
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn aqueous phase TEMPO mediated electrooxidation of 2-thiophenemethanol using MnO2–Pi dispersed nanocarbon spheres on a carbon fiber paper electrode†
Agnus T. Mathewa,
Supriya Sb,
Akshaya K. B.a,
Anitha Varghese *a and
Gurumurthy Hegde*b
*a and
Gurumurthy Hegde*b
aDepartment of Chemistry, CHRIST (Deemed to be University), Hosur Road, Bengaluru 560029, India. E-mail: anitha.varghese@christuniversity.in
bCentre for Nano-Materials and Displays, B.M.S. College of Engineering, Bull Temple Road, Basavanagudi, Bengaluru 560019, India. E-mail: murthyhegde@gmail.com
First published on 7th January 2021
Abstract
An environmentally benign and economic method was developed for the electrocatalytic oxidation of 2-thiophenemethanol in an aqueous acidic medium. Nanocarbon spheres (NCS) coated on carbon fiber paper (CFP) were used as a host matrix to disperse manganese dioxide nanoparticles from phosphate buffer solution through electrochemical deposition. The developed electrode (MnO2–Pi–NCS/CFP) was used as a working electrode for electrochemical oxidation of 2-thiophenemethanol in the presence of a mediator TEMPO in 0.01 M H2SO4 medium. Different analytical methods were used to characterize the modified electrodes. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were used to study the electrochemical properties of the modified electrodes. The electrochemically active surface area values calculated for bare CFP, NCS coated CFP and MnO2–Pi–NCS/CFP electrodes were found to be 1.43 cm2, 2.86 cm2, and 6.72 cm2 respectively for the geometric area of 0.7 cm2 of the electrodes. Coating of NCS and MnO2–Pi resulted in porosity and roughness of the CFP electrode which enhances the surface area. MnO2–Pi–NCS/CFP demonstrated higher electrocatalytic activity for oxidation of 2-thiophenemethanol to 2-thiophenemethanal in aqueous acidic media with a TEMPO mediator compared to unmodified electrodes.
Introduction
The application of electrochemical methods to oxidize organic molecules is attractive due to low cost, fast response, low energy consumption, easy operation, and an environmentally benign process. The replacement of redox chemical reagents by electricity is an inevitable step towards green chemical processes.1,2 The general methods of oxidation include named reactions such as Corey–Kim, Dess–Martin, Oppenauer oxidation, aerobic oxidation, dehydrogenation by Cu/Ag at 573 K, and electro-catalytic oxidation. Most of these methods make use of various oxidizing reagents such as KMnO4, K2Cr2O7, pyridinium chlorochromate, pyridinium dichromate, etc.2 Of all oxidation methods, the electrochemical technique has received great attention for the advantage of employing a clean reagent (electron) for oxidation reaction, which has the environmental benefit of reducing secondary pollutants.3 Therefore, from the point of environmental applications, it is a challenge to prepare an electrode with good physical, chemical and electrochemical stability, high selectivity, excellent conductivity, and great efficiency, which can produce substantial yield in a single step.Notably, the electroactive molecules such as alcohols, phenols, heterocyclic alcohols, methyl benzene, etc. can be oxidized by electrochemical method.4–7 But, when the bare electrode is used, the oxidation of these substrates occurs approximately at similar potential values, resulting in poor selectivity and less current density. Therefore, the development of a modified electrode which exhibits better selectivity, higher catalytic activity, and economically viable, is in demand.8–12
In recent years, various electrode materials have been developed with electrochemically active large surface area, higher selectivity, and current densities. In this regard, various materials are being used to modify the bare electrodes, like conducting polymers, metal nanoparticles, carbon nanomaterials, metal oxides, and semiconductors.13–15 Presently, carbon-based materials such as charcoal, activated carbon, carbon nanotubes, carbon nanofibers, fullerenes, graphene, and porous nanocarbons are receiving significance due to low density, large surface area, good flexibility, high mechanical strength, and stability.16 Among carbon-based materials, the nanocarbon spheres (NCS) exhibit distinguishing advantages, like simple preparation methods, uniform particle size, consistent surface morphology, larger porosity, and specific surface area, good dispersity, higher chemical and thermal stability, excellent biocompatibility, larger ionic and electronic conductivities and higher packing density, when compared to the other forms of carbons.17,18 NCS has been used as a substrate for electrode materials in the manufacture of polymer electrolyte membrane fuel cells,19 energy storage devices, such as supercapacitors.20–22
The general methods of synthesizing NCS include arc discharge, chemical vapor deposition (CVD), nano-casting, hydrothermal carbonization of sugars, and polymeric aerogels.23 But these methods follow harsh synthetic conditions and demand a large consumption of energy. Therefore, in this work, NCS is prepared from Sago bark waste in a clean, environmentally friendly, catalyst-free, and economically favorable one-pot pyrolysis process.
Electrodeposition of transition-metal oxides is a salient technique followed for the surface modification of electrode materials; it is also used to prepare nanostructured films for catalytic, biomedical, and electrochemical applications.24,25 The transition metal oxides/hydroxides coated electrode materials have been found to exhibit high capacitance and current densities, good conductivity, and large electrochemically active surface area. The films of metal oxide nanoparticle could be obtained on the electrode surface using anodic oxidation or cathodic reduction methods.24 Several metal oxides such as MnO2, Co3O4, Fe2O3, SnO2, RuO2, etc., are being studied for electrochemical applications. MnO2 is considered as the most favorable transition-metal oxide because of its abundant reserves, environmentally benign nature, less toxicity, low cost, and excellent electrochemical performances.26
Literatures reveal that various crystalline structures of MnO2 nanoparticles are of great importance as catalysts, lithium batteries, alkaline Zn/MnO2 cells, electrochemical supercapacitors, sensors, and electrochemical oxidation.24 Electrodeposition of metal ions such as Mn2+, in phosphate buffer solutions on substrate electrodes, contained substantial amounts of anions from the buffer medium. Such materials are clusters of interrelated metal-oxo/hydroxo cubane's with terminal ligation to the electrolytic species. These catalysts can operate under neutral or near-neutral pH conditions and have displayed long term catalytic activity. Many applications of MnO2 combined with carbon-based materials (carbon nanotubes, active carbon, graphene, carbon fibers, and carbon spheres) have great advantages.27–29 Of all carbon-based materials, the NCS has gained special attention due to unique morphology, large voids, high chemical and thermal stability, and large surface area.27
Cyclic voltammetry is a versatile technique of investigating the redox processes of chemical species.30,31 Generally, these experiments are performed using three-electrode cells and scanning the potential of the working electrode linearly while recording the resulting current. When compared to conventional synthesis of organic compounds, the electrochemical method stands as the most favorable and environment-friendly approach because it makes use of electrons rather than conventional toxic reducing and oxidizing reagents.32 Reactions can be carried out either directly or indirectly with the use of electron as the reagent.33,34 2,2,6,6-Tetramethylpiperidinyl-l-oxyl (TEMPO) is a prominent catalyst for the oxidation of organic compounds as it acts a major source of nitroxyl radicals. The stability of TEMPO in aqueous and non-aqueous media is because of steric hindrance around the nitroxyl moiety, which makes the most important electron providing radical in the electrochemical process. The catalytic system is effective in an acidic medium; the disproportionation of TEMPO to oxoammonium salt (TEMPO+) is catalyzed by the acidic medium (eqn (1)).35 TEMPO can be oxidized either chemically by using oxidants such H2O2, O2, and metal halides or by electrochemical methods.36 TEMPO is considered as a selective oxidant for the oxidation of primary and secondary alcohols to the corresponding aldehydes and ketones due to the prevention of over-oxidation of synthesized products.37–39
In the current study, MnO2–Pi nanoparticles scattered on the NCS layer coated carbon fiber paper (CFP) electrode is applied for the electrocatalytic oxidation of 2-thiophenemethanol to 2-thiophenemethanal, in a TEMPO mediated acidic medium. The methods reported, so far, regarding the oxidation process include several steps; whereas, the present work introduces a novel method of oxidizing 2-thiophenemethanol in one step, using MnO2–Pi based solid nanocomposite modified electrode. This method can be scaled up easily due to facile nature and low-cost. A schematic illustration of the current work is represented in Scheme 1.
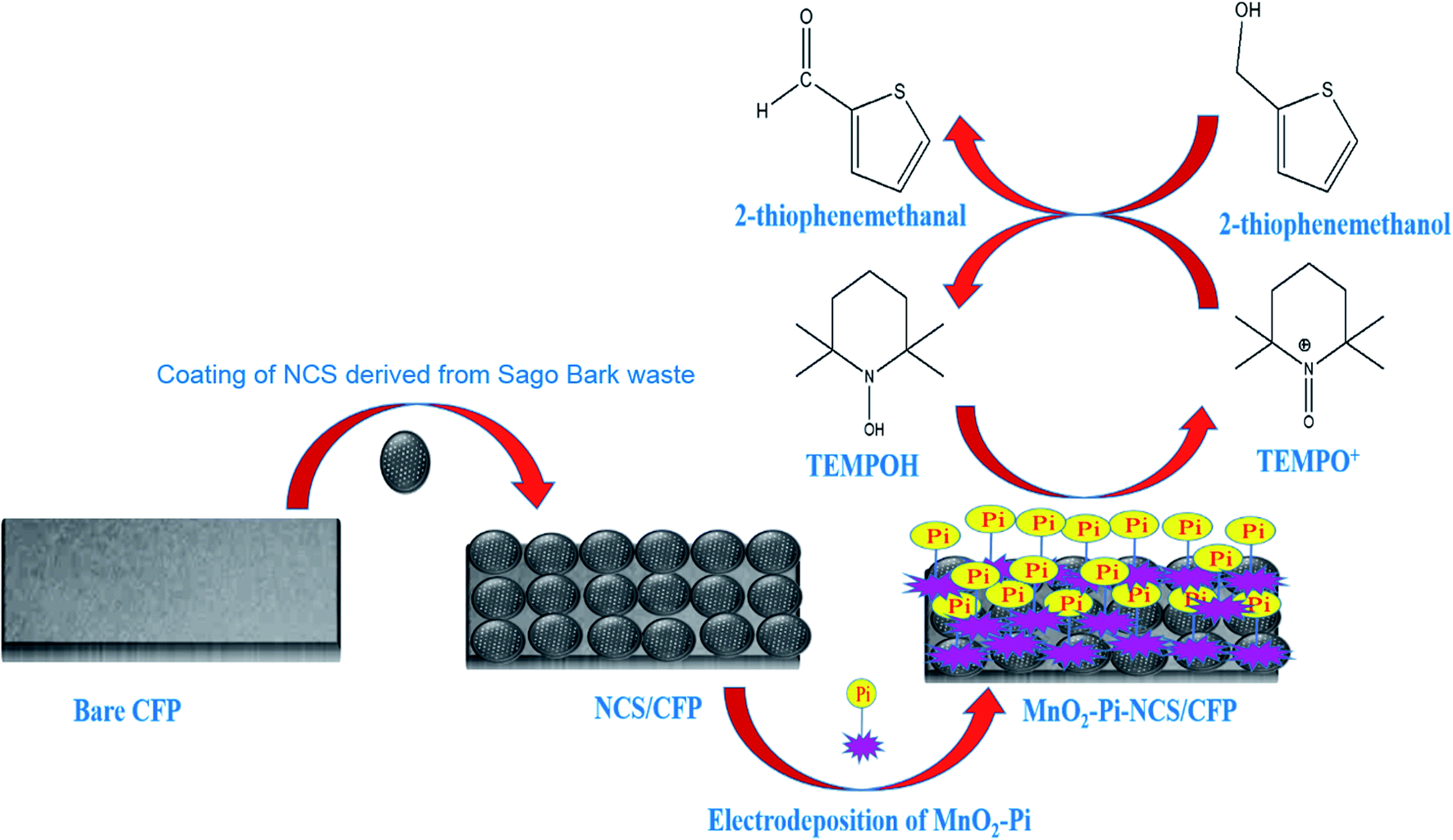 | ||
| Scheme 1 Schematic illustration for the application of MnO2–Pi–NCS/CFP electrode, for electrocatalytic oxidation of 2-thiophenemethanol. | ||
Results and discussion
Characterization of Sago bark waste-derived NCS
The carbon nanomaterials were produced by one-step pyrolysis of waste barks of the Sago tree. The products were analyzed using various characterization techniques to demonstrate their morphological and structural properties.The XRD pattern (Fig. 1a) showed characteristic peaks of carbon at 2θ = 27° and 43°. Adding to that, the broad peak in 20–30°, with a sharp peak at 27° indicated the material to be turbostratic in nature (JCPDS: 10750410). The other peaks with lower intensities occurred due to the traces of calcium oxides or carbonates. Further, the Raman spectroscopic analysis was conducted to understand the structural properties. The spectrum (Fig. 1b) showed two prominent peaks at 1336 and 1595 cm−1, which were called D band and G band, respectively. Generally, D band hints at the disorder in the structure; whereas, G band gives information regarding the graphitic nature of the material. In the spectrum of NCS, the intensity of D band was larger than that of the G band; this could be attributed to the strain caused at the curvatures due to the bending of lattice planes of graphitic layers in nanospheres.
 | ||
| Fig. 1 Microscopic images of the Sago bark derived NCS obtained from (a) SEM and (b) TEM; (c) XRD pattern; (d) Raman spectrum of NCS showing D and G band. | ||
The microscopic images obtained from SEM and TEM (Fig. 1c and d, respectively) clearly showed the formation of spherical structures, with their size in nanoscale, hence called nanocarbon spheres.
Electrodeposition of MnO2–Pi nanoparticles on NCS/CFP electrode
The efficiency of electrochemical methods in controlling the homogeneity and thickness was utilized for electrochemical deposition of MnO2–Pi nanoparticles on NCS/CFP electrode surface. The porous structure of NCS magnifies its surface area for the deposition of MnO2–Pi nanoparticles on NCS/CFP electrode. The MnO2 nanoparticles were deposited on the NCS/CFP electrode by cyclic voltammetry from the solutions of KMnO4 (0.025 M), which is purchased from sigma Aldrich-Merck and PBS (pH 7.0) for 25 cycles as represented in ESI 1 (Fig. S1†). The cathodic reduction peak appeared at 0.1274 V (corresponding to Mn7+) implies the deposition of MnO2–Pi (eqn (1)). After the deposition process, the electrodes were rinsed with distilled water and dried; the dried electrodes were used for the electrochemical oxidation of 2-thiophenemethanol.| MnO4− + 2H2O + 3e− → MnO4 + 4OH− | (1) |
Electrochemical characteristics of the modified electrodes
Cyclic voltammetric studies were performed for redox reactions of K4[Fe(CN)6]/K3[Fe(CN)6] at bare CFP and modified CFP electrodes (Fig. 2) electrochemically active surface area of the three electrodes was determined using Randles–Sevcik equation.40 Anodic peak currents were plotted against the square root of the scan rates and the slopes of the plots were used for the surface area calculation. The active surface area determined for a geometric area of 0.7 cm2 in the bare CFP, NCS coated CFP and MnO2–Pi–NCS/CFP electrodes were 1.43 cm2, 2.86 cm2, and 6.72 cm2, respectively. The results showed that the electroactive surface area of the bare electrode is smaller than that of the modified electrodes. The roughness factor was determined by the ratio of the electroactive surface area to the geometrical area of the electrode. The roughness factor of the MnO2–Pi–NCS/CFP electrode was 9.6, which was higher than that of the NCS/CFP (4.085) and the bare CFP electrode (2.042). These studies highlighted the enhancement of surface features by coating a thin layer of NCS on CFP and the MnO2–Pi nanoparticles on NCS/CFP. The results were also substantiated by the highest redox current density obtained for MnO2–Pi–NCS/CFP electrode compared to the other two electrodes.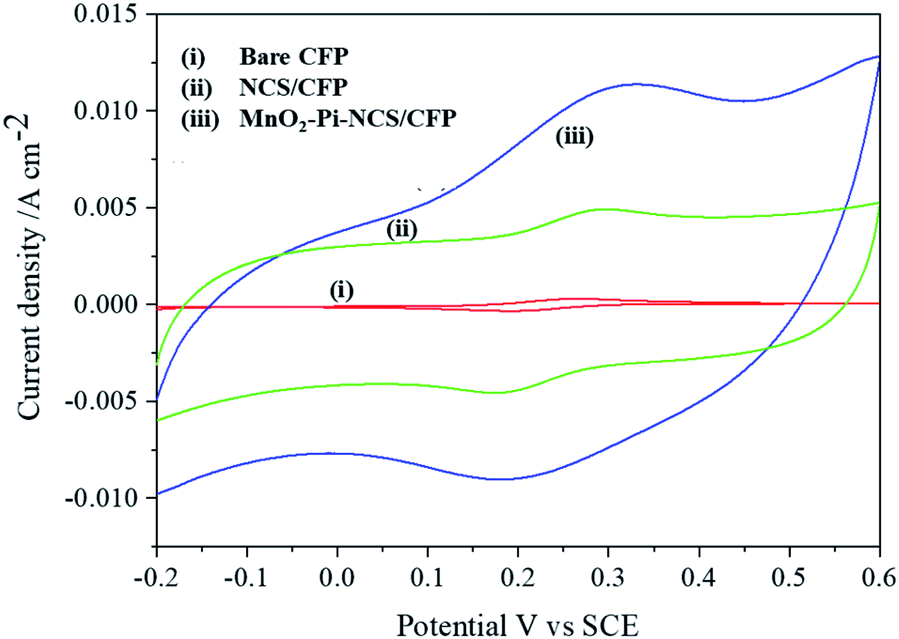 | ||
| Fig. 2 (i) Bare CFP (ii) NCS/CFP and (iii) MnO2–Pi–NCS/CFP cyclic voltammograms obtained for 5 mM K4[Fe(CN)6]/K3[Fe(CN)6] in 0.1 M KCl at scan rate 0.05 V s−1. | ||
EIS is a popular method to monitor the charge transfer processes at electrode–electrolyte interface; it gives a fast and sensitive response to the electrode surface changes.41 In (ESI 2†), Fig. S2† represents Nyquist plots of the bare and modified electrodes, which are obtained in 5 mM K4[Fe(CN)6]/K3[Fe(CN)6] taken in 0.1 M KCl (supporting electrolyte). The impedance data is fitted and the corresponding Randles equivalence circuit model is given as inset in Fig. S2.†42 The circuit indicates resistance to the charge transfer reaction (Rct) at the electrode surface and the Warburg coefficient (W) in parallel to the double-layer capacitance (Cdl), and electrolyte solution resistance (RS). The parallel structure of Rct and Cdl gave rise to a semicircle in the Nyquist plot of Z′ against Z′′. Electron transfer kinetics in the redox process of K4[Fe(CN)6]/K3[Fe(CN)6] at electrode interface is controlled by charge transfer reaction at the surface of the electrode (Rct), which varies with adsorption on modified electrode surface. Rct could be estimated by the semicircular region in the Nyquist plot. The increase in the linear portion (Warburg impedance) of the slope indicates favorable diffusion of K4[Fe(CN)6]/K3[Fe(CN)6] towards the modified MnO2–Pi–NCS/CFP electrode. The Rct determined for bare CFP is 93.71 Ω; it represents high charge transfer resistance at the interface due to poor conductivity and the less sensitivity of the electrode. Rct (54.31 Ω) of NCS/CFP electrode was observed to decrease, which is attributed to the electronic conducting pathway formed between the electrode and the electrolyte solution. The deposition of MnO2 nanoparticles on NCS/CFP electrode resulted in a further reduction of Rct value (14.72 Ω), which confirms the increased conductivity in the modified electrode. From the results, it is evident that the modification of bare CFP with NCS and MnO2–Pi enhances the electrical conductivity and electron transfer kinetics. The optimized conductivity of the modified electrode was appreciated in the determination of [Fe(CN)6]3−/4− ions; it also ensures an excellent platform for the electrocatalytic oxidation of 2-thiophenemethanol.
Physicochemical studies of modified electrodes
 | ||
| Fig. 3 SEM images of (a) NCS/CFP and (b) MnO2–Pi–NCS/CFP electrodes. | ||
The morphological nature of MnO2–Pi nanoparticles deposited on NCS/CFP was further investigated using TEM and the images are shown in Fig. S6a in (ESI 4†). MnO2–Pi nanostructures dispersed on NCS will facilitate TEMPO mediated oxidation of 2-thiophene methanol at the surface of MnO2–Pi–NCS/CFP electrode leading to effective oxidation. TEM studies of MnO2–Pi–NCS/CFP (Fig. S6†) reveals that MnO2 nanoparticles are dispersed on NCS coated CFP as lengthy nano needle shaped structures. Fig. S6b† represents the HRTEM image of MnO2–Pi–NCS/CFP electrode which represents the presence of metal nanoparticles in its crystal lattice.
 | ||
| Fig. 4 XRD graphs obtained for (a) NCS/CFP and (b) MnO2–Pi–NCS/CFP electrodes. | ||
XPS analysis of MnO2–Pi–NCS/CFP electrodes
XPS analysis of MnO2–Pi–NCS/CFP electrodes was conducted before and after the oxidation process. The core-level XPS spectra of C 1s, before and after oxidation of 2-thiophene methanol are given in Fig. 5a and b, respectively. The peaks at 284.0 eV, 284.3 eV, 284.7 eV, and 285.6 eV observed in the C 1s core-level spectrum, before oxidation, could be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or C–C or C–H bonds present in graphitic carbon of either CFP or NCS43,44 respectively. The peak observed at 286.0 eV confirms the presence of C–S from 2-thiophenemethanol.45 At 286.9 eV and 289.7 eV, peaks belonging to C–O of alcoholic OH group of 2-thiophenemethanol was observed (Fig. 5a).45
C or C–C or C–H bonds present in graphitic carbon of either CFP or NCS43,44 respectively. The peak observed at 286.0 eV confirms the presence of C–S from 2-thiophenemethanol.45 At 286.9 eV and 289.7 eV, peaks belonging to C–O of alcoholic OH group of 2-thiophenemethanol was observed (Fig. 5a).45
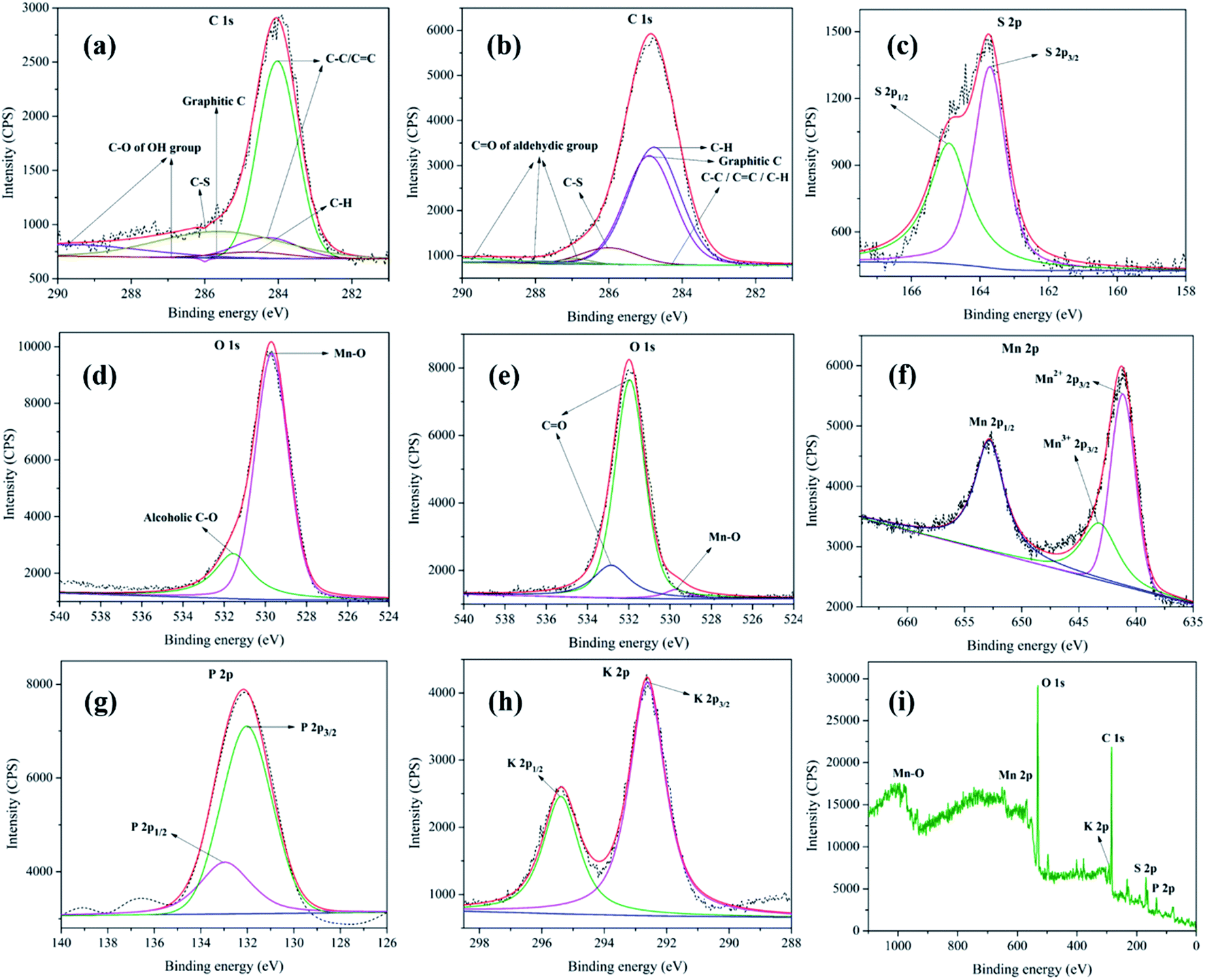 | ||
| Fig. 5 XPS analysis of MnO2–Pi–NCS/CFP electrodes, showing C 1s spectrum (a) before and (b) after oxidation of thiophene methanol; (c) S 2p core level spectrum; O 1s spectra (d) before and (e) after oxidation; (f), (g) and (h) represent core level spectrum of Mn 2p, P 2p, and K 2p, respectively. (i) Wide XPS spectrum of MnO2–Pi–NCS/CFP electrode. The original spectra are indicated by dotted lines; the sum of deconvoluted peaks is represented by solid red lines. | ||
The C 1s core-level XPS spectrum of MnO2–Pi–NCS/CFP electrode, after oxidation, is given in Fig. 5b. The peaks specific for C 1s were seen at 284.1 eV and 284.3 eV, which belong to C–C bond; the peaks at 284.5 eV and 284.7 eV can be attributed to C–H bond.46 The peak at 286.0 eV suggests C–S bond of 2-thiophenemethanal.45 The peaks appeared at 287.0 eV, 288.0 eV, and 289.8 eV belong to CO of aldehyde group; thereby, confirming the formation of 2-thiophenemethanal by the oxidation of its corresponding alcohol.47 The S 2p core-level XPS spectra obtained for MnO2–Pi–NCS/CFP electrode used before and after oxidation of 2-thiophenemethanol was identical and is given in Fig. 5c. The S 2p spectrum composed of 2p3/2 peak at 163.7 eV and 2p1/2 peak at 164.9 eV; the intensity ratio of these peaks was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as found theoretically based on spin–orbit splitting effect.48
:1, as found theoretically based on spin–orbit splitting effect.48
The O 1s peak in the core-level XPS spectrum is captured for the MnO2–Pi–NCS/CFP electrode, before (Fig. 5d) and after (Fig. 5e) the oxidation process. The peak appeared at 529.6 eV in these graphs correspond to the oxygen bonded to manganese in MnO2 nanocrystals.49 The pristine electrode shows a peak at 531.5 eV in (Fig. 5d), which suggests the alcoholic C–O bond in 2-thiophenemethanol.46 Further, the electrode used for oxidation showed two prominent peaks at higher binding energies, 531.9 eV and 532.8 eV which were attributed to the aldehyde group in the oxidized product.47 The Mn 2p spectrum of MnO2–Pi–NCS/CFP electrode displayed in Fig. 5f, shows peaks related to Mn2+ 2p3/2 (641.1 eV) and Mn 2p1/2 (652.8 eV). The spin energy separation of 11.7 eV in these binding energies was found to be in concordant with the literature value for MnO2.50 Moreover, in the high-resolution spectrum centered on Mn 2p3/2, Mn3+ 2p3/2 peak was observed at 643.2 eV.51
The P 2p core-level XPS spectrum (Fig. 5g) exhibits P 2p1/2 and P 2p3/2 at 132.9 eV and 132.0 eV, respectively. It suggests the presence of weak interaction between Pi and MnO2 nanoparticles.52 The K 2p XPS core-level high resolution spectrum (Fig. 5h) exhibits K 2p1/2 and K 2p3/2 doublet at 295.3 eV and 292.6 eV, respectively, with the spin–orbit splitting (2.7 eV).53,54 The XPS wide spectrum of MnO2–Pi–NCS/CFP is shown in Fig. 5i. The XPS peak shifts clearly indicate effective deposition of MnO2–Pi on NCS coated CFP. Thus, the XPS analysis of MnO2–Pi–NCS/CFP electrodes before and after the oxidation process demonstrates the conversion of 2-thiophenemethanol to 2-thiophenemethanal.
Surface morphology and roughness of the bare and modified CFP electrodes
The surface morphology and roughness of bare CFP and modified electrodes is analyzed by optical profilometry. The two dimensional (2D) and three dimensional (3D) images were captured using an optical profilometer at 50× magnification are given in Fig. 6. The zeta 3D images obtained are used to deduce the dimensional and roughness features. Fig. 6a–d, show the intertwined morphology of fibers in the bare CFP and NCS/CFP, respectively. The roughness of the surfaces was analyzed on 12 different areas and the root mean square value with standard deviation was reported (Table 1). Root mean square roughness obtained for the bare CFP, NCS/CFP, and MnO2–Pi–NCS/CFP were 1.746 ± 0.151 μm, 3.975 ± 0.318 μm, and 9.312 ± 0.753 μm, respectively, as given in Table 1. The higher roughness of NCS/CFP than the bare CFP could be ascribed to the formation of a rough NCS film on the CFP fibers. The MnO2–Pi–NCS/CFP electrode surface showed aggregates of nanoparticles (Fig. 6e and f). The significant increase in the roughness of this electrode surface could be attributed to the deposition of MnO2–Pi nanoparticles on the NCS coated CFP electrode surface.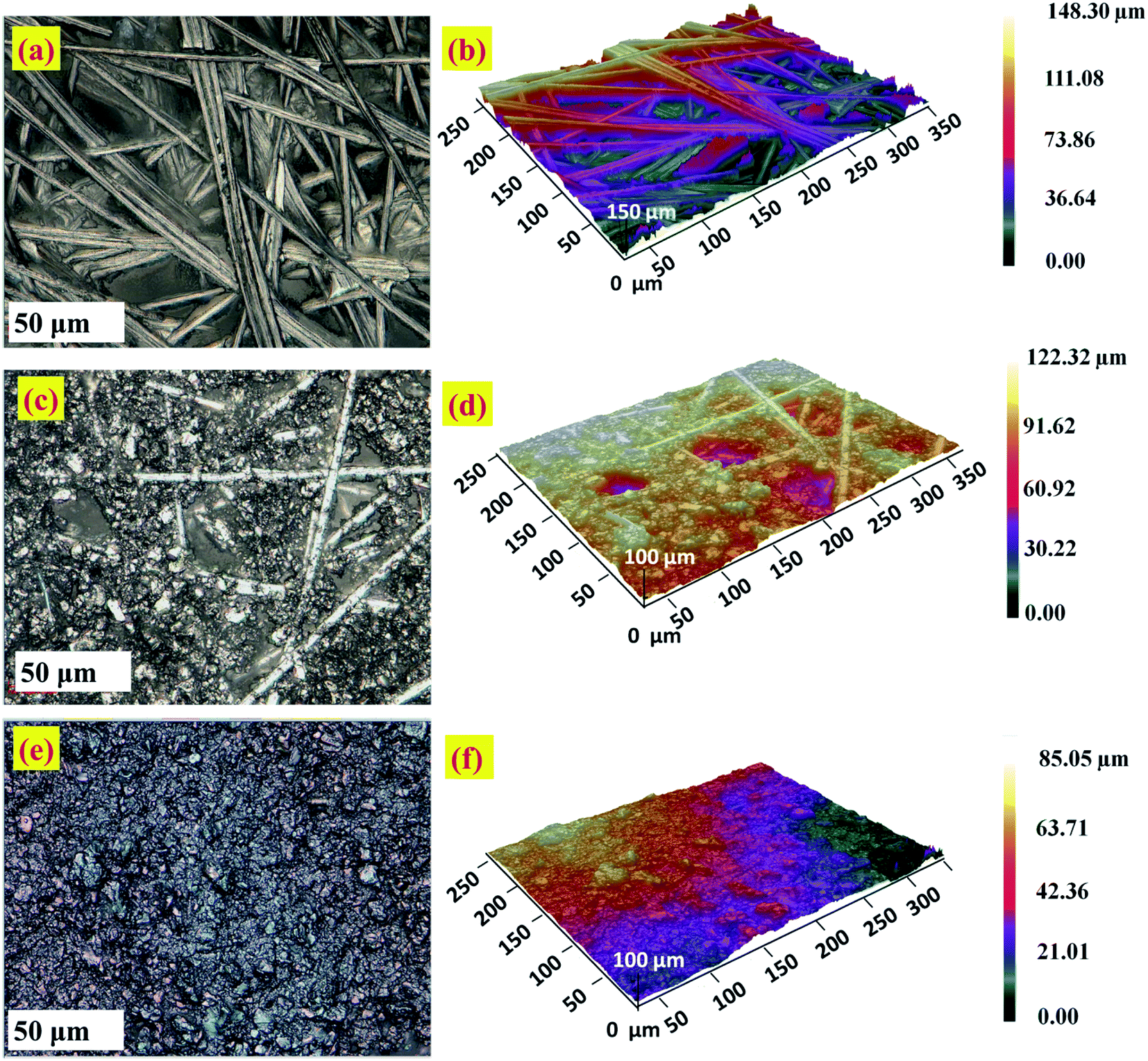 | ||
| Fig. 6 Optical profilometer images at higher magnification 50×: 2D and 3D images of (a and b) the bare CFP, (c and d) NCS/CFP and (e and f) MnO2–Pi–NCS/CFP electrodes. | ||
| Electrode | Root mean square roughness (μm) | Standard deviation |
|---|---|---|
| Bare CFP | 1.746 | 0.151 |
| NCS/CFP | 3.975 | 0.318 |
| MnO2–Pi–NCS/CFP | 9.312 | 0.753 |
Electrocatalytic oxidation of 2-thiophenemethanol at bare and modified CFP electrodes
Cyclic voltammetric experiments were conducted to investigate the electrocatalytic oxidation of 2-thiophenemethanol at the bare CFP, NCS/CFP, and MnO2–Pi–NCS/CFP electrodes (Fig. 7). | ||
| Fig. 7 Cyclic voltammograms obtained for 0.01 M TEMPO in 0.01 M H2SO4 at scan rate of 0.05 V s−1, (a) in the absence of 2-thiophenemethanol (b) in the presence of 10 mM 2-thiophenemethanol in the acid medium. | ||
The study was conducted at a scan rate of 0.05 V s−1 in TEMPO medium with a potential range between −0.6 and 0.8 V vs. SCE, in solutions of 0.01 M H2SO4. No characteristic peak was observed in the cyclic voltammograms obtained for the bare and modified CFP electrodes. But when TEMPO solution (0.01 M) was added to the above reaction medium, redox peaks were appeared at 0.4491 V and 0.5283 V, which could be attributed to the transfer of an electron from TEMPO forming TEMPO+; the peak currents of bare CFP, NCS/CFP and MnO2–Pi–NCS/CFP were found to be (−8.479 × 10−6 A cm−2 and 1.155 × 10−5 A cm−2), (1.77 × 10−5 A cm−2 and 2.269 × 10−5 A cm−2) and (−5.3535 × 10−5 A cm−2 and 7.1613 × 10−5 A cm−2), respectively, as shown in Fig. 7a. On adding 2-thiophenemethanol (10 mM) to the reaction medium, the peaks were shifted towards negative potential, −0.3631 V and 0.2533 V (Fig. 7b); the corresponding peak current was increased to −0.00128 A cm−2 and 0.02009 A cm−2 for the finally modified electrodes. The results indicate a marginally small current flow through bare CFP and NCS/CFP electrodes (curves i and ii). At MnO2–Pi–NCS/CFP electrode, the high energy active sites of MnO2–Pi nanoparticles attract substrate molecules largely towards the electrode surface, leading to increased electron transfer kinetics and higher current flow (curve iii). Molecules of 2-thiophenemethanol were greatly attracted to the electrode surface.
Oxo-ammonium salt (TEMPO+) is the actual oxidizing agent in the experiment; it is formed by the disproportionation of TEMPO in an acidic medium. TEMPO+ selectively acts on 2-thiophenemethanol and converts it into 2-thiophenemethanal and also prevents the further oxidation of aldehyde into thiophene carboxylic acid. TEMPO is regenerated from the hydroxyl amine compound, TEMPOH, which is formed by the reduction of TEMPO+ in a 2e-transfer process at the electrode surface. Scheme 2 illustrates the plausible mechanism for TEMPO mediated oxidation of 2-thiophene methanol at MnO2–Pi–NCS/CFP electrode. After 48 hours, the reaction mixture was extracted with ethyl acetate; the product is isolated using column chromatography and confirmed by FTIR and 1H NMR spectroscopy as discussed in ESI 5 (Fig. S7) and ESI 6 (Fig. S8†), respectively. The isolated yield of 2-thiophenemethanal was 81% and the conversion percentage as per gas chromatography is 85%.
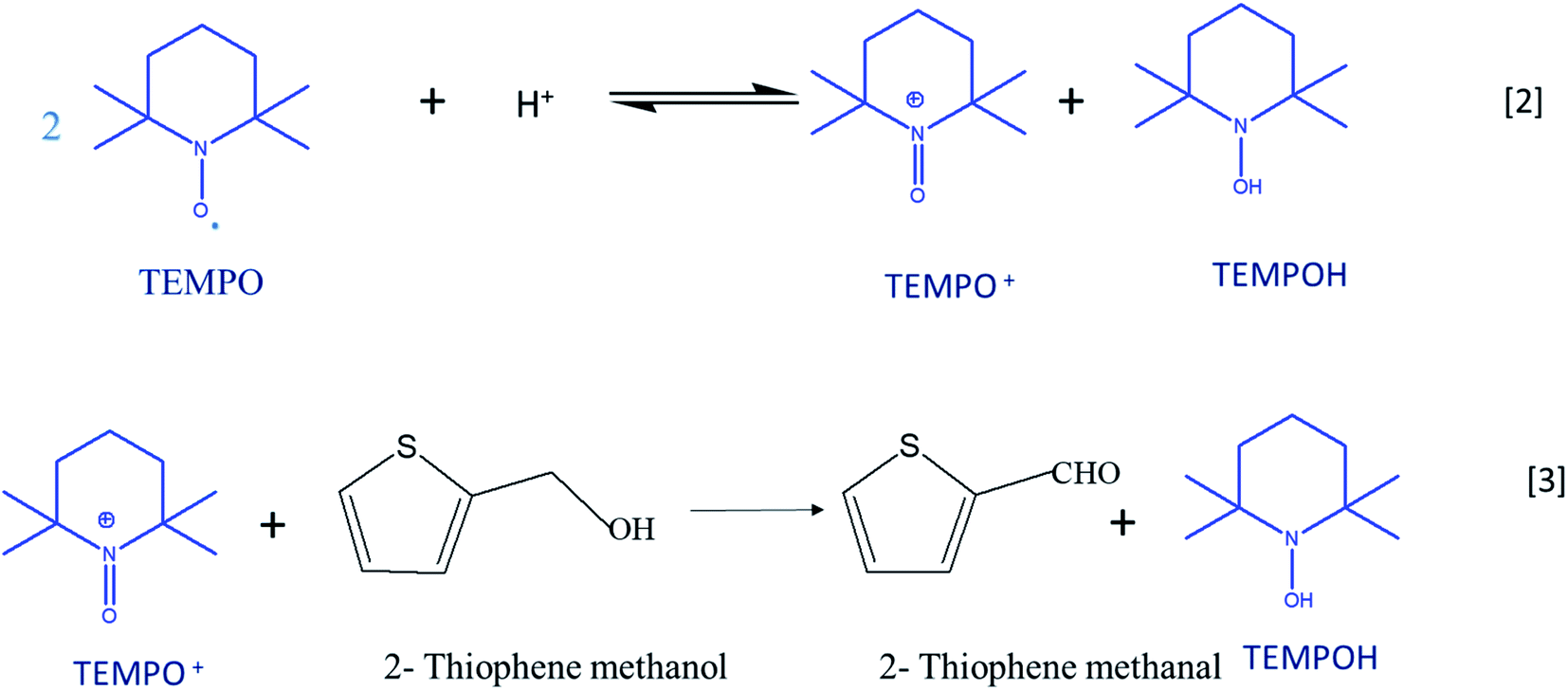 | ||
| Scheme 2 Plausible mechanism for the TEMPO mediated electrochemical oxidation of thiophene methanol at MnO2–Pi–NCS/CFP electrode. | ||
Optimization of experimental conditions for the electrocatalytic oxidation of thiophenemethanol
Several parameters including the concentration of functional monomer, number of cycles used for the electropolymerization process of conducting polymer and the effect of scan rate were optimized in order to obtain the best electrochemical response for oxidation of 2-thiophenemethanol. The details for each of these optimization processes were explained in ESI 7 and ESI 8.†The optimum number of cycles employed during CV experiments towards the electropolymerization process is also found to affect the sensitivity and efficiency of the modified electrode for electrochemical oxidation. Electrodeposition MnO2–Pi nanoparticles on NCS/CFP were examined between a range of 5 to 25 number of cycles used and the corresponding cyclic voltammograms were recorded. The redox peak current values, obtained from cyclic voltammetry (Fig. S9†), show an increasing trend with an increase in the number of scanning potential cycles for MnO2 deposition, and the optimum number of cycles is found to be 25.
To study the effect of scan rate on the oxidation of 2-thiophene methanol at MnO2 Pi–NCS/CFP electrode, CVs were recorded at various scan rates from 0.01 to 0.13 V s−1 in 10 mM 2-thiophene methanol in an aqueous acidic medium, and the corresponding cyclic voltammograms are given in Fig. S10a.† A linear relationship between the anodic and cathodic peak current and the square root of the scan rate in the range of 0.01 to 0.13 V s−1 is observed in Fig. S10b and c† illustrating that the oxidation of 2-thiophenemethanol is adsorption controlled rather than diffusion-controlled process.
Repeatability, reproducibility and stability
Repeatability of the data was studied by repeating the electrochemical experiments up to 6 cycles emphasizing no significance change either in the potential or anodic currents.The reproducibility of electrode response was tested by performing the TEMPO mediated electrooxidation of 2-thiophenemethanol using six different electrodes. The stability of the MnO2–Pi–NCS/CFP electrode was tested for 50 continuous cycles using CV in the absence of TEMPO and 2-thiophenemethanol and is depicted in Fig. 8 showing no significant changes. The current responses were checked daily up to 10 days, and the current response was still retained 98% value of the initial response. Therefore, the results confirmed good repeatability, reproducibility, and excellent stability of the MnO2–Pi–NCS/CFP electrode.
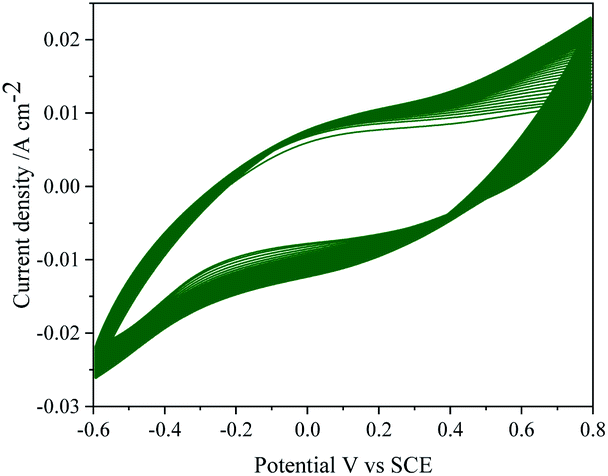 | ||
| Fig. 8 CV response of MnO2–Pi–NCS/CFP electrode up to 50 continuous cycles, in the absence of TEMPO and 2-thiophenemethanol in 0.01 M H2SO4 at scan rate 0.05 V s−1. | ||
Experimental
Material and methods
Thiophene methanol, potassium permanganate (KMnO4), TEMPO and polyvinylidene difluoride (PVDF) were procured from Sigma Aldrich-Merck. Potassium ferrocyanide (K4[Fe(CN)6]·3H2O), potassium ferricyanide (K3[Fe(CN)6]), monopotassium phosphate (KH2PO4), dipotassium hydrogen phosphate (K2HPO4), N-methyl-2-pyrrolidone (NMP) and sulphuric acid (H2SO4) were purchased from SD Fine-Chem Pvt. Ltd., India. The phosphate buffer solution (PBS, 0.1 M, pH 7.0) was prepared by mixing the stock solutions of K2HPO4 and KH2PO4. All the aqueous solutions were prepared by using distilled water (DW). The raw material, wastes of Sago bark, used for the preparation of NCS, was collected from the South East Asian regions. For the electrochemical studies, CH Instruments, Inc. USA (model CHI608E) was used. Electrochemical measurements were performed using a three-electrode system. Saturated calomel electrode (SCE) as the reference electrode and a platinum foil was used as a counter electrode (CE). Three different working electrodes were prepared; (i) MnO2–Pi nanoparticles electrochemically deposited on the Sago bark waste-derived NCS coated CFP; (ii) NCS/CFP, in the absence of MnO2–Pi nanoparticles; (iii) bare CFP. The working electrodes were prepared using 0.2 mm thick Toray carbon fiber paper obtained from M s−1 Vinpro Technologies, Hyderabad, India. The CFP papers of size 2 cm × 0.7 cm were taken for the experiments; one end of the paper with an area 0.70 cm2 was immersed in the electrolyte solution, a conducting ink was used to establish electrical contact with the copper wire. The experiments were carried out at room temperature.Modification of electrodes
The wastes of Sago bark were used as raw material for the preparation of porous NCS. The wastes collected from south-east Asian regions were initially dried under the sun, subsequently in an oven overnight. It was then crushed to a fine powder and sieved using a mesh of pore size ∼60 μm. The Sago bark powder was heated at a rate of 10 °C min−1 and pyrolyzed at 850 °C (where entire weight loss taking place in the presence of inert gas) for one hour under a nitrogen environment, in the absence of any catalysts. The carbonized product was washed using 0.1 M HCl, followed by distilled water, before electrocatalytic experiments.MnO2–Pi nanoparticles were electrochemically deposited on NCS coated CFP electrode from 0.1 M phosphate buffer solution (PBS, pH 7.0), containing 20 mL of 0.025 M KMnO4. The electrodeposition of MnO2–Pi nanoparticles on NCS/CFP electrode was performed in the potential range −0.1 to 0.6 V at a sweep rate of 0.05 V s−1 up to 25 cycles.
Electrocatalytic oxidation of 2-thiophenemethanol at MnO2–Pi–NCS/CFP electrode by TEMPO mediator
The electrocatalytic oxidation of 2-thiophenemethanol was performed using a modified MnO2–Pi–NCS/CFP electrode. The TEMPO mediated electrochemical oxidation process was monitored by cyclic voltammetry, between −0.6 and 0.8 V potentials at a sweep rate of 0.05 V s−1. The electrolyte solution used in the study contained 10 mM 2-thiophenemethanol, TEMPO (0.1 M), H2SO4 (0.01 M), and distilled water.Instrumentation
The cyclic voltammetric studies, electrochemical oxidation behavior of bare CFP, NCS/CFP and MnO2–Pi–NCS/CFP electrodes, and electrochemical impedance spectroscopy (EIS) measurements were conducted using an electrochemical analyzer. The SEM (Zeiss Gemini, model Ultra 55) clubbed with EDAX was used to capture microscopic images and elemental composition, respectively. XRD patterns were captured using Bruker AXS D8 Advance X-ray Diffractometer. X-ray photoelectron spectroscopy (XPS) studies (Kratos Axis Ultra X-ray photoelectron spectrometer) employed polychromatic MgKα X-rays (hν = 1253.6 eV). The surface analysis of bare and modified CFP electrodes was conducted using ZETA-20, Z10002-1-1 optical profilometer (KLA-Tencor, US-CA). Images were taken using 50× and 20× lens, with a working distance of 1.00 mm, and analysis of the images was done using Zeta 3D software. The roughness of the surfaces was analyzed at 12 different areas on CFP and the average value with the standard deviation was reported. ATR- FTIR spectra (Shimadzu, IR spirit-00107) were recorded in the frequency range of 400–4000 cm−1. The product formed was analyzed using 1H NMR spectroscopy (Bruker 400 MHz).Conclusions
A promising electrode was prepared by uniformly depositing MnO2–Pi nanoparticles on NCS coated CFP, for the electrocatalytic oxidation of 2-thiophenemethanol. NCS acts as a good host material for the dispersion of MnO2 nanoparticles. The results confirmed that the modified electrode could be extended to the oxidation of a large number of organic molecules. The excellent conductivity of the MnO2–Pi coated electrode was favorable for the electron transfer between its surface and the organic molecules. Thiophenemethanol was electrochemically oxidized to thiophenemethanal by a simple, clean, and environmentally benign process in TEMPO mediated acidic environment. Further, the newly designed electrode demonstrated high sensitivity and excellent selectivity. The electrocatalytic activity of MnO2–Pi–NCS/CFP electrode towards the oxidation of thiophene methanol was studied by cyclic voltammetric technique, which indicated that the modified electrode is several times more efficient than the bare CFP electrode towards the TEMPO mediated electrocatalytic oxidation of thiophene methanol. The direct oxidation of 2-thiophenemethanol to 2-thiophenemethanal could be achieved in a single step at MnO2–Pi–NCS/CFP electrode using TEMPO as a mediator.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to KSTEPS, DST, Govt. of Karnataka, for provided assistance. One of the authors, Dr Gurumurthy Hegde would like to thank DST-Nanomission, Govt. of India for providing a grant with file no SR/NM/NT-1026/2017.References
- E. Salih, M. Mekawy, R. Y. Hassan and I. M. El-Sherbiny, J. Nanostruct. Chem., 2016, 6, 137–144 CrossRef.
- S. Li, Y. Ma, Y. Liu, G. Xin, M. Wang, Z. Zhang and Z. Liu, RSC Adv., 2019, 9, 2997–3003 RSC.
- A. Khoobi, A. M. Attaran, M. Yousofi and M. A. Enhessari, J. Nanostruct. Chem., 2019, 9, 29–37 CrossRef CAS.
- Z. Zhang, C. Zhang, J. Sun, T. Kou and C. Zhao, RSC Adv., 2012, 2, 11820–11828 RSC.
- A. T. Mathew, K. B. Akshaya, T. P. Vinod, A. Varghese and L. George, ChemistrySelect, 2020, 5, 3283–3294 CrossRef CAS.
- E. K. Joice, S. Rison, K. B. Akshaya and A. Varghese, J. Appl. Electrochem., 2019, 49, 937–947 CrossRef CAS.
- E. K. Joice, A. Varghese, Y. N. Sudhakar, B. Ganesh and J. Selvaraj, J. Electrochem. Soc., 2018, 165, H399–H406 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 2000, 45, 2377–2385 CrossRef CAS.
- A. S. Bandarenka, E. Ventosa, A. Maljusch, J. Masa and W. Schuhmann, Analyst, 2014, 139, 1274–1291 RSC.
- G. Chen, Sep. Purif. Technol., 2004, 38, 11–41 CrossRef CAS.
- Y. J. Feng and X. Y. Li, Water Res., 2003, 37, 2399–2407 CrossRef CAS.
- A. Dominguez-Ramos, R. Aldaco and A. Irabien, Ind. Eng. Chem. Res., 2008, 47, 9848–9853 CrossRef CAS.
- K. B. Akshaya, T. P. Vinod, M. Nidhin, A. Varghese and L. George, J. Electrochem. Soc., 2018, 165, B582–B595 CrossRef CAS.
- A. Kumar, G. Hegde, S. A. B. A. Manaf, Z. Ngaini and K. V. Sharma, Chem. Commun., 2014, 50, 12702–12705 RSC.
- G. M. Jacob and I. Zhitomirsky, Appl. Surf. Sci., 2008, 254, 6671–6676 CrossRef CAS.
- B. K. Balan and S. Kurungot, Inorg. Chem., 2012, 51, 9766–9774 CrossRef CAS.
- K. B. Akshaya, V. S. Bhat, A. Varghese, L. George and G. Hegde, J. Electrochem. Soc., 2019, 166, B1097–B1106 CrossRef CAS.
- A. T. Mathew, V. S. Bhat, S. Supriya, T. Maiyalagan, A. Varghese and G. Hegde, Electrochim. Acta, 2020, 136624 CrossRef CAS.
- J. Marie, S. Berthon-Fabry, P. Achard, M. Chatenet, A. Pradourat and E. Chainet, J. Non-Cryst. Solids, 2004, 350, 88–96 CrossRef CAS.
- G. Hegde, S. A. Abdul Manaf, A. Kumar, G. A. Ali, K. F. Chong, Z. Ngaini and K. V. Sharma, ACS Sustainable Chem. Eng., 2015, 3, 2247–2253 CrossRef CAS.
- A. Divyashree and G. Hegde, RSC Adv., 2015, 5, 88339–88352 RSC.
- G. A. Ali, A. Divyashree, S. Supriya, K. F. Chong, A. S. Ethiraj, M. V. Reddy, H. Algarni and G. Hegde, Dalton Trans., 2017, 46, 14034–14044 RSC.
- G. Singh, K. S. Lakhi, S. Sil, S. V. Bhosale, I. Kim, K. Albahily and A. Vinu, Carbon, 2019, 148, 164–186 CrossRef CAS.
- G. M. Jacob and I. Zhitomirsky, Appl. Surf. Sci., 2008, 254, 6671–6676 CrossRef CAS.
- F. Shi, X. L. Wang, C. D. Gu and J. P. Tu, RSC Adv., 2014, 4, 41910–41921 RSC.
- J. Du, G. Shao, X. Qin, G. Wang, Y. Zhang and Z. Ma, Mater. Lett., 2012, 84, 13–15 CrossRef CAS.
- Q. Zhang, L. Li, Y. Wang, Y. Chen, F. He, S. Gai and P. Yang, Electrochim. Acta, 2015, 176, 542–547 CrossRef CAS.
- L. Shen, L. Du, S. Tan, Z. Zang, C. Zhao and W. Mai, Chem. Commun., 2016, 52, 6296–6299 RSC.
- T. Liu, C. Jiang, W. You and J. Yu, J. Mater. Chem. A, 2017, 5, 8635–8643 RSC.
- H. Rahman, I. Hussain and S. M. Haque, Pharma Chem., 2015, 7, 12 Search PubMed.
- G. A. Mabbott, J. Chem. Educ., 1983, 60, 697 CrossRef CAS.
- S. Johnston, B. H. Suryanto and D. R. MacFarlane, Electrochim. Acta, 2019, 297, 778–783 CrossRef CAS.
- N. N. Lu, S. J. Yoo, L. J. Li, C. C. Zeng and R. D. Little, Electrochim. Acta, 2014, 142, 254–260 CrossRef CAS.
- K. Y. Zhang, N. N. Lu, S. J. Yoo, L. M. Hu, R. D. Little and C. C Zeng, Electrochim. Acta, 2016, 199, 357–365 CrossRef CAS.
- A. Cecchetto, F. Fontana, F. Minisci and F. Recupero, Tetrahedron Lett., 2001, 42, 6651–6653 CrossRef CAS.
- A. C. Herath and J. Y. Becker, Electrochim. Acta, 2008, 53, 4324–4330 CrossRef CAS.
- J. M. Bobbitt and M. L. Flores, Heterocycles, 1988, 27, 509–533 CrossRef CAS.
- J. J. Lu, J. Q. Ma, J. M. Yi, Z. L. Shen, Y. J. Zhong, C. A. Ma and M. C. Li, Electrochim. Acta, 2014, 130, 412–417 CrossRef CAS.
- M. L. Wang, Y. Y. Zhang, Q. J. Xie and S. Z. Yao, Electrochim. Acta, 2005, 51, 1059–1068 CrossRef CAS.
- C. C. Tsai and G. J. Wang, J. Electrochem. Soc., 2012, 160, B1–B5 CrossRef.
- W. Yang, J. J. Gooding and D. B. Hibbert, J. Electroanal. Chem., 2001, 516, 10–16 CrossRef CAS.
- H. O. Finklea, D. A. Snider, J. Fedyk, E. Sabatani, Y. Gafni and I. Rubinstein, Langmuir, 1993, 9, 3660–3667 CrossRef CAS.
- K. P. Chaithra, B. S. Vinay, K. B. Akshaya, T. Maiyalagan, G. Hegde, A. Varghese and L. George, ACS Biomater. Sci. Eng., 2020, 6, 5264–5273 CrossRef.
- A. Bhaumik, A. Haque, M. F. N. Taufique, P. Karnati, R. Patel, M. Nath and K. Ghosh, J. Mater. Sci. Eng., 2017, 6, 1–11 Search PubMed.
- J. Noh, E. Ito, K. Nakajima, J. Kim, H. Lee and M. Hara, J. Phys. Chem. B, 2002, 106, 7139–7141 CrossRef CAS.
- O. Nibel, S. M. Taylor, A. Pătru, E. Fabbri, L. Gubler and T. J. Schmidt, J. Electrochem. Soc., 2017, 164, A1608–A1615 CrossRef CAS.
- J. V. Rojas, M. Toro-Gonzalez, M. C. Molina-Higgins and C. E. Castano, J. Mater. Sci. Eng. B, 2016, 205, 28–35 CrossRef CAS.
- F. Buckel, F. Effenberger, C. Yan, A. Gölzhäuser and M. Grunze, Adv. Mater., 2000, 12, 901–905 CrossRef CAS.
- Q. Qu, P. Zhang, B. Wang, Y. Chen, S. Tian, Y. Wu and R. Holze, J. Phys. Chem. C, 2009, 113, 14020–14027 CrossRef CAS.
- Z. Lei, F. Shi and L. Lu, ACS Appl. Mater. Interfaces, 2012, 4, 1058 CrossRef CAS.
- M. B. Zakaria, C. Li, M. Pramanik, Y. Tsujimoto, M. Hu, V. Malgras, S. Tominaka and Y. Yamauchi, J. Mater. Chem. A, 2016, 4, 9266–9274 RSC.
- A. Irshad and N. Munichandraiah, ACS Appl. Mater. Interfaces, 2015, 7, 15765–15776 CrossRef CAS.
- W. Kuch, M. Schulze, W. Schnurnberger and K. Bolwin, Surf. Sci., 1993, 287, 600–604 CrossRef.
- Y. Zhang, A. Savara and D. R. Mullins, J. Phys. Chem. C, 2017, 121, 23436–23445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09488a |
| This journal is © The Royal Society of Chemistry 2021 |