
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen bond mediated intermolecular magnetic coupling in mononuclear high spin iron(III) Schiff base complexes: synthesis, structure and magnetic study with theoretical insight†
Tanmoy Basaka,
Carlos J. Gómez-García b,
Rosa M. Gomilac,
Antonio Fronterad and
Shouvik Chattopadhyay*a
b,
Rosa M. Gomilac,
Antonio Fronterad and
Shouvik Chattopadhyay*a
aDepartment of Chemistry, Inorganic Section, Jadavpur University, Kolkata-700032, India. E-mail: shouvik.chem@gmail.com
bDepartamento de Química Inorgánica, Instituto de Ciencia Molecular, Universidad de Valencia, 46980 Paterna, Valencia, Spain
cServeis Científico-Tècnics, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Plama de Mallorca, Baleares, Spain
dDepartament de Química, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain
First published on 15th January 2021
Abstract
The crystal structure and magnetic properties of two mononuclear iron(III) Schiff base complexes, [FeL1(NCS)2] (1), HL1 = 2-[1-[[2-[(2-aminoethyl)amino]ethyl]imino]ethyl]phenol and [FeL2(N3)Cl] (2), HL2 = 2-(-1-(2-(2-aminoethylamino)ethylimino)ethyl)-4-methylphenol are reported. Each complex contains a Fe(III) ion surrounded by a N3O Schiff base ligand and two NCS− ligands (in 1) or one N3− and one Cl− ligands (in 2). The magnetic properties can be well reproduced with zero field splittings in the high spin S = 5/2 Fe(III) ions and weak intermolecular Fe–Fe interactions mediated by hydrogen bonds. This intermolecular antiferromagnetic interaction has been validated by using DFT calculations in complex 2. Moreover, the interaction energies of the H-bonded dimers in both complexes have been estimated using DFT calculations and characterized using a combination of QTAIM and NCI plot computational tools. Complexes 1 and 2 constitute two rare examples of Fe(III) complexes with magnetic interactions through H-bonds.
Introduction
The rational design and synthesis of iron(III) Schiff base complexes have attracted extensive interest due to their structural diversities and potential applications in molecular magnetism. These complexes represent an important class of spin crossover (SCO) systems because of the possibility of the co-existence of two different magnetic states under the same conditions.1,2 The phenomenon of spin cross-over is induced by external perturbations, such as pH, temperature, pressure, light irradiation, etc.3–7 The iron(III) sites in some heme proteins are also reported to exhibit SCO behaviors, which play key roles in their biological functions.8–10 Several iron(III) complexes of various geometries have been prepared using different chelating ligand,11–13 of which Schiff bases are special choices due to the easy routes of their synthesis, the wide range of thermal stability and their very good chelating ability.14–19 Iron(III) complexes with N3O2-donor Schiff base (SB) ligands present a family of coordination compounds in which investigation of the impact of the structural influences on the resulting magnetic behavior, spin states and SCO properties is very appealing.20,21In the present work, two tetradentate N3O half-salen type mono-condensed Schiff base ligands, {HL1 = 2-[1-[[2-[(2-aminoethyl)amino]ethyl]imino]ethyl]phenol and HL2 = 2-(-1-(2-(2-aminoethylamino)ethylimino)ethyl)-4-methylphenol}, have been used for the synthesis of two mononuclear iron(III) complexes, [Fe(L1)(NCS)2] (1) and [Fe(L2)(N3)Cl] (2), respectively. Both complexes have been characterized by elemental and spectral analysis and the structures of the complexes have been confirmed by single crystal X-ray diffraction studies. The solid-state supramolecular interactions of both complexes have also been explored. Magnetic studies show that both complexes are essentially high spin S = 5/2 paramagnets with a zero field splitting and a very weak intermolecular interaction. This intermolecular interaction has been also proved by using DFT calculations and the broken symmetry approach. Moreover, the dimerization energies of the H-bonded dimers used to compute the magnetic coupling have been calculated and the interactions characterized using the quantum theory of “atoms-in-molecules” and the noncovalent interaction plot (NCI plot) index.
Experimental
Materials
All chemicals were of reagent grade and used as purchased from Sigma-Aldrich without further purification.Caution!!! Although no problems were encountered in this work, organic ligands in the presence of azide are potentially explosive. Only a small amount of the material should be prepared and it should be handled with care.
Preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in 15 mL of acetonitrile and refluxed during 1 hour to prepare the corresponding Schiff base ligands: 2-[1-[[2-[(2-aminoethyl)amino]ethyl]imino]ethyl]phenol (HL1) and 2-[1-[[2-[(2-aminoethyl)amino]ethyl]imino]ethyl]-4-methylphenol (HL2). The ligands, HL1 and HL2, were not isolated and the acetonitrile solutions of these ligands were used for the syntheses of complexes 1 and 2, respectively.
:1 molar ratio in 15 mL of acetonitrile and refluxed during 1 hour to prepare the corresponding Schiff base ligands: 2-[1-[[2-[(2-aminoethyl)amino]ethyl]imino]ethyl]phenol (HL1) and 2-[1-[[2-[(2-aminoethyl)amino]ethyl]imino]ethyl]-4-methylphenol (HL2). The ligands, HL1 and HL2, were not isolated and the acetonitrile solutions of these ligands were used for the syntheses of complexes 1 and 2, respectively.[Fe(L1)(NCS)2] (1). An acetonitrile solution (10 mL) of iron(III) perchlorate hexahydrate (∼1 mmol, 0.360 g) was added to the previously prepared acetonitrile solution of the Schiff base ligand (HL1) under refluxing condition. An aqueous solution (15 mL) of sodium thiocyanate (2 mmol, 0.180 g) was then added to the resulting solution and refluxed further for ca. 30 min. The solution was then filtered and kept in open atmosphere at room temperature for 2 days. A dark crystalline product was collected by filtration. X-ray quality single crystals were collected from this crystalline product.
Yield: 0.282 g, ∼72% (based on iron). Anal. calc. for C14H18FeN5OS2 (F.W. 392.32 g mol−1): C, 42.86; H, 4.62; N, 17.85%. Found: C, 42.7; H, 4.5; N, 17.9%. FT-IR (KBr, cm−1): 3225 (νN−H); 2884–2955 (νC–H); 2075 (νNCS); 2042 (νNCS); 1580 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). λmax (nm) [εmax (M−1 cm−1)] (acetonitrile): 515 (2 × 103), 329 (5 × 103), 268 (1.3 × 104), 230 (2.4 × 104).
N). λmax (nm) [εmax (M−1 cm−1)] (acetonitrile): 515 (2 × 103), 329 (5 × 103), 268 (1.3 × 104), 230 (2.4 × 104).
[Fe(L2)(N3)Cl] (2). An acetonitrile solution (10 mL) of iron(III) chloride hexahydrate (1 mmol, 0.170 g) was added to the acetonitrile solution of the Schiff base ligand (HL2) under refluxing condition. An aqueous solution (5 mL) of sodium azide (1 mmol, 0.070 g) was then added to the resulting solution and refluxed further for ca. 30 min. The solution was then filtered and kept in open atmosphere at room temperature for 2 days. A dark crystalline product was collected by filtration. X-ray quality single crystals were collected from this crystalline product.
Yield: 0.261 g, ∼71% (based on iron). Anal. calc. for C13H20ClFeN6O (F.W. 367.65 g mol−1): C, 42.47; H, 5.48; N, 22.86%. Found: C, 42.3; H, 5.3; N, 22.9%. FT-IR (KBr, cm−1): 3225 (νN−H); 2893–2936 (νC–H); 2041 (νN3); 1578 (νCN). λmax (nm) [εmax (lit mol−1 cm−1)] (acetonitrile): 520 (2.5 × 103), 330 (5.4 × 103), 269 (1.3 × 104), 231 (2.5 × 104).
Physical measurements
Elemental analysis (C, H and N) was performed using a Perkin-Elmer 240C elemental analyser. IR spectrum in KBr (4500–500 cm−1) was recorded with a Perkin-Elmer Spectrum Two spectrophotometer. Electronic spectra in DMF were recorded on a Shimadzu UV-1700 spectrophotometer. Variable temperature magnetic susceptibility measurements were performed on polycrystalline samples of both complexes (with masses of 37.447 and 36.265 mg, for 1 and 2, respectively) with a Quantum Design MPMS-XL-5 SQUID susceptometer in the temperature range 2–400 K with an applied magnetic field of 0.1 T. The data were corrected for the sample holder and for the diamagnetic contribution of the salts using Pascal's constants (χdia = −197.3 × 10−6 and −204.7 × 10−6 cm3 mol−1 for 1 and 2, respectively).22X-ray crystallography
Suitable crystals of both complexes were used for data collection using a ‘Bruker D8 QUEST area detector’ diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Molecular structures were solved by direct methods and refined by full-matrix least squares on F2 using the SHELX-14 package.23a Non-hydrogen atoms were refined with anisotropic thermal parameters. The hydrogen atoms attached to nitrogen atoms were located by difference Fourier maps and were kept at fixed positions. All other hydrogen atoms were placed in their geometrically idealized positions and constrained to ride on their parent atoms. Multi-scan empirical absorption corrections were applied to the data using the program SADABS.23b In complex 2 there are two disordered chlorine atoms taking two close positions which were refined with occupation factors x and 1 − x, with x refining to 0.52(3). Details of crystal data and refinement are given in Table 1. Important bond lengths and bond angles are gathered in Tables 2 and S1 (ESI†) respectively.| Complex | 1 | 2 |
|---|---|---|
| Formula | C14H18FeN5OS2 | C13H20ClFeN6O |
| Formula weight | 392.32 | 367.65 |
| Temperature (K) | 273 | 273 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| a (Å) | 11.6962(10) | 11.143(3) |
| b (Å) | 12.7639(10) | 13.276(3) |
| c (Å) | 12.7562(10) | 11.981(3) |
| α (°) | 90 | 90 |
| β (°) | 112.592(2) | 108.225(6) |
| γ (°) | 90 | 90 |
| Z | 4 | 4 |
| dcal (g cm−3) | 1.482 | 1.451 |
| μ (mm−1) | 1.105 | 1.064 |
| F(000) | 812 | 764 |
| Total reflection | 24912 |
16196 |
| Unique reflections | 3891 | 2923 |
| Observe data [I > 2σ(I)] | 3167 | 2234 |
| R(int) | 0.036 | 0.074 |
| R1, wR2 (all data) | 0.0498, 0.0371 | 0.1011, 0.0787 |
| R1, wR2 [I > 2σ(I)] | 0.1144, 0.1053 | 0.2336, 0.2050 |
| Complex | 1 | 2 |
|---|---|---|
| Fe1–O1 | 1.8602(19) | 1.868(4) |
| Fe1–N1 | 2.164(3) | 2.177(7) |
| Fe1–N2 | 2.162(2) | 2.174(5) |
| Fe1–N3 | 2.143(2) | 2.162(5) |
| Fe1–N4 | 2.067(2) | 2.070(7) |
| Fe1–N5 | 2.036(3) | — |
| Fe1–Cl1 | — | 2.367(4) |
Theoretical methods
All calculations included in this study have been performed using the ORCA program, version 4.1.2.24a For the calculation of the magnetic coupling constants we have used the TPPSh functional24b combined with the def2-TZVP basis set.24c This level of theory has been recommended for the calculation of J values accurately.24d For the interaction energies, we have used the B3LYP/def2-TZVP level of theory. The B3LYP/def2-TZVP wave function obtained from the ORCA program was used to generate a spin density cube that was represented using the Gaussview software.24e The QTAIM/NCI plot representation has been performed at the same level using the ORCA wave function and the AIMAll program.24fResults and discussion
Synthesis
Schiff base ligands, HL1 and HL2, were prepared by 1:1 condensation of diethyltriamine with 2-hydroxy-acetophenone and 2-hydroxy-5-methylacetophenonein acetonitrile, respectively.25b Acetonitrile solution of both ligands, HL1 and HL2, were reacted with iron(III) perchlorate and iron(III) chloride hydrate under reflux. Around 2 mmol of sodium thiocyanate was used as coligand in complex 1 and 1 mmol of sodium azide was used for complex 2. Both complexes are stable at room temperature. The synthetic routes for both complexes have been shown in Scheme 1.
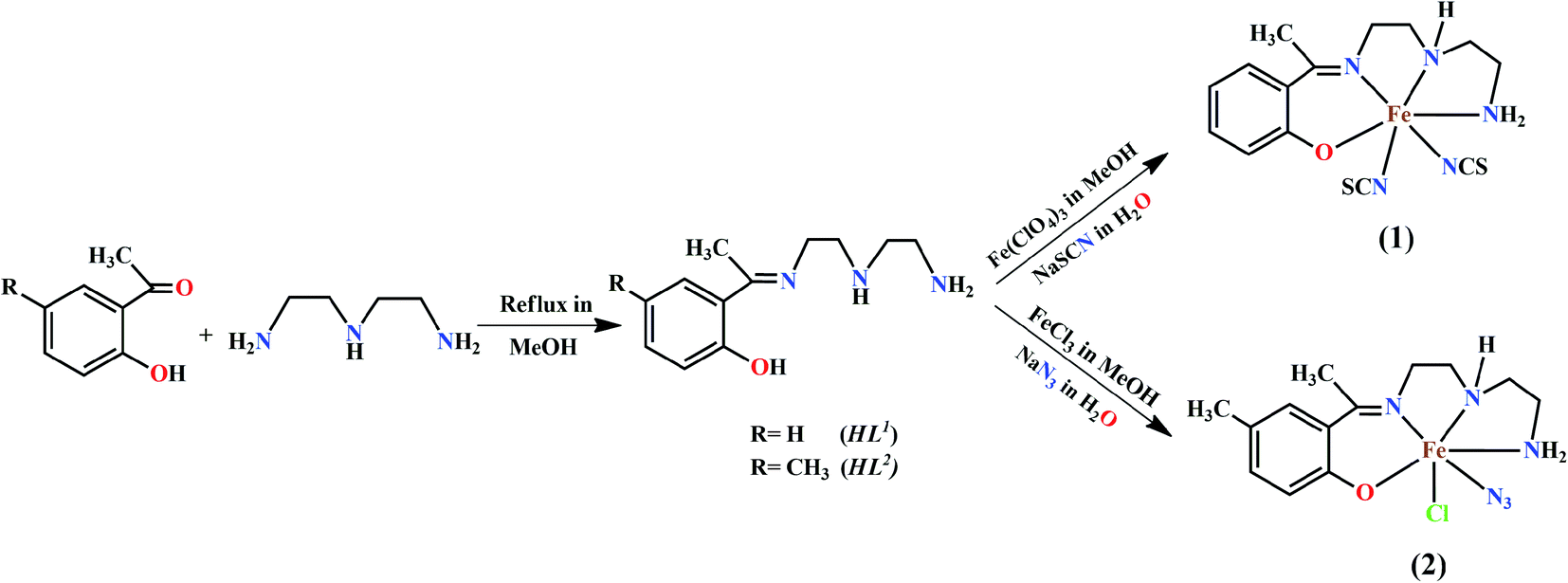 | ||
| Scheme 1 Synthetic route for ligand and complexes 1 and 2. | ||
Crystal structure of [Fe(L1)(NCS)2] (1)
Complex 1 crystallizes in the monoclinic space group P21/n. The asymmetric unit consists of a discreet mononuclear unit [Fe(L1)(NCS)2]. A perspective view of this complex with selected atom-numbering scheme is shown in Fig. 1. The iron(III) center, Fe1, is octahedrally coordinated by a deprotonated Schiff base ligand and two terminal thiocyanate group. The equatorial plane consists of two amine nitrogen atoms, N1 and N2, one phenoxido oxygen atom, O1 of a deprotonated Schiff base ligand (L1)− and one nitrogen atom, N4 of a terminal thiocyanate group. The axial positions are occupied by one imine nitrogen atom, N3 of the deprotonated Schiff base and one nitrogen atom, N5 of another terminal thiocyanate group. The deviations of the coordinating atoms N1, N2, N4 and O1 from the least-square mean plane through them are −0.175(3) Å, 0.198(2) Å, −0.152(2) Å and −0.184(16) Å, respectively, and that of Fe1 from the same plane is −0.055(4) Å. The trans angles, N1–Fe1–N4, N2–Fe1–O1 and N3–Fe1–N5, around Fe1 are 165.1(1)°, 163.15(9)° and 175.36(9)°, respectively (Table S1, ESI†). Both five-membered chelate rings [Fe1–N1–C1–C2–N2] and [Fe1–N2–C3–C4–N3] have half-chair conformation, with puckering parameters,26 q = 0.489(3) Å, φ = 66.1(3)° and q = 0.465(3) Å, φ = 53.4(3)°, respectively.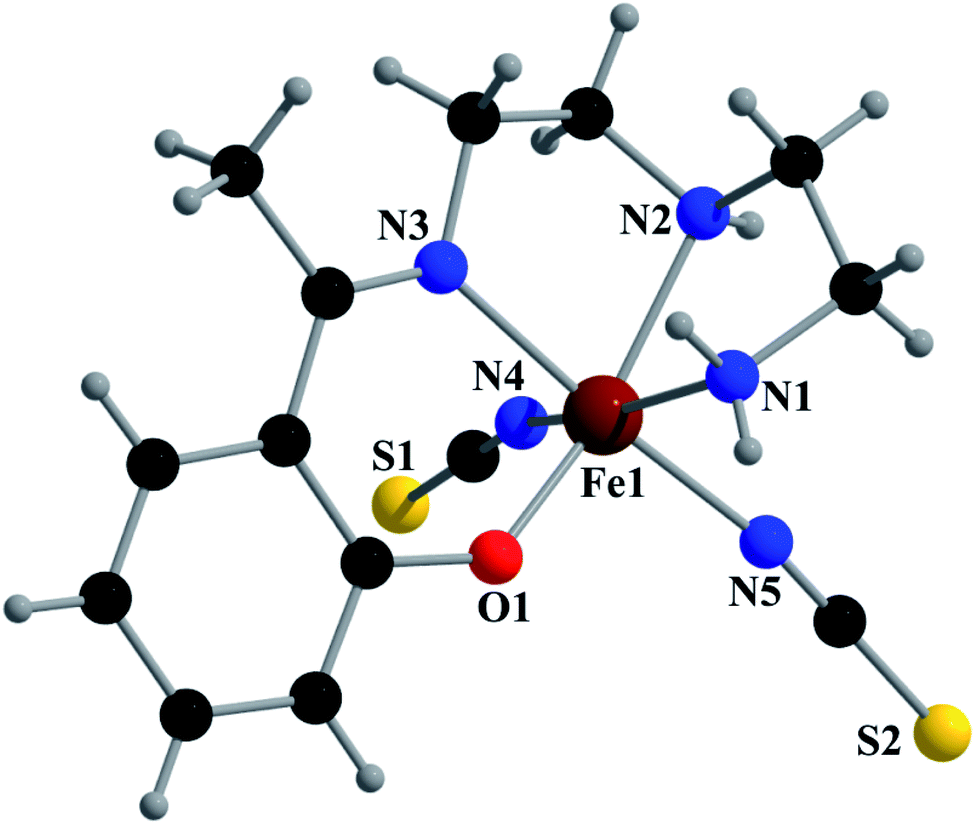 | ||
| Fig. 1 Perspective view of complex 1 with selected numbering scheme. | ||
Complex 1 presents some interesting non-covalent interactions that give rise to a supramolecular network. Two types of non-covalent interactions have been observed: (i) hydrogen bonding and (ii) C–H⋯π interactions. There are two unconventional hydrogen bonding interactions in 1. On one side, the hydrogen atom H1N, attached to N1, is involved in intermolecular hydrogen bonding with the sulphur atom S2. On the other side, the hydrogen atom H2N, attached to N2, is involved in intermolecular hydrogen bonding with the sulphur atom S1. These inter-molecular hydrogen bonding interactions originate a one-dimensional array as shown in Fig. 2. The second type of interaction ((C–H⋯π) is formed between the hydrogen atom H9, attached to C3, and the symmetry related (1/2 + x, 3/2−y, 1/2 + z) chelate ring, Cg4 [C7–C8–C9–C10–C11–C12] and between the atom H14, attached to C6, and the symmetry related (1 − x, 1 − y, −z) chelate ring, Cg4 [C7–C8–C9–C10–C11–C12]. These intermolecular C–H⋯π interactions give rise to layers as shown in Fig. 3. Details of the geometric features of the C–H⋯π interactions are given in Table 3. Details of H-bonding interactions are given in Table 4.
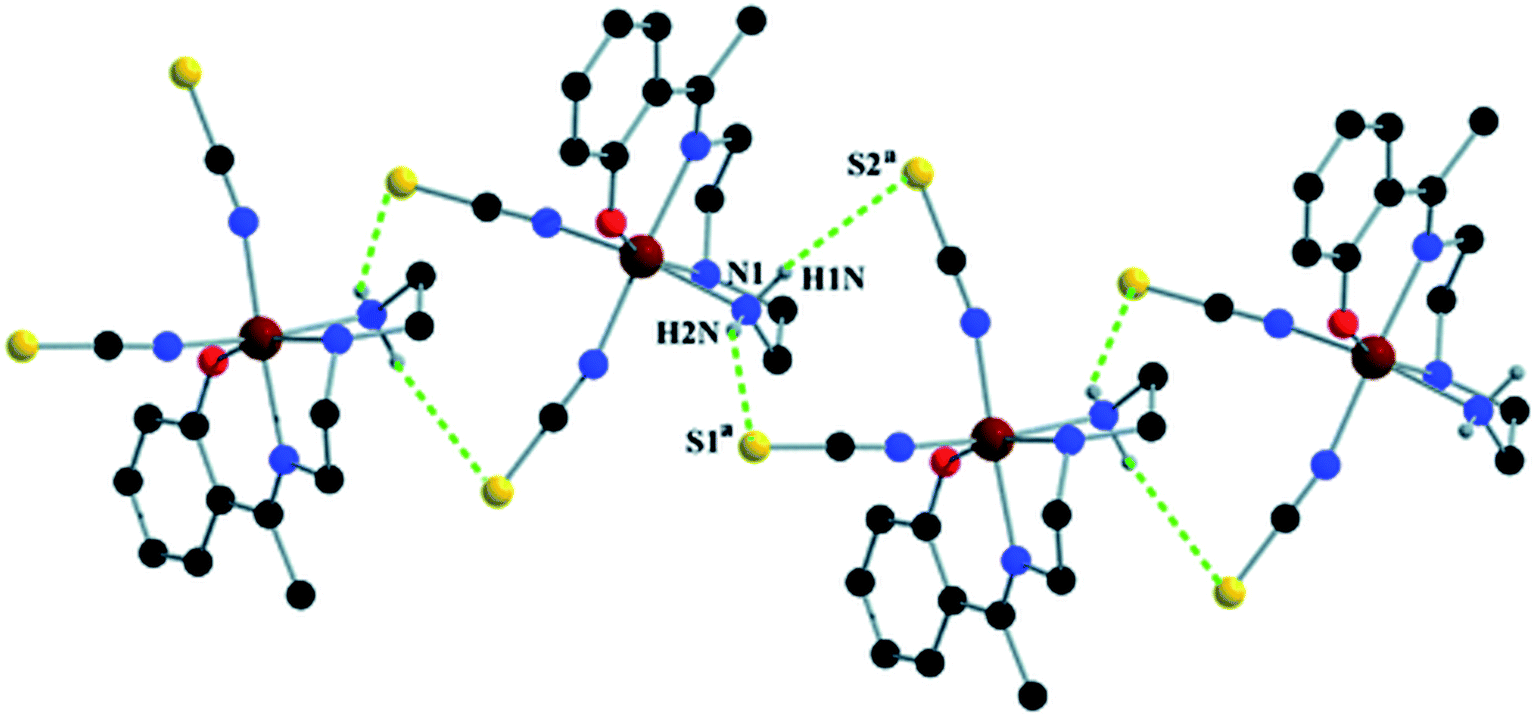 | ||
| Fig. 2 1D supramolecular array in 1 formed by N–H⋯S hydrogen bonding interactions. Symmetry transformations: a = 1/2 − x, 1/2 + y, 3/2 − z. | ||
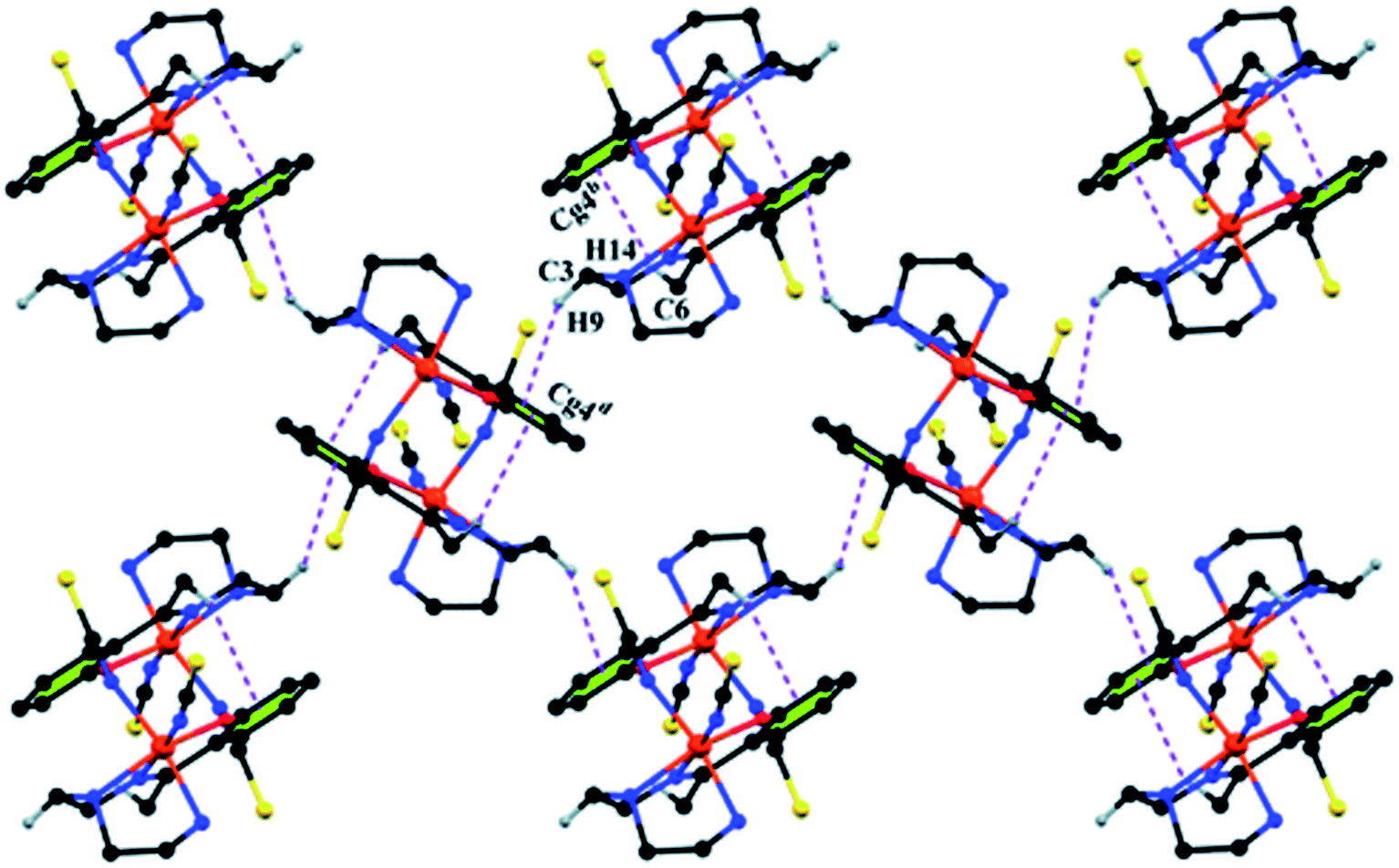 | ||
| Fig. 3 Supramolecular 2D structure in 1 formed by intermolecular C–H⋯π interactions. Symmetry transformations: b = 1 − x, 1 − y, − z; d = 1/2 + x, 3/2 − y, 1/2 + z. | ||
| Complex | D–H⋯A | D–H | H⋯A | D⋯A | ∠D–H⋯A |
|---|---|---|---|---|---|
| a D = donor; H = hydrogen; A = acceptor. Symmetry transformations: a = 1/2 − x,1/2 + y,3/2 − z; b = 1 − x,1 − y,−z and c = 3/2 − x,−1/2 + y,1/2 − z. | |||||
| 1 | N(1)–H(1N)–S(2)a | 0.96(3) | 2.85(3) | 3.787(3) | 165(3) |
| N(1)–H(2N)–S(1)a | 0.76(4) | 2.87(4) | 3.551(4) | 151(4) | |
| 2 | N(2)–H(2)–Cl(1)b | 0.9800 | 2.48(5) | 3.391(5) | 154(1) |
| N(1)–H(2N)–N(6)c | 0.91(8) | 2.21(8) | 3.062(9) | 155(7) | |
| C–H⋯Cg(ring) | H⋯Cg (Å) | C–H⋯Cg (°) | C⋯Cg (Å) |
|---|---|---|---|
| a Symmetry transformations: d = 1/2 + x,3/2 − y,1/2 + z; b = 1 − x,1 − y,−z; Cg(4) = centre of gravity of the ring [C7–C8–C9–C10–C11–C12]. | |||
| C3–H9⋯Cg(4)d | 3.00 | 138 | 3.773(3) |
| C6–H14⋯Cg(4)b | 2.98 | 143 | 3.787(4) |
Crystal structure of [Fe(L2)(N3)Cl] (2)
Complex 2 crystallizes in the monoclinic space group P21/n. The asymmetric unit consists of a discrete mononuclear unit [Fe(L2)(N3)Cl]. A perspective view of this complex with selected atom-numbering scheme is shown in Fig. 4.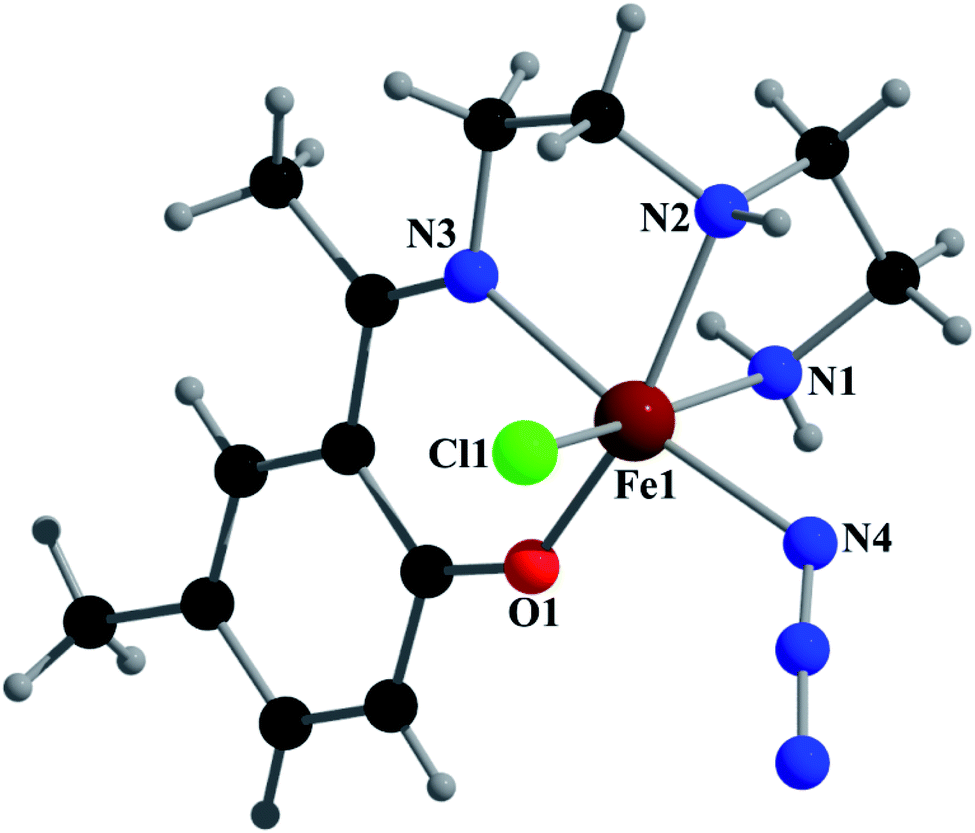 | ||
| Fig. 4 Perspective view of complex 2 with selected atom numbering scheme. | ||
The iron(III) center, Fe1, is octahedrally coordinated by a deprotonated Schiff base ligand, a Cl atom and a terminal azide ligand. The equatorial plane contains two amine nitrogen atoms, N1 and N2, one phenoxido oxygen atom, O1, of a deprotonated Schiff base ligand (L2)− and one chlorine atom, Cl1. The axial positions are occupied by one imine nitrogen atom, N3, of the deprotonated Schiff base ligand (L2)− and by one nitrogen atom, N4, of a terminal azide group. The deviations of the coordinated atoms N1, N2, O1 and Cl1 from the least-square mean plane through them are 0.216(8) Å, −0.225(5) Å, −0.217(5) Å and 0.170(3) Å, respectively. Fe1 deviation from the same plane is −0.055(9) Å. The trans angles N1–Fe1–Cl1, N2–Fe1–O1 and N3–Fe1–N4, around Fe1 are 167.3(2)°, 162.3(2)° and 172.5(2)°, respectively (Table S1†). Both saturated five-membered chelate rings [Fe1–N1–C1–C2–N2] and [Fe1–N2–C3–C4–N3] have envelop conformation, with puckering parameters,26 q = 0.485(8) Å, φ = 245.1(8)° and q = 0.471(6) Å, φ = 227.1(7)°, respectively.
Complex 2 shows two types of hydrogen bonds: on one side, hydrogen atom H2, attached to N1, is involved in intermolecular hydrogen bonding with Cl1 and on the other side, hydrogen atom H2N, attached to N2, is involved in intermolecular hydrogen bonding with N6. These inter-molecular hydrogen bonding interactions give rise to a two-dimensional structure as shown in Fig. 5. Details of the geometric features of these hydrogen bonding interactions are given in Table 4.
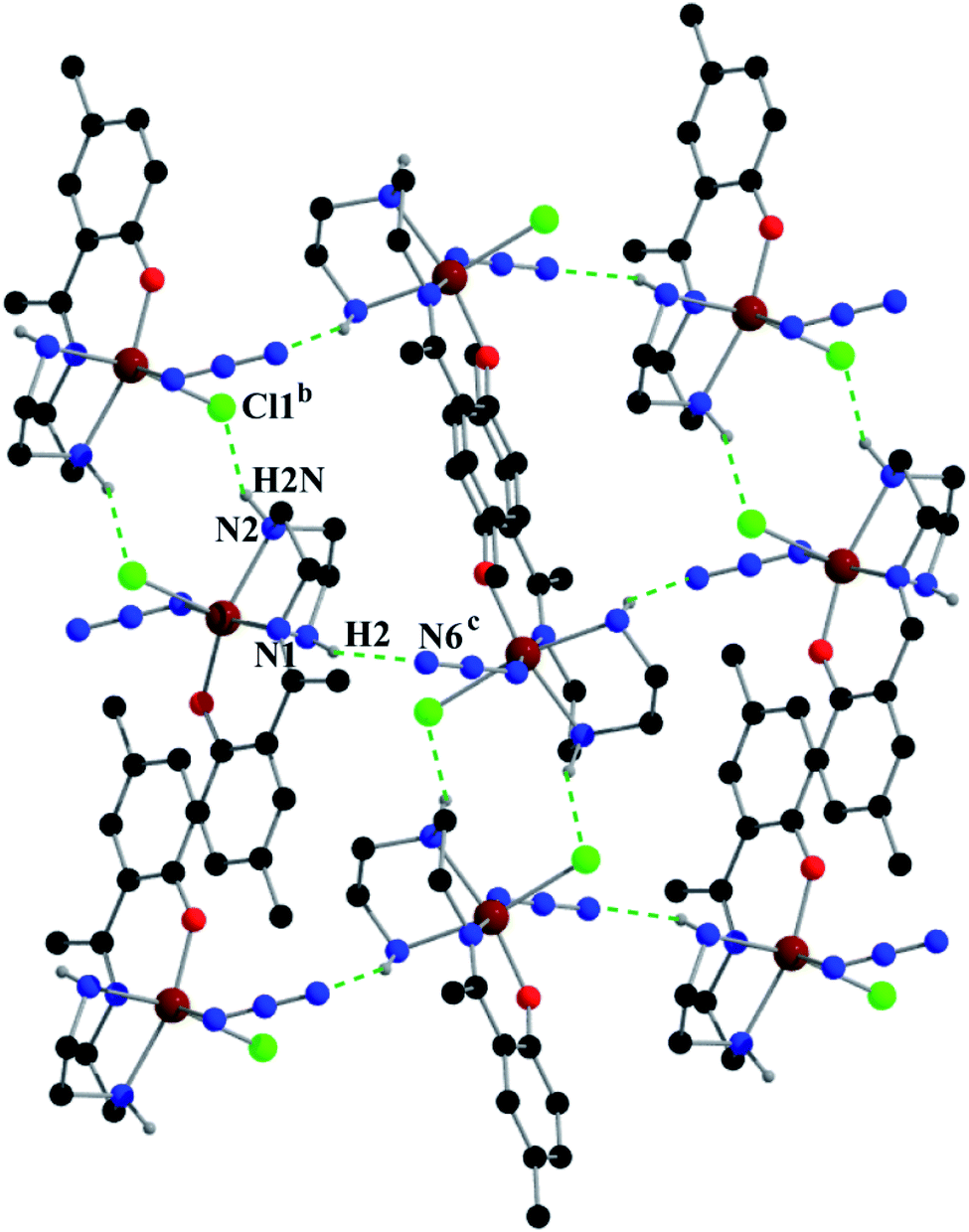 | ||
| Fig. 5 Supramolecular 2D structure in 2 formed by intermolecular hydrogen bonding interactions. Symmetry transformations: b = 1 − x,1 − y,−z; c = 3/2 − x,−1/2 + y,1/2 − z. | ||
Magnetic properties
The thermal variation of the product of the molar magnetic susceptibility times the temperature (χmT) per Fe(III) ions in both complexes is, as expected, very similar. They show room temperature values of ca. 4.4 cm3 K mol−1 (μeff ≈ 5.93 μB), very close to the spin only value for a S = 5/2 Fe(III) ion. When the temperature decreases, χmT remains constant in the range down to ca. 30 K in complex 1 and ca. 80 K in complex 2 (Fig. 6). Below these temperatures χmT shows a progressive decrease to reach values of ca. 3.1 and 1.2 cm3 K mol−1 in 1 and 2, respectively. This behaviour indicates that both complexes behave essentially as paramagnets with a weak antiferromagnetic intermolecular interaction and/or a zero Field Splitting (ZFS) of the S = 5/2 spin ground state. Accordingly, we have fit the magnetic properties with a S = 5/2 monomer including a ZFS and an inter-molecular exchange interaction (zJ) using the PHI software.27 This model reproduces very satisfactorily the magnetic properties of both complexes in the whole temperature range with g = 2.043(6), |D| = 1.23(3) cm−1 and zJ = −0.03(1) cm−1 in 1 and g = 2.126(2), |D| = 1.6(1) cm−1 and zJ = −0.23(1) cm−1 in 2 (solid lines in Fig. 6). Note that the sign of D cannot be determined with powder magnetic measurements.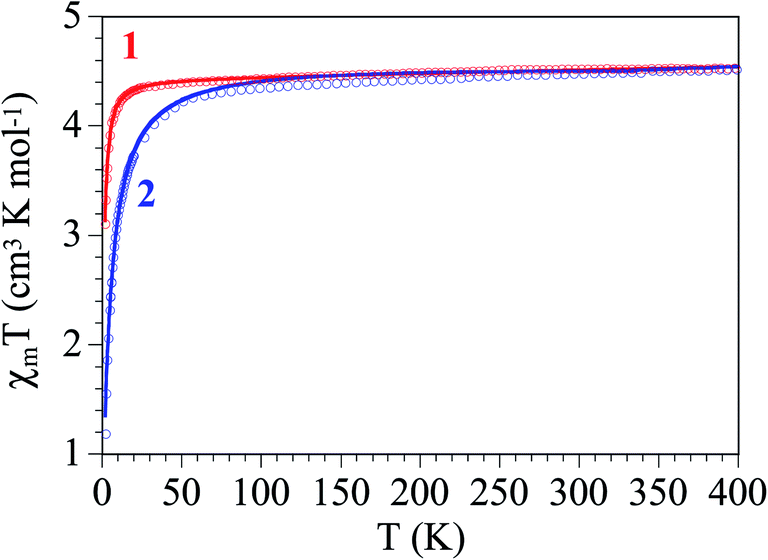 | ||
| Fig. 6 Thermal variation of χmT for complexes 1 and 2. Solid lines are the best fit to the model (see text). | ||
As expected, both complexes show very weak antiferromagnetic inter-molecular exchange interactions. The larger absolute value found in complex 2 agrees with the most important decrease observed in χmT in this complex and can be easily explained by the stronger H-bonds observed in complex 2, with much shorter D⋯A distances (3.062 and 3.391 Å in 2 compared to 3.551 and 3.787 Å in 1) and also shorter H⋯A distances (2.21 and 2.48 Å in 2 compared to 2.85 and 2.87 Å in 1, Table 4). On the other side, the D values are within the normal range observed for other high spin S = 5/2 Fe(III) monomers.28
Note that, although there are a few examples of ferromagnetic interactions and even long range ferro- and antiferromagnetic ordering, in most cases the magnetic interactions through hydrogen bonds are weak and antiferromagnetic (Table 5). Theoretical calculations performed by several groups also confirm this experimental observation.29 Interestingly, most of the reported complexes30–53 showing magnetic interactions through H-bonds are Cu(II) complexes with Cu–O–H⋯O–Cu pathways (Table 5). As can be seen in Table 5, there are very few examples containing other metals as Ni(II), Mn(II), Mn(III) and Fe(III). Therefore, complexes 1 and 2 are expected to show very weak antiferromagnetic interactions through the H-bonds and C–H–π interactions, in agreement with the experimental data.
| Complex | CSD code/CCDC no. | J (cm−1) | M | H-Bond type | D–H⋯A (Å) | Ref. |
|---|---|---|---|---|---|---|
| a A zero field splitting of the Fe(III) ion is included in the fit, resulting in reduced J value.b La–w = ligand names have been given in Table S2 (ESI). | ||||||
| [Cu2(La)2] | — | ≈−100 | Cu(II) | O–H⋯O | 2.29 | 30 |
| [Cu2(La)2] | HEAICU10 | −94 | Cu(II) | O–H⋯O (x2) | 2.31 | 31 |
| O–H⋯O (x2) | 2.33 | |||||
| [Cu3(Lb)2(C6H5COO)2Cl]Cl | DEPSAT | −0.4(2) | Cu(II) | N–H⋯Cl | 3.163 | 32 |
| [Cu2(μ2-H2O)(Lc)2(H2O)2](ClO4)2·2H2O | KEDNIR | −13.23(1) | Cu(II) | O–H⋯O (x2) | 2.627 | 33 |
| [Cu2(Ld)2(H2O)2(ClO4)](ClO4)·H2O | FUTCON | −19.8(2) | Cu(II) | O–H⋯O | 2.685 | 34 |
| O–H⋯O | 2.601 | |||||
| [Cu(Le)2(H2O)] | BEYRAY | +0.23(1) | Cu(II) | O–H⋯O (x2) | 2.695 | 35 |
| [ZnII(H2O)6][CuII(Lf)2(H2O)2] | HULMOQ | −0.220(1) | Cu(II) | O–H⋯O (x2) | 2.771 | 36 |
| [Cu2(Lg)2(H2O)2]·2H2O | MATLOJ | −1.6(1) | Cu(II) | O–H⋯O (x2) | 2.721 | 37 |
| [Cu(Lh)(H2O)(NO3)] | NUQKOZ01 | −26.6 | Cu(II) | O–H⋯O (x2) | 2.685 | 38 |
| [Cu(Li)2(H2O)2]n | FAHNAE | −3.26 | Cu(II) | O–H⋯O (x2) | 2.71 | 39 |
| [Cu(Lj)(H2O)]·4H2O | SAGLAC | −2.19(2) | Cu(II) | O–H⋯O (x2) | 2.757 | 40 |
| [NiCl2(Lk)2] | FUJQOQ | −1.72(1) | Ni(II) | N–H⋯Cl (x2) | 3.256 | 41 |
| −0.22(1) | N–H⋯Cl | 3.275 | ||||
| [Cu(HLl)(Ll)(H2O)]2NO3 | AETCUB | −70 | Cu(II) | O–H⋯O (x2) | 2.516 | 42 |
| [Cu(HLm)(Lm)]2(NO3)2 | AETCUA | −56 | Cu(II) | O–H⋯O (x2) | 2.452 | 42 |
| O–H⋯O (x2) | 2.434 | |||||
| [{Cu(H2Ln)}{Cu(Ln)}]BF4 | ODALAG | −7 | Cu(II) | O–H⋯O (x2) | 2.44 | 43 |
| O–H⋯O (x2) | 2.66 | |||||
| [{Cu(H2Ln)}2](BF4)2 | ODALEK | −21 | Cu(II) | O–H⋯O (x2) | 2.60 | 43 |
| [Cu(HLo)(Lo)]PF6 | MASQIJ | −9.91(2) | Cu(II) | O–H⋯O | 2.412 | 44 |
| [Cu(HLp)(Lp)]BF4·2H2O | YUKCOX | −23.1 | Cu(II) | O–H⋯O | 2.425 | 45 |
| [Cu(Lq)2(Lr)(H2O)2] | BUQLIJ | −6.25 | Cu(II) | O–H⋯O (x2) | 2.692 | 46 |
| cis-[Cu(Ls)2(H2O)2] | NEDPAO | −3.0 | Cu(II) | O–H⋯O (x2) | 2.767 | 47 |
| O–H⋯O (x2) | 2.713 | |||||
| trans-[Cu(Ls)2(H2O)2]·H2O | PAXTUE | −4.0 | Cu(II) | O–H⋯O (x2) | 2.651 | 47 |
| [Ni3(Lt)(CO3)(H2O)(py)7] | GIDNAK | −39.6 | Ni(II) | O–H⋯O (x2) | 2.71 | 48 |
| 2.73 | ||||||
| [{Mn(bpy)(H2O)}(Lu)2(μ-O){Mn(bpy)(ClO4)}]ClO4 | AGOJOY | −9.2 | Mn(II) | O–H⋯O | 2.074 | 49 |
| 2.797 | ||||||
| [{Mn(bpy)(H2O)}(Lu)2(μ-O){Mn(bpy)(NO3)}]NO3 | AGOJUE | −27.3 | Mn(II) | O–H⋯O | 2.746 | 49 |
| 2.843 | ||||||
| [Fe(Lv)Cl(H2O)]·MeOH | AZOXAO | TN = 3.2 K | Fe(III) | O–H⋯O | 2.582 | 50 |
| O–H⋯O | 2.610 | |||||
| O–H⋯O | 2.827 | |||||
| N–H⋯O | 2.901 | |||||
| [(Ni(Lw)2)3(Fe(CN)6)2]·7H2O | ROQCAB | TC = 23 K | FeIII3NiII2 | O–H⋯N (x4) | 2.76–3.09 | 51 |
| O–H⋯O (x4) | 2.78–3.08 | |||||
| {[Mn(OH)(OAc)2]·AcOH·H2O}n | HUWHOW | TN = 6.1 K | Mn(III) | O–H⋯O | 2.550 | 52 |
| O–H⋯O | 2.691 | 53 | ||||
| [FeL1(NCS)2] | 2036380 | −0.03(1)a | Fe(III) | N–H⋯S (x2) | 3.551 | This work |
| 3.787 | ||||||
| [FeL2(N3)Cl] | 2036381 | −0.23(1)a | Fe(III) | N–H⋯Cl | 3.391 | This work |
| N–H⋯N | 3.062 | |||||
In order to verify this explanation (stronger H-bonded dimer in 2) we have performed a DFT study using the RIJCOSX-B3LYP/def2-TZVP level of theory. Fig. 7 shows the combined QTAIM/NCI Plot analysis of the H-bonded dimers commented above (see Fig. 2 and 5 for details) and the dimerization energies. In the dimer of 1, the QTAIM shows an intricate combination of bond critical points and bond paths connecting the monomers. That is, in addition to the N–H⋯S H-bonds (marked with asterisk symbols in Fig. 7a), the combined QTAIM/NCI plot analysis also reveals the existence of several C–H⋯S and C–H⋯N H-bonds (marked with arrows). All these H-bonds are characterized by a bond critical point (CP) and bond path connecting the H-atom to the electron rich atom (S, N). These interactions are further characterized by green NCI plot isosurfaces located between the interacting atoms. The green colour is indicative of weak interaction,54 which is further validated by the modest dimerization energy (ΔE1 = −8.3 kcal mol−1) obtained for 1, taking into consideration the number of H-bonds that are established between the monomers. The QTAIM distribution of bond CPs and bond paths in complex 2 (see Fig. 7b) shows two symmetrically related bond CPs and bond paths that connect the N–H groups to the chlorido ligands. Moreover, two additional and symmetrically related bond CPs and bond path connect two C–H bonds two the N-atoms of the azido ligands. The N–H⋯Cl bond are also characterized by bluish isosurfaces, indicated moderately strong H-bonds. Consequently, the H-bonded dimer of complex 2 presents a stronger interaction (ΔE2 = −25.6 kcal mol−1) compared to 1, in agreement with the stronger antiferromagnetic coupling constant observed experimentally.
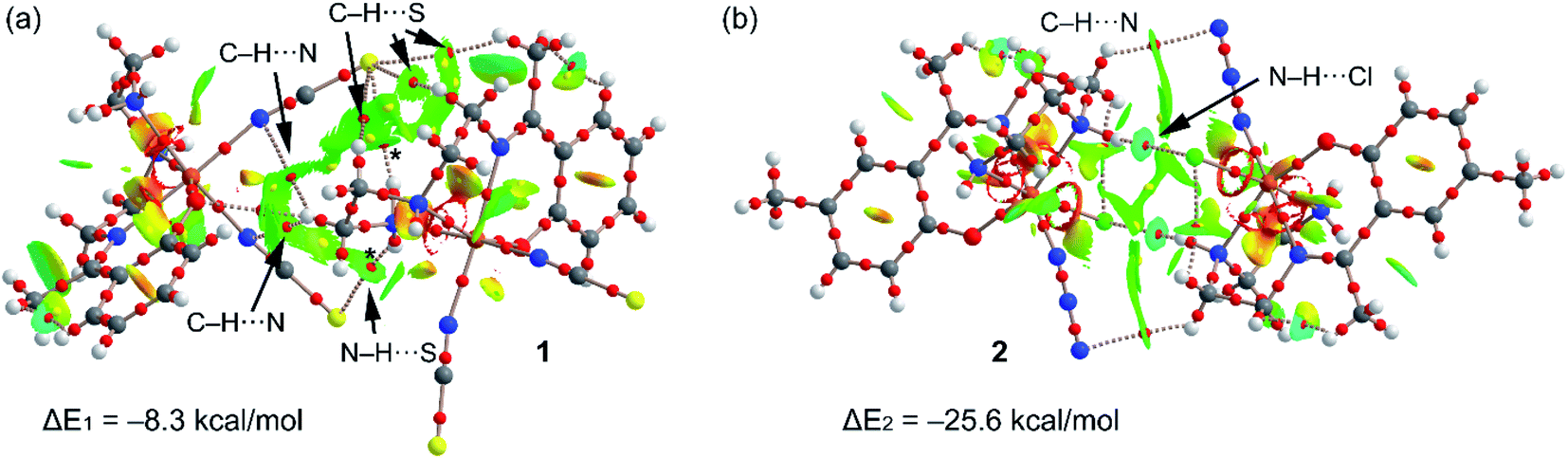 | ||
| Fig. 7 Combined QTAIM (bond CPs in red and ring CP in yellow) and NCIPlot analyses of the H-bonded dimers of 1 (a) and 2 (b) at the RIJCOSX-B3LYP/def2-TZVP level of theory. The dimerization energies are also indicated. | ||
We have also computed the theoretical zJ value using the broken symmetry (BS) approach and the equation proposed by Ruiz and coworkers (J = −(EHS − EBS)/(Smax(Smax + 1))), where the energies of the high spin (HS) and low spin (BS) states are used,55 as implemented in the ORCA package. In case of 1, the experimental J value is too small to be reliably reproduced by the DFT calculations (−0.03 cm−1 is within the accuracy of the DFT method). Therefore, we have focused the DFT study on the calculation of complex 2. Interestingly, the calculation predicts a zJ value of −0.18 cm−1, which is in reasonable agreement with the experimental value (−0.23 cm−1) and confirms the very weak antiferromagnetic coupling of the Fe metal centers in the H-bonded dimer of 2. However, the |D| value is somewhat underestimated by the DFT calculation (0.53 cm−1). The spin density plot of the high spin configuration of the dimer of 2 is represented in Fig. 8. The observed spherical distribution of the spin density around the Fe atoms is typical of a d5 configuration with a single occupation of each d orbital. It can be also observed that some spin is delocalized onto the atoms of the ligands directly bonded to the metal. It is also worth mentioning that some of the delocalized spin density is located to the heteroatoms involved in the strong N–H⋯Cl hydrogen bonds.
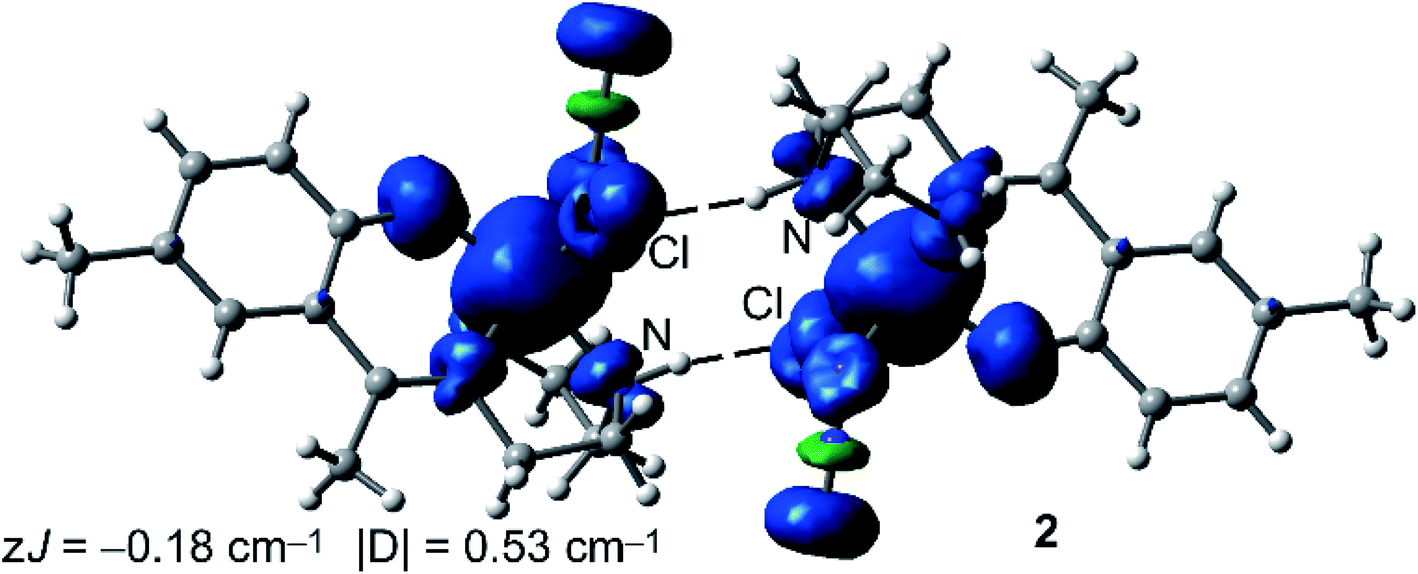 | ||
| Fig. 8 Spin density plot (isosurface = 0.004 a.u.) of the high spin configuration of the H-bonded dimer of 2 at the RIJK-TPSSh/def2-TZVP level of theory. | ||
Conclusion
We have shown a convenient way to prepare two high spin Fe(III) complexes with two different Schiff base ligands and different pseudohalides as counterions. The magnetic properties of both complexes can be very well reproduced with a weak inter-molecular antiferromagnetic interaction (mediated through the inter-molecular H-bonds) and a zero field splitting of the high spin S = 5/2 ground state. The DFT study shows that H-bonded dimer of complex 2 is significantly stronger than 1, in agreement with its larger antiferromagnetic coupling zJ value. The antiferromagnetic coupling in 2 has been confirmed by DFT calculations and rationalized by the spin density plot.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. B. thanks the CSIR, India, for awarding a Senior Research Fellowship [Sanction No. 09/096(0861)/2016-EMR-I]. S. C. gratefully acknowledges UGC-CAS II program, Department of Chemistry, Jadavpur University, for financial support under the head [Chemicals/Consumables/Glassware]. We also thank the Spanish MINECO (project CTQ2017-87201-P AEI/FEDER, UE) and the Generalidad Valenciana (Prometeo/2019/076) for financial support.References
- Top. Curr. Chem., ed. P. Gütlich and H. A. Goodwin, 2004, pp. 233–235 Search PubMed.
- S. Jana, A. Bhattacharyya, B. N. Ghosh, K. Rissanen, S. Herrero, R. Jiménez-Aparicio and S. Chattopadhyay, Inorg. Chim. Acta, 2013, 453, 715–723 CrossRef.
- H. A. Goodwin, Coord. Chem. Rev., 1976, 18, 293–325 CrossRef CAS.
- E. König, Prog. Inorg. Chem., 1987, 35, 527–622 Search PubMed.
- P. Gütlich, Y. Garcia and T. Woike, Coord. Chem. Rev., 2001, 219–221, 839–879 CrossRef.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed., 1994, 33, 2024–2054 CrossRef.
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC.
- A. H. Ewald, R. L. Martin, I. G. Ross and A. H. White, Proc. R. Soc. London, Ser. A, 1984, 280, 235–257 Search PubMed.
- G. Harris, Theor. Chim. Acta, 1966, 5, 379–397 CrossRef CAS.
- M. Zerner, M. Gouterman and H. Kobayashi, Theor. Chim. Acta, 1966, 6, 363–400 CrossRef CAS.
- T. Basak, K. Ghosh, C. J. Gómez-García and S. Chattopadhyay, Polyhedron, 2018, 146, 42–54 CrossRef CAS.
- S. Jana, S. Chatterjee and S. Chattopadhyay, Inorg. Chim. Acta, 2012, 48, 189–198 CAS.
- K. Santo, M. Hirotsu and I. Kinoshita, Dalton Trans., 2015, 44, 4155–4166 RSC.
- T. Basak, D. Das, P. P. Ray, S. Banerjee and S. Chattopadhyay, CrystEngComm, 2020, 22, 5170–5181 RSC.
- T. Basak, A. Frontera and S. Chattopadhyay, Inorg. Chim. Acta, 2021, 516, 120081 CrossRef CAS.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- T. Basak, A. Frontera and S. Chattopadhyay, Dalton Trans., 2020, 22, 5731 CAS.
- T. Basak, K. Ghosh and S. Chattopadhyay, Polyhedron, 2018, 146, 81–92 CrossRef CAS.
- M. Orio, O. Jarjayes, H. Kanso, C. Philouze, F. Neese and F. Thomas, Angew. Chem., Int. Ed., 2010, 49, 4989–4992 CrossRef CAS.
- C. Faulmann, K. Jacob, S. Dorbes, S. Lampert and I. Malfant, Inorg. Chem., 2007, 46, 8548–8559 CrossRef CAS.
- C. Faulmann, J. Chahine, L. Valade, G. Chastanet, J.-F. Létard and D. de Caro, Eur. J. Inorg. Chem., 2013, 1058–1067 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS; (b) G. M. Sheldrick, SADABS, V2014/5, Software for Empirical Absorption Correction, University of Göttingen, Institute fur Anorganische Chemie der Universitat GottingenGermany, 1999–2003 Search PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed; (b) V. N. Staroverova and G. E. Scuseria, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef; (c) A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef; (d) M. Orio, D. A. Pantazis, T. Petrenko and F. Neese, Inorg. Chem., 2009, 48(15), 7251–7260 CrossRef CAS; (e) R. Dennington, T. Keith and J. Millam, GaussView, Version 6.1.1, Semichem Inc., Shawnee Mission, KS, 2019 Search PubMed; (f) T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019, http://aim.tkgristmill.com Search PubMed.
- (a) P. Masarova, P. Zoufaly, J. Moncol, I. Nemec, J. Pavlik, M. Gembicky, Z. Travnicek, R. Boca and I. Salitros, New J. Chem., 2015, 39, 508–519 RSC; (b) T. Basak, D. Das, P.P Ray, S. Banerjee and S. Chattopadhyay, CrystEngComm, 2020, 22, 5170–5181 RSC; (c) T. Basak, A. Frontera and S. Chattopadhyay, CrystEngComm, 2020, 22, 5731–5742 RSC.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS.
- R. Boca, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef CAS.
- (a) N. A. G. Bandeira and B. Le Guennic, J. Phys. Chem. A, 2012, 116, 3465–3473 CrossRef CAS; (b) C. Desplanches, E. Ruiz, A. Rodríguez-Fortea and S. Alvarez, Exchange Coupling of Transition-Metal Ions through Hydrogen Bonding: A Theoretical Investigation, J. Am. Chem. Soc., 2002, 124, 5197–5205 CrossRef CAS; (c) M. Perić, M. Zlatar, S. Grubišić and M. Gruden-Pavlović, Polyhedron, 2012, 42, 89–94 CrossRef; (d) J. Tercero, C. Diaz, J. Ribas, E. Ruiz, J. Mahía and M. Maestro, Inorg. Chem., 2002, 41, 6780–6789 CrossRef CAS.
- J. A. Bertrand and F. T. Helm, J. Am. Chem. Soc., 1973, 95, 8184–8185 CrossRef CAS.
- J. A. Bertrand, T. D. Black, P. Eller, P. Gary and F. T. Helm, Inorg. Chem., 1976, 15, 2965–2970 CrossRef CAS.
- S. Sarkar, A. Mondal, D. Chopra, J. Ribas and K. K. A. Rajak, Eur. J. Inorg. Chem., 2006, 3510–3516 CrossRef CAS.
- P. Talukder, S. Sen, S. Mitra, L. Dahlenberg, C. Desplanches and J. P. Sutter, Eur. J. Inorg. Chem., 2006, 329–333 CrossRef CAS.
- J. Tang, J. S. Costa, A. Golobic, B. Kozlevcar, A. Robertazzi, A. V. Vargiu, P. Gamez and J. Reedijk, Inorg. Chem., 2009, 48, 5473–5479 CrossRef CAS.
- C. L. Klein, R. J. Majeste, L. M. Trefonas and C. J. Oconnor, Inorg. Chem., 1982, 21, 1891–1897 CrossRef CAS.
- Y. Rodriguez-Martin, J. Sanchiz, C. Ruiz-Perez, F. Lloret and M. Julve, CrystEngComm, 2002, 631–637 RSC.
- H. Núñez, L. Soto, J. J. Server-Carrió, J. García-Lozano, A. Sancho, R. Acerete and E. Escrivá, Inorg. Chem., 2005, 44, 4644–4655 CrossRef.
- P. Baran, R. Boca, M. Breza, H. Elias, H. Fuess, V. Jorik, R. Klement and I. Svoboda, Polyhedron, 2002, 21, 1561–1571 CrossRef CAS.
- S. Salunke-Gawali, S. Y. Rane, V. G. Puranik, C. Guyard-Duhayon and F. Varret, Polyhedron, 2004, 23, 2541–2547 CrossRef CAS.
- W. Estes and W. E. Hatfield, Inorg. Chem., 1978, 17, 3226–3231 CrossRef CAS.
- R. D. Willett, C. J. Gómez-García, B. Twamley, S. Gomez-Coca and E. Ruiz, Inorg. Chem., 2012, 51, 5487–5493 CrossRef CAS.
- J. A. Bertrand, E. Fujita and D. G. VanDerveer, Inorg. Chem., 1980, 19, 2022–2028 CrossRef CAS.
- W. Plass, A. Pohlmann and J. Rautengarten, Angew. Chem., Int. Ed., 2001, 40, 4207–4210 CrossRef CAS.
- T. Hamaguchi, T. Nagata, S. Hayami, S. Kawata and I. Ando, Dalton Trans., 2017, 46, 6196–6201 RSC.
- A. Okazawa and T. Ishida, Chem. Phys. Lett., 2009, 480, 198–202 CrossRef CAS.
- Z. Vasková, J. Moncol, M. Korabik, D. Valigura, J. Švorec, T. Lis, M. Valko and M. Melník, Polyhedron, 2010, 29, 154–163 CrossRef.
- B. Kozlevčar, N. Kitanovski, Z. Jagličić, N. A. G. Bandeira, V. Robert, B. Le Guennic and P. Gamez, Inorg. Chem., 2012, 51, 3094–3102 CrossRef.
- G. A. Craig, O. Roubeau, J. Ribas-Ariño, S. J. Teat and G. Aromí, Polyhedron, 2013, 52, 1369–1374 CrossRef CAS.
- J. M. Pages, L. Escriche-Tur, M. Font-Bardia, G. Aullon and M. Corbella, CrystEngComm, 2018, 20, 6629–6639 RSC.
- R. J. Staples, W. M. Reiff and E. V. Rybak-Akimova, Inorg. Chem., 2004, 43, 3930–3941 CrossRef.
- K. Van Langenberg, S. R. Batten, K. J. Berry, D. C. R. Hockless, B. Moubaraki and K. S. Murray, Inorg. Chem., 1997, 36, 5006–5015 CrossRef CAS.
- D. J. Price, S. R. Batten, B. Moubaraki and K. S. Murray, Polyhedron, 2003, 22, 2161–2167 CrossRef CAS.
- A. J. Tasiopoulos, N. C. Harden, K. A. Abboud and G. Christou, Polyhedron, 2003, 22, 133–143 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391–1400 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2036380 and 2036381 contain the supplementary crystallographic data for the complex. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09425k |
| This journal is © The Royal Society of Chemistry 2021 |