
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodonium salts as efficient iodine(III)-based noncovalent organocatalysts for Knorr-type reactions†
Sevilya N. Yunusovaa,
Alexander S. Novikov a,
Natalia S. Soldatovaa,
Mikhail A. Vovkb and
Dmitrii S. Bolotin*a
a,
Natalia S. Soldatovaa,
Mikhail A. Vovkb and
Dmitrii S. Bolotin*a
aInstitute of Chemistry, Saint Petersburg State University, Universitetskaya Nab. 7/9, Saint Petersburg, 199034, Russian Federation. E-mail: d.s.bolotin@spbu.ru
bCenter for Magnetic Resonance, Saint Petersburg State University, Universitetskii Pr., 26, Saint Petersburg, 198504, Russian Federation
First published on 22nd January 2021
Abstract
Hypervalent iodine(III)-derivatives display higher catalytic activity than other aliphatic and aromatic iodine(I)– or bromine(I)-containing substrates for a Knorr-type reaction of N-acetyl hydrazides with acetyl acetone to give N-acyl pyrazoles. The highest activity was observed for dibenziodolium triflate, for which 10 mol% resulted in the generation of N-acyl pyrazole from acyl hydrazide and acetyl acetone typically at 50 °C for 3.5–6 h with up to 99% isolated yields. 1H NMR titration data and DFT calculations indicate that the catalytic activity of the iodine(III) is caused by the binding with a ketone.
Introduction
In the past decade, organocatalysis has been the focus of extensive studies owing to its significant advantages over catalysis by metal-containing species including lower toxicity, reduced environmental footprint, and low to negligible sensitivity to air and moisture.1,2 In general, these organocatalysts can function either through a covalent or noncovalent bonding activation. A covalent activation mode involves the formation of covalent bond(s) between a substrate and catalyst (such as amines,1–5 heterocyclic carbenes,1,6,7 or phosphines8), whereas a noncovalent mode involves the activation of substrates through noncovalent linkages to the catalyst.2,9–18For noncovalent catalysis, an organic catalyst typically interacts with a substrate through hydrogen bonding (HB), and many important results were achieved for the reactions based on such HB donors as ureas,2,19–24 squaramides,24–26 and other Brønsted acids,27–31 whereas catalytic reactions involving halogen (XB)9,10,15,32–35 or chalcogen bonding (ChB)34,36,37 are far less explored. Although XB has been established as a valuable tool in solid-state chemistry and crystal engineering,38–41 over the last five years XB has been studied in solution33–35,42–44 and applied to homogeneous catalytic transformations.9,15,16,34,37
Recently Huber and coworkers reported that iodine(III) derivatives such as diaryliodonium salts can serve as efficient organocatalysts and utilize XBs for which the catalytic activity is similar or even higher than other well-proven organocatalysts that are based on iodine(I) derivatives, such as oligodentate 2-iodoimidazolium species.45,46 Indeed, diaryliodonium salts exhibited a high catalytic activity in reactions in which a halide abstraction occurred46 (including their application in living cationic polymerization of olefins;47 Fig. 1(i) and (ii)) and in the synthetic transformations of carbonyl compounds, namely Diels–Alder cycloaddition46 (Fig. 1(iii)) and Mannich reaction48 (Fig. 1(iv)).
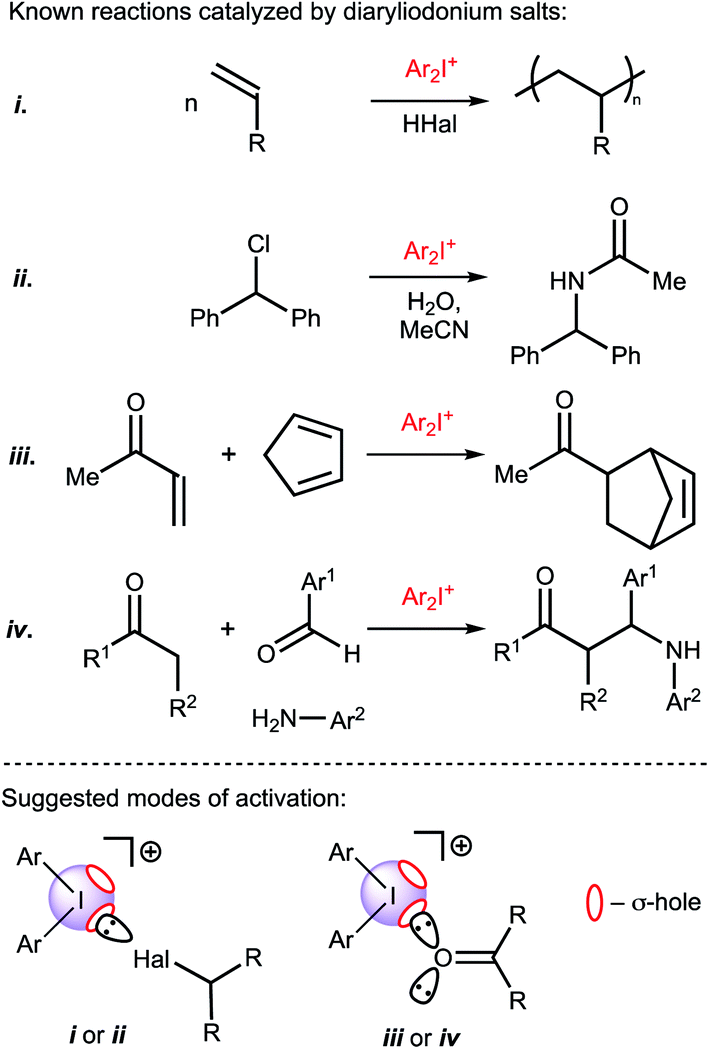 | ||
| Fig. 1 Previously studied reactions catalyzed by diaryliodonium salts and plausible modes of activation of substrates. σ-hole is a region of positive electrostatic potential on an atom.49 | ||
Considering the promising catalytic properties of diaryliodonium salts and the small number of reported examples for their application in homogeneous organocatalysis, further studies are worthwhile. In this study, we report on the catalytic activity of a series of diaryliodonium salts and provide a comparison of their activity with other XB donors in a model reaction that requires carbonyl activation. In agreement with previous suggestions that the catalytic activity of diaryliodonium salts is caused by the electrophilic activation of the electrophile,46,47 their catalytic activity in the studied Knorr-type cyclization is caused by binding with a ketone.
Results and discussion
Knorr-type syntheses50,51 conventionally proceed under mild conditions even in the absence of a catalyst if amines and carbonyl compounds are employed as the reactants.52 However, for less nucleophilic hydrazine derivatives, e.g. 2-pyridine hydrazines50 or acyl hydrazides,53,54 a catalyst is desirable to conduct the reaction under mild conditions. Thus, the reaction of N-acyl hydrazides with 1,3-dicarbonyl compounds, which leads to N-acyl pyrazoles, typically requires an additional activation and is usually conducted in the presence of a Brønsted or a Lewis acid under reflux for several hours.55–57 This reaction was chosen as a model to verify the catalytic effect of diaryliodonium salts.Comparison of the catalytic activity for the halogen-containing organic species
Benzoyl hydrazide (BH) was treated with acetyl acetone (AA) in CD3OD (50 °C, 5 h), which led to only a trace amount of the corresponding N-benzoyl pyrazole 1 (Fig. 2 and Table 1, entry 1). The addition of 1 equivalent (equiv.) of iodine(I)- or bromine(I)-containing species A–E, which feature expressed σ-holes at their halogen atoms,58–60 resulted in negligible or no catalytic effect (entries 1–6). In contrast, the utilization of the dibenziodolium species F, G, and H led to a significant acceleration and a half-conversion was achieved after approximately 70 min (entries 7–9). Notably, other types of iodine(III) species (I and J) appeared to be catalytically inactive in the model reaction (entries 10–11) and the results obtained for the activity of F–I are fully consistent with the recently reported relative Lewis acidity of the iodine(III)-based XB donors.61 | ||
| Fig. 2 Halogen-containing species tested as organocatalysts. | ||
| Entry | Catalyst | Catalyst load (mol%) | AA load (equiv.) | t (min) | Yield (%) |
|---|---|---|---|---|---|
| 1 | None | — | 1 | 300 | Traces |
| 2 | A | 100 | 1 | 300 | 32 |
| 3 | B | 100 | 1 | 300 | Traces |
| 4 | C | 100 | 1 | 300 | Traces |
| 5 | D | 100 | 1 | 300 | 30 |
| 6 | E | 100 | 1 | 300 | 29 |
| 7 | F | 100 | 1 | 70 | 50 |
| 8 | G | 100 | 1 | 70 | 46 |
| 9 | H | 100 | 1 | 70 | 52 |
| 10 | I | 100 | 1 | 70 | 3 |
| 11 | J | 100 | 1 | 70 | Traces |
| 12 | H | 100 | 1.5 | 70 | 98 |
| 13 | H | 100 | 1.2 | 70 | 95 |
| 14 | H | 50 | 1.2 | 100 | 98 |
| 15 | H | 10 | 1.2 | 220 | 98 |
Having examined dibenziodolium triflate H, whose catalytic activity was the highest among all the studied halogen-containing species, we further optimized the reaction conditions. Based upon 1H NMR monitoring of the reaction performed with various relative quantities of AA, we found that the optimal quantity of the carbonyl reactant was 1.2 equiv. (Fig. 3; Table 1, entries 9, 12, 13).
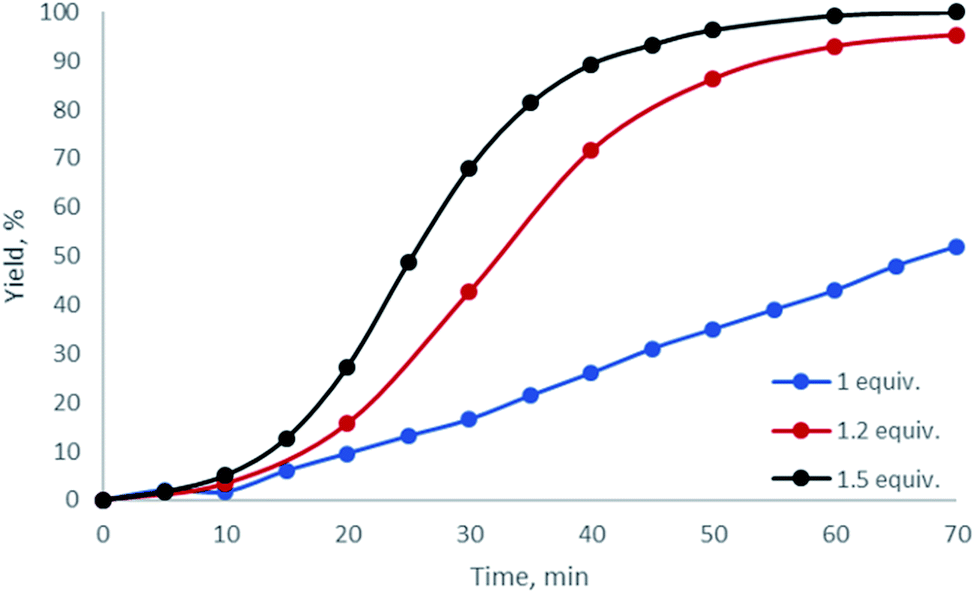 | ||
| Fig. 3 1H NMR monitoring of the reaction of benzoyl hydrazide with various amounts of acetylacetone in the presence of 1 equiv. H in CD3OD at 50 °C (entries 9, 12, 13). NMR yield of 1 is given on the y-axis. | ||
The S-shape of the kinetic curves was caused by the initial accumulation of the intermediate MeC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NNHBz)CH2COMe in the reaction mixture, which was then transformed to 1. The overlap of the proton resonances of MeC(NNHBz)CH2COMe with those of the reactants and with the signals of 1 did not allow the accurate calculation of its relative concentration; therefore, the corresponding plot is not provided.
NNHBz)CH2COMe in the reaction mixture, which was then transformed to 1. The overlap of the proton resonances of MeC(NNHBz)CH2COMe with those of the reactants and with the signals of 1 did not allow the accurate calculation of its relative concentration; therefore, the corresponding plot is not provided.
Next, the relative quantity of organocatalyst H was varied (Fig. 4, Table 1, entries 1, 9, 14, 15) and we found that even 10 mol% of H effectively catalyzed the reaction and 1 could be obtained within 1 d (Table 1, entry 15).
 | ||
| Fig. 4 1H NMR monitoring of the reaction of benzoyl hydrazide with acetylacetone with variable amount of H (CD3OD, 50 °C; entries 1, 9, 14, 15). | ||
The optimal conditions for the studied reaction were 10 mol% of H, 1.2 equiv. of the 1,3-dicarbonyl component, and 50 °C. Under these conditions the substrate scope was verified.
Binding of H with the reaction substrates
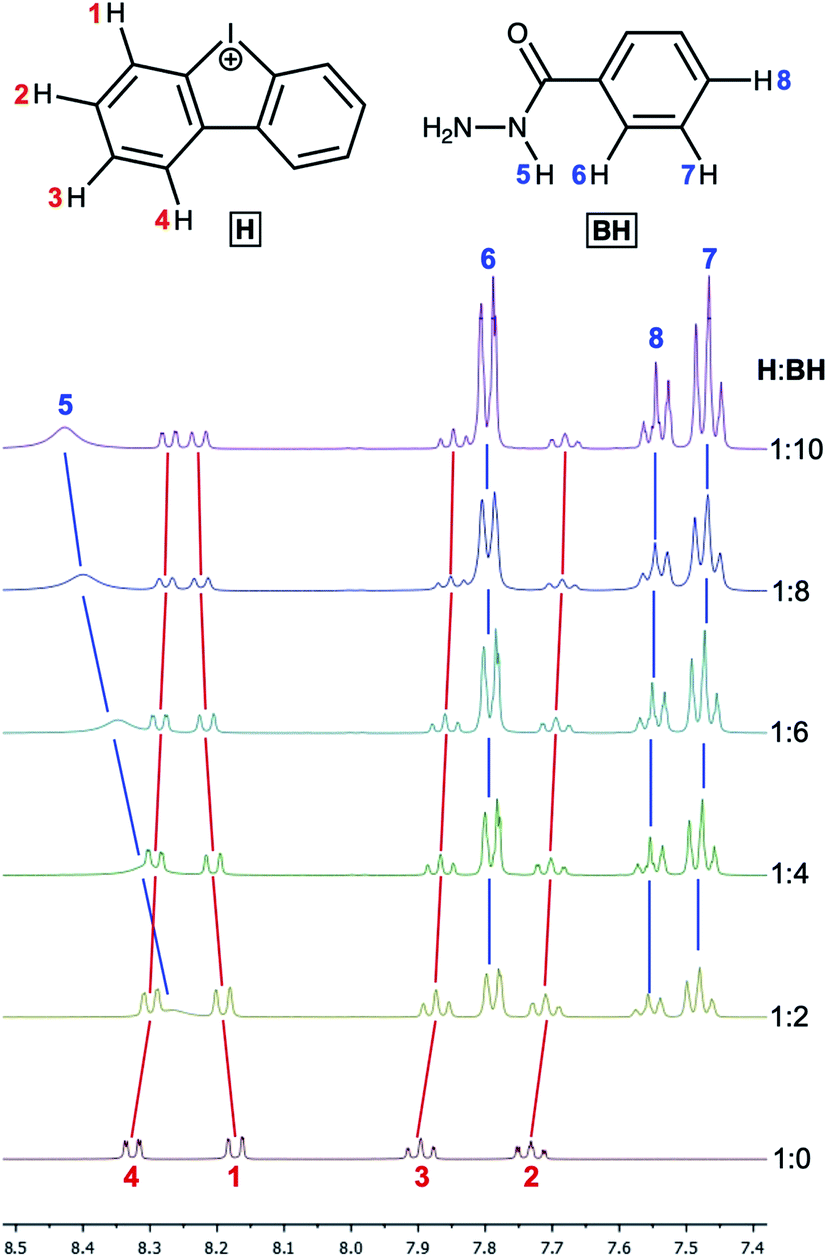 | ||
| Fig. 5 The 1H NMR titration of H by benzoyl hydrazide. | ||
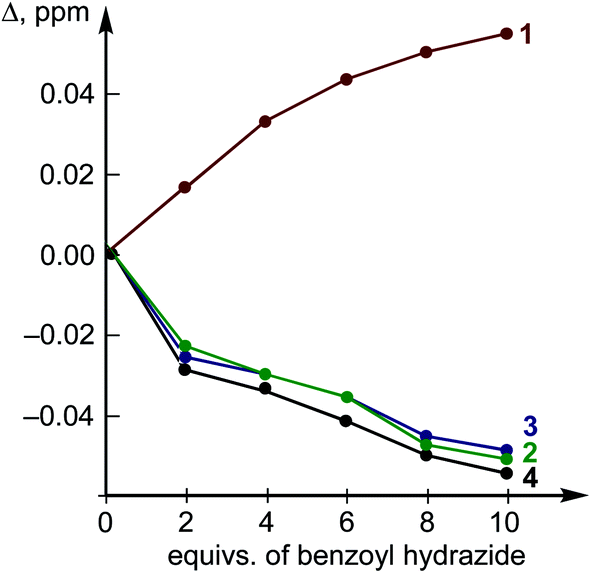 | ||
| Fig. 6 The changes in chemical shifts of the H proton resonances upon the titration. | ||
The titration of H by AA resulted in negligible changes in the chemical shifts (within 0.002 ppm) for the dibenziodolium cation, even with an excess of AA up to 25-fold; therefore, the corresponding binding constant could not be estimated. This observation might indicate either a low value of the binding constant or similar chemical shifts of the H atoms in the H–AA complex and separated substrates H and AA.
| Entry | Reaction | ΔG (kJ mol−1) | ΔH (kJ mol−1) |
|---|---|---|---|
| a H – dibenziodolium triflate; BH – benzoyl hydrazide; AA – acetyl acetone. | |||
| 1 | H + BH → H·BH (binding by O) | 25.5 | −20.9 |
| 2 | H + BH → H·BH (binding by N) | 24.7 | −25.1 |
| 3 | H + AA → H·AA | 28.0 | −27.2 |
| 4 | i | −1.3 | 23.8 |
| 5 | ii | 1.7 | 22.2 |
| 6 | iii | 1.3 | 18.0 |
Considering that the enthalpies of all the reactions are negative, these results indicate the significant impact of entropy change in the model systems. Accordingly, we included MeOH solvent molecules, which are the subject of substitution during the binding of the substrates, in the explicit form in the model systems (Scheme 1).
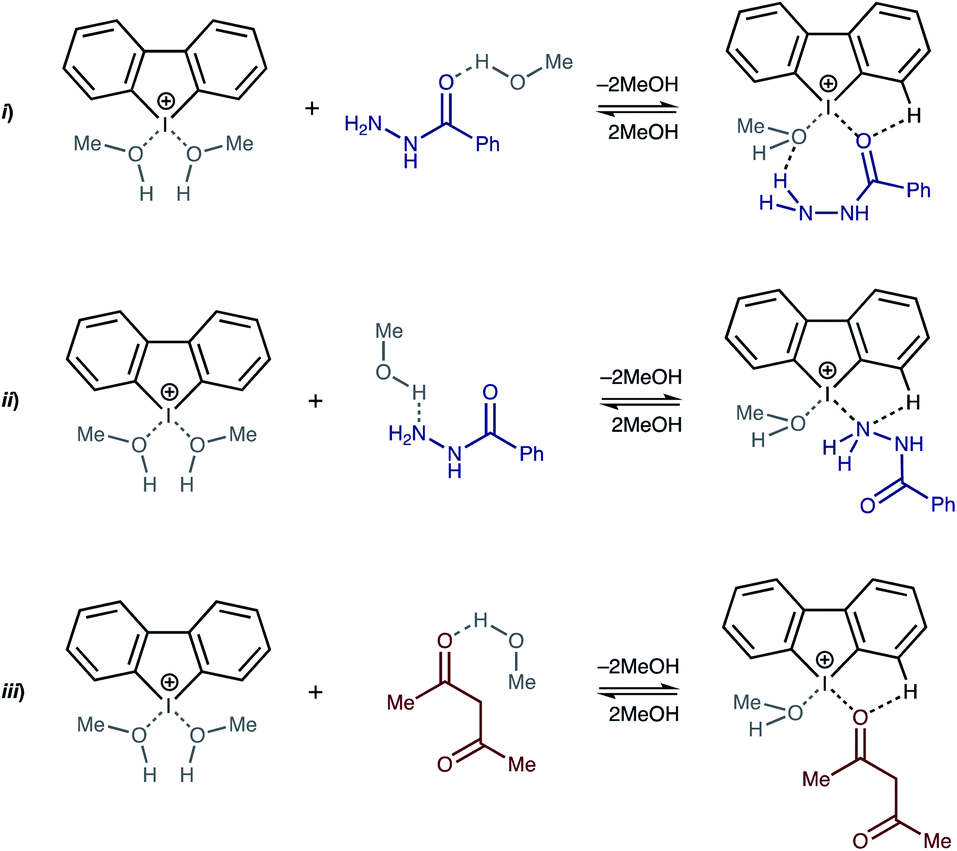 | ||
| Scheme 1 Model binding reactions. | ||
The obtained results (Table 2, entries 4–6) appeared to be coherent with the titration data, i.e. the experimentally observed small negative ΔG for the binding of BH (approx. −2.5(5) kJ mol−1 within 3 st. dev. based on K = 2.8(2) L mol−1 at 298 K) and the absence AA binding. Moreover, the theoretical calculations indicated that the binding of the O atom in BH to the I atom in H is more energetically favorable in MeOH than coordination by the Nsp3 atom.
Theoretical study of the catalytic mechanism
To understand the catalytic mechanism of H in the studied reaction (BH + AA → 1 + 2H2O), DFT calculations were performed for the first (and apparently rate-limiting) step. Initial calculations indicated that the direct nucleophilic attack of the Nsp3 atom in BH on the C atom in the carbonyl moiety of AA was impossible in the absence of H. All attempts to fix on the potential energy surface of the zwitterionic intermediate PhC(O)NHN+H2C(–O−)MeCH2COMe and the corresponding transition state (TS) for a nucleophilic attack were unsuccessful in the absence of bound H (the geometry optimization procedure resulted in the collapse of the relevant model structures to the initial reactants). Nevertheless, the zwitterionic intermediate formation is possible then H is bound to AA (Scheme 2, a–c; accordingly to the processes on Scheme 1, an additional four MeOH solvent molecules were taken into account in the explicit form).
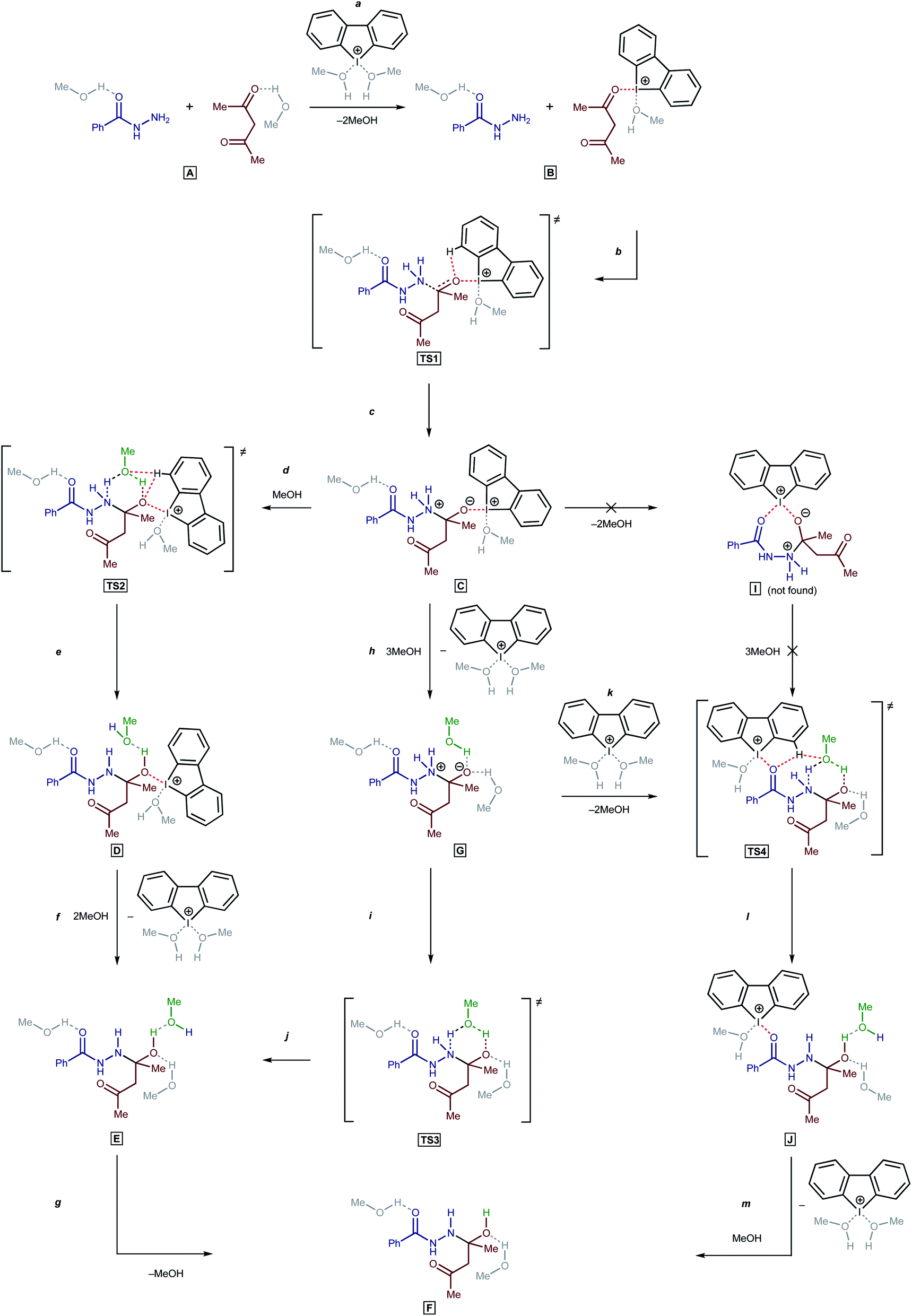 | ||
| Scheme 2 Possible reaction paths. | ||
For the proton transfer from the N atom to the O atom, the inclusion of one MeOH solvent molecule in the reaction mechanism was realized (TS2, TS3, and TS4).62 Incorporation of the solvent molecule in the transition state of the rate-limiting reaction step was successfully employed by us in previous theoretical studies.63 For aprotic solvents, at least in the case of hydrazine derivatives, an additional molecule of a protic solvent can be changed by a second molecule of the nucleophile.53
Next, three reaction paths for the proton transfer were studied, i.e., the catalyzed route provided by the coupling of H with AA (C to D via TS2), the non-catalyzed route (G to E via TS3), and the coupling of H with the BH (G to H via TS4). The binding of H with an electrophilic substrate (AA) resulted in stabilization of the zwitterionic intermediate and reduced the Gibbs free energy of activation for the reaction (Fig. 7, TS2). The binding of H with BH (acting in this reaction as a nucleophile) resulted in a decrease in the energy of the transition state TS4 in compare with TS2 and TS3, but taking into account that cyclic intermediate I was not found, this reaction path was ruled out.
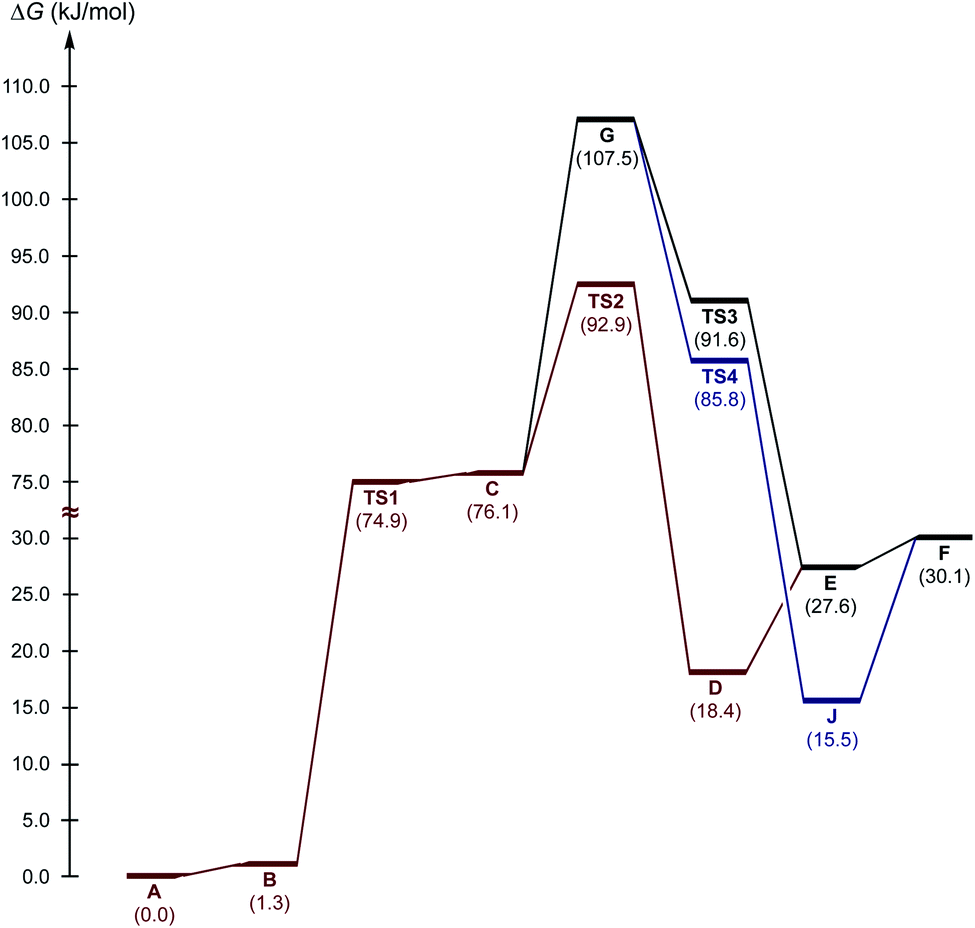 | ||
| Fig. 7 The energy profile for the studied reaction paths. | ||
Based on the kinetic measurements, 1H NMR titration data, and DFT calculations, the plausible mechanism of the studied reaction includes the binding of the organocatalyst H with AA (Scheme 2, a), a nucleophilic attack of the Nsp3 atom on the C atom of the carbonyl group of AA (b and c), incorporation of a MeOH solvent molecule for the proton transfer (d and e), and regeneration of H (f).
To better understand how organocatalyst H binds with the reaction substrates, a theoretical estimation of the strength of the hydrogen and halogen bonds in the optimized equilibrium model structures was performed using the topological analysis of the electron density distribution technique (QTAIM analysis, see Table 1S in ESI for details†). The calculations indicated that in all the structures, H forms typical noncovalent bonds with ligands and the bond energies of H⋯O and I⋯O are 9.2–15.8 kJ mol−1 and 19.6–35.7 kJ mol−1, respectively.
Investigation of covalent catalysis by H
Another type of catalysis—through the formation of covalent bonds of the O atom of the AA carbonyl moiety—was also checked theoretically. All attempts to fix on the potential energy surface of the different kinds of minima for the Meisenheimer complex-like intermediates were unsuccessful (Fig. 8). Thus, geometry optimization for the appropriate model species obtained through the formation of O–C–I, O–C–H1, O–C–H2, O–C–H3, and O–C–H4 (accordingly to the numeration on Fig. 4) led to the initial reactants, and these hypothetical structures were excluded from consideration. These results excluded the possibility of covalent catalysis.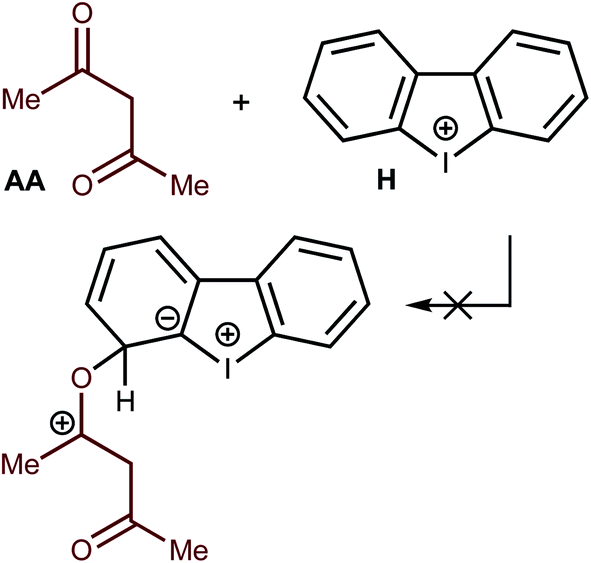 | ||
| Fig. 8 One of the five hypothetical Meisenheimer complex-like intermediate. | ||
Reaction scope
Dibenzidolium triflate catalyzes the generation of N-acyl pyrazoles from AA and a series of aromatic and aliphatic acyl hydrazides (Scheme 3). For the aromatic acyl hydrazides, the reaction typically proceeded for 3.5 h, but for the substrates featuring strong EWG NO2 functionality (5) or bulky Rs in the ortho-position (6, 7, 10) heating for 6 h was required. The reaction with the aliphatic acyl hydrazides also required a prolonged reaction time (12–15). In all studied cases, the reactions initially were monitored by 1H NMR and each reaction was stopped when the concentration of 1–15 was maximal, rather than at the maximal conversion. This is caused by the slow solvolysis of 1–15 under the reaction conditions to give 3,5-dimethyl pyrazole and the corresponding RCO2Me. Our experiments indicated that isolation of the products after 24 h resulted in reduced yields of the pyrazoles by approximately 25% (except 15: isolated yield 53% after 24 h).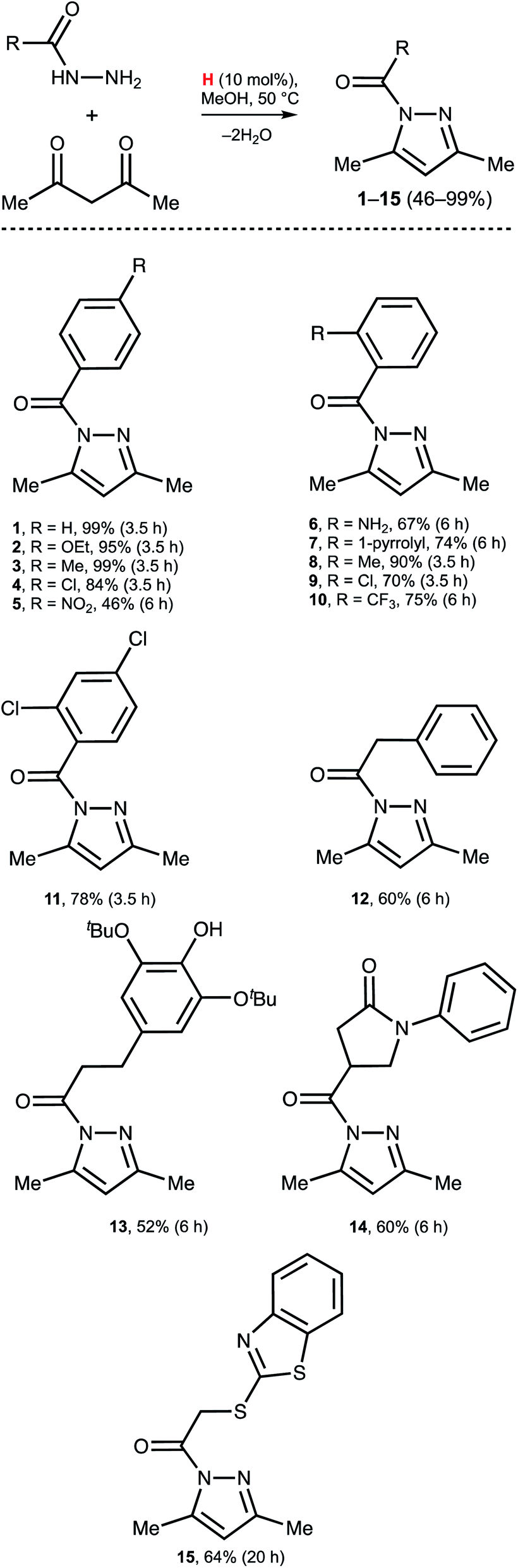 | ||
| Scheme 3 Substrate scope. Isolated yields are given. | ||
Conclusions
We found that dibenziodolium salts are convenient iodine(III)-based noncovalent organocatalysts, which effectively catalyze the Knorr-type reactions of N-acetyl hydrazides with AA to give N-acyl pyrazoles. The reactivity of these salts only slightly depends on the identity of the cation, but, as expected,61 the reactivity of the triflate appears to be higher than the other studied salts. 1H NMR titration data and DFT calculations provided collateral evidence that in MeOH, the catalyst binds to the hydrazide species, which serve as a nucleophile, and this binding proceeds through halogen and hydrogen bonds. Our study indicates that for correct study of binding of iodonium salts with substrates, solvent molecules should be taken into account in the explicit form.The catalytic effect of the iodonium salt is caused by binding with the ketone, which is less energetically favorable than binding with the hydrazide. Binding to the electrophile results in the reduction of the Gibbs free energy of the transition state of a rate-limiting step of the reaction and thus causes the catalytic effect of the iodonium salt.
Experimental section
Materials and instrumentation
Solvents and acetylacetone were obtained from commercial sources and used as received. Acyl hydrazides64 and iodine(III) derivatives65–67 were synthesized by the known methods. All syntheses were conducted in air. Chromatographic separation was carried out on Macherey-Nagel silica gel 60 M (0.063–0.2 mm). Analytical TLC was performed on unmodified Merck ready-to-use plates (TLC silica gel 60 F254) with UV detection. Melting points were measured on a Stuart SMP30 apparatus in capillaries and are not corrected. Electrospray ionization mass-spectra were obtained on a Bruker micrOTOF spectrometer equipped with an electrospray ionization (ESI) source. The instrument was operated in positive ion mode using an m/z range 50–1200. The nebulizer gas flow was 1.0 bar and the drying gas flow 4.0 L min−1. For HRESI+, the studied compounds were dissolved in MeOH. 1H, 13C{1H} and 19F NMR spectra were measured on a Bruker Avance 400 in CD3CN, (CD3)2SO, or CD3OD at 298 K; the residual solvent signal was used as the internal standard.Preparation procedure for benzoyl hydrazide
A heterogeneous mixture of methyl benzoate (341 mg, 250 mmol) and hydrazine monohydrate (15.0 g, 300 mmol) was refluxed for 18 h during which time a clear colorless solution was obtained. After cooling, all volatile components were evaporated in vacuo, and the resulting colorless crystalline solid was dried at room temperature. Recrystallization from benzene/ethanol (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) gave analytically pure benzohydrazide as colorless needles (27.1 g, 79%).
:1) gave analytically pure benzohydrazide as colorless needles (27.1 g, 79%).
Preparation procedure for F
2-Iodobiphenyl (0.898 mmol, 251.0 mg) and TFA (1.39 mL) were dissolved in CH2Cl2 (5 mL) and CH2Cl2 was added until a volume of 10 mL was reached. Then the reagent mixture was pumped through an Oxone filled column with a syringe pump (flow rate of 0.11 mL min−1). After the addition of the reagent, the syringe was immediately replaced with a syringe containing CH2Cl2 (10 mL). CH2Cl2 was pumped for 30 min (flow rate of 0.11 mL min−1) and then the flow rate was increased (0.5 mL min−1) until no CH2Cl2 was left in the syringe. The solvent was removed from the resulting solution in vacuo and the obtained residue was diluted with water (5 mL). The product was extracted with CH2Cl2 (3 × 5 mL). The organic layer was dried over Na2SO4 and the solvent was removed in vacuo. Then Et2O (5 mL) was added to the residue and formation precipitate formation was observed. The suspension was stirred for 10 min and the product was filtered and washed with Et2O (5 mL) and hexane (2 × 5 mL). The product was dried in vacuo. Dibenzoiodonium trifluoroacetate F (96% yield, 379 mg) was obtained as a beige crystalline solid.Preparation procedure for G
m-CPBA (77%, 0.736 g, 3.2 mmol) and a solution of HNTf2 (1.26 g, 4.5 mmol) in CH2Cl2 (5.0 mL) were added to a stirred solution of 2-iodobiphenyl (0.56 g, 0.35 mL, 2.0 mmol) in anhydrous CH2Cl2 (6.0 mL). The solution was stirred for 24 h at room temperature, then the solvent was removed on a rotary evaporator. Et2O (10 mL) was added to the remaining solid. The mixture was stirred for 20 min, then filtered. The obtained solid was washed with Et2O three times and dried in vacuo to produce dibenziodolium bis(trifluoromethane)sulfonimide G (0.84 g, 75%) as a pale-yellow solid.Preparation procedure for H
m-CPBA (77%, 0.736 g, 3.2 mol) and triflic acid (0.57 mL, 6.5 mmol) were added to a stirred solution of 2-iodobiphenyl (0.56 g, 0.35 mL, 2.0 mmol) in anhydrous CH2Cl2 (6 mL) and stirred for 1 h at room temperature. Then CH2Cl2 was removed in vacuo. Et2O (4 mL) was added to the remaining solid. The mixture was stirred for 20 min and then filtered. The obtained solid was washed with Et2O three times and dried in vacuo to provide dibenziodolium trifluoromethanesulfonate H (0.781 g, 91%) as a white solid.Preparation procedure for I
Mixture 1 was obtained by dissolving iodobenzene (0.898 mmol, 183 mg, 100 μL) and TFA (1.39 mL) to in CH2Cl2 to achieve a total volume of 10 mL. Mixture 2 was obtained by dissolving 1,3,5-trimethoxybenzene (1.037 mmol) and TFA (2.92 mL) in CH2Cl2 to achieve a total volume of 11 mL.Mixture 1 was pumped through an Oxone-filled cartridge using a syringe pump (flow rate of 0.11 mL min−1). After 15.5 min, mixture 2 was added using a second syringe pump (flow rate of 0.11 mL min−1). After the addition of mixture 1, the syringe was immediately replaced with a syringe containing CH2Cl2 (10 mL) and pumped (flow rate of 0.11 mL min−1) until mixture 2 was completely added. Then the flow rate was increased (0.22 mL min−1) for 5 min, then increased again (0.5 mL min−1) until the syringe was empty. The solvent was removed from the resulting solution in vacuo and the residue was diluted with water (5 mL). The product was extracted using CH2Cl2 (3 × 5 mL). The organic layer was dried over Na2SO4 and the solvent was removed in vacuo. Then diethyl ether (5 mL) was added to the residue and a precipitate was formed, the suspension was stirred for 10 min, then the product was filtered and washed with diethyl ether (5 mL) and hexane (10 mL). The obtained product was dried in vacuo.
Preparation procedure for J
Finely-crushed, solid 2-iodobenzoic acid (1.0 mmol) was mixed with powdered Oxone (400 mg, 0.65 mmol) in a round-bottomed flask (50 mL) and stirred without a solvent for 5 min using a magnetic stirrer until a homogeneous reaction mass was formed. Then the reaction mixture was cooled with ice to 5 °C and, under magnetic stirring and pre-cooled H2SO4 (5 °C, total 0.8 mL) was added in four portions (0.2 mL) to the center of the reaction mixture. After the addition of each portion, the reaction mass was mechanically shaken to achieve thorough mixing—the color of the resulting mass varied from pale yellow to brown depending on the intensity of mixing. After the addition of H2SO4, the reaction was continuously stirred for 30 min at room temperature. The mixture was then cooled to 5 °C, CH2Cl2 (4 mL) and 1,3,5-trimethylbenzene (3.0 mmol) were added, and was then continuously stirred at room temperature for 3 h. The reaction mixture was re-cooled to 5 °C and CH2Cl2 (5 mL) and a saturated aqueous solution of NaHCO3 were added in small portions until pH 8.0 was reached. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (5 mL). The organic extracts were combined, dried with Na2SO4, the solvent evaporated, and the crystalline product was dried in vacuo. Additional purification of products can be performed by crystallization from water.Syntheses of 1–15
Powder of a benzoyl hydrazide (1.0 mmol) was added to a solution of the iodonium salt (0.1 mmol) in methanol (5 mL) placed in a 10 mL round-bottomed flask, whereupon acetylacetone (1.2 mmol) was added to the mixture. The flask was closed and the obtained homogeneous solution (suspension for 12) was kept at 50 °C for 3.5 h (for 1–8), 6 h (for 9–14), or 20 h (for 15). For 1–15, crude product was subjected to column chromatography on silica gel (eluted with hexane:ethyl acetate = 4:1, v/v) and after collection of the fracture with the product the solvent was evaporated in vacuo at 50 °C. A light yellow oil (1–4, 6, 8–11, and 13) and a colorless crystalline solid (5, 7, 12, 14, and 15) were obtained.
Compounds 1–15 were characterized by HRESI+-MS, IR, 1H and 13C{1H} NMR spectroscopies. N-Acyl pyrazoles 1–5,54,68–71 7,70 9,54 14,72 and 15 (ref. 73) were previously reported and the spectra of these species prepared by our procedure fully agree with the previously published spectral data. The ESI† file contains characterization and spectra of the compounds 1–15.
1H NMR monitoring
Computational details
The full geometry optimization of all model structures in methanol solution was carried out at the DFT level of theory using the M06-2X functional74 with the help of Gaussian-09 program package.75 The quasi-relativistic pseudopotential MWB46 (ref. 76) that described 46 core electrons and appropriate contracted basis set were used for the iodine atom and standard 6-31G* basis sets were used for other atoms. The solvent effects were taken into account using the SMD (Solvation Model based on Density) continuum solvation model suggested by Truhlar and coworkers.77 No symmetry restrictions have been applied during the geometry optimization procedure. The Hessian matrices were calculated analytically for all optimized model structures to prove the location of correct minimum or saddle point on the potential energy surface (no imaginary frequencies or only one imaginary frequency, respectively) and to estimate the thermodynamic parameters, the latter being calculated at 25 °C. The topological analysis of the electron density distribution (QTAIM analysis)78 was performed by using the Multiwfn program (version 3.7).79 The Cartesian atomic coordinates for all optimized equilibrium model structures are presented in the attached xyz-files.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (grant 20-73-10013). Dr Pavel S. Postnikov and Prof. Dr Mekhman S. Yusubov (Tomsk Polytechnic University) are thanked for their valuable advices on the syntheses of the iodine(III) compounds conducted by N. S. S. Physicochemical studies were performed at the Center for Magnetic Resonance, Center for X-ray Diffraction Studies, Chemistry Educational Centre, and Center for Chemical Analysis and Materials Research (all belonging to Saint Petersburg State University).References
- V. Oliveira, M. Cardoso and L. Forezi, Catalysts, 2018, 8, 605 CrossRef.
- Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS.
- T. Chanda and J. C. G. Zhao, Adv. Synth. Catal., 2018, 360, 2–79 CrossRef CAS.
- S. Vellalath and D. Romo, Angew. Chem., Int. Ed., 2016, 55, 13934–13943 CrossRef CAS.
- L. Klier, F. Tur, P. H. Poulsen and K. A. Jorgensen, Chem. Soc. Rev., 2017, 46, 1080–1102 RSC.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS.
- S. Mondal, S. R. Yetra, S. Mukherjee and A. T. Biju, Acc. Chem. Res., 2019, 52, 425–436 CrossRef CAS.
- H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS.
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS.
- D. Bulfield and S. M. Huber, Chem.–Eur. J., 2016, 22, 14434–14450 CrossRef CAS.
- X. Zhang, J. Ren, S. M. Tan, D. Tan, R. Lee and C.-H. Tan, Science, 2019, 363, 400–404 CrossRef CAS.
- C. Bolm, A. Bruckmann and M. Pena, Synlett, 2008, 2008, 900–902 CrossRef.
- S. M. Walter, F. Kniep, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2011, 50, 7187–7191 CrossRef CAS.
- F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028–7032 CrossRef CAS.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- M. Breugst, D. von der Heiden and J. Schmauck, Synthesis, 2017, 49, 3224–3236 CrossRef CAS.
- C. M. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- M. N. Grayson and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 9041–9044 CrossRef CAS.
- M. Puripat, R. Ramozzi, M. Hatanaka, W. Parasuk, V. Parasuk and K. Morokuma, J. Org. Chem., 2015, 80, 6959–6967 CrossRef CAS.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS.
- X. Fang and C. J. Wang, Chem. Commun., 2015, 51, 1185–1197 RSC.
- O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051–7071 RSC.
- F. E. Held and S. B. Tsogoeva, Catal. Sci. Technol., 2016, 6, 645–667 RSC.
- X. Han, H. B. Zhou and C. Dong, Chem. Rec., 2016, 16, 897–906 CrossRef CAS.
- B. L. Zhao, J. H. Li and D. M. Du, Chem. Rec., 2017, 17, 994–1018 CrossRef CAS.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS.
- T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS.
- T. James, M. van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388–9409 CrossRef CAS.
- J. Merad, C. Lalli, G. Bernadat, J. Maury and G. Masson, Chem.–Eur. J., 2018, 24, 3925–3943 CrossRef CAS.
- F. E. Held, D. Grau and S. B. Tsogoeva, Molecules, 2015, 20, 16103–16126 CrossRef CAS.
- S. Schindler and S. M. Huber, in Halogen Bonding II, ed. P. Metrangolo and G. Resnati, Springer, 2015, pp. 167–204 Search PubMed.
- M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS.
- D. von der Heiden, A. Vanderkooy and M. Erdélyi, Coord. Chem. Rev., 2020, 407, 213147 CrossRef CAS.
- L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS.
- J. Bamberger, F. Ostler and O. G. Mancheno, ChemCatChem, 2019, 11, 5198–5211 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- L. Brammer, G. Mínguez Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC.
- K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC.
- M. A. Kryukova, D. M. Ivanov, M. A. Kinzhalov, A. S. Novikov, A. S. Smirnov, N. A. Bokach and V. Yu Kukushkin, Chem.–Eur. J., 2019, 25, 13671–13675 CrossRef CAS.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- S. M. Walter, F. Kniep, L. Rout, F. P. Schmidtchen, E. Herdtweck and S. M. Huber, J. Am. Chem. Soc., 2012, 134, 8507–8512 CrossRef CAS.
- S. Libri, N. A. Jasim, R. N. Perutz and L. Brammer, J. Am. Chem. Soc., 2008, 130, 7842–7844 CrossRef CAS.
- F. Heinen, E. Engelage, C. J. Cramer and S. M. Huber, J. Am. Chem. Soc., 2020, 142, 8633–8640 CrossRef CAS.
- F. Heinen, E. Engelage, A. Dreger, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2018, 57, 3830–3833 CrossRef CAS.
- R. Haraguchi, T. Nishikawa, A. Kanazawa and S. Aoshima, Macromolecules, 2020, 53, 4185–4192 CrossRef CAS.
- Y. Zhang, J. Han and Z.-J. Liu, RSC Adv., 2015, 5, 25485–25488 RSC.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS.
- M. Nozari, A. W. Addison, G. T. Reeves, M. Zeller, J. P. Jasinski, M. Kaur, J. G. Gilbert, C. R. Hamilton, J. M. Popovitch, L. M. Wolf, L. E. Crist and N. Bastida, J. Heterocycl. Chem., 2018, 55, 1291–1307 CrossRef CAS.
- M. J. Kim, S. M. Gaube, M. H. R. Beh, C. D. Smith and A. Thompson, RSC Adv., 2019, 9, 31773–31780 RSC.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- V. K. Burianova, D. S. Bolotin, A. S. Mikherdov, A. S. Novikov, P. P. Mokolokolo, A. Roodt, V. P. Boyarskiy, D. Dar’in, M. Krasavin, V. V. Suslonov, A. P. Zhdanov, K. Y. Zhizhin and N. T. Kuznetsov, New J. Chem., 2018, 42, 8693–8703 RSC.
- G.-P. Yang, X. He, B. Yu and C.-W. Hu, Appl. Organomet. Chem., 2018, 32, e4532 CrossRef.
- B. Balandis, G. Ivanauskaite, J. Smirnoviene, K. Kantminiene, D. Matulis, V. Mickevicius and A. Zubriene, Bioorg. Chem., 2020, 97, 103658 CrossRef CAS.
- A. Mumtaz, K. Saeed, A. Mahmood, S. Zaib, A. Saeed, J. Pelletier, J. Sevigny and J. Iqbal, Bioorg. Chem., 2020, 101, 103996 CrossRef CAS.
- L. A. Baeva, R. M. Nugumanov, R. R. Gataullin and A. A. Fatykhov, Chem. Heterocycl. Compd., 2020, 56, 548–554 CrossRef CAS.
- Z. M. Bikbaeva, D. M. Ivanov, A. S. Novikov, I. V. Ananyev, N. A. Bokach and V. Y. Kukushkin, Inorg. Chem., 2017, 56, 13562–13578 CrossRef CAS.
- K. E. Riley, J. S. Murray, J. Fanfrlik, J. Rezac, R. J. Sola, M. C. Concha, F. M. Ramos and P. Politzer, J. Mol. Model., 2011, 17, 3309–3318 CrossRef CAS.
- C. B. Aakeroy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem.–Eur. J., 2013, 19, 16240–16247 CrossRef.
- R. J. Mayer, A. R. Ofial, H. Mayr and C. Y. Legault, J. Am. Chem. Soc., 2020, 142, 5221–5233 CrossRef CAS.
- E. Erdtman, E. A. C. Bushnell, J. W. Gauld and L. A. Eriksson, Comput. Theor. Chem., 2011, 963, 479–489 CrossRef CAS.
- D. S. Bolotin, V. K. Burianova, A. S. Novikov, M. Y. Demakova, C. Pretorius, P. P. Mokolokolo, A. Roodt, N. A. Bokach, V. V. Suslonov, A. P. Zhdanov, K. Y. Zhizhin, N. T. Kuznetsov and V. Y. Kukushkin, Organometallics, 2016, 35, 3612–3623 CrossRef CAS.
- M. H. Klingele and S. Brooker, Eur. J. Org. Chem., 2004, 2004, 3422–3434 CrossRef.
- P. S. Postnikov, O. A. Guselnikova, M. S. Yusubov, A. Yoshimura, V. N. Nemykin and V. V. Zhdankin, J. Org. Chem., 2015, 80, 5783–5788 CrossRef CAS.
- M. S. Yusubov, N. S. Soldatova, P. S. Postnikov, R. R. Valiev, D. Y. Svitich, R. Y. Yusubova, A. Yoshimura, T. Wirth and V. V. Zhdankin, Eur. J. Org. Chem., 2018, 2018, 640–647 CrossRef CAS.
- N. S. Soldatova, P. S. Postnikov, M. S. Yusubov and T. Wirth, Eur. J. Org. Chem., 2019, 2019, 2081–2088 CrossRef CAS.
- H. D. Mkoyi, S. O. Ojwach, I. A. Guzei and J. Darkwa, J. Organomet. Chem., 2013, 724, 95–101 CrossRef CAS.
- J. M. Ovian, C. B. Kelly, V. A. Pistritto and N. E. Leadbeater, Org. Lett., 2017, 19, 1286–1289 CrossRef CAS.
- P. A. Channar, S. Afzal, S. A. Ejaz, A. Saeed, F. A. Larik, P. A. Mahesar, J. Lecka, J. Sevigny, M. F. Erben and J. Iqbal, Eur. J. Med. Chem., 2018, 156, 461–478 CrossRef CAS.
- H. Sharghi, J. Aboonajmi, M. Mozaffari, M. M. Doroodmand and M. Aberi, Appl. Organomet. Chem., 2017, 32, e4124 CrossRef.
- V. Mickevičius and R. Vaickelioniené, Chem. Heterocycl. Compd., 2008, 44, 170–172 CrossRef.
- A. Rutavicius, S. Valiulene and Z. Kuodis, Chem. Heterocycl. Compd., 1995, 31, 629–633 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 2006, 80, 1431–1441 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09640g |
| This journal is © The Royal Society of Chemistry 2021 |