
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysical insights into protection effect of conjugated polymers by natural antioxidants†
Mingcai Xie ,
Yan Hua,
Daocheng Hong,
Sushu Wan and
Yuxi Tian*
,
Yan Hua,
Daocheng Hong,
Sushu Wan and
Yuxi Tian*
Key Laboratory of Mesoscopic Chemistry of MOE, School of Chemistry and Chemical Engineering, Jiangsu Key Laboratory of Vehicle Emissions Control, Nanjing University, Nanjing, China. E-mail: tyx@nju.edu.cn
First published on 5th January 2021
Abstract
Conjugated polymers (CPs) known as organic semiconductors have been broadly applied in photovoltaic and light emitting devices due to their easy fabrication and flexibility. However, one of the bottlenecks limiting the application of CPs is their poor photostability upon continuous excitation which is one of the crucial parameters of CPs. How to improve the photostability of CPs is always one of the key questions in this field. In this work, we found that the photostability of poly(3-hexylthiophene-2,5-diyl) (P3HT) molecules can be largely improved by addition of vitamin E (VE) in bulk solution, solid films and single molecules. In solution and films, VE can not only significantly retard the photodegradation of P3HT but also enhance the fluorescence intensity. For individual P3HT molecules, with increasing VE concentrations, the on-time duration increases and the off-time duration becomes shorter. VE as natural antioxidants can not only donate electrons to the long-lived charged species but also quench the triplet states of CPs via energy transfer accelerating the depopulation process back to the ground state. The short duration time of the charged species and the triplet states provides higher fluorescence intensity. Furthermore, VE can also directly react with singlet oxygen or other reactive oxygen species (ROS) preventing them from reacting with CPs. These results not only provide an efficient strategy for improving the photostability of conjugated polymers in solution and films, but also shed light on better understanding the photophysics of conjugated polymers at single-molecule level.
Introduction
Conjugated polymers (CPs) have continuously attracted attention from physicists, chemists, and materials scientists due to their excellent optical and electrical properties with potential applications in optoelectronic devices, such as organic light emitting devices (OLEDs),1 solar cells,2,3 field-effect transistors,4 and chemical sensors.5,6 As organic semiconductors, CPs have both the optoelectronic properties of semiconductors and the flexibility of organic molecules. This unique combination allows CPs to be very promising materials for soft optoelectronic devices. However, as organic molecules, it is well acknowledged that CPs are easily suffered from photodegradation,7,8 photobleaching9,10 and photoblinking11,12 under continuous light illumination including complex processes such as conformation changes,13,14 inter- or intra-molecular charge transfer,15,16 photogenerated hole polarons,17,18 and photooxidized defects.19,20 Thus, the photostability of CPs becomes the bottleneck problem which limits their applications.In order to improve the photostability of CPs for better application, people have tried different methods. For example, Gonçalves et al.21 reported that the photostability of PPV derivatives could be enhanced by coating a layer of hydrolyzed poly(vinyl alcohol) (PVA) reducing the permeability of oxygen. Švrček et al.22 found that incorporation of boron doped freestanding silicon nanocrystals (Si-ncs) could significantly improve the stability of (poly(2-methoxy-5-(2′-ethylhexyloxy)-1,4-phenylenevinylene)) (MEH-PPV) by increasing the carrier mobility and inhibiting oxygen diffusion. Reduced graphene oxide (r-GO) was also used as additive to form MEH-PPV/r-GO composite, which could efficiently facilitate photo-induced excitation electron transfer from MEH-PPV to r-GO and thus protect MEH-PPV from further chemical photodegradation.23 Singlet oxygen quencher triphenylamine (TPA) and triplet quencher trans-stilbene (TSB) also showed effective improvement of the fluorescence intensity and stability of poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(1,4-benzo-{2,1′,3}-thiadiazole)] (PFBT).24 It is clear that introducing additives into the system to either isolate the CPs from oxygen or help to release the extra energy of active electrons is an effective way to improve the photostability of CPs.
Single-molecule spectroscopy (SMS) provides the opportunity to study individual molecules, which has received great attention in biology, physics, chemistry and materials science due to its unique superiority to uncover the photophysical properties and chemical kinetics beyond ensemble-averaged measurements.25–33 SMS has been used for revealing unexpected results of CPs such as fluorescence blinking and quenching.34,35 Poly(3-hexylthiophene-2,5-diyl) (P3HT) has shown promising properties for solar cells and is commonly used as the model conjugated polymer for photophysical studies. Here in this work, we investigated the effect of natural antioxidant vitamin E on the photostability of P3HT in solution and films as well as the fluorescence blinking behaviors at single-molecule level to understand the fundamental mechanisms of photostability enhancement in P3HT molecules via addition of VE. We found the photostability and fluorescence intensity of P3HT with addition of VE can be enhanced significantly and explained VE as natural antioxidants with reducibility to donate electrons can repair photooxidized trap sites and quench the triplet state as well as singlet oxygen. Our results not only pave the way of using conjugated polymers (CPs) with better photostability for single-molecule imaging and labeling, but also provide an effective method to improve photostability of CPs.
Experimental
Chemicals and materials
Regiorandom poly(3-hexylthiophene-2,5-diyl) (P3HT, average molecular weight Mn ≈ 25–50 kDa), poly(methyl methacrylate) (PMMA, average molecular weight Mn ≈ 996![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and alpha tocopherol which is a component of vitamin E (VE) family were purchased from Sigma-Aldrich without further purification (the chemical structure of P3HT as well as VE is shown in Fig. S1 in the ESI†), and toluene (AR, ≥98.5%) was purchased from Shanghai Lingfeng Chemical Reagent Co., Ltd.
000) and alpha tocopherol which is a component of vitamin E (VE) family were purchased from Sigma-Aldrich without further purification (the chemical structure of P3HT as well as VE is shown in Fig. S1 in the ESI†), and toluene (AR, ≥98.5%) was purchased from Shanghai Lingfeng Chemical Reagent Co., Ltd.
Sample preparations
A stock solution of 8.19 × 10−5 M P3HT and 10 mg mL−1 PMMA matrix polymers were prepared by dissolving P3HT or PMMA in toluene. A series of different concentrations of VE in toluene (0.02 M, 0.06 M, 0.08 M, and 0.22 M) were also prepared. To measure the effect of VE additives on the photostability of P3HT, we also prepared P3HT solution with addition of VE by equivalent volume mixing of P3HT solutions and VE solution (different concentrations for comparison). For comparison, we also prepared a control sample by equivalent volume mixing of P3HT solution and toluene.For solid films and single molecule samples, we diluted the P3HT stock solution with 10 mg mL−1 PMMA to appropriate concentrations and added VE of different concentrations by equivalent volume mixing to investigate VE effect on the photostability of P3HT. 25 μL of the as-prepared solution was dropped on the glass cover slips and spin-coated at 820 rpm. The spin-coated films were then annealed at 70 °C for 2 minutes and stored in dark atmosphere for several hours to remove residual solvent completely. The prepared films were then performed photostability measurements. All the sample preparations were performed under ambient conditions.
Photostability measurements
For fluorescence photostability measurements, a home-built wide-field fluorescence microscope (see Fig. S2 in the ESI†) based on Olympus IX73 was used with 532 nm CW diode laser as excitation light source. The fluorescence of P3HT in solutions was collected by air objective lens (Olympus LUCPlanFI 40×, NA = 0.6) and detected by an EMCCD camera (iXon Ultra 888, Andor) after passing through a 550 nm long-pass filter (ET550lp, Chroma). Oil immersion objective (Olympus LUCPlanFI 60×, NA = 1.25) was used for P3HT films and single-molecule measurements. A transmission grating (Newport, 150 lines per mm) was put in front of the camera using 405 nm CW diode laser as excitation light source for the fluorescence spectra measurement. PL lifetime measurements were carried out using time-correlated single photon counting system (TCSPC, Picoharp 300) with 532 nm pulse excitation source from super continuous laser (Fianium SC-400, 40 MHz). All the measurements were performed under ambient conditions unless indicated otherwise.Results and discussion
Photostability measurements of P3HT in solution and films
The fluorescence intensity traces of P3HT in solution with addition of VE (CVE = 0.22 M) are shown in Fig. 1a (The raw data is provided in Fig. S3a in the ESI†). Clear photodegradation of P3HT without VE can be observed. Interestingly, the photodegradation is completely suppressed by addition of VE. The fluorescence intensity keeps constant for about 4 minutes under continuous light irradiation without any photodegradation and the fluorescence intensity is slightly enhanced. Such effect is more significant for P3HT in solid films. As shown in Fig. 1b (the raw data is provided in Fig. S3b in the ESI†), the fluorescence intensity decays very fast in P3HT films without VE and almost completely degrades within one minute while the photodegradation process becomes much slower after a short period of fast decay in P3HT films with addition of VE (CVE = 0.22 M) achieving a retarding effect against the photodegradation. For quantitative comparison, we fitted the fluorescence intensity traces by a bi-exponential function. The average decay time of P3HT in films is elongated from 5 s to 97 s by adding VE which is almost a 20-fold improvement. More importantly, not only the photostability is improved, the fluorescence intensity is also enhanced by 4 times as shown in the initial intensity in Fig. S3b in the ESI.† There are mainly two reasons explaining this phenomenon: (1) the lifetime of P3HT films with VE is 2-fold longer than that of P3HT without VE as shown in Fig. 2b, which means the photobleaching paths can be suppressed. (2) From single-molecule blinking measurements as shown in Fig. 3–6 we found that the off-time duration as well as total off-time duration become shorter with increasing VE concentrations, which indicates that the long-lived charged species and triplet states as well as photodegradation can be reduced or suppressed with the help of VE as natural antioxidants, triplet quencher and singlet oxygen quencher.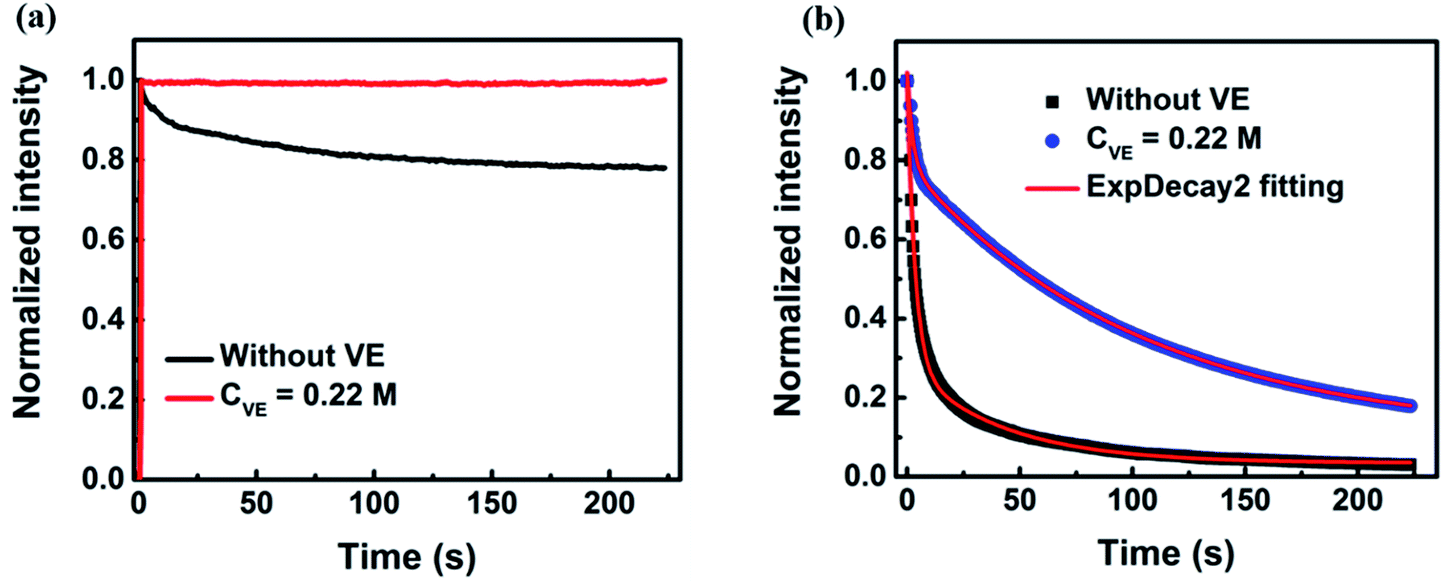 | ||
| Fig. 1 (a) The normalized fluorescence intensity traces of P3HT in solution with and without addition of VE. (b) The normalized fluorescence intensity traces of P3HT in solid films with (blue solid circles) and without (black solid square) addition of VE. The red solid lines are the fitting curves of the fluorescence decay using a bi-exponential function. The power density of the excitation light of the 532 nm laser was 3.11 and 4.30 W cm−2 for P3HT in solutions and in films, respectively. The exposure time was 0.3 s. | ||
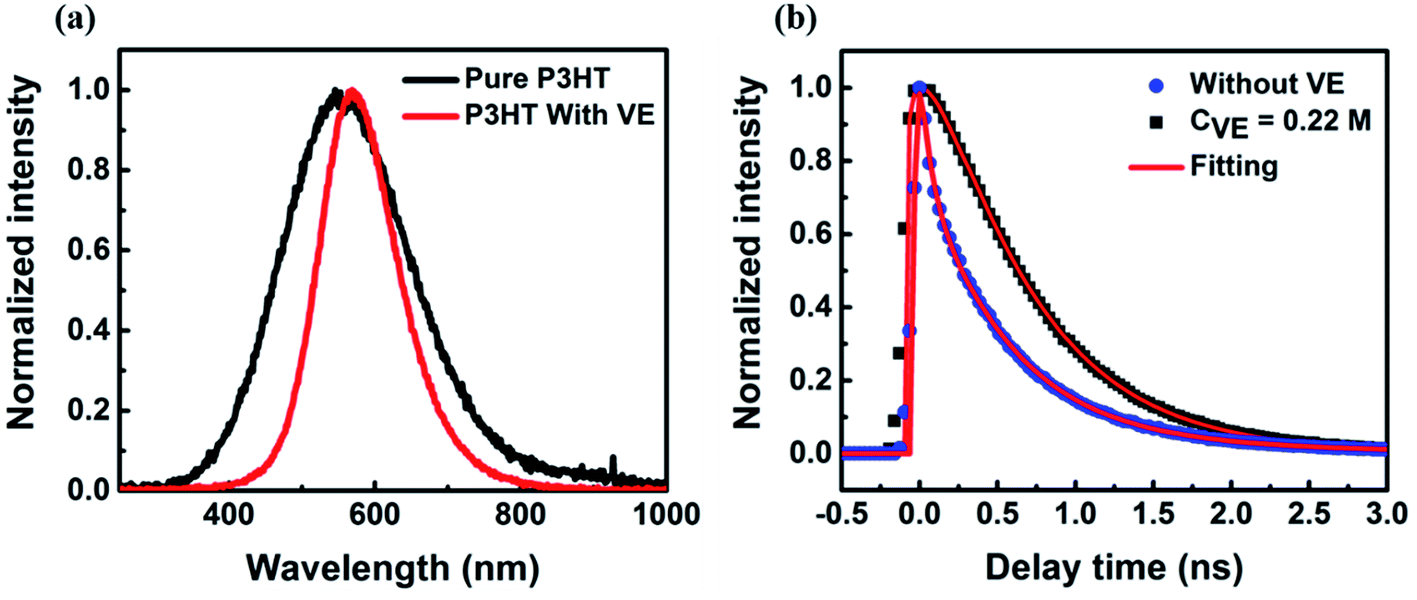 | ||
| Fig. 2 (a) The normalized fluorescence spectra and (b) lifetime of P3HT films with and without addition of VE. The concentration of VE is 0.22 M. The 405 nm CW laser and 532 nm pulse laser was used as excitation source for fluorescence spectra and lifetime measurements, respectively. | ||
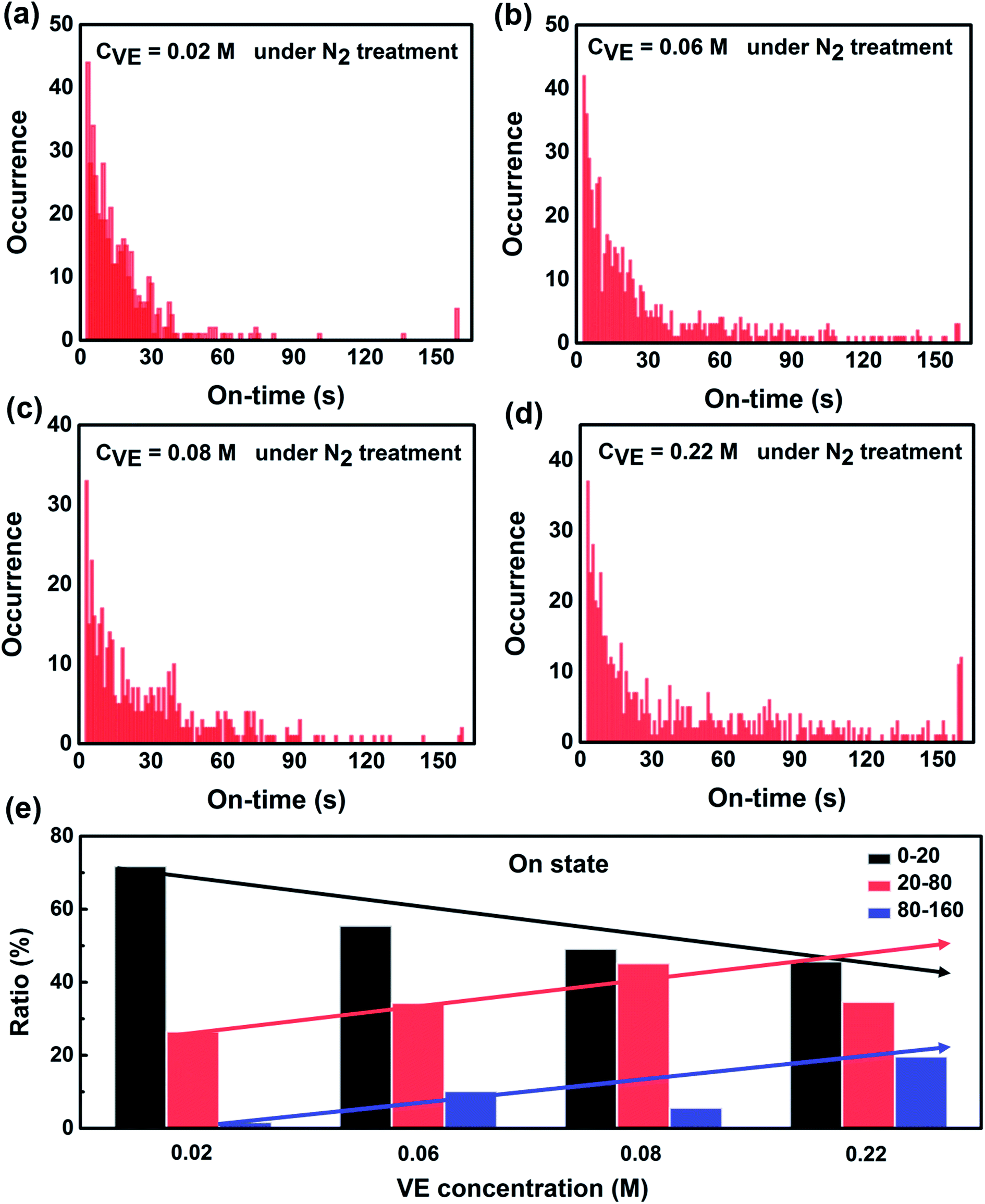 | ||
| Fig. 3 Statistics of on-state duration time of single P3HT molecules under continuous light irradiation for 160 s with addition of different VE concentrations. (a) 300 molecules with CVE = 0.02 M. (b) 331 molecules with CVE = 0.06 M. (c) 224 molecules with CVE = 0.08 M. (d) 272 molecules with CVE = 0.22 M. (e) Proportion of different on-time periods dependence on VE concentrations. The 532 nm laser with power density of 7.23 W cm−2 was used as excitation source and the exposure time was 1 s. The solid arrows are for guiding the eyes. | ||
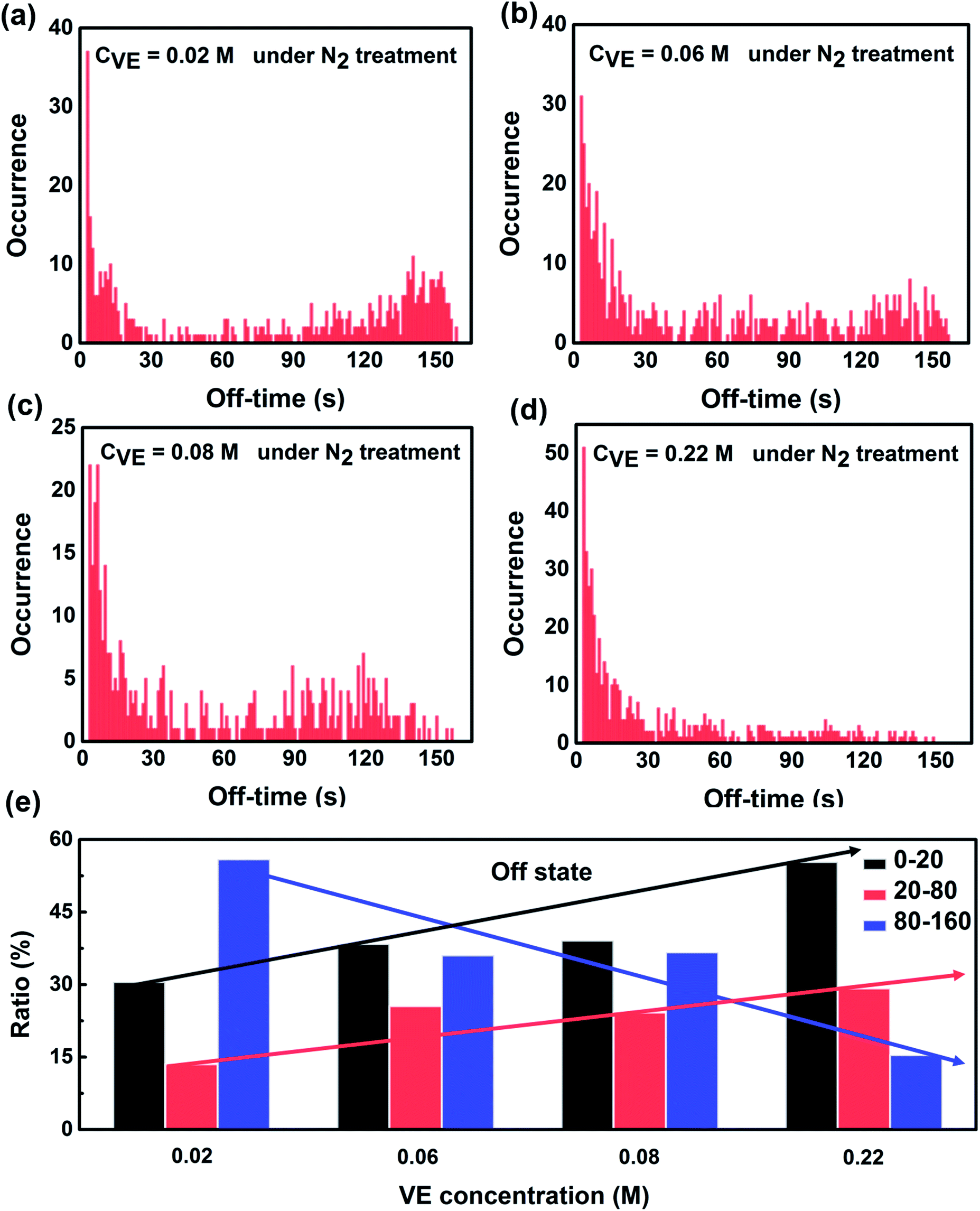 | ||
| Fig. 4 Statistics of off-state duration time of single P3HT molecules under continuous light irradiation for 160 s with addition of different VE concentrations. The number of molecules and experimental condition are the same as Fig. 3. The solid arrows are for guiding the eyes. | ||
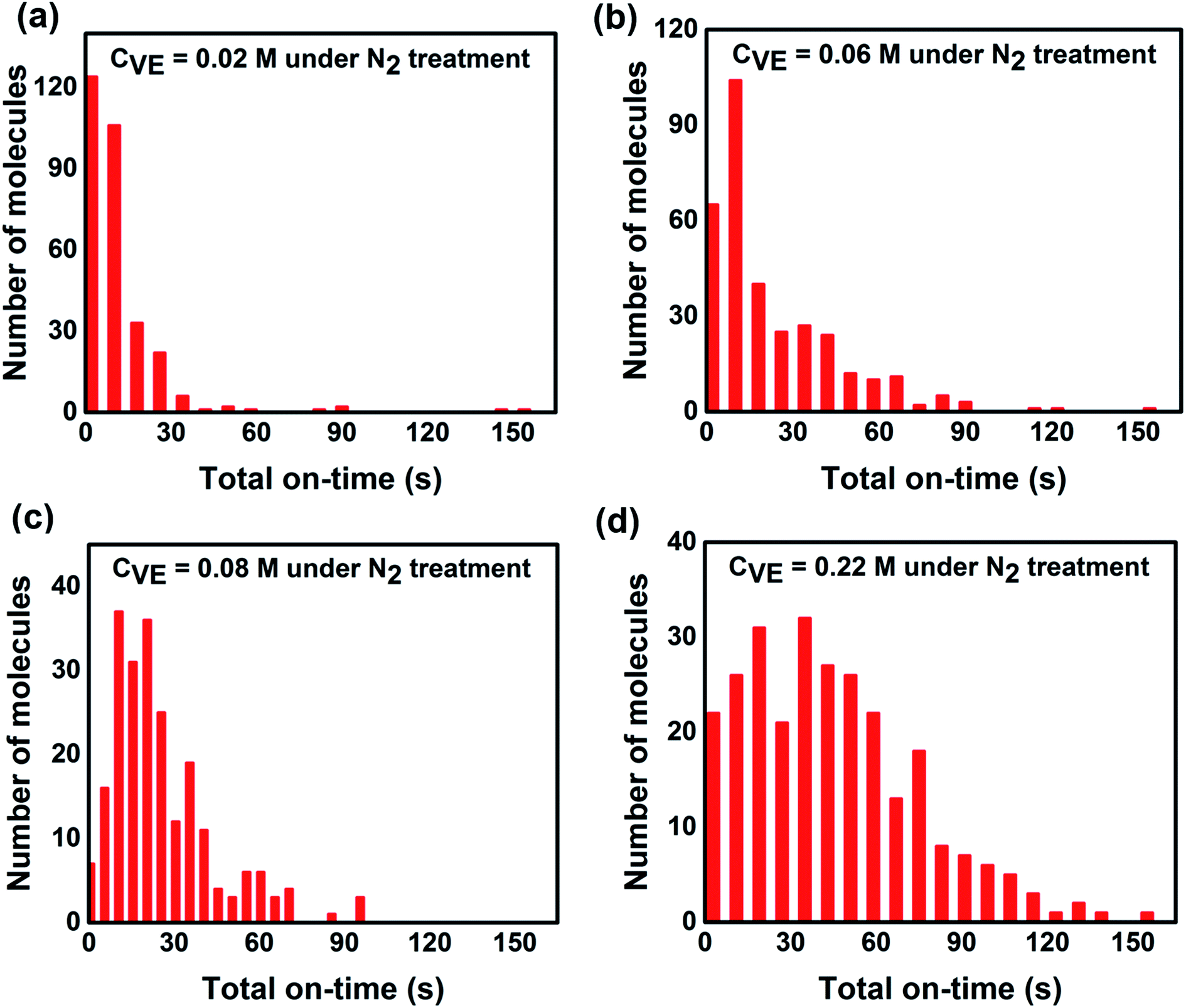 | ||
| Fig. 5 Statistics of total on-state duration time of individual P3HT molecules under continuous light irradiation for 160 s with different VE concentrations. The number of molecules and experimental condition are the same as Fig. 3. | ||
 | ||
| Fig. 6 Statistics of total off-state duration time of each single P3HT molecules under continuous light irradiation for 160 s with addition of different VE concentrations. The number of molecules and experimental condition are the same as Fig. 3. | ||
Spectra and lifetime measurements of P3HT in films
We also checked the fluorescence spectra and lifetime of P3HT in films with and without VE as shown in Fig. 2. The spectra with VE is narrower and slightly red-shifted compared to that of P3HT without VE, as indicated in Fig. 2a. It is previously reported that the spectrum of CPs in films is normally broader compared to that in solution due to strong interaction among CP molecules.36–38 Here, the fluorescence spectrum of P3HT becomes narrower with VE compared to that without VE is because VE can prevent the interaction between different P3HT molecules. To be accurate, the addition of VE can provide more friendly environment for P3HT polymer chains to form extended conformations with less intrachain or interchain interactions (such as electron or energy transfer). As shown in Fig. 2b, the fluorescence lifetime is prolonged after adding VE into the films suggesting that some non-radiative channels are blocked. The lifetime results can partially explain the enhancement of the fluorescence intensity as shown Fig. S3b.† However, the 2-times enhancement of the lifetime cannot fully explain the 4-fold enhancement of fluorescence intensity. Other processes have to be considered to fully understand the mechanism of the improved fluorescence intensity and photostability by addition of VE.Statistics of on/off-state duration time of individual molecules
To further reveal the fundamental mechanism of the enhanced photostability and fluorescence intensity, we investigated the effect of VE addition on individual P3HT molecules by single-molecule spectroscopy that has been shown to be a promising tool in studying photophysics of single conjugated polymer chains.14,20,39 The typical fluorescence image and intensity traces of single P3HT molecules are shown in Fig. S4 in the ESI.† Obvious fluorescence blinking and photobleaching behaviors can be seen from all the samples similar as reported.12,40,41 To compare the photoblinking behaviors and photostability quantitatively, we analyzed the fluorescence intensity traces with four different parameters: on-time duration (the duration time a molecule stays at the on-state before it switches to off-state), off-time duration (the duration time a molecule stays at the off-state before it switches to on-state), total on-time duration (the total time of a molecule stays at on-state before photobleaching) and total off-time duration (the total time of a molecule stays at off-state before photobleaching). The detailed analysis and calculated methods are provided in Fig. S5 in the ESI.†Fig. 3 shows the distributions of on-time duration of 300, 331, 224 and 272 individual P3HT molecules with corresponding VE concentrations of 0.02 M, 0.06 M, 0.08 M and 0.22 M under continuous light irradiation for 160 s. We can clearly see that the on-time duration becomes longer with higher concentrations of VE. We also calculated the proportion of different on-time periods (0–20 s, 20–80 s, 80–160 s) as shown in Fig. 3e. It is clear that the proportion of the long on-time duration (20–80 s, 80–160 s) increases and proportion of the short on-time duration (0–20 s) decreases with increasing of VE concentration, which means the transition from on-state to off-state is significantly suppressed.
On the other hand, we analyzed the distributions of the off-time duration of individual P3HT molecules as shown in Fig. 4. It can be seen clearly that the off-time duration becomes shorter with higher concentration of VE. The proportion of the long off-time events is significantly suppressed with the increasing VE concentrations. The off-state of the CPs is reported to be due to the charged states or radical ion states which cannot absorb or emit light.42,43 The short off-time duration suggests that VE can accelerate the recovery of P3HT from these dark states, which is in good agreement with the on-time duration results.
Statistics of total on/off-state duration time of individual molecules
From single molecule point of view, the fluorescence intensity of a single P3HT molecule is controlled not only by the time duration of each on-time period but also by the total time that the molecule stays at the on-state. In addition, the total time of the on-state indicates the photostability of individual molecule. Thus we analyzed the total on/off-time duration time of individual molecules (the detailed explanation of the total on/off-time duration is provided in Fig. S5 in the ESI†). As shown in Fig. 5 and 6, it is obvious that the total on-time duration significantly increases while the total off-time duration clearly decreases with higher concentration of VE. From these results, we can directly conclude that with addition of VE, P3HT molecules can emit more photons before they are photobleached showing better photostability and higher fluorescence intensity.It is well accepted that photobleaching and photodegradation of fluorophores come from two primary pathways.43–45 For one thing, the fluorophores will transfer to triplet states via intersystem crossing upon continuous excitation, which will generate singlet oxygen or other reactive oxygen species (ROS) via photosensitization of energy or electron transfer.46–48 Such active ROS can react with fluorophores leading to photodegradation. In addition, fluorophores can also be directly photooxidized or photoionized by photo-excitation and form reactive radical intermediates leading to their photodegradation.49–51 For another thing, photooxidized defects can act as non-emissive traps, which is often observed in most fluorescent conjugated polymers (CPs). These traps will quench the fluorescence of neighboring-tightly folded chains via energy migration52,53 or electron transfer54,55 leading to decreased fluorescence intensity and severe photobleaching and photodegradation. To avoid the photodegradation/photobleaching of fluorophores, antioxidants,56 triplet quenchers57 as well as singlet oxygen scavengers58 are introduced to prevent the above mentioned photodegradation pathways. Calver et al.42 reported that addition of the antioxidant/triplet quenchers ascorbic acid (AA) and trolox (TX) in PBS solution can significantly enhance the photostability of a 49-mer long poly(phenylene-ethynylene) conjugated polymer bearing carboxylate side groups (PPE-CO2-49). However, addition of AA or TX in solution with an enzymatic oxygen scavenging system will decrease the photostability of PPE-CO2-49 due to reactive radical anions produced by quenching the triplet states, which may open an additional photodegradation channel leading to reduced photostability.
In this work, vitamin E (alpha tocopherol) as a popular natural antioxidants with electron-donating ability plays multiple roles which are antioxidant,59–63 triplet quenchers64 and singlet oxygen quenchers.65 First of all, once the radical or charged states of P3HT is formed, VE can donate electrons to P3HT and restore it back to ground state leading to decreased lifetime of the dark states. Thus, the total fluorescence intensity of the molecule will be enhanced. On the other hand, the triplet state of P3HT can also be recovered by VE via energy transfer avoiding the formation of active reactive oxygen species (ROS). In addition, VE can also eliminate the singlet oxygen or ROS produced by photosensitization preventing the photodegradation of P3HT. These will not only prolong the on time duration but also improve the photostability of P3HT.
Conclusions
In conclusion, we found that the photostability and fluorescence intensity of P3HT molecules in solution and films can be significantly improved with addition of VE. Based on the experimental results from bulk sample to individual molecule level, we proposed that VE simultaneously play multiple roles to prevent photobleaching and photoblinking processes of P3HT. Firstly, VE as natural antioxidants with electron-donating ability can donate electrons to the photooxidized defects or the radical cations, which can heal these trap sites or recover the radical cations back to the ground state decreasing the chance of photodegradation. Secondly, VE as triplet state quencher can depopulate the triplet states of P3HT by energy transfer, which can prevent the formation of singlet oxygen or reactive oxygen species (ROS). Finally, VE can also directly react with singlet oxygen or ROS protecting P3HT molecules from photodegradation. To our knowledge, this is for the first time that the protection mechanism of conjugated polymer by antioxidants is investigated in detail from bulk sample to single-molecule level. The results provide an effective way to improve the photostability and the fluorescence intensity of conjugated polymers which gives suggestions to optimize the fabrication processes of OLEDs and polymer solar cells with higher efficiency and stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC no. 21673114 and 22073046).References
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121–128 CrossRef CAS.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CAS.
- L. Chen, D. W. McBranch, H.-L. Wang, R. Helgeson, F. Wudl and D. G. Whitten, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12287–12292 CrossRef CAS.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS.
- H. Neugebauer, C. Brabec, J. C. Hummelen and N. S. Sariciftci, Sol. Energy Mater. Sol. Cells, 2000, 61, 35–42 CrossRef CAS.
- B. Louis, S. Caubergh, P. O. Larsson, Y. Tian and I. G. Scheblykin, Phys. Chem. Chem. Phys., 2018, 20, 1829–1837 RSC.
- W. R. Salaneck and J. L. Brédas, Solid State Commun., 1994, 92, 31–36 CrossRef CAS.
- D. Sahoo, Y. Tian, G. Sforazzini, H. L. Anderson and I. G. Scheblykin, J. Mater. Chem. C, 2014, 2, 6601–6608 RSC.
- J. Vogelsang, T. Adachi, J. Brazard, D. A. Vanden Bout and P. F. Barbara, Nat. Mater., 2011, 10, 942–946 CrossRef CAS.
- H. Lin, S. R. Tabaei, D. Thomsson, O. Mirzov, P. O. Larsson and I. G. Scheblykin, J. Am. Chem. Soc., 2008, 130, 7042–7051 CrossRef CAS.
- G. Bounos, S. Ghosh, A. K. Lee, K. N. Plunkett, K. H. Dubay, J. C. Bolinger, R. Zhang, R. A. Friesner, C. Nuckolls, D. R. Reichman and P. F. Barbara, J. Am. Chem. Soc., 2011, 133, 10155–10160 CrossRef CAS.
- J. Vogelsang, J. Brazard, T. Adachi, J. C. Bolinger and P. F. Barbara, Angew. Chem., 2011, 123, 2305–2309 CrossRef.
- J. Yu, D. Hu and P. F. Barbara, Science, 2000, 289, 1327–1330 CrossRef CAS.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef.
- Z. Tian, J. Yu, C. Wu, C. Szymanski and J. McNeill, Nanoscale, 2010, 2, 1999–2011 RSC.
- J. Yu, C. Wu, Z. Tian and J. Mcneill, Nano Lett., 2012, 12, 1300–1306 CrossRef CAS.
- D. A. Vanden Bout, W. Yip, D. Hu, D. K. Fu, T. M. Swager and P. F. Barbara, Science, 1997, 277, 1074–1077 CrossRef CAS.
- D. Hu, J. Yu, K. Wong, B. Bagchi, P. J. Rossky and P. F. Barbara, Nature, 2000, 405, 1030–1033 CrossRef CAS.
- V. C. Gonçalves, A. J. Carvalho and D. T. Balogh, Polym. Int., 2010, 59, 637–641 CrossRef.
- V. Švrček, H. Fujiwara and M. Kondo, Appl. Phys. Lett., 2008, 92, 1–4 CrossRef.
- C. Ran, M. Wang, W. Gao, J. Ding, Y. Shi, X. Song, H. Chen and Z. Ren, J. Phys. Chem. C, 2012, 116, 23053–23060 CrossRef CAS.
- Z. Tian, J. Yu, X. Wang, L. C. Groff, J. L. Grimland and J. D. McNeill, J. Phys. Chem. B, 2013, 117, 4517–4520 CrossRef CAS.
- M. J. Fernée, P. Tamarat and B. Lounis, Chem. Soc. Rev., 2014, 43, 1311–1337 RSC.
- M. Orrit, T. Ha and V. Sandoghdar, Chem. Soc. Rev., 2014, 43, 973–976 RSC.
- W. E. Moerner, J. Phys. Chem. B, 2002, 106, 910–927 CrossRef CAS.
- W. E. Moerner and M. Orrit, Science, 1999, 283, 1670–1676 CrossRef CAS.
- P. Chen, X. Zhou, H. Shen, N. M. Andoy, E. Choudhary, K.-S. Han, G. Liu and W. Meng, Chem. Soc. Rev., 2010, 39, 4560–4570 RSC.
- S. Habuchi, N. Satoh, T. Yamamoto, Y. Tezuka and M. Vacha, Angew. Chem., 2010, 49, 1418–1421 CrossRef CAS.
- E. Barkai, Y. Jung and R. Silbey, Annu. Rev. Phys. Chem., 2004, 55, 457–507 CrossRef CAS.
- J. B. Sambur and P. Chen, Annu. Rev. Phys. Chem., 2014, 65, 395–422 CrossRef CAS.
- W. Wang, Chem. Soc. Rev., 2018, 47, 2485–2508 RSC.
- O. Mirzov, F. Cichos, C. V. Borczyskowski and I. Scheblykin, J. Lumin., 2005, 112, 353–356 CrossRef CAS.
- H. Lin, Y. Tian, K. Zapadka, G. Persson, D. Thomsson, O. Mirzov, P.-O. Larsson, J. Widengren and I. G. Scheblykin, Nano Lett., 2009, 9, 4456–4461 CrossRef CAS.
- Y. Tian, V. Sheinin, O. Kulikova, N. Mamardashvili and I. G. Scheblykin, Chem. Phys. Lett., 2014, 599, 142–145 CrossRef CAS.
- S. Onda, H. Kobayashi, T. Hatano, S. Furumaki, S. Habuchi and M. Vacha, J. Phys. Chem. Lett., 2011, 2, 2827–2831 CrossRef CAS.
- C. Szymanski, C. Wu, J. Hooper, M. A. Salazar, A. Perdomo, A. Dukes and J. Mcneill, J. Phys. Chem. B, 2005, 109, 8543–8546 CrossRef CAS.
- T. Adachi, J. Brazard, P. Chokshi, J. C. Bolinger, V. Ganesan and P. F. Barbara, J. Phys. Chem. C, 2010, 114, 20896–20902 CrossRef CAS.
- S. Habuchi, S. Onda and M. Vacha, Chem. Commun., 2009, 32, 4868–4870 RSC.
- P. A. Dalgarno, C. A. Traina, J. C. Penedo, G. C. Bazan and I. D. W. Samuel, J. Am. Chem. Soc., 2013, 135, 7187–7193 CrossRef CAS.
- C. F. Calver, B. A. Lago, K. S. Schanze and G. Cosa, Photochem. Photobiol. Sci., 2017, 16, 1821–1831 RSC.
- J. Vogelsang, R. Kasper, C. Steinhauer, B. Person, M. Heilemann, M. Sauer and P. Tinnefeld, Angew. Chem., 2008, 47, 5465–5469 CrossRef CAS.
- T. Cordes, J. Vogelsang and P. Tinnefeld, J. Am. Chem. Soc., 2009, 131, 5018–5019 CAS.
- J. Vogelsang, T. Cordes, C. Forthmann, C. Steinhauer and P. Tinnefeid, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8107–8112 CrossRef CAS.
- R. Katahira, A. Mittal, K. McKinney, X. Chen, M. P. Tucker, D. K. Johnson and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 1474–1486 CrossRef CAS.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 CAS.
- R. D. Scurlock, B. Wang, P. R. Ogilby, J. R. Sheats and R. L. Clough, J. Am. Chem. Soc., 1995, 117, 10194–10202 CrossRef CAS.
- T. Cordes, J. Vogelsang, M. Anaya, C. Spagnuolo, A. Gietl, W. Summerer, A. Herrmann, K. Müllen and P. Tinnefeld, J. Am. Chem. Soc., 2010, 132, 2404–2409 CrossRef CAS.
- E. J. Sanchez, L. Novotny, G. R. Holtom and X. S. Xie, J. Phys. Chem. A, 1997, 101, 7019–7023 CrossRef CAS.
- C. Steinhauer, C. Forthmann, J. Vogelsagn and P. Tinnefeld, J. Am. Chem. Soc., 2008, 130, 16840–16841 CrossRef CAS.
- L. Romaner, A. Pogantsch, P. S. De Freitas, U. Scherf, M. Gaal, E. Zojer and E. J. W. List, Adv. Funct. Mater., 2003, 13, 597–601 CAS.
- J. C. Bolinger, M. C. Traub, J. Brazard, T. Adachi, P. F. Barbara and D. A. Vanden Bout, Acc. Chem. Res., 2012, 45, 1992–2001 CrossRef CAS.
- S. A. Jenekhe and D. Zhu, Polym. Chem., 2013, 4, 5142–5143 RSC.
- R. E. Palacios, F. R. F. Fan, J. K. Grey, J. Suk, A. J. Bard and P. F. Barbara, Nat. Mater., 2007, 6, 680–685 CrossRef CAS.
- M. A. Van Dijk, L. C. Kapitein, J. Van Mameren, C. F. Schmidt and E. J. G. Peterman, J. Phys. Chem. B, 2004, 108, 6479–6484 CrossRef CAS.
- J. Widengren, A. Chmyrov, C. Eggeling, P. Å. Löfdahl and C. A. M. Seidel, J. Phys. Chem. A, 2007, 111, 429–440 CAS.
- D. M. Jerina, J. W. Daly and B. Witkop, J. Am. Chem. Soc., 1967, 90, 6527–6528 Search PubMed.
- G. W. Burton, T. Doba, E. J. Gabe, L. Hughes, F. L. Lee, L. Prasad and K. U. Ingold, J. Am. Chem. Soc., 1985, 107, 7053–7065 CrossRef CAS.
- C. Dockendorff, O. Aisiku, L. Verplank, J. R. Dilks, D. A. Smith, S. F. Gunnink, L. Dowal, J. Negri, M. Palmer, L. MacPherson, S. L. Schreiber and R. Flaumenhaft, ACS Med. Chem. Lett., 2012, 3, 232–237 CrossRef CAS.
- G. W. Burton, Annu. Rev. Nutr., 1990, 10, 357–382 CrossRef CAS.
- K. Mukai, A. Tokunaga, S. Itoh, Y. Kanesaki, K. Ohara, S. I. Nagaoka and K. Abe, J. Phys. Chem. B, 2007, 111, 652–662 CrossRef CAS.
- M. G. Traber and J. Atkinson, Free Radicals Biol. Med., 2007, 43, 4–15 CrossRef CAS.
- L. Shen and H. F. Ji, J. Photochem. Photobiol., A, 2008, 199, 119–121 CrossRef CAS.
- S. Dad, R. H. Bisby, I. P. Clark and A. W. Parker, Free Radical Res., 2006, 40, 333–338 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical structure of P3HT and VE; experimental setup of a home-built wide-field fluorescence microscope; the typical single-molecule fluorescence image and intensity traces of single P3HT molecules; the raw fluorescence intensity traces of P3HT in solution and films with and without addition of VE; the statistic and calculation methods of on/off-state and total on/off-state duration time. See DOI: 10.1039/d0ra09657a |
| This journal is © The Royal Society of Chemistry 2021 |