
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence activation, patterning and enhancement with photogenerated radicals, a prefluorescent probe and silver nanostructures†
Karol P. Golian ,
Aviya S. Akari,
Gregory K. Hodgson and
Stefania Impellizzeri*
,
Aviya S. Akari,
Gregory K. Hodgson and
Stefania Impellizzeri*
Laboratory for Nanomaterials and Molecular Plasmonics, Department of Chemistry and Biology, Ryerson University, Toronto, ON, Canada. E-mail: simpellizzeri@ryerson.ca
First published on 28th January 2021
Abstract
We synthesized a dual fluorogenic system based on a boron-dipyrromethene (BODIPY) dye coupled with a paramagnetic 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) nitroxide. Electron exchange facilitates the non-radiative relaxation of the singlet state of the chromophore, partially quenching the fluorescence from the BODIPY moiety. Nonetheless, the system can be ‘activated’ in solution or thin polymer films by ultraviolet (UVA) light in the presence of a free radical initiator. UVA illumination promotes the decomposition of the initiator, resulting in the release of a carbon-centered free radical. The dye-TEMPO probe can concomitantly trap the radical or undergo H-abstraction. Both processes consequently turn on the fluorescence and are reversible in nature. We used this protocol to optically imprint a fluorescent pattern on a surface. Moreover, we investigated metal-enhanced fluorescence (MEF) effects by silver nanostructures (AgNP) as a means to improve the performance of our molecular strategy for fluorescence activation. Overall, this work contributes toward the development of improved nanoparticle–dye systems for applications of light-activated fluorescence such as multicolour fluorescence patterning and microscopy, where shorter reaction times, milder conditions, reversibility and a more diverse selection of both excitation sources and emission wavelengths are beneficial.
Introduction
Molecular strategies for writing of data and patterning of functional images on thin polymer films have attracted considerable attention for the fabrication of new functional materials for information technology.1 Such strategies often rely on irreversible chemical transformations that molecules are subjected to upon photoexcitation,2,3 owing to the fact that light irradiation can be performed rapidly and accurately. In addition, methods that rely upon photochemical processes to fabricate a pattern into a material (e.g., optical lithography)4,5 allow for the creation of desired patterns with spatial and temporal control. For such purposes, the ability to write and encode information in the form of fluorescence can greatly facilitate the implementation of optical techniques and devices. For example, writing and reading using fluorescence-controlled molecular systems can be a viable and relatively inexpensive technology for the fabrication of displays with well-defined blue, green, and red fluorescence patterns or practical tools for information storage.1 In this context, the identification and development of mechanisms to photoactivate fluorescence – that is, the custom engineering of organic chromophores with the ability to photochemically switch from a nonemissive state to a fluorescent one under optical control6–10 – is a key aspect of facilitating the implementation of methods for ‘writing’ with fluorescence. This strategy relies on the illumination of a quenched or weakly emissive reactant at an appropriate wavelength, which induces the formation of a fluorescent product. The latter can be accomplished either by intramolecular photochemical transformation of the reactant, or by initiating an intermolecular process that yields an emissive product from the nonemissive reactant. In both instances, only a small, selective population of molecules are allowed to fluoresce at a given time (those that have been ‘activated’). In turn, external control over the ability of chromophores to emit enables advanced light-based applications, such as sensing and super-resolution microscopy.11–17 The same principles of photoactivation of fluorophores can be applied for the development of lithographic-like features through the precise and rationally designed generation of functional images. This ingenious approach ultimately delivers a viable, practical hybridization between imaging and lithography. To date, various methods for preparing fluorescent patterns in thin polymer films have been reported. For the most part, these examples rely on the utilization of photoacid generators (PAGs).18–26 PAGs are photoactive molecules that can release acids either in solution or at the solid state;27–29 upon light irradiation, typically in the ultraviolet regime, a fluorogenic species can be encouraged to emit fluorescence by protonation induced by the PAGs. This design not only allows for photogeneration of a fluorescent pattern wherever the acid has been formed, but also facilitates the study of the mechanism and efficiency of photoacid generation, delivering valuable insights on the spatial distribution of, for example, chemically amplified polymerization reactions. In principle, the same approach can be applied with free radical initiators coupled with nonfluorescent species that are activated by the presence of radicals.30–34 In this design, prefluorescent radical probes can be used synergistically with free radical photoinitiators in the very same manner as halochromic (acid-sensitive) dyes are used with photoacid generators. Upon the basis of these considerations, we have designed a system composed of a switch-on fluorophore that can be selectively activated by ultraviolet light in the presence of a radical initiator (Scheme 1) and investigated its spectroscopic behaviour in solution and in thin polymeric films. Moreover, we tested the ability of our system to optically imprint fluorescence images into synthetic materials. Metal-enhanced fluorescence (MEF) by silver nanoplates (AgNP) was achieved in accordance with the principles of radiative decay engineering.26,35–40 The presence of AgNP allowed the observation of a strong fluorescent output with shorter reaction times, lower irradiance, and increased signal to noise.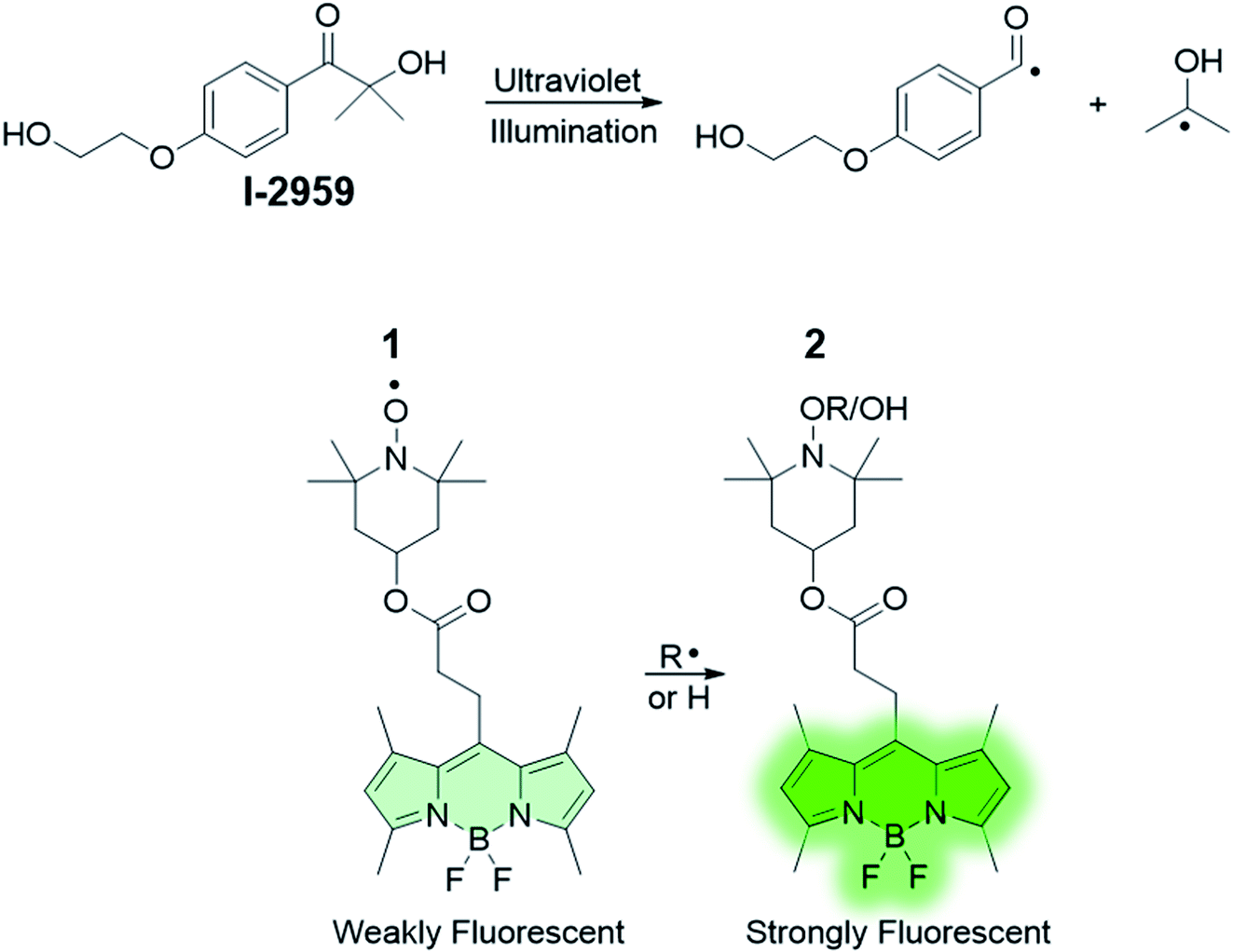 | ||
| Scheme 1 Structures of the fluorogenic probe 1 and the free radical initiator (2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, Irgacure®-2959) used in this work. | ||
Experimental
Synthesis
All reagents were purchased from Sigma Aldrich and Fisher Scientific. Solvents were purchased from ACP Chemicals Inc. Ultrapure deionized water (MilliΩ, 18.2 MΩ) was obtained from a Millipore Purification System. Reactions were monitored by thin layer chromatography using aluminum backed sheets coated with 200 μm silica (60, F254). SiliaFlash® P60, 40–63 mm (230–400 mesh) silica gel from SiliCycle was used for purification of compounds by column chromatography. The synthetic scheme is illustrated in the ESI.† All synthetic protocols were performed using standard personal protective equipment (lab coat, safety glasses and gloves) as well as face masks for additional protection. Safety Data Sheets were consulted prior the usage of all chemical precursors. Silver nanoplates were synthesized according to our previously published protocol.26 The synthesis of the organic probes 10 and 1 afforded milligram quantities of the probes in spectroscopic grade purity: for the purpose of this work, no further effort was made to improve the percentage yield of the synthetic process.Synthesis of 10
2,4-Dimethylpyrrole (1 mL, 9.7 mmol) was dissolved in CH2Cl2 (40 mL) into a cleaned 250 mL 2-neck round bottom flask equipped with a stir bar. The flask was subsequently put under N2 atmosphere and the solution was stirred for 10 minutes. A colour change from clear to red was observed. Succinyl chloride (0.28 mL, 2.54 mmol) was added under inert atmosphere resulting in a colour change from red to green. The solution was refluxed for 30 minutes, then left to cool to room temperature. Triethylamine (1.57 mL, 11.24 mmol) was added to the reaction mixture, which was stirred for 30 minutes. To avoid the possibility of amine degradation due to light exposure, the reaction flask was kept in the dark. Boron trifluoride diethyl etherate (3.2 mL, 25.92 mmol) was added, and the solution refluxed for 2 hours. Note that BF3·OEt2 reacts promptly with water and moisture in the air to liberate hydrogen fluoride gas: as such, it is essential to maintain the reaction under controlled atmosphere and avoid exposure to air. The content was then transferred to a separatory funnel and washed with 0.1 M HCl (3 × 20 mL) and saturated NaCl (3 × 20 mL). The organic phase was dried under Na2SO4 and filtered, and the solvent evaporated under reduced pressure. The crude product was then purified using column chromatography [SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexanes/EtOAc 4:1 (v/v)] to yield 400 mg (12%) of 10. 1H NMR (400 MHz, CDCl3): δ 6.07 (2H, s), 3.3–3.34 (2H, m), 2.63–2.67 (2H, m), 2.52 (6H, s), 2.44 (6H, s). 13C {1H} NMR (CDCl3, 400 MHz): δ 176.5, 154.8, 142.8, 140.4, 131.2, 122.1, 35.2, 23.4, 16.4, 14.5. FTIR-ATR: 3310 cm−1 (νs, O–H); 2960, 2920, 2865 cm−1 (νs, C–H); 1700 cm−1 (νs, C
:hexanes/EtOAc 4:1 (v/v)] to yield 400 mg (12%) of 10. 1H NMR (400 MHz, CDCl3): δ 6.07 (2H, s), 3.3–3.34 (2H, m), 2.63–2.67 (2H, m), 2.52 (6H, s), 2.44 (6H, s). 13C {1H} NMR (CDCl3, 400 MHz): δ 176.5, 154.8, 142.8, 140.4, 131.2, 122.1, 35.2, 23.4, 16.4, 14.5. FTIR-ATR: 3310 cm−1 (νs, O–H); 2960, 2920, 2865 cm−1 (νs, C–H); 1700 cm−1 (νs, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1546, 1504 cm−1 (νs, CC); 1405 cm−1 (νb, CH3); 1301, 1196 cm−1 (νs, C–N); 1060 cm−1 (νs, C–O); 947 cm−1 (νb, O–H); 803 cm−1 (νb, C–H).
O), 1546, 1504 cm−1 (νs, CC); 1405 cm−1 (νb, CH3); 1301, 1196 cm−1 (νs, C–N); 1060 cm−1 (νs, C–O); 947 cm−1 (νb, O–H); 803 cm−1 (νb, C–H).
Synthesis of 1
In a clean 250 mL round bottom flask equipped with a stir bar, 10 (200 mg, 0.62 mmol) was dissolved in 10 mL CH2Cl2. The flask was placed in an ice bath and wrapped in aluminum foil. 4-Hydroxy-2,2,6,6-tetramethyl-piperidinoxy (106.9 mg, 0.62 mmol), 4-(dimethylamino)pyridine (15.2 mg, 0.12 mmol) and 1-(3-dimethylaminopropyl)-3-ethylcabodiimide hydrochloride (190.4 mg, 0.99 mmol) were added to the solution, which was stirred for 30 minutes at 0 °C. The ice bath was removed, and the solution was left stirring at room temperature for 72 hours. The solution was then transferred to a separatory funnel and washed with deionized water (6 × 10 mL). The organic phase was dried under Na2SO4, filtered and the solvent was removed under reduced pressure. The crude was then purified using column chromatography [SiO2:hexanes/EtOAc 9:1 (v/v)]. The combined fractions were dried to yield 30 mg (10.2%) of 1, which was stored in the dark. FTIR-ATR: 2975, 2925, 2854 cm−1 (νs, C–H); 1730 cm−1 (νs, CO), 1548, 1510 cm−1 (νs, CC); 1410 cm−1 (νb, CH3); 1307, 1228, 1200 cm−1 (νs, C–N); 1285 cm−1 (νs, N–O); 1066 cm−1 (νs, C–O); 879 cm−1 (νs, C–O); 879, 800 cm−1 (νb, C–H). In addition to the typical N–O (nitroxide) stretching, successful esterification of 1 with TEMPO is confirmed by the disappearance of the O–H stretching at 3310 cm−1 and the increase in the stretching frequency of the carbonyl group from 1700 cm−1 in 10 to 1730 cm−1 in 1.
Materials and methods
NMR spectra were recorded at room temperature with a Bruker Avance 400 spectrometer. FTIR Diamond ATR spectra were recorded with a Cary 630 spectrometer by Agilent Technologies. Steady-state absorption spectra were recorded with an Agilent Cary 60 UV-visible spectrometer, using quartz cells with a path length of 1 cm. Steady-state emission spectra were recorded with an Agilent Cary Eclipse spectrometer with right angle geometry in aerated solutions. Slit widths and detector voltage were chosen as 5nm and 600 V. Mass spectral analysis was performed with a 7890B GC System equipped with a 5977 mass selective detector from Agilent Technologies. Illumination at 365 nm was performed using a laboratory TLC lamp (Mineralight UVGL 25, 0.4 mW cm−2) for solutions or an in-house designed illumination setup consisting of light-emitting diodes (LEDs, LEDEngin LZ4-UV00) connected to a heat sink and diaphragm-based active cooling system (Nuventix SynJet) for thin polymer films. A DC power supply provided a current of 700 mA. According to manufacturer specifications these conditions deliver an average radiant flux of 3.3 W per LED. Illumination at 505 nm was performed using Cyan Luxeon Rebel LEDs mounted onto a 3-Up Endor Star MCPCB (LEDSupply 07007-PE000-K). Films for spectrometry were prepared by spin coating (1000 rpm, 45 s) 75 μL of a solution of 1 and 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure®-2959, or I-2959) in 10% PMMA/CH3CN on glass slides (Fisher Scientific) two times. The coated slides were used immediately. The same protocol was employed to deposit polymer films on glass slides pre-coated with silver nanoparticles. The percentage increase in fluorescence was calculated by subtracting the starting value from the final value and dividing this amount by the absolute starting value, × 100.
Results and discussion
Photochemical and photophysical properties
The absorption spectrum of 1 in CH3CN (Fig. 1) shows a band attributable to the boron dipyrromethene (BODIPY) fluorophore at λabs = 497 nm. Nonetheless, the fluorescence of 1 (Fig. 1) is significantly quenched by the presence of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO), which has been covalently linked to the BODIPY chromophore. Paramagnetic nitroxides covalently linked to fluorophores are well known to quench the excited states of the latter.41–59 In these systems, intramolecular quenching occurs through electron exchange between the two units,43,44,46,50,60 which causes relaxation of the local singlet state to the ground state of the fluorophore. Quenching by TEMPO is also evidenced by comparing the emission spectra of the BODIPY precursor 10 and 1 (Fig. S1, ESI†). However, radical trapping by TEMPO leads to the formation of a diamagnetic product (2 in Scheme 1), thereby disabling the quenching pathway and resulting in increased fluorescence quantum yield. Based upon these considerations, activation of the fluorescence of compound 1 can be used to report on and quantify radical reaction from the nitroxide counterpart in real time with fluorescence spectroscopy. Indubitably, from a synthetic standpoint, the chemical coupling of a chromophore to nitroxides provides a prudent and reliable strategy toward using free radicals as activators of fluorescence. In the presence of 10 equivalents of the free radical initiator 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure®-2959, or I-2959, Scheme 1), we observed a fast and dramatic increase of the emission band at 507 nm (Fig. 1) upon illumination of the same solution with ultraviolet light. I-2959 can concomitantly generate a benzoyl and a ketyl radical via photochemically induced α-cleavage (Scheme 1).61 While the benzoyl radical is known to quickly oxidize to the corresponding benzoic acid in aerated solutions, the ketyl radical is a strong reductant and can be easily intercepted when an appropriate acceptor becomes available.61–63 In our system, illumination at 365 nm decomposes I-2959 and encourages the generation of such a radical, which is then trapped by the nitroxide to activate the emission of the fluorophore-TEMPO dyad 1. In parallel, disproportionation to form TEMPO–H can also occur (see ESI†), similarly producing a fluorescent N-hydroxylamine. A bench scale investigation of the photochemical species produced was conducted by GC-MS analysis of an irradiated solution of I-2959 and an equimolar amount of 4-hydroxy-TEMPO (Fig. S2 and S3† and related discussion). Mass spectral analysis demonstrates that both pathways are likely to occur, suggesting the existence of a balance between N-hydroxylamine and N-alkoxyamine (both fluorescent) formation. For convenience, we will refer to both species as emissive product 2. The absorbance changes observed in Fig. 1 at wavelengths <350 nm, where the photoinitiator dominates the spectral profile, are consistent with those observed during the photoinduced cleavage of I-2959 alone (Fig. S4†).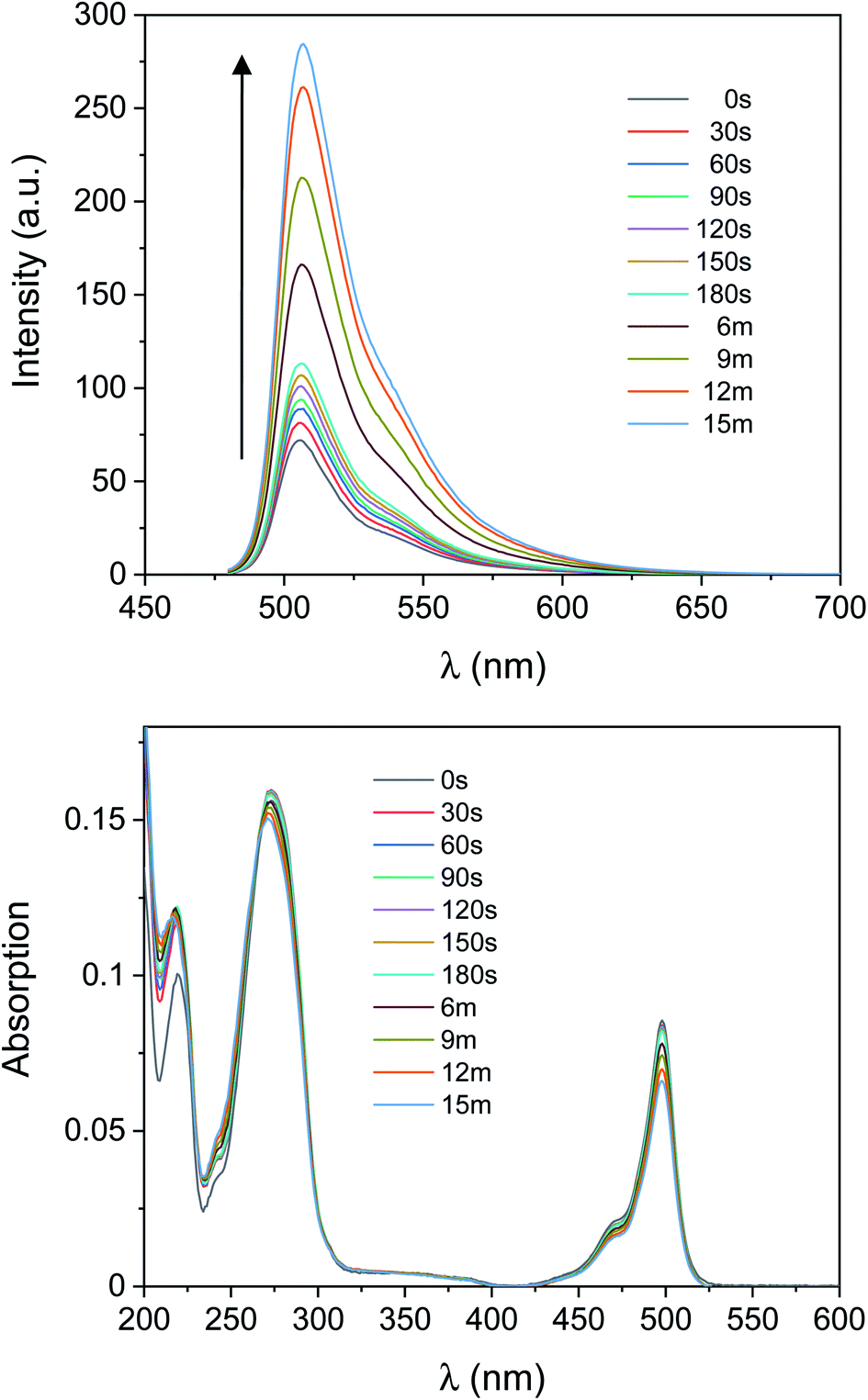 | ||
| Fig. 1 Emission (top, λex = 470 nm) and absorption (bottom) spectra of a CH3CN solution of 1 (1 μM, 20 °C) and I-2959 (10 μM) before and after ultraviolet irradiation (365 nm, 0–15 min, 0.4 mW cm−2) under air. | ||
In comparison, UVA irradiation of 1 alone does not produce a significant enhancement of the fluorescence. In fact, the same solution exposed to ultraviolet light but without the photoinitiator (b in Fig. S5†) experiences a negligible increase in fluorescence intensity (300% vs. 11% increase after 15 min with and without I-2959, respectively), confirming that the radical initiator is required to turn on the fluorescence. Importantly, the absorption band in the visible region is not affected by ultraviolet illumination (Fig. 1 and a in Fig. S5†), demonstrating that the BODIPY chromophore does not bleach under the relatively mild irradiation conditions that are nonetheless sufficient to photochemically cleave I-2959. However, some photodecomposition is observed after 30 min of UVA irradiation, with the fluorescence intensity decreasing by ca. 10% from the maximum value (obtained after 15 min of exposure).
In the past, it has been noted that N-alkoxyamines (RC-ONR′) decompose in aerated or oxygenated solutions via homolysis of the C–O bond,64–69 and examples of reversible trapping and release of nitroxides have been reported for chromophore-TEMPO dyads.49,70,71 Concurrently, the O–H bond in the hydrogenated derivative of 2 is weak, and nitroxides can easily reform by treatment of their corresponding hydroxylamines with mild oxidants (including air and molecular oxygen) even in the absence of a catalyst.72 This behaviour is caused by the low bond dissociation energies (BDE) of the NO–C bond in TEMPO-alkoxyamines,67,68 and of the NO–H bond in TEMPO–hydroxylamines.72,73 Under anaerobic conditions, decomposition is followed by rapid radical–radical recombination into the diamagnetic species 2; however, molecular oxygen (if present) is capable of scavenging carbon-centred radicals to form a peroxyl radical, meanwhile regenerating the stable radical nitroxide 1. In this scenario, the resulting peroxyl-radical species is also able to abstract a hydrogen atom from the dye–TEMPO–hydroxylamine product,74 further promoting the regeneration of 1 in the presence of oxygen.
To investigate the relative stability of the species photogenerated herein, the aerated solution of 1 and I-2959 of Fig. 1 irradiated with ultraviolet light was capped to avoid evaporation of the solvent and stored in the dark. Emission spectra were then recorded over the course of a week in otherwise identical conditions. Results (yellow bins in Fig. 2 and S6†) show that the fluorescence intensity post-activation begins to decrease after only 1 hour, and the original pre-irradiation value is reobtained after 3 days, suggesting that the photoinduced formation of 2 is spontaneously reversible. Absorption values remained constant during the resting period. We exploited the photochemical reversibility of our reaction to imprint information on a compact surface (vide infra). Interestingly, the deactivation process can also be accelerated by exposing the activated fluorescent solution to a 505 nm light-emitting diode (LED, Fig. S7†). Indeed, 30 min of exposure with green light can turn off the emission to a value comparable to spontaneous, in-the-dark deactivation after ∼24 h (compare the green traces in both Fig. S6 and S7†). This process is likely due to a combination of photobleaching and intramolecular transfer of the absorbed energy from 1 to TEMPO, which sensitizes cleavage of 2 in its N-alkoxyamine form and oxidation in its N-hydroxylamine form, respectively, regenerating 1.70,71 Even more remarkably, a second cycle of UVA irradiation increases the emission of the same solution up to half of the original activation (∼150% net increase, violet trace in Fig. S6†). The excess of photoinitiator favours the reactivation of 1, thus illustrating the potential of our system to be ‘rewritten’ after an active writing/erasing cycle.
 | ||
| Fig. 2 Evolution of the post-irradiation fluorescence intensity at λem = 507 nm for a solution stored in the dark under aerated conditions (yellow bins) or N2 atmosphere (purple bins). The solutions were photoactivated for 30 min (aerated) or 15 min (degassed), respectively. | ||
As discussed above, dye–TEMPO alkoxyamines and hydroxylamines decompose at higher rates in aerated or oxygenated solutions, while they can remain stable for longer times if oxygen is absent.64,66,70–72,75 We thus investigated the photochemical behaviour of 1 under N2 atmosphere with two independent experiments. First, we repeated the irradiation experiment of Fig. 1 for a degassed mixture of 1 and I-2959, where we verified that the maximum increase in the emission intensity observed upon 15 min of UVA illumination (b in Fig. S8†) is 305%, in perfect agreement with the results obtained in aerated solutions. Second, we verified the stability of a fresh solution of 1 and I-2959 irradiated under air but stored in the dark under N2 atmosphere (purple bins in Fig. 2 and S9†). Results prove that our fluorescent TEMPO-derived photoproducts can be preserved for a longer period of time under anaerobic environment.
The spectroscopic changes shown in Fig. 1 are reproducible in a 1% poly(methyl methacrylate) (PMMA) solution in CH3CN. Upon irradiation at 365 nm, the emission spectrum of 1 in the presence of I-2959 and PMMA (b in Fig. S10†) shows a progressive growth in intensity, consistent with the light-induced radical trapping and H-abstraction of compound 1 observed in pure CH3CN. Although at shorter wavelengths (<250 nm) the absorption spectrum is obscured by the polymer absorption, the spectral changes of I-2959 at 270 nm are nonetheless detectable (a in Fig. S10†). Addition of the polymer causes a substantial increase of the emission intensity of the photogenerated adducts due to the increased viscosity of the medium and reduced rotational motion of the free dye;76 overall, the fluorescence rises by ∼550% after only 6 minutes of UVA exposure (note from Fig. S10† that we observed some bleaching of the luminescence from 9 to 15 minutes of irradiation, up to 465% net increase in fluorescence at completion of the experiment). Indeed, the higher fluorescence output obtained in the presence of the polymer, at lower irradiation time, is a promising result for the application of this system in fluorescence patterning on surfaces and thin films.
Photoactivated fluorescence and patterning in thin polymer films
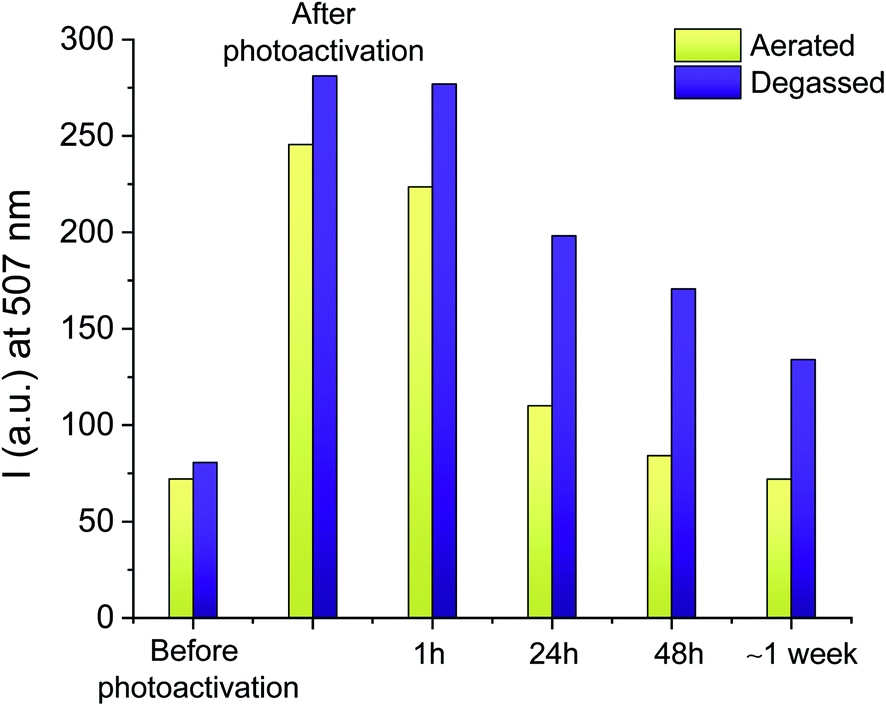
Information technology strives to move away from paper-based record-keeping approaches toward sustainable platforms for cataloging and sharing protected information. However, current data communication techniques still suffer from severe shortcomings in resolution, loading capacity, and security (e.g., erasing a message, or making it temporarily inaccessible). Writing data with fluorescence allows for information retrieval and extraction (‘reading’) simply by illuminating the prewritten samples with a suitable light-source. With this strategy, stored information can be secured, and becomes accessible exclusively under correct illumination conditions.To this end, we reproduced the photoinduced radical activation of 1 assisted by I-2959 in a fixed polymer matrix in the absence of any solvent. The absorption spectrum of a thin PMMA film (a in Fig. S11†) doped with 1 and I-2959 spin coated on glass slides reveals the absorption of the quenched BODIPY chromophore at 502 nm, while the corresponding emission band (b in Fig. S11†) is equally bathochromically shifted (∼5 nm) with respect to the freely-diffusing species in solution. The small red-shift in absorption and emission wavelengths is consistent with environmental differences around the chromophore, and it is expected for film samples.76 To start, we prepared the films using the same ratio between the fluorogenic probe and the photoinitiator adopted for solution experiments (1 and 10 equivalents of 1 and I-2959, respectively). Following irradiation at 365 nm of these films for 5 minutes, we observed an increase in fluorescence intensity of 140% (Fig. S11† and green bins in Fig. 3), consistent with the photogeneration of free radicals by I-2959 and subsequent activation of 1. However, PMMA films containing 1 without the photoinitiator also experience a change in fluorescence upon irradiation (48% increase after 5 minutes, Fig. S12† and yellow bins in Fig. 3). Such behaviour of fluorophore–nitroxide adducts trapped in polymer films has been observed previously, and it can be attributed to the well-known hydrogen abstraction by an excited state of the nitroxide from PMMA, the latter being a better H-donor substrate than MeCN. Nevertheless, this side reaction becomes negligible upon increasing the amount of I-2959 to 100 equivalents (blue bins in Fig. 3), so that proximity between 1 and the photogenerated radicals can be ensured even in a rigid, diffusion-restricted matrix, and fluorescence can be activated to higher values (285% increase) while keeping the irradiation time short.
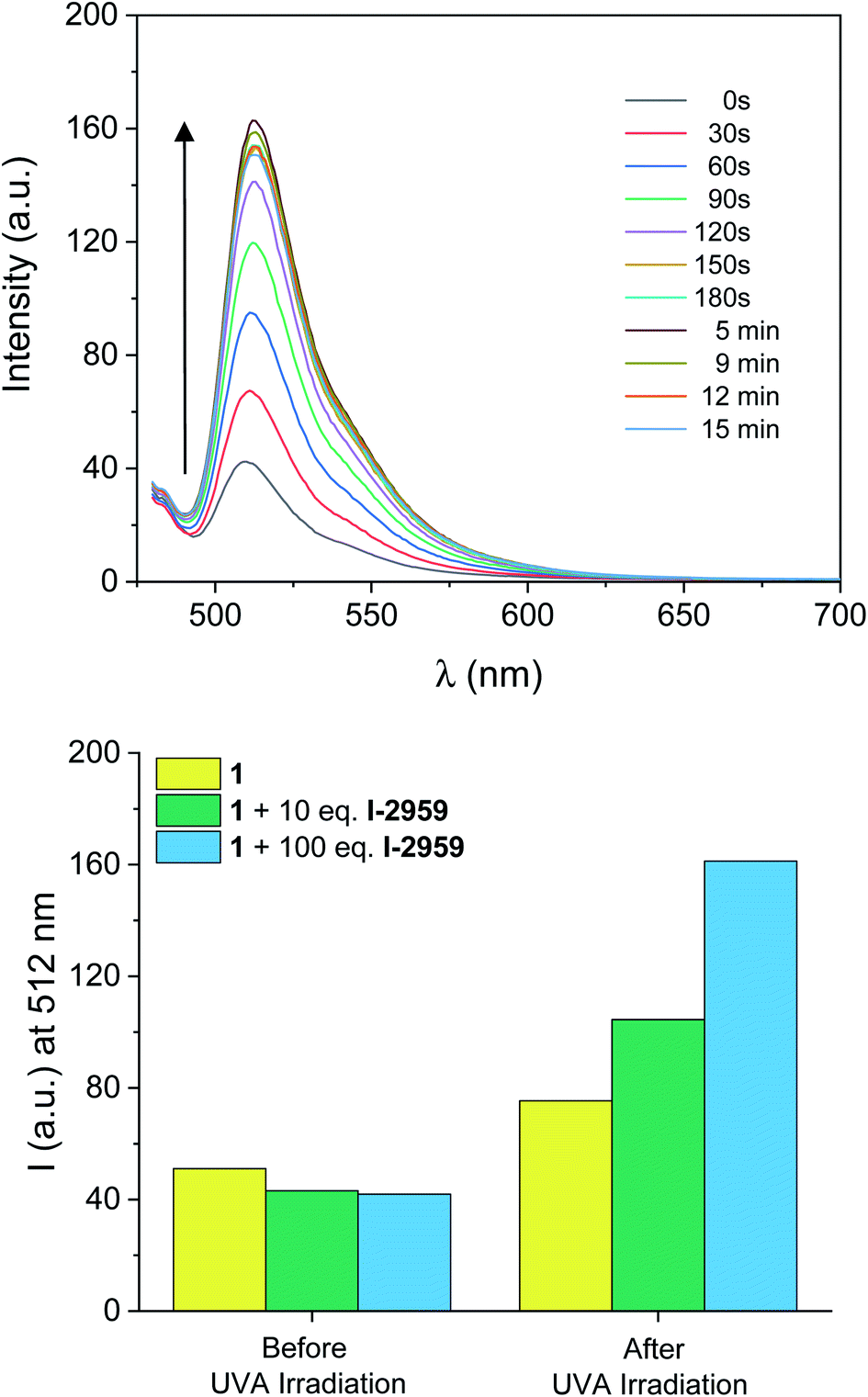 | ||
| Fig. 3 (Top) Emission spectra of a PMMA (10% w/v in MeCN) film containing 1 (0.5 mM, λex = 470 nm) and I-2959 (50 mM) before and after ultraviolet irradiation (LED365, 0–15 min). (Bottom) Evolution of the fluorescence intensity at λem = 512 nm under identical ultraviolet illumination (5 min) of polymer films containing 1 (yellow bins), 1 + 10 eq. of I-2959 (green bins) and 1 + 100 eq. of I-2959 (blue bins). | ||
The ability to turn on the fluorescence within a defined illumination area offers the opportunity to imprint a desired pattern on a substrate. Irradiation of a PMMA film of 1 and I-2959 at 365 nm through a 3D printed mask for 3 minutes encourages the formation of radicals exclusively in the irradiated area, meanwhile producing a fluorescent image (b in Fig. 4). In contrast, no pattern is formed under the same illumination conditions in the absence of the photoinitiator (Fig. S13†).
 | ||
| Fig. 4 Photographs of PMMA films doped with 1 (0.5 mM) and I-2959 (50 mM) before (a), immediately (b) and 48 hours (c) after exposure at 365 nm for 3 min through a RU-shaped 3D printed mask. | ||
Chemical reversibility further improves the flexibility and usefulness of fluorescent patterning methods. For example, materials that can store graphical or textual information for a restricted period of time are very attractive for applications in secure communications and controlled information management. The reversibility of the ‘turn-on’ system described here permits self-erasing of the written information by spontaneous deactivation (Fig. S6†) as noticed by the slow, but progressive fading of the written pattern (c in Fig. 4).
Nanoparticle-enhanced fluorescence
Metal nanoparticles can alter the photophysical properties of nearby organic fluorophores and enhance their emission through near-field interactions.26,35–38,40,76–79 The phenomenon can be envisioned as an active response of the electronic cloud of metal surfaces to the oscillating dipole of radiating species in the nanoparticles' proximity; in turn, the rates of excitation and emission of the chromophores and/or their quantum yield can be modified, and the overall steady-state brightness improved. This process, known as metal-enhanced fluorescence (MEF), can be engineered around the relative overlap between absorption and emission wavelengths of the fluorophore and the scattering component of the extinction spectrum of the metal nanoparticle. In earlier work, we studied MEF of a modified BODIPY fluorophore coupled to triangular silver nanoplates (AgNP).26 Herein, we adopted the same methodology for sample preparation to boost the fluorescence output of the photogenerated species 2. The absorption and emission spectra of the BODIPY used previously and compound 1 (or 2) are similar in both shape and wavelength, with 1 being slightly hypsochromically shifted (∼10 nm) due to the lack of the aryl substituent at the meso position (C8). Thin films for MEF experiments were prepared as described above, with the exception that slides were first functionalized with AgNP prior to spin-coating 1 and I-2959 in PMMA and that the solution was spin coated only once (to reduce film thickness and enhance MEF). We adopted our previously reported method26 to estimate the thickness of the PMMA layer using specular reflectance. The latter was used to verify that the PMMA layer is between 59 and 104 nm thick (Fig. S14† and associated discussion). The absorption spectrum of a thin PMMA film containing 1 and I-2959 a top AgNP (Fig. S15†) shows the broad extinction of AgNP dominating the visible wavelength regime; nonetheless, the absorption band for 1 at λabs = 507 nm is clearly identifiable. Moreover, emission spectra of the same film (Fig. 5) unequivocally show the expected fluorescence band of the fluorochrome at λem = 512 nm. Exposure of this film sample to ultraviolet light (Fig. 5) reveals that the photoconversion of I-2959 and the subsequent radical trapping by 1 can also be performed in the presence of the nanostructures. With our sample preparation method, it is reasonable to expect that fluorophores are distributed throughout the polymer layer and are thus located at various distances from AgNP, up to a theoretical maximum nanoparticle–fluorophore distance equal to the film thickness. Although fluorochromes up to about 200 nm away from AgNP may experience MEF, fluorescence enhancement at the macroscopic (bulk) level can be better appreciated at smaller distances from the metal surfaces. Indeed, the fluorescence intensity of films atop AgNP is enhanced with respect to the same experiment conducted in the absence of the metallic nanoparticles. After 5 minutes of irradiation, we observe an overall net increase in fluorescence of 460% in the presence of AgNP vs. 195% in the absence of the nanostructures for single-coated films (Fig. 5). Notably, the fluorescence output obtained after only 90 seconds of irradiation in the presence of AgNP is higher than that achieved after irradiating for 5 min in the absence of nanoparticles, drastically reducing the necessary irradiation time.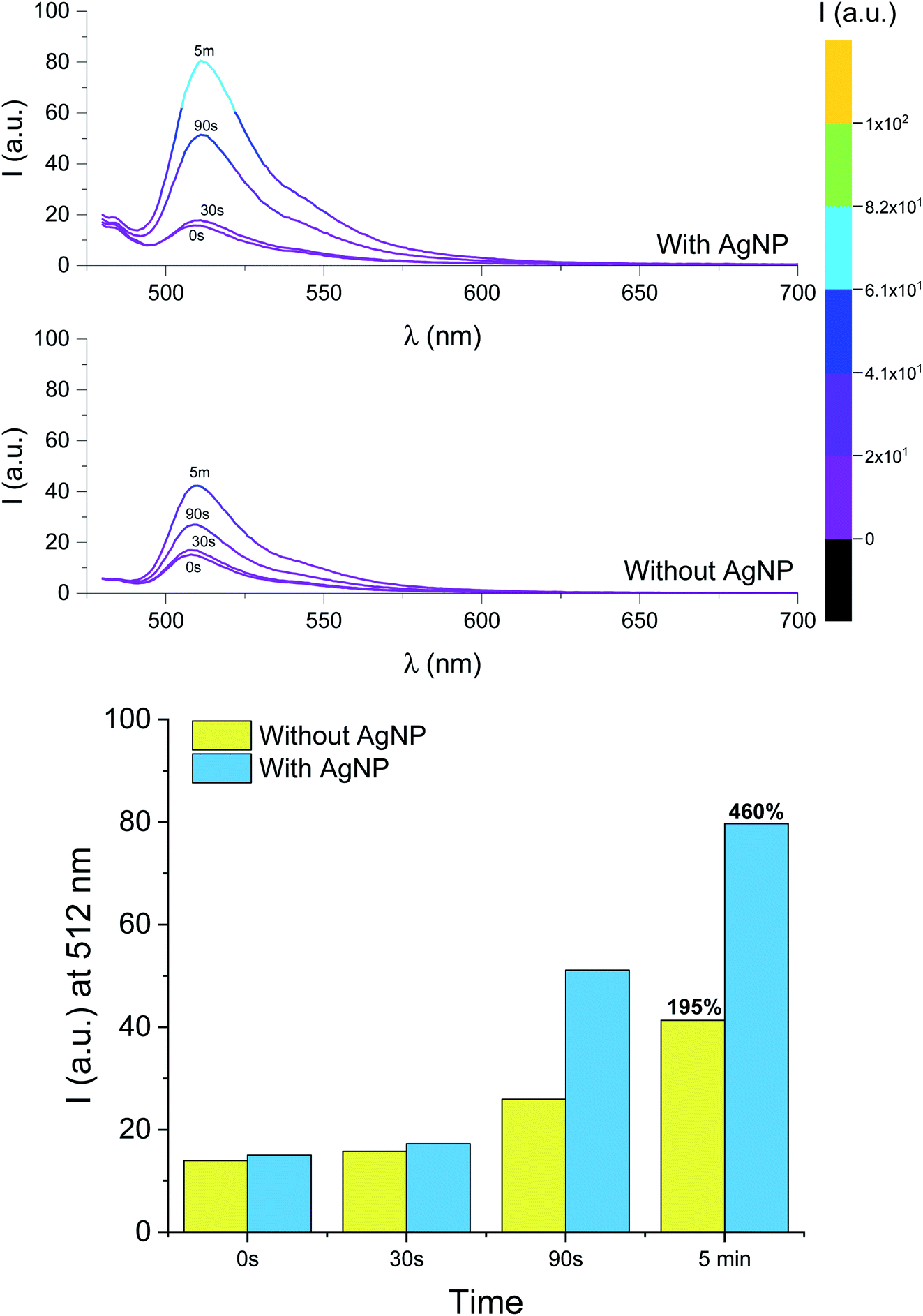 | ||
| Fig. 5 (Top) Emission spectra of a PMMA (10% w/v in MeCN) single-coated film containing 1 (0.5 mM, λex = 470 nm) and I-2959 (50 mM) before and after ultraviolet irradiation (LED365, 0–5 min) in the presence or absence of AgNP. (Bottom) Fluorescence intensity recorded at 512 nm for the same films. | ||
Conclusions
We have designed a switchable molecular system composed of a fluorogenic BODIPY–TEMPO dyad that can be selectively activated under mild illumination conditions upon photochemical decomposition of a radical photoinitiator, and investigated its spectroscopic behaviour in solution and in polymer films. A fluorescent species is formed upon the concomitant trapping of a carbon-centred radical by TEMPO or H-abstraction. Therefore, patterned illumination of a film sample through a mask offers the opportunity to imprint a pattern on the photoresponsive substrate that can be conveniently read by exciting the fluorescence using a portable UV lamp. Moreover, the formation of the fluorescent product is reversible, and the fluorescence can be “switched off” spontaneously in the dark causing a progressive fading of the pattern. This investigation also demonstrated that MEF by AgNP can be employed to improve the macroscopic performance of emissive molecular systems, by increasing the steady state fluorescence intensity output and reducing experimental duration required. We were pleased to see that, at spectroscopic concentrations, the presence of excess free radicals did not affect the ability of AgNP to induce MEF. We foresee that the implementation of MEF strategies will facilitate the development of enhanced operating principles for optical writing with relatively low illumination intensities, and lead to practical improvements to the fields of fluorescence spectroscopy, microscopy, and lithography. Within these fields, applications of fluorescence activation will benefit from the types of advances discussed herein, such as milder conditions, shorter reaction times and a more diverse selection of both excitation sources and emission wavelengths.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada (Discovery Grant), Ryerson University (start-up funds) and Ryerson University Faculty of Science Dean's Research Fund and Discovery Accelerator Grant for supporting our research program.References
- H. Wang, X. Ji, Z. A. Page and J. L. Sessler, Mater. Chem. Front., 2020, 4, 1024–1039 RSC.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of molecular photochemistry: an introduction, University Science Books, Herndon, 2009 Search PubMed.
- V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics: Concepts, Research, Applications, Wiley-VCH, Weinheim, 2014 Search PubMed.
- C. Mack, Fundamental Principles of Optical Lithography, John Wiley & Sons, Ltd, Chichester, UK, 2007 Search PubMed.
- B. J. Lin, Optical Lithography, SPIE, 1000 20th Street, Bellingham, WA 98227-0010 USA, 2010 Search PubMed.
- L. M. Wysocki and L. D. Lavis, Curr. Opin. Chem. Biol., 2011, 15, 752–759 CrossRef CAS PubMed.
- D. Puliti, D. Warther, C. Orange, A. Specht and M. Goeldner, Bioorg. Med. Chem., 2011, 19, 1023–1029 CrossRef CAS PubMed.
- F. M. Raymo, J. Phys. Chem. Lett., 2012, 3, 2379–2385 CrossRef CAS PubMed.
- W. H. Li and G. Zheng, Photochem. Photobiol. Sci., 2012, 11, 460–471 CrossRef CAS PubMed.
- F. M. Raymo, Phys. Chem. Chem. Phys., 2013, 15, 14840–14850 RSC.
- P. J. Wu, J. L. Chen, C. P. Chen and Y. H. Chan, Chem. Commun., 2013, 49, 898–900 RSC.
- K. Hwang, P. Wu, T. Kim, L. Lei, S. Tian, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 13798–13802 CrossRef CAS PubMed.
- V. Grenier, A. S. Walker and E. W. Miller, J. Am. Chem. Soc., 2015, 137, 10894–10897 CrossRef CAS PubMed.
- J. M. Goldberg, F. Wang, C. D. Sessler, N. W. Vogler, D. Y. Zhang, W. H. Loucks, T. Tzounopoulos and S. J. Lippard, J. Am. Chem. Soc., 2018, 140, 2020–2023 CrossRef CAS PubMed.
- H. Li and J. C. Vaughan, Chem. Rev., 2018, 118, 9412–9454 CrossRef CAS PubMed.
- E. Kozma and P. Kele, Org. Biomol. Chem., 2019, 17, 215–233 RSC.
- L. Wang, M. S. Frei, A. Salim and K. Johnsson, J. Am. Chem. Soc., 2019, 141, 2770–2781 CrossRef CAS PubMed.
- A. Sekiguchi, J. Photopolym. Sci. Technol., 2012, 25, 473–480 CrossRef CAS.
- J. L. Jessop, S. N. Goldie, A. B. Scranton, G. J. Blanchard, B. Rangarajan, L. Capodieci, R. Subramanian and M. K. Templeton, Advances in Resist Technology and Processing XVI, 1999, vol. 3678, pp. 914–922 Search PubMed.
- G. Pohlers, S. Virdee, J. C. Scaiano and R. Sinta, Chem. Mater., 1996, 8, 2654–2658 CrossRef CAS.
- G. Pistolis, S. Boyatzis, M. Chatzichristidi and P. Argitis, Chem. Mater., 2002, 14, 790–796 CrossRef CAS.
- M. Frenette, C. Coenjarts and J. C. Scaiano, Macromol. Rapid Commun., 2004, 25, 1628–1631 CrossRef CAS.
- G. D. Feke, D. Hessman, R. D. Grober, B. Lu and J. W. Taylor, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2000, 18, 136–139 CrossRef CAS.
- G. D. Feke, R. D. Grober, G. Pohlers, K. Moore and J. F. Cameron, Anal. Chem., 2001, 73, 3472–3480 CrossRef CAS PubMed.
- M. D. Mason, K. Ray, G. D. Feke, R. D. Grober, G. Pohlers and J. F. Cameron, in Proceedings of SPIE - The International Society for Optical Engineering, ed. D. J. Herr, 2003, vol. 5038, p. 473 Search PubMed.
- N. P. Dogantzis, G. K. Hodgson and S. Impellizzeri, Nanoscale Adv., 2020, 2, 1956–1966 RSC.
- Frontiers of Nanoscience - Materials and Processes for Next Generation Lithography, ed. A. Robinson and R. Lawson, Elsevier, 2016, vol. 11 Search PubMed.
- C. J. Martin, G. Rapenne, T. Nakashima and T. Kawai, J. Photochem. Photobiol., C, 2018, 34, 41–51 CrossRef CAS.
- N. A. Kuznetsova, G. V Malkov and B. G. Gribov, Russ. Chem. Rev., 2020, 89, 173–190 CrossRef CAS.
- R. Popielarz, A. M. Sarker and D. C. Neckers, Macromolecules, 1998, 31, 951–954 CrossRef CAS.
- C. Coenjarts, O. García, L. Llauger, J. Palfreyman, A. L. Vinette and J. C. Scaiano, J. Am. Chem. Soc., 2003, 125, 620–621 CrossRef CAS PubMed.
- A. Aspée, O. García, L. Maretti, R. Sastre and J. C. Scaiano, Macromolecules, 2003, 36, 3550–3556 CrossRef.
- J. Kabatc, B. Jdrzejewska and J. Pączkowski, J. Appl. Polym. Sci., 2006, 99, 207–217 CrossRef CAS.
- I. Kamińska, J. Ortyl and R. Popielarz, Polym. Test., 2015, 42, 99–107 CrossRef.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171–194 CrossRef CAS PubMed.
- K. Aslan, Z. Leonenko, J. R. Lakowicz and C. D. Geddes, J. Fluoresc., 2005, 15, 643–654 CrossRef CAS PubMed.
- P. Anger, P. Bharadwaj and L. Novotny, Phys. Rev. Lett., 2006, 96, 113002 CrossRef PubMed.
- Y. Fu, J. Zhang and J. R. Lakowicz, J. Fluoresc., 2007, 17, 811–816 CrossRef CAS PubMed.
- J. R. Lakowicz, K. Ray, M. Chowdhury, H. Szmacinski, Y. Fu, J. Zhang and K. Nowaczyk, Analyst, 2008, 133, 1308–1346 RSC.
- J. W. Liaw, H. Y. Wu, C. C. Huang and M. K. Kuo, Nanoscale Res. Lett., 2016, 11, 1–9 CrossRef CAS PubMed.
- C. Aliaga, M. C. Rezende and C. Tirapegui, Tetrahedron, 2009, 65, 6025–6028 CrossRef CAS.
- Y. Liu, M. Zhu, J. Xu, H. Zhang and M. Tian, Analyst, 2011, 136, 4316–4320 RSC.
- N. V. Blough and D. J. Simpson, J. Am. Chem. Soc., 1988, 110, 1915–1917 CrossRef CAS.
- D. J. Kieber and N. V. Blough, Anal. Chem., 1990, 62, 2275–2283 CrossRef CAS.
- G. I. Likhtenstein, K. Ishii and S. Nakatsuji, Photochem. Photobiol., 2007, 83, 871–881 CrossRef CAS PubMed.
- G. I. Likhtenshtein, Pure Appl. Chem., 2008, 80, 2125–2139 CAS.
- L. Cao, Q. Wu, Q. Li, S. Shao and Y. Guo, J. Fluoresc., 2014, 24, 313–318 CrossRef CAS.
- S. A. Green, D. J. Simpson, G. Zhou, P. S. Ho and N. V. Blough, J. Am. Chem. Soc., 1990, 112, 7337–7346 CrossRef CAS.
- S. E. Bottle, J. L. Clement, M. Fleige, E. M. Simpson, Y. Guillaneuf, K. E. Fairfull-Smith, D. Gigmes and J. P. Blinco, RSC Adv., 2016, 6, 80328–80333 RSC.
- C. Aliaga, P. Fuentealba, M. C. Rezende and C. Cárdenas, Chem. Phys. Lett., 2014, 593, 89–92 CrossRef CAS.
- N. B. Yapici, S. Jockusch, A. Moscatelli, S. R. Mandalapu, Y. Itagaki, D. K. Bates, S. Wiseman, K. M. Gibson, N. J. Turro and L. Bi, Org. Lett., 2012, 14, 50–53 CrossRef CAS PubMed.
- F. Mito, K. Kitagawa, T. Yamasaki, C. Shirahama, T. Oishi, Y. Ito, M. Yamato and K. I. Yamada, Free Radical Res., 2011, 45, 1103–1110 CrossRef CAS PubMed.
- C. Bueno, L. Mikelsons, L. Maretti, J. C. Scaiano and A. Aspée, Photochem. Photobiol., 2008, 84, 1535–1542 CrossRef CAS PubMed.
- A. Kaur, J. L. Kolanowski and E. J. New, Angew. Chem., Int. Ed., 2016, 55, 1602–1613 CrossRef CAS PubMed.
- S. Impellizzeri, K. G. Stamplecoskie and J. C. Scaiano, Phys. Chem. Chem. Phys., 2013, 15, 14873–14878 RSC.
- S. Sato, M. Suzuki, T. Soma and M. Tsunoda, Spectrochim. Acta, Part A, 2008, 70, 799–804 CrossRef PubMed.
- S. Sato, M. Tsunoda, M. Suzuki, M. Kutsuna, K. Takido-uchi, M. Shindo, H. Mizuguchi, H. Obara and H. Ohya, Spectrochim. Acta, Part A, 2009, 71, 2030–2039 CrossRef PubMed.
- C. Aliaga, F. Celis, S. Lühr and R. Oñate, J. Fluoresc., 2015, 25, 979–983 CrossRef CAS PubMed.
- C. Xu and L. Cai, Luminescence, 2014, 29, 36–41 CrossRef CAS PubMed.
- S. E. Herbelin and N. V. Blough, J. Phys. Chem. B, 1998, 102, 8170–8176 CrossRef CAS.
- S. Jockusch, M. S. Landis, B. Freiermuth and N. J. Turro, Macromolecules, 2001, 34, 1619–1626 CrossRef CAS.
- J. C. Scaiano, K. G. Stamplecoskie and G. L. Hallett-Tapley, Chem. Commun., 2012, 48, 4798–4808 RSC.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS.
- S. A. F. Bon, G. Chambard and A. L. German, Macromolecules, 1999, 32, 8269–8276 CrossRef CAS.
- S. Marque, H. Fischer, E. Baier and A. Studer, J. Org. Chem., 2001, 66, 1146–1156 CrossRef CAS PubMed.
- S. Marque, C. Le Mercier, P. Tordo and H. Fischer, Macromolecules, 2000, 33, 4403–4410 CrossRef CAS.
- A. Gaudel-Siri, D. Siri and P. Tordo, ChemPhysChem, 2006, 7, 430–438 CrossRef CAS PubMed.
- M. V. Ciriano, H. G. Korth, W. B. Van Scheppingen and P. Mulder, J. Am. Chem. Soc., 1999, 121, 6375–6381 CrossRef CAS.
- P. Vasileva, B. Donkova, I. Karadjova and C. Dushkin, Colloids Surf., A, 2011, 382, 203–210 CrossRef CAS.
- A. Goto, J. C. Scaiano and L. Maretti, Photochem. Photobiol. Sci., 2007, 6, 833–835 CrossRef CAS PubMed.
- J. Su, X. Liu, M. Li, T. Zhang and Y. Cui, Int. J. Polym. Sci., 2016, 2016, 1–8 CrossRef.
- The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids, ed. Z. Rappoport and J. F. Liebman, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
- P. S. Billone, P. A. Johnson, S. Lin, J. C. Scaiano, G. A. Dilabio and K. U. Ingold, J. Org. Chem., 2011, 76, 631–636 CrossRef CAS PubMed.
- M. Griesser, R. Shah, A. T. Van Kessel, O. Zilka, E. A. Haidasz and D. A. Pratt, J. Am. Chem. Soc., 2018, 140, 3798–3808 CrossRef CAS.
- B. Maillard, K. U. Ingold and J. C. Scaiano, J. Am. Chem. Soc., 1983, 105, 5095–5099 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 2006 Search PubMed.
- M. H. Chowdhury, K. Aslan, S. N. Malyn, J. R. Lakowicz and C. D. Geddes, J. Fluoresc., 2006, 16, 295–299 CrossRef CAS PubMed.
- J. R. Lakowicz, K. Ray, M. Chowdhury, H. Szmacinski, Y. Fu, J. Zhang and K. Nowaczyk, Analyst, 2008, 133, 1308–1346 RSC.
- S. D. Choudhury, R. Badugu, K. Ray and J. R. Lakowicz, J. Phys. Chem. C, 2012, 116, 5042–5048 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09565f |
| This journal is © The Royal Society of Chemistry 2021 |