSC.
- A. Dondoni and A. Marra, Eur. J. Org. Chem., 2014, 3955–3969 CrossRef CAS.
- (a) M. Kodomari, G. Saitoh and S. Yoshitomi, Bull. Chem. Soc. Jpn., 1991, 64, 2485–2487 CrossRef CAS; (b) P. D. Halphen and T. C. Owen, J. Org. Chem., 1973, 38, 3507–3510 CrossRef CAS; (c) Y. Sarrafi, M. Sadatshahabi, K. Alimohammadi and M. Tajbakhsh, Green Chem., 2011, 13, 2851–2858 RSC.
- H.-Y. Shiu, T.-C. Chan, C.-M. Ho, Y. Liu, M.-K. Wong and C.-M. Che, Chem.–Eur. J., 2009, 15, 3839–3850 CrossRef CAS.
- (a) O. Daglar, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2019, 52, 3558–3572 CrossRef CAS; (b) C. K. W. Jim, A. Qin, J. W. Y. Lam, F. Mahtab, Y. Yu and B. Z. Tang, Adv. Funct. Mater., 2010, 20, 1319–1328 CrossRef CAS; (c) A. B. Lowe, Polymer, 2014, 55, 5517–5549 CrossRef CAS; (d) O. Daglar, E. Cakmakci, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2020, 53(8), 2965–2975 CrossRef CAS.
- U. S. Gunay, M. Cetin, O. Daglar, G. Hizal, U. Tunca and H. Durmaz, Polym. Chem., 2018, 9, 3037–3054 RSC.
- (a) V. X. Truong and A. P. Dove, Angew. Chem., Int. Ed., 2013, 52, 4132–4136 CrossRef CAS; (b) O. Daglar, S. Luleburgaz, E. Baysak, U. S. Gunay, G. Hizal, U. Tunca and H. Dumaz, Eur. Polym. J., 2020, 137, 109926 CrossRef CAS.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2020, 59, 3609–3617 CrossRef CAS.
- G. T. Crisp and M. J. Millan, Tetrahedron, 1998, 54, 637–648 CrossRef CAS.
- (a) C. Yuan, J. Chen, S. Yu, Y. Chang, J. Mao, Y. Xu, W. Luo, B. Zeng and L. Dai, Soft Matter, 2015, 11, 2243–2250 RSC; (b) Y. Arntz, N. Kharouf and V. Ball, Colloids Surf., A, 2020, 593, 124624 CrossRef CAS.
- (a) Z. Li, S. Zhao, Z. Wang, S. Zhang and J. Li, J. Hazard. Mater., 2020, 396, 122722 CrossRef CAS; (b) K. Wei, B. Senturk, M. T. Matter, X. Wu, I. K. Herrmann, M. Rottmar and C. Toncelli, ACS Appl. Mater. Interfaces, 2019, 11, 47707–47719 CrossRef CAS.
- (a) F. Nador, E. Guisasola, A. Baeza, M. A. Moreno-Villaécija, M. Vallet-Regí and D. Ruiz-Molina, Chem.–Eur. J., 2016, 23, 2753–2758 CrossRef; (b) J. Cui, Y. Yan, G. K. Such, K. Liang, C. J. Ochs, A. Postma and F. Caruso, Biomacromolecules, 2012, 13, 2225–2228 CrossRef CAS.
- (a) M.-A. Moreno-Villaécija, J. Sedó-Vegara, E. Guisasola, A. Baeza, M. V. Regí, F. Nador and D. Ruiz-Molina, ACS Appl. Mater. Interfaces, 2018, 10, 7661–7669 CrossRef; (b) K. G. Malollari, P. Delparastan, C. Sobek, S. J. Vachhani, T. D. Fink, R. H. Zha and P. B. Messersmith, ACS Appl. Mater. Interfaces, 2019, 11, 43599–43607 CrossRef CAS.
- (a) M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS; (b) N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS.
- (a) F. Nador, F. Novio and D. Ruiz-Molina, Chem. Commun., 2014, 50, 14570–14572 RSC; (b) F. Nador, K. Wnuk, J. García-Pardo, J. Lorenzo, R. Solorzano, D. Ruiz-Molina and F. Novio, ChemNanoMat, 2018, 4, 183–193 CrossRef CAS.
- (a) I. Y. Chukicheva, I. V. Timusheva, L. V. Spirikhin and A. V. Kuchin, Chem. Nat. Compd., 2007, 43, 245–249 CrossRef CAS; (b) E. Findik, M. Ceylan and M. Elmastas, Eur. J. Med. Chem., 2011, 46, 4618–4624 CrossRef CAS; (c) J. Duan, W. Wu, Z. Wei, D. Zhu, J. Tu and A. Zhang, Green Chem., 2018, 20, 912–920 RSC; (d) Y. Kuninobu, T. Matsuki and K. Takai, J. Am. Chem. Soc., 2009, 131, 9914–9915 CrossRef CAS; (e) X. Liu, Y. Ou, S. Chen, X. Lu, H. Cheng, X. Jia, D. Wang and G.-C. Zhou, Eur. J. Med. Chem., 2010, 45, 2147–2153 CrossRef CAS.
- J. Mancebo-Aracil, C. Casagualda, M. A. Moreno-Villaécija, F. Nador, J. García-Pardo, A. Franconetti-García, F. Busqué, R. Alibés, M. J. Esplandiu, D. Ruiz-Molina and J. Sedó-Vegara, Chem.–Eur. J., 2019, 25, 12367–12379 CrossRef CAS.
- J. Mancebo-Aracil, J. Sedó-Vegara and D. Ruiz-Molina, WO2019025498, 2019.
- W. J. Yang, W. Xu, X. Tao, W. Wang, Y. Hu, X. Li, E.-T. Kang and L. Wang, Polym. Chem., 2020, 11, 2986–2994 RSC.
- I. Kaminska, W. Qi, A. Barras, J. Sobczak, J. Niedziolka-Jonsson, P. Woisel, J. Lyskawa, W. Laure, M. Opallo, M. Li, R. Boukherroub and S. Szunerits, Chem.–Eur. J., 2013, 19, 8673–8678 CrossRef CAS.
- Y. Yang, W. Dong, Y. Guo and R. M. Rioux, Green Chem., 2013, 15, 3170–3175 RSC.
- Y. Yang and R. M. Rioux, Green Chem., 2014, 16, 3916–3925 RSC.
- C. Cao, L. R. Fraser and J. A. Love, J. Am. Chem. Soc., 2005, 127, 17614–17615 CrossRef CAS.
- N. Shida, C. Kabuto, T. Niwa, T. Ebata and Y. Yamamoto, J. Org. Chem., 1994, 59, 4068–4075 CrossRef CAS.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2019, 58, 696–714 CrossRef CAS.
- (a) M. Kodomari, G. Saitoh and S. Yoshitomi, Bull. Chem. Soc. Jpn., 1991, 64, 3485–3487 CrossRef; (b) C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2006, 45, 2176–2203 CrossRef CAS.
- (a) F. Nador, L. Fortunato, Y. Moglie, C. Vitale and G. Radivoy, Synthesis, 2009, 4027–4031 CAS; (b) F. Alonso, Y. Moglie and G. Radivoy, Acc. Chem. Res., 2015, 46, 2516–2528 CrossRef; (c) F. Nador, M. A. Volpe, F. Alonso, A. Feldhoff, A. Kirschning and G. Radivoy, Appl. Catal., A, 2013, 455, 39–45 CrossRef CAS.
- (a) S. Suárez-García, N. Arias-Ramos, C. Frias Botella, A. P. Candiota, C. Arús, J. Lorenzo, D. Ruiz-Molina and F. Novio, ACS Appl. Mater. Interfaces, 2018, 10, 38819–38832 CrossRef; (b) M. Borges, S. Yu, A. Laromaine, A. Roig, S. Suárez-García, J. Lorenzo, D. Ruiz-Molina and F. Novio, RSC Adv., 2015, 5, 86779–86783 RSC.
- R. Solórzano, O. Tort, J. García-Pardo, T. Escribà, J. Lorenzo, M. Arnedo, D. Ruiz-Molina, R. Alibés, F. Busqué and F. Novio, Biomater. Sci., 2019, 7, 178–186 RSC.
- (a) F. Novio, J. Lorenzo, F. Nador, K. Wnuk and D. Ruiz-Molina, Chem.–Eur. J., 2014, 20, 15443–15450 CrossRef CAS; (b) A. Nayarassery, C. Frias, T. M. Ponnoth Lohidakshan, J. Lorenzo, F. Novio, J. Garcia-Pardo and D. Ruiz-Molina, Chem.–Eur. J., 2014, 20, 15443–15450 CrossRef.
- I. Imaz, D. Maspoch, C. Rodríguez-Blanco, J. M. Pérez-Falcón, J. Campo and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2008, 47, 1857–1860 CrossRef CAS.
- D. Strübing, H. Neumann, S. Klaus, S. Hübner and M. Beller, Tetrahedron, 2005, 61, 11333–11344 CrossRef.
- E. H. Huntress, T. E. Lesslie and J. Bornstein, Org. Synth., 1952, 32, 55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM and STEM images, EDX analysis and NMR spectra. See DOI: 10.1039/d0ra09687c |
| This journal is © The Royal Society of Chemistry 2021 |