
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed Tsuji–Trost-type reaction of benzofuran-2-ylmethyl acetates with nucleophiles†
Antonio Arcadi a,
Giancarlo Fabrizib,
Andrea Fochettib,
Francesca Ghirgac,
Antonella Goggiamani*b,
Antonia Iazzetti*b,
Federico Marroneb,
Giulia Mazzoccantib and
Andrea Serraioccob
a,
Giancarlo Fabrizib,
Andrea Fochettib,
Francesca Ghirgac,
Antonella Goggiamani*b,
Antonia Iazzetti*b,
Federico Marroneb,
Giulia Mazzoccantib and
Andrea Serraioccob
aDipartimento di Scienze Fisiche e Chimiche, Università degli Studi di L'Aquila, Via Vetoio, 67100 Coppito, AQ, Italy
bDipartimento di Chimica e Tecnologie del Farmaco, Sapienza, Università di Roma, P.le A. Moro 5, 00185 Rome, Italy. E-mail: antonella.goggiamani@uniroma1.it
cCenter for Life Nano Science@Sapienza, Istituto Italiano di Tecnologia, Viale Regina Elena 291, 00161 Rome, Italy
First published on 4th January 2021
Abstract
The palladium-catalyzed benzylic-like nucleophilic substitution of benzofuran-2-ylmethyl acetate with N, S, O and C soft nucleophiles has been investigated. The success of the reaction is dramatically influenced by the choice of catalytic system: with nitrogen based nucleophiles the reaction works well with Pd2(dba)3/dppf, while with sulfur, oxygen and carbo-nucleophiles [Pd(η3-C3H5)Cl]2/XPhos is more efficient. The regiochemical outcome shows that the nucleophilic substitution occurs only on the benzylic position of the η3-(benzofuryl)methyl complex. The high to excellent yields and the simplicity of the experimental procedure make this protocol a versatile synthetic tool for the preparation of 2-substituted benzo[b]furans.
The benzo[b]furan core is a key structural feature present in several natural and unnatural pharmacologically active compounds. Members of this class of compound exhibit various biological properties including anti-inflammatory, anti-oxidant, anti-arrhythmic, hemostatic, antimicrobial, anti-viral, antifungal, and anti-tumor activities and are antagonists for the H3 receptor and angiotensin II.1 Some of them are promising drugs against Parkinson's2 and Alzheimer's disease.3
Because of this, benzo[b]furans are an attractive synthetic target, and, in this context, transition metal catalysis has played a remarkable role. Particularly, palladium catalyzed reactions have been widely employed in the de novo construction of benzo[b]furan ring and in the selective functionalization of the preformed benzo[b]furan system providing functional group tolerance, simplified procedures, and improved yields.4–9
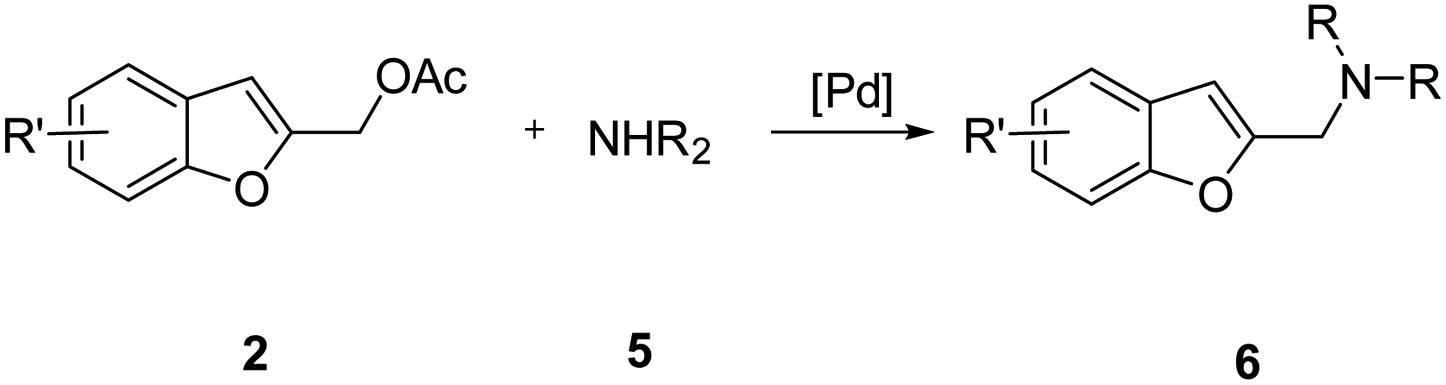
Since 1-(benzofuran-2-ylmethyl)-4-benzylpiperazine has been selected as lead compound for σ1 receptor affinity and selectivity over the σ2 receptor,10 we decided to study a new and efficient protocol for the preparation of 2-(aminomethyl)benzo[b]furans.
As part of our continuing interest in the reactivity of propargyl carbonates,11 and in the development of new approaches for the synthesis of heterocycles, we previously reported the palladium and/or copper catalyzed construction of 2-(aminomethyl)indoles starting from ethyl 3-(o-trifluoroacetamidophenyl)-1-propargylic carbonates12 or 3-(o-trifluoroacetamidoaryl)-1-propargylic alcohols13 and amines (Scheme 1a).
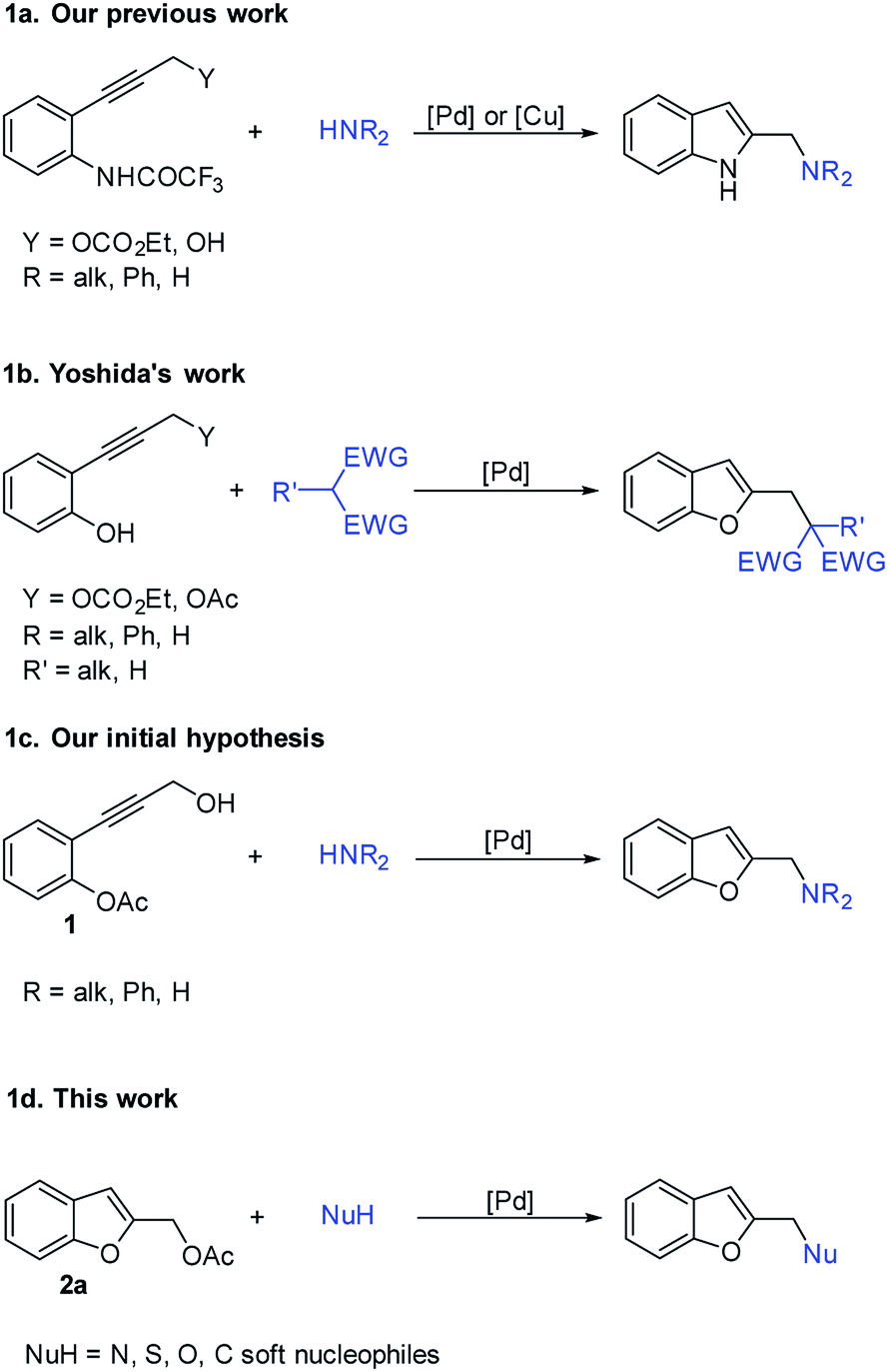 | ||
| Scheme 1 (a and b) Previous works; (c and d) work hypotheses. | ||
Furthermore, Yoshida showed that benzo[b]furan system could be synthesized through the palladium-catalyzed reaction of phenols bearing an ortho propargyl carbonate or acetate and carbon nucleophiles (Scheme 1b); other nucleophiles such as phenols failed because the reactive phenolic hydroxy group would also act as an additional nucleophile leading to complex mixtures.14
Based on this background, we hypothesized that the palladium catalyzed reaction of 2-(3-hydroxyprop-1-yn-1-yl)phenyl acetate 1 with nitrogen nucleophiles, could be a good strategy for producing a variety of 2-(aminomethyl)benzo[b]furans (Schema 1c).
However, in our initial attempts, the reactions of 1 with various amines led to the formation of only traces of the desired products together with benzofuran-2-ylmethanol 4a and polymerized byproducts. These results prompt us to explore the use of the benzofuran-2-ylmethyl acetate 2a as a more suitable building bock to afford our target derivatives through the palladium-catalyzed benzylic-like nucleophilic substitutions with amines and, more generally, with soft nucleophiles (Scheme 1d).
It is well-known that this type of substrate could generate the intermediate η3-heterocyclic complex A (Fig. 1).
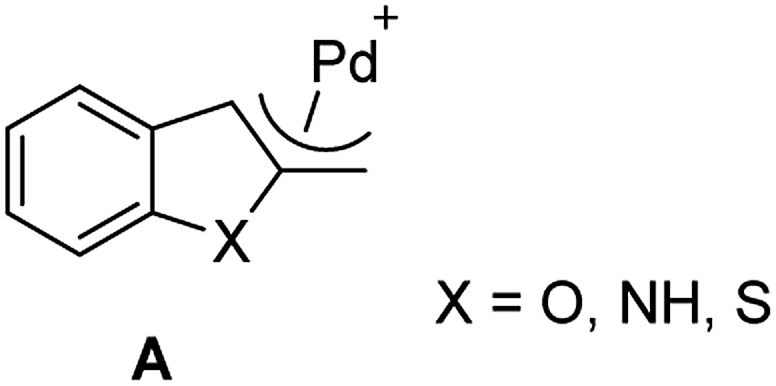 | ||
| Fig. 1 Structure of the η3-heterocyclic complex. | ||
Although the Tsuji–Trost-type reactions of benzylic derivatives with C, N, O, S soft nucleophiles have been widely studied,15 only few examples of the related functionalization of (heteroaryl)methyl acetates, carbonates and pivalates have been reported and with benzofuran based substrates the reactions are limited to dimethyl malonate anions.16
Hereafter we report the results of our investigation.
Results and discussion
The starting benzofuran-2-ylmethyl acetate 2a was prepared in excellent overall yield from commercially available benzofuran-2-carboxylic acid according to the two-step sequence outlined in Scheme 2.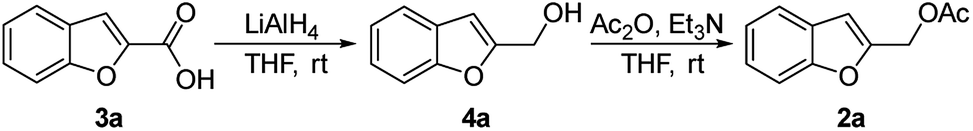 | ||
| Scheme 2 Preparation of 2a. | ||
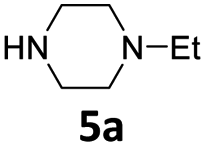
The reaction of 2a with 1-ethylpiperazine 5a was initially examined as the model system. Part of our optimization work using different ligands and solvents is shown in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Catalyst system | Solvent | Time (h) | Yieldb (%) |
| a Unless otherwise stated, reactions were carried out on a 0.4 mmol scale under an argon atmosphere at 120 °C using 0.025 equiv. of Pd2(dba)3, 0.05 equiv. of phosphine ligand, 2 equiv. of 5a, 2 equiv. of K2CO3 in 2.0 mL of solvent.b Yields are given for isolated products.c 2a was recovered in 91% yield.d 2a was recovered in 50% yield.e 4a was isolated in 39% yield.f 0.10 equiv. of phosphine ligand. | ||||
| 1 | — | MeCN | 24 | —c |
| 2 | — | DMSO | 24 | —d,e |
| 3 | Pd2(dba)3/P(o-fur)3f | MeCN | 24 | — |
| 4 | Pd2(dba)3/dppe | DMF | 18 | 34 |
| 5 | Pd2(dba)3/dppe | MeCN | 24 | 60 |
| 6 | Pd2(dba)3/dppf | MeCN | 20 | 87 |
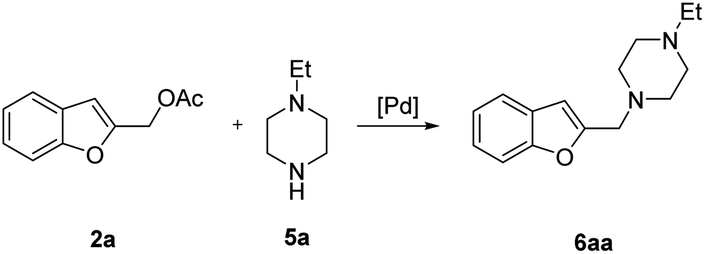
No evidence of the product 6aa was observed performing the reaction without any catalyst (Table 1, entries 1 and 2), or with palladium complexes containing a monodentate phosphine ligand (Table 1, entry 3). Instead, the product 6aa was isolated in 60% and 87% yield, switching to bidentate bisphoshine ligands bearing an appropriate bite angle such us dppe and dppf17 and performing the reaction in MeCN at 120 °C in presence of K2CO3 (Table 1, entries 5 and 6).15a,d
We next examined the reaction using various benzofuran-2-ylmethyl acetates 2 and nitrogen-based nucleophiles under the optimized conditions [Pd2(dba)3, dppf, K2CO3, MeCN, 120 °C] in order to determine the scope and limitations of this process. The results are listed in Table 2. Usually, the reaction gave 2-(aminomethyl)benzofurans 6 in good to excellent yields with a variety of 1-alkyl, aryl and benzyl piperazine (Table 2, entries 1–6, 11 and 12) as well as mono and dialkyl amines (Table 2, entries 7–9) and N-alkylanilines (Table 2, entry 10).
|
||||
|---|---|---|---|---|
| Entry | 1 | Amine 5 | Time (h) | Yieldb (%) |
| a Unless otherwise stated, reactions were carried out on a 0.4 mmol scale under an argon atmosphere at 120 °C using 0.025 equiv. of Pd2(dba)3, 0.05 equiv. of dppf, 2 equiv. of 5, 2 equiv. of K2CO3 in 2.0 mL of MeCN.b Yields are given for isolated products. | ||||
| 1 | 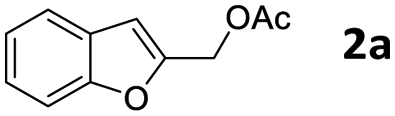 |
 |
20 | 87 (6aa) |
| 2 |  |
16 | 78 (6ab) | |
| 3 |  |
24 | 76 (6ac) | |
| 4 | 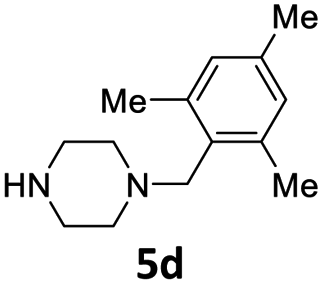 |
8 | 88 (6ad) | |
| 5 | 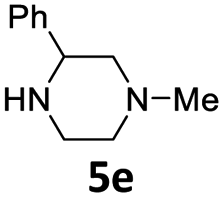 |
24 | 94 (6ae) | |
| 6 | 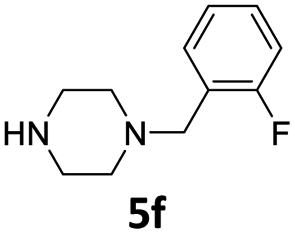 |
20 | 91 (6af) | |
| 7 | 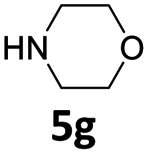 |
5 | 75 (6ag) | |
| 8 | 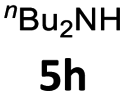 |
27 | 75 (6ah) | |
| 9 | 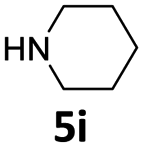 |
24 | 92 (6ai) | |
| 10 | 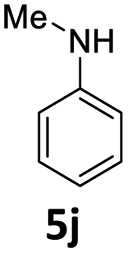 |
48 | 48 (6aj) | |
| 11 | 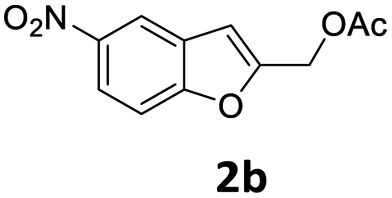 |
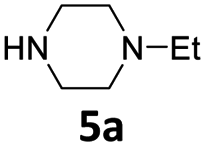 |
3 | 84 (6ba) |
| 12 | 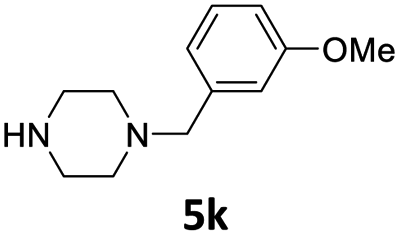 |
3 | 84 (6bk) | |
Encouraged by these results, we decided to investigate the reactivity of 2a with other soft nucleophiles. Because of the presence of the aryl sulfone fragment in a number of compounds exhibiting important biological activities,18 a great deal of attention has been devoted to their synthesis.19
We therefore selected as member of the sulfur nucleophilic class the commercially available sodium p-toluenesulfinate 7a.
When the sulfonylation of 2a with 7a was attempted under the reaction conditions that were successfully employed with nitrogen nucleophiles [Pd2(dba)3, dppf, K2CO3, MeCN, 120 °C] for 48 h, a dramatic decrease in efficiency was observed and benzofuran 8aa was obtained only in 30% yield (Table 3, entry 3).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst system | Solvent | Time (h) | Yieldb 8aa (%) | Yieldb 8′aa (%) |
| a Unless otherwise stated, reactions were carried out on a 0.4 mmol scale under an argon atmosphere at 120 °C using 0.05 equiv. of Pd, 0.05 equiv. of phosphine ligand, 2 equiv. of 7a, 2 equiv. of K2CO3 in 2.0 mL of anhydrous solvent.b Yields are given for isolated products.c 2a was recovered in almost quantitative yield.d 2a was recovered in 70% yield.e 2a was recovered in 60% yield.f 2a was recovered in 33% yield.g 4a was isolated in 17% yield.h 2a was recovered in almost quantitative yield.i 2a was recovered in 38% yield.j 0.10 equiv. of phosphine ligand.k Carried out in 2.0 mL of anhydrous MeCN and 0.5 mL of anhydrous THF. | |||||
| 1 | — | MeCN | 24 | —c | — |
| 2 | Pd2(dba)3/dppf | MeCN | 24 | 5d | — |
| 3 | Pd2(dba)3/dppf | MeCN | 48 | 30e | — |
| 4 | Pd2(dba)3/dppf | DMSO | 48 | —f,g | — |
| 5 | Pd(PPh3)4 | MeCN | 24 | —h | — |
| 6 | Pd2(dba)3/P(o-fur)3 | MeCN | 48 | 30i, j | — |
| 7 | Pd2(dba)3/DavePhos | MeCN | 2 | 63 | — |
| 8 | Pd2(dba)3/SPhos | MeCN | 7 | 82 | — |
| 9 | [Pd(η3-C3H5)Cl]2/SPhos | MeCN | 2 | 86 | 12 |
| 10 | [Pd(η3-C3H5)Cl]2/SPhos | THF | 20 | 58 | 8 |
| 11 | [Pd(η3-C3H5)Cl]2/SPhos | MeCN/THF | 2 | 92k | 5 |
| 12 | [Pd(η3-C3H5)Cl]2/RuPhos | MeCN/THF | 3 | 70k | 23 |
| 13 | [Pd(η3-C3H5)Cl]2/XPhos | MeCN/THF | 1.5 | 98k | Traces |
| 14 | [Pd(η3-C3H5)(XPhos)Cl] | MeCN/THF | 1 | 72k | 5 |
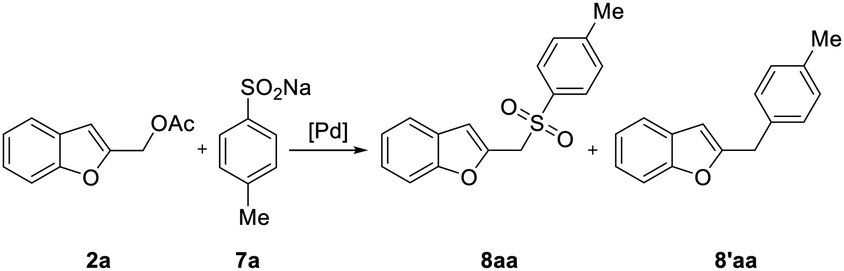
Reexamining the influence of some variables, such as ligands, palladium precatalyst, temperature and equivalents of sulfinate salt on the reaction outcome (Table 3), we found that the employment of Buchwald dialkylmonophosphine ligands20 led to a significant improvement. For example, when the model reaction was carried out with Pd2(dba)3 and DavePhos or SPhos in presence of K2CO3 in MeCN at 120 °C, 8aa was isolated in 63% and 82% yield, respectively (Table 3, entry 7 and 8).
The employment of [Pd(η3-C3H5)Cl]2 as a precatalyst, previously used in sulfonylation15d,e and phosphonylation15i of benzylic carbonates with bidentate bisphosphine ligands such as DPPF and DPEphos, was also attempted.
Because recently Colacot21 and O'Connor22 described the preparation and characterization of neutral Pd(ally)LCl complexes containing Buchwald-type ligands that are high reactive precatalyst for coupling reactions, we thought that the in situ formation of this type of precatalyst could deserve advantages.
Indeed, compound 8aa was isolated in excellent 92% yield after 2 h along with the desulfination product23 2-(4-methylbenzyl)benzofuran 8′aa by generating the active palladium complex Pd(allyl)(Sphos)Cl in THF and performing the reaction in MeCN (Table 3, entry 11). Interestingly, employing XPhos under the same conditions afforded the desired sulfone in 98% yield after 1.5 h (Table 3, entry 13).
To verify the greater effectiveness of our procedure, we carried out a comparative experiment with the isolated complex Pd(allyl)(XPhos)Cl; the result demonstrated the greater efficiency of the in situ generated complex (compare entries 13 and 14, Table 3).
The best result in terms of yield, reaction time, and excess of sulfinate was therefore obtained when the reaction was carried out using [Pd(η3-C3H5)Cl]2/XPhos, 2 equiv. of 7a at 120 °C in a mixture of MeCN/THF solvents. Consequently, these conditions were employed when the procedure was extended to include functionalized benzofurans and benzene sulfinate 7b (Table 4). No benzofuran-2-ylmethyl arylsulfinate resulting from the competitive O-attack of the ambident sulfinate anion was observed in all experiments,24 while little amount of 8′ was usually isolated.
|
|||||
|---|---|---|---|---|---|
| Entry | 2 | Ar | Time (h) | Yieldb 8 (%) | Yieldb 8′ (%) |
| a Unless otherwise stated, reactions were carried out on a 0.4 mmol scale under an argon atmosphere at 120 °C using 0.025 equiv. of [Pd(η3-C3H5)Cl]2, 0.05 equiv. of XPhos, 2 equiv. of 7, 2 equiv. of K2CO3 in 2 mL anhydrous MeCN and 0.5 mL of anhydrous THF.b Yields are given for isolated products. | |||||
| 1 |  |
4-MeC6H4 7a | 1.5 | 98 (8aa) | Traces |
| 2 | C6H5 7b | 1.5 | 89 (8ab) | 5 (8′ab) | |
| 3 |  |
4-MeC6H4 7a | 3 | 84 (8ba) | |
| 4 | C6H5 7b | 3 | 84 (8bb) | 9 (8′bb) | |
| 5 |  |
4-MeC6H4 7a | 1.5 | 84 (8ca) | |
| 6 | C6H5 7b | 1 | 91 (8cb) | 6 (8′cb) | |

The potential of this strategy for the preparation of 2-polysubstituted benzo[b]furans is further demonstrated by the formation of 2-(aryloxymethyl)benzofuran 10 in good to excellent yields by reaction of 2 with many neutral, electron-rich, and electron-poor phenols 9 (Table 5). The experimental conditions tolerate a variety of functional groups including ether, keto, ester, and cyano, groups.
|
||||
|---|---|---|---|---|
| Entry | 2 | 9 | Time (h) | Yieldb (%) |
| a Unless otherwise stated, reactions were carried out on a 0.4 mmol scale under an argon atmosphere at 120 °C using 0.025 equiv. of [Pd(η3-C3H5)Cl]2, 0.05 equiv. of XPhos, 2 equiv. of 9, 2 equiv. of K2CO3 in 2 mL anhydrous MeCN and 0.5 mL of anhydrous THF.b Yields are given for isolated products.c 11a and 11b were isolated respectively in 6 and 8% yield. | ||||
| 1 |  |
4-OMeC6H4 9a | 1 | 90 (10aa) |
| 2 | 3-CO2MeC6H4 9b | 1 | 92 (10ab) | |
| 3 | 4-FC6H4 9c | 1 | 98 (10ac) | |
| 4 | 2,3,5-Me3C6H2 9d | 3 | 75 (10ad)c | |
| 5 | 4-CNC6 H4 9e | 1.5 | 84 (10ae) | |
| 6 | 4-PhC6H4 9f | 1.5 | 87 (10af) | |
| 7 | 3-(C15H31) C6H4 9g | 0.75 | 82 (10ag) | |
| 8 | 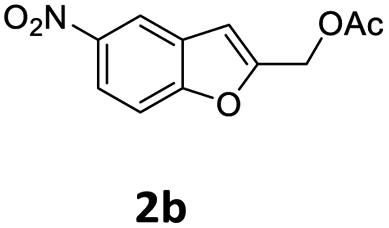 |
4-OMeC6H4 9a | 0.75 | 87 (10ba) |
| 9 | 4-tBuC6H4 9h | 1 | 93 (10bh) | |
| 10 | 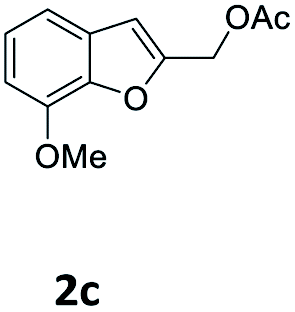 |
4-OMeC6H4 9a | 2 | 90 (10ca) |
| 11 | 3-COMeC6H4 9i | 1.5 | 85 (10ci) | |
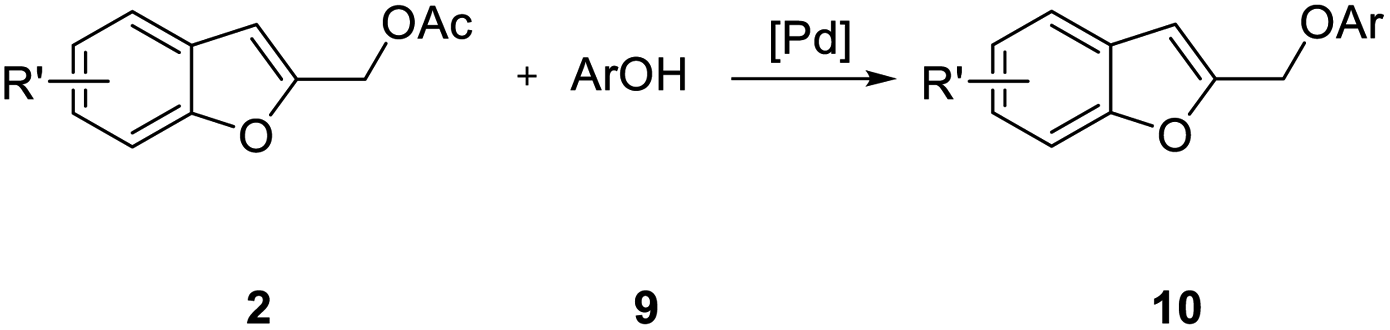
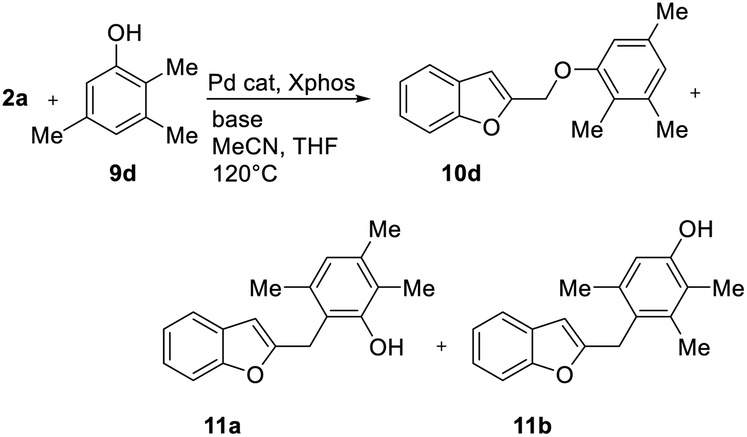
With phenol 9d, C-alkylated compounds 11a and 11b were isolated together with the expected O-alkylated main product 10d. Since with bidentate anions C/O-alkylated ratio is affected by the degree of aggregation, we briefly investigated the influence of the cation in the M2CO3 bases (Table 6, entries 1–5). As expected, the O/C-alkylation ratio correlates with the M+ size: larger is the cation, higher O/C resulted.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | Atomic radius (Å) | Yieldb 10d (%) | Yieldb 11a (%) | Yieldb 11b (%) | 10d/(11a + 11b) |
| a Unless otherwise stated, reactions were carried out on a 0.4 mmol scale under an argon atmosphere at 120 °C using 0.025 equiv. of [Pd(η3-C3H5)Cl]2, 0.05 equiv. of XPhos, 2 equiv. of 9, 2 equiv. of base in 2 mL anhydrous MeCN and 0.5 mL of anhydrous THF.b Yields are given for isolated products. | ||||||
| 1 | Li2CO3 | 0.76 | — | — | — | — |
| 2 | Na2CO3 | 1.02 | 22 | 13 | 16 | 44/56 |
| 3 | K2CO3 | 1.38 | 75 | 6 | 8 | 84/16 |
| 4 | Rb2CO3 | 1.52 | 86 | 8 | 4 | 87/13 |
| 5 | Cs2CO3 | 1.67 | 98 | Traces | Traces | ≤99/1 |
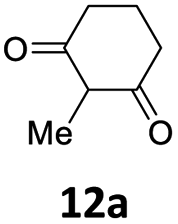
Subsequent studies were directed toward soft carbon nucleophiles derived from compounds with activated methylene group, a target of obvious interest for academic and industrial applications. We also observed very high yields with this class of pro-nucleophiles (Table 7); furthermore, to the best of our knowledge, we are reporting the first example of the Tsuji–Trost-type reaction of heterobenzylic compounds with Meldrum's acid derivatives, whose reactivity in palladium-catalyzed nucleophilic substitution of propargylic carbonates we previously reported.25
|
||||
|---|---|---|---|---|
| Entry | 2 | 12 | Time (h) | Yieldb (%) |
| a Unless otherwise stated, reactions were carried out on a 0.4 mmol scale under an argon atmosphere at 120 °C using 0.025 equiv. of [Pd(η3-C3H5)Cl]2, 0.05 equiv. of XPhos, 2 equiv. of 12, 2 equiv. of K2CO3 in 2 mL anhydrous MeCN and 0.5 mL of anhydrous THF.b Yields are given for isolated products.c Diethyl 2,2-bis(benzofuran-2-ylmethyl)malonate 13′ae was isolated in 15% yield. | ||||
| 1 | 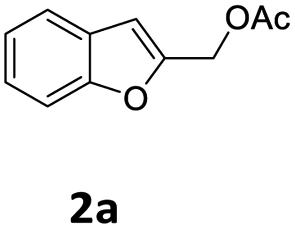 |
 |
0.25 | 87 (13aa) |
| 2 |  |
2 | 86 (13ab) | |
| 3 | 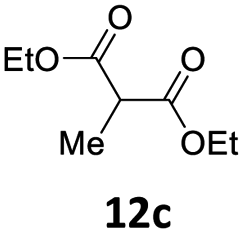 |
2 | 70 (13ac) | |
| 4 | 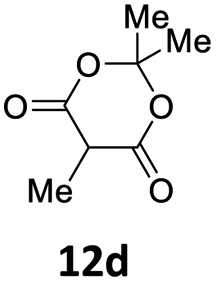 |
1 | 97 (13ad) | |
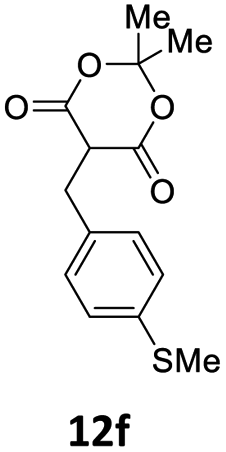
| 5 | 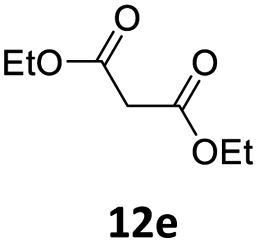 |
1 | 57 (13ae) c | |
| 6 | 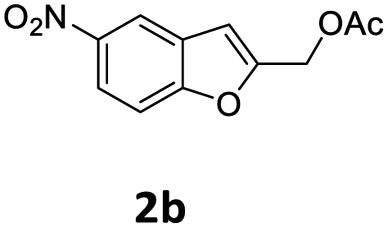 |
 |
1 | 98 (13bf) |
According to literature,18a–d the key intermediate of the functionalization of 2-benzofuranyl methyl acetates was suggested to be the η3-benzofurylmethyl complexes A, which undergoes the nucleophilic attack of the added nucleophile. In our experiments, regardless of the nature of the nucleophiles, the nucleophilic attack was found to occur exclusively at the benzylic carbon, the less sterically hindered position; no evidence was ever obtained of products derived from nucleophilic attack at the C3-position of the benzofuran ring.
Conclusions
In conclusion, we have developed a regioselective palladium-catalyzed benzylic-like nucleophilic substitution of benzofuran-2-ylmethyl acetates with N, S, O and C-nucleophiles to afford 2-substituted benzofurans.The usually high to excellent yields and the simplicity of the experimental procedure make this method particularly convenient for the preparation of this class of compounds.
Experimental
A list of chemicals and instrumentation is provided in the ESI.†Typical procedure for the preparation of benzofuran-2-ylmethanol 4a
In a flame dried two-necked round bottom flask charged with a stir bar, LiAlH4 (2 M, 3.4 mL, 6.787 mmol, 1.1 equiv.) was added drop to drop to a solution of benzofuran-2-carboxylic acid 3a (1.0 g, 6.170 mmol, 1 equiv.) at 0 °C in anhydrous THF (20 mL) under Ar. The mixture was allowed to warm to room temperature and stirred for 2 hours. After the complete consumption of the starting material (TLC, hexane/EtOAc 90/10 v/v), the reaction was cooled down to 0 °C and quenched by slow addition of an 80 percent aqueous MeOH solution. The mixture was extracted with AcOEt, washed with brine and the combined organic phase was dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica gel, n-hexane/EtOAc 85/15 v/v, Rf = 0.24) to afford 0.713 g of benzofuran-2-ylmethanol 4a as a white solid (80% yield).Typical procedure for the preparation of benzofuran-2-ylmethyl acetate 2a
To a stirred solution of benzofuran-2-ylmethanol 4a (0.700 g, 4.7 mmol) in THF (10 mL) was successively added acetic anhydride (280 μL, 5.170 mmol, 1.1 equiv.) and triethylamine (350 μL, 5.640 mmol, 1.2 equiv.) at 0 °C. The mixture was allowed to warm to room temperature and stirred for 24 h. After fully consumption of substrate 4a, the reaction was quenched with a solution of KHSO4 (10% w/w), diluted with AcOEt and washed with a saturated NaHCO3 solution and with brine. The combined organic layer was dried over Na2SO4, filtered and concentrated under vacuum. The crude product 2a was pure enough to be used directly in the next step (quantitative yield).Typical procedure for the preparation of 1-(benzofuran-2-ylmethyl)-4-ethylpiperazine 6aa
In a 50 mL Carousel Tube Reactor (Radely Discovery Technology) containing a magnetic stirring bar Pd2dba3 (9.2 mg, 0.010 mmol, 0.025 equiv.) and dppf (11.1 mg, 0.020 mmol, 0.05 equiv.) were dissolved at room temperature with 1.0 mL of anhydrous MeCN. Then, benzofuran-2-ylmethyl acetate 2a (76.0 mg, 0.4 mmol, 1.0 equiv.), N-ethylpiperazine 5a (152 mL, 0.80 mmol, 2.0 equiv.), K2CO3 (165.6 mg, 1.20 mmol, 2.0 equiv.), and 1.0 mL of solvent were added. The mixture was stirred for 24 h at 100 °C under Ar. After this time, the reaction mixture was cooled to room temperature, diluted with Et2O, washed with a saturated NaHCO3 solution and with brine. The organic extract was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by chromatography on SiO2 (25–40 μm), eluting with a 80/20 (v/v) n-hexane/AcOEt mixture (Rf = 0.22) to obtain 84.9 mg (87% yield) of 1-(benzofuran-2-ylmethyl)-4-ethylpiperazine 6aa.Typical procedure for the preparation of 2-(tosylmethyl)benzofuran 8aa
In a 50 mL Carousel Tube Reactor (Radely Discovery Technology) containing a magnetic stirring bar [Pd(η3-C3H5)Cl]2 (3.7 mg, 0.010 mmol, 0.025 equiv.) and XPhos (9.5 mg, 0.020 mmol, 0.05 equiv.) were dissolved at room temperature with 0.5 mL of anhydrous THF under Ar. Then, benzofuran-2-ylmethyl acetate 2a (76.0 mg, 0.4 mmol, 1.0 equiv.), sodium 4-tolylsulphinate 7a (142.5 mg, 0.80 mmol, 2.0 equiv.), K2CO3 (110.4 mg, 0.80 mmol, 2.0 equiv.), and 1.0 mL of anhydrous MeCN were added. The mixture was stirred for 1 h at 120 °C under Ar. After this time, the reaction mixture was cooled to room temperature, diluted with Et2O, washed with brine. The organic extract was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by chromatography on SiO2 (25–40 μm), eluting with a 70/30 (v/v) n-hexane/AcOEt mixture (Rf = 0.24) to obtain 112.2 mg (98% yield) of 2-(tosylmethyl)benzofuran 8aa.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work carried out in the framework of PRIN 2017 (2017SXBSX$) and the University “Sapienza”, Rome.Notes and references
- For recent review, see: (a) H. Khanam and Shamsuzzaman, Eur. J. Med. Chem., 2015, 97, 483 CrossRef CAS; (b) Y.-H. Miao, Y.-H. Hu, J. Yang, T. Liu, J. Sun and X.-J. Wang, RSC Adv., 2019, 9, 27510 RSC. For specific paper, see: (c) D. B. Judd, M. D. Dowle, D. Middlemiss, D. I. C. Scopes, B. C. Ross, T. I. Jack, M. Pass, E. Tranquillini, J. E. Hobson, T. A. Panchal, P. G. Stuart, J. M. S. Paton, T. Hubbard, A. Hilditch, G. M. Drew, M. J. Robertson, K. L. Clark, A. Travers, A. A. E. Hunt, J. Polley, P. J. Eddershaw, M. K. Bayliss, G. R. Manchee, M. D. Donnelly, D. G. Walker and S. A. Richards, J. Med. Chem., 1994, 37, 3108 CrossRef CAS; (d) D. Middlemiss, S. P. Watson, B. C. Ross, M. D. Dowle, D. I. C. Scopes, J. G. Montana, P. Shah, G. C. Hirst, T. A. Panchal, J. M. S. Paton, M. Pass, T. Hubbard, J. Hamblett, K. S. Cardwell, T. I. Jack, G. Stuart, S. Coote, J. Bradshaw, G. M. Drew, A. Hilditch, K. L. Clark, M. J. Robertson, M. K. Bayliss, M. Donnelly, E. Palmer and G. R. M. Manchee, Bioorg. Med. Chem. Lett., 1993, 3, 589 CrossRef CAS; (e) P. R. Halfpenny, D. C. Horwell, J. C. Hunter and D. C. Rees, J. Med. Chem., 1990, 33, 286 CrossRef CAS; (f) W.-J. Song, X.-D. Yang, X.-H. Zeng, X.-L. Xu, G.-L. Zhang and H.-B. Zhang, RSC Adv., 2012, 2, 4612 RSC; (g) S. Butini, S. Gemma, G. Campiani, S. Franceschini, F. Trotta, M. Borriello, N. Ceres, S. Ros, S. S. Coccone, M. Bernetti, M. D. Angelis, M. Brindisi, V. Nacci, I. Fiorini, E. Novellino, A. Cagnotto, T. Mennini, K. Sandager-Nielsen, J. T. Andreasen, J. Scheel-Kruger, J. D. Mikkelsen and C. Fattorusso, J. Med. Chem., 2009, 52, 151 CrossRef CAS; (h) S. A. Galal, A. S. Abd El-All, M. M. Abdallah and H. I. El Diwani, Bioorg. Med. Chem. Lett., 2009, 19, 2420 CrossRef CAS; (i) S. M. Bakunova, S. A. Bakunov, T. Wenzler, T. Barszcz, K. A. Werbovetz, R. Brun, J. E. Hall and R. R. Tidwell, J. Med. Chem., 2007, 50, 5807 CrossRef CAS; (j) A. E. Kozikowsky, WO 010008, 2006 (Chem. Abstr., 2006, 144, 164278) Search PubMed; (k) R. E. Ziegert, K. Knepper and S. Brease, J. Comb. Chem., 2005, 7, 147 CrossRef CAS; (l) J. J. Marugan, C. Manthey, B. Anaclerio, L. Lafrance, T. Lu, T. Markotan, K. A. Leonard, C. Crysler, E. Eisennagel, M. Dasgupta and B. Tomczuk, J. Med. Chem., 2005, 48, 926 CrossRef CAS; (m) G. A. Gfesser, R. Faghih, Y. L. Bennani, M. P. Curtis, T. A. Esbenshade, A. A. Hancock and M. D. Cowart, Bioorg. Med. Chem. Lett., 2005, 15, 2559 CrossRef CAS; (n) M. Wahab Khan, M. Jahangir Alam, M. A. Rashid and R. Chowdhury, Bioorg. Med. Chem., 2005, 13, 4796 CrossRef CAS; (o) M. Cowart, J. K. Pratt, A. O. Stewart, Y. L. Bennani, T. A. Esbenshade and A. A. Hancock, Bioorg. Med. Chem. Lett., 2004, 14, 689 CrossRef CAS; (p) M. D. Collini, D. H. Kaufman, E. S. Manas, H. A. Harris, R. A. Henderson, Z. B. Xu, R. J. Unwalla and C. P. Miller, Bioorg. Med. Chem. Lett., 2004, 14, 4925 CrossRef CAS; (q) K. Kawasaki, M. Masabuchi, K. Morikami, S. Sogabe, T. Aoyama, H. Ebiike, S. Niizuma, M. Hayase, T. Fujii, K. Sakata, H. Shindoh, Y. Shiratori, Y. Aoki, T. Ohtsuka and N. Shimma, Bioorg. Med. Chem. Lett., 2003, 13, 87 CAS; (r) R. Fleischer and M. Wiese, J. Med. Chem., 2003, 46, 4988 CrossRef CAS; (s) L. Bettinetti, K. Schlotter, H. Hubner and P. Gmeiner, J. Med. Chem., 2002, 45, 4594 CrossRef CAS; (t) J.-L. Peglion, B. Goument, N. Despaux, V. Charlot, H. Giraud, C. Nisole, A. N. Tancredi, A. Dekeine, M. Bertrand, P. Genissel and M. J. Millan, J. Med. Chem., 2002, 45, 165 CrossRef CAS; (u) S. Yoo, S.-H. Kim, S.-H. Lee, N.-J. Kim and D.-W. Lee, Bioorg. Med. Chem., 2000, 8, 2311 CrossRef CAS; (v) T. Nagahara, Y. Yokoyama, K. Inamura, S. Katakura, S. Komoriya, H. Yamaguchi, T. Hara and M. Iwamoto, J. Med. Chem., 1994, 37, 1200 CrossRef CAS; (w) M. Masubuchi, H. Ebiike, K. Kawasaki, S. Sogabe, K. Morikami, Y. Shiratori, S. Tsujii, T. Fujii, K. Sakata, M. Hayase, H. Shindo, Y. Aoki, T. Ohtuska and N. Shimma, Bioorg. Med. Chem., 2003, 11, 4463 CrossRef CAS; (x) A. Gangjee, R. Devraj, J. J. McGuire and R. L. Kisliuk, J. Med. Chem., 1995, 38, 3798 CrossRef CAS.
- O. Saku, M. Saki, M. Kurokawa, K. Ikeda, T. Takizawa and N. Uesaka, Bioorg. Med. Chem. Lett., 2010, 20, 1090 CrossRef CAS.
- (a) S. Rizzo, C. Riviare, L. Piazzi, A. Bisi, S. Gobbi, M. Bartolini, V. Andrisano, F. Morroni, A. Tarozzi, J.-P. Monti and A. Rampa, J. Med. Chem., 2008, 51, 2883 CrossRef CAS; (b) Z. Lu, G. R. Ott, R. Anand, R.-Q. Liu, M. B. Covington, K. Vaddi, M. Qian, R. C. Newton, D. D. Christ, J. Trzaskos and J. J.-W. Duan, Bioorg. Med. Chem. Lett., 2008, 18, 1958 CrossRef CAS; (c) H. J. Ha, D. W. Kang and H. M. Kim, J. Med. Chem., 2018, 61, 396 CrossRef CAS.
- (a) A. P. Kozikowski, D. Ma, L. Du, N. E. Lewin and P. M. Blumberg, Pure Appl. Chem., 1994, 66, 2087 CAS; (b) T. Sakamoto, Y. Kondo and H. Yamanaka, Heterocycles, 1988, 27, 2225 CrossRef CAS; (c) C. P. Dell, Sci. Synth., 2001, 10, 11 CAS; (d) S. Cacchi, G. Fabrizi and A. Goggiamani, Heterocycles, 2002, 56, 613 CrossRef CAS; (e) X.-L. Hou, Z. Yang and H. N. C. Wong, Prog. Heterocycl. Chem., 2003, 15, 167 CAS; (f) S. Schröter, C. Stock and T. Bach, Tetrahedron, 2005, 61, 2245 CrossRef; (g) J. J. Li and G. W. Gribble, Palladium in Heterocyclic Chemistry, Pergamon, New York, 2000 Search PubMed; (h) S. Cacchi, G. Fabrizi and A. Goggiamani, Curr. Org. Chem., 2006, 10, 1423 CrossRef CAS; (i) S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Biomol. Chem., 2011, 9, 641 RSC; (j) S. Priyadarshini, P. J. Amal Joseph, P. Srinivas, H. Maheswaran, M. Lakshmi Kantam and S. Bhargava, Tetrahedron Lett., 2011, 52, 1615 CrossRef CAS; (k) C.-H. Lin, Y.-W. Wang and C.-F. Lee, Eur. J. Org. Chem., 2010, 23, 4368 Search PubMed; (l) E. A. Jaseer, D. J. C. Prasad and G. Sekar, Tetrahedron, 2010, 66, 2077 CrossRef CAS; (m) J. Liu, W. Chen, Y. Ji and L. Wanga, Adv. Synth. Catal., 2012, 354, 1585 CrossRef CAS; (n) A. Carrüer, D. Brinet, J.-C. Florent, P. Rousselle and E. Bertounesque, J. Org. Chem., 2012, 77, 1316 CrossRef; (o) B. Yin, C. Cai, G. Zeng, R. Zhang, X. Li and H. Jiang, Org. Lett., 2012, 14, 1098 CrossRef CAS; (p) M. Leibeling, M. Pawliczek, D. Kratzert, D. Stalke and D. B. Werz, Org. Lett., 2012, 14, 346 CrossRef CAS; (q) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Comb. Chem. High Throughput Screening, 2012, 15, 451 CrossRef CAS; (r) C. Li, Y. Zhang, P. Li and L. Wang, J. Org. Chem., 2011, 76, 4692 CrossRef CAS; (s) S. Wang, P. Li, Y. Lin and L. Wang, Org. Lett., 2011, 13, 5968 CrossRef CAS; (t) Y. Luo and J. Wu, Org. Lett., 2011, 13, 5558 Search PubMed; (u) L. Wu, X. Shi, X. Xu, F. Liang and G. Huang, J. Chem. Sci., 2011, 123, 697 CrossRef CAS; (v) V. Guilarte, M. P. Castroviejo, E. Alvarez and R. Sanz, Beilstein J. Org. Chem., 2011, 7, 1255 CrossRef CAS; (w) E. Rasolofonjatovo, A. Hamze, O. Provot, J. Joanna Wdzieczak-Bakala, J. Dubois, J.-D. Brion and M. Alami, Eur. J. Org. Chem., 2011, 4868 Search PubMed; (x) A. Arcadi, F. Blesi, S. Cacchi, G. Fabrizi, A. Goggiamani and F. Marinelli, J. Org. Chem., 2013, 78, 4490 CrossRef CAS.
- For functionalization of benzo[b]furan system via benzofuryl halides or triflates, see: (a) T. Bach and M. Bartels, Synlett, 2001, 1284 CrossRef CAS; (b) T. Bach and M. Bartels, Synthesis, 2003, 925 CrossRef CAS; (c) R. L. Hudkins, J. L. Diebold and F. D. Marsh, J. Org. Chem., 1995, 60, 6218 CrossRef CAS; (d) T. R. Kelly, A. Szabados and Y.-J. Lee, J. Org. Chem., 1997, 62, 428 CrossRef CAS; (e) A. Arcadi, S. Cacchi, G. Fabrizi, F. Marinelli and L. Moro, Synlett, 1999, 1432 CrossRef CAS; (f) F. Kerrigan, C. Martin and G. H. Thomas, Tetrahedron Lett., 1998, 39, 2219 CrossRef CAS; (g) A. Arcadi, F. Blesi, S. Cacchi, G. Fabrizi and A. Goggiamani, Tetrahedron Lett., 2011, 52, 5149 CrossRef CAS.
- For functionalization of benzo[b]furan system via benzofurylstannanes, see: (a) J. F. Einhorn, P. Demerseman and R. Royer, Synthesis, 1984, 978 CrossRef CAS; (b) H. Nakamura, M. Aizawa, D. Takeuchi, A. Murai and O. Shimoura, Tetrahedron Lett., 2000, 41, 2185 CrossRef CAS ; “Palladium in Heterocyclic Chemistry 26, Second Edition: A Guide For The Synthetic Chemist (Tetrahedron Organic Chemistry)”.
- For functionalization of benzo[b]furan system via benzofurylboronic acids, see: (a) B. Joseph, B. Malapel and J.-Y. Mérour, Synth. Commun., 1996, 26, 3289 CrossRef CAS; (b) A. S. Ionkin and W. J. Marshall, Organometallics, 2004, 23, 3276 CrossRef CAS.
- For functionalization of benzo[b]furan system via benzofurylzinc compound, see: I. S. Mann, D. A. Widdowson and J. M. Clough, Tetrahedron, 1991, 47, 7981 CrossRef CAS; A. Seggio, A. Jutand, G. Priem and F. Mongin, Synlett, 2008, 2955 Search PubMed.
- For direct CH activation, see: (a) A. Ohta, Y. Akita, T. Ohkuwa, M. Chiba, R. Fukunaga, A. Miyafuji, T. Nakata, N. Tani and Y. Aoyagi, Heterocycles, 1990, 31, 195 Search PubMed; (b) T. A. Dwight, N. R. Rue, D. Charyk, R. Josselyn and B. DeBoef, Org. Lett., 2007, 9, 3137 CrossRef CAS; (c) S.-D. Yang, C.-L. Sun, Z. Fang, B.-J. Li, Y.-Z. Li and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1473 CrossRef CAS; (d) R. Cano, J. M. Perez, D. J. Ramon and G. P. McGlacken, Tetrahedron, 2016, 72, 1043 CrossRef CAS.
- I. A. Moussa, S. D. Banister, C. Beinat, N. Giboureau, A. J. Reynolds and M. Kassiou, J. Med. Chem., 2010, 53, 6228 CrossRef CAS.
- S. Cacchi, G. Fabrizi, E. Filisti, A. Goggiamani, A. Iazzetti and L. Maurone, Org. Biomol. Chem., 2012, 10, 4699 RSC.
- I. Ambrogio, S. Cacchi and G. Fabrizi, Org. Lett., 2006, 8, 2083 CrossRef CAS.
- (a) S. Cacchi, G. Fabrizi, A. Iazzetti, C. Molinaro, R. Vediglione and A. Goggiamani, Adv. Synth. Catal., 2015, 357, 1053 CrossRef CAS; (b) S. Cacchi, G. Fabrizi, A. Goggiamani, C. Molinaro and R. Vediglione, J. Org. Chem., 2014, 79, 1053 CrossRef; (c) S. Cacchi, G. Fabrizi, A. Goggiamani, A. Iazzetti and R. Vediglione, Synthesis, 2017, 49, 4163 CrossRef CAS.
- (a) M. Yoshida, Y. Morishita, M. Fujita and M. Ihara, Tetrahedron Lett., 2004, 45, 1861 CrossRef CAS; (b) M. Yoshida, Y. Morishita, M. Fujita and M. Ihara, Tetrahedron, 2005, 61, 4381 CrossRef CAS.
- (a) R. Kuwano, Y. Kondo and Y. Matsuyama, J. Am. Chem. Soc., 2003, 125, 12104 CrossRef CAS; (b) A. M. Johns, J. W. Tye and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 16010 CrossRef CAS; (c) R. Kuwano and Y. Kondo, Org. Lett., 2004, 6, 3545 CrossRef CAS; (d) R. Kuwano, Y. Kondo and T. Shirahama, Org. Lett., 2005, 7, 2973 CrossRef CAS; (e) M. Yokogi and R. Kuwano, Tetrahedron Lett., 2007, 48, 6109 CrossRef CAS; (f) A. Najib, K. Hirano and M. Miura, Org. Lett., 2017, 19, 2438 CrossRef CAS; for review, see: (g) B. Liégault, J.-L. Renaud and C. Bruneau, Chem. Soc. Rev., 2008, 37, 290 RSC; (h) R. Kuwano, Synthesis, 2009, 7, 1049 CrossRef; (i) J. Le Bras and J. Muzart, Eur. J. Org. Chem., 2016, 2565 CrossRef CAS; (j) R. Kuwano and H. Kusano, Chem. Lett., 2007, 36, 528 CrossRef CAS; (k) R. Kuwano and H. Kusano, Org. Lett., 2008, 10, 1979 CrossRef CAS; (l) Y. Makida, K. Usui, S. Ueno and R. Kuwano, Chem. Lett., 2017, 46, 1814 CrossRef CAS.
- (a) G. Primault, J.-Y. Legros and J.-C. Fiaud, J. Organomet. Chem., 2003, 687, 353 CrossRef CAS; (b) J.-Y. Legros, G. l. Primault, M.-A. Toffano, M. Rivière and J.-C. Fiaud, Org. Lett., 2000, 2, 433 CrossRef CAS; (c) A. Boutros, J. Y. Legros and J. C. Fiaud, Tetrahedron Lett., 1999, 40, 7329 CrossRef CAS; (d) S. Tabuchi, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2016, 55, 6973 CrossRef CAS.
- Bite angles of DPPE and DPPF in the palladium dichloride complexes are 86° and 99°, respectively: (a) W. L. Steffen and G. J. Palenik, Inorg. Chem., 1976, 15, 2432 CrossRef CAS; (b) T. Hayashi, M. Konishi, Y. Kobori, M. Kumada, T. Higuchi and K. Hirotsu, J. Am. Chem. Soc., 1984, 106, 158 CrossRef CAS.
- (a) I. Ahmad and W. Shagufta, Int. J. Pharmacol. Pharm. Sci., 2015, 7, 19 CAS; (b) D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793 CrossRef CAS; (c) M. Butawan, L. R. Benjamin and J. R. Bloomer, Nutrients, 2017, 9, 290 CrossRef; (d) X. Chen, S. Hussain and S. Parveen, Curr. Med. Chem., 2012, 19, 3578 CrossRef CAS.
- (a) N. S. Simpkins, Sulfones in Organic Synthesis, Pergamon Press, Oxford, 1993 Search PubMed; (b) G. Le Duc, E. Bernoud, G. Prestat, S. Cacchi, G. Fabrizi, A. Iazzetti, D. Madec and G. Poli, Synlett, 2011, 20, 2943 Search PubMed; (c) A. Shavnya, K. D. Hesp, V. Mascitti and A. C. Smith, Angew. Chem., Int. Ed., 2015, 54, 13571 CrossRef CAS; (d) A. Shavnya, S. B. Coffey, A. C. Smith and V. Mascitti, Org. Lett., 2013, 15, 6226 CrossRef CAS; (e) M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew.Chem. Int. Ed., 2014, 53, 4404 (Angew.Chem., 2014, 126, 4493) CrossRef CAS; (f) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 53, 10204 CrossRef CAS; (g) C. S. Richards-Taylor, D. C. Blakemore and M. C. Willis, Chem. Sci., 2014, 5, 222 RSC.
- (a) D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722 CrossRef CAS; see also: (b) X. Huang, K. W. Anderson, D. Zim, L. Jiang, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 6653 CrossRef CAS; (c) T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS; (d) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS; (e) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS.
- A. J. DeAngelis, P. G. Gildner, R. Chow and T. J. Colacot, J. Org. Chem., 2015, 80, 6794 CrossRef CAS.
- K. R. McGarry, M. McDaniel, B. C. Chan and A. R. O'Connor, Polyhedron, 2016, 114, 101 CrossRef CAS.
- (a) S. G. Modha, V. P. Mehtazb and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 5042 RSC; (b) D. H. Ortgies, A. Hassanpour, F. Chen, S. Woo and P. Forgione, Eur. J. Org. Chem., 2016, 408 CrossRef CAS; (c) X. Zhou, J. Luo, J. Liu, S. Peng and D. Deng, Org. Lett., 2011, 13, 1432 CrossRef CAS; (d) B. Liu, Q. Guo, Y. Cheng, J. Lan and J. You, Chem.–Eur. J., 2011, 17, 13415 CrossRef CAS; (e) F. Zhao, Q. Tan, F. Xiao, S. Zhang and G. Deng, Org. Lett., 2013, 15, 1520 CrossRef CAS.
- For the competitive O/S-attack of the ambident sulfinate anion in palladium catalyzed cross coupling reactions with aryl halides see: (a) S. Cacchi, G. Fabrizi, A. Goggiamani and L. M. Parisi, Org. Lett., 2002, 4, 4719 CrossRef CAS; (b) S. Cacchi, G. Fabrizi, A. Goggiamani and L. M. Parisi, Synlett, 2003, 3, 361 CrossRef; (c) S. Cacchi, G. Fabrizi, A. Goggiamani and L. M. Parisi, J. Org. Chem., 2004, 69, 5608 CrossRef CAS.
- I. Ambrogio, S. Cacchi, G. Fabrizi, A. Goggiamani and A. Iazzetti, Eur. J. Org. Chem., 2015, 3147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data of all compounds, and copies of 1H and 13C NMR spectra (PDF). See DOI: 10.1039/d0ra09601f |
| This journal is © The Royal Society of Chemistry 2021 |