
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArenesulfonyl indole: new precursor for diversification of C-3 functionalized indoles
Banni Preet Kaur
a,
Jasneet Kaur
b and
Swapandeep Singh Chimni
 *a
*a
aDepartment of Chemistry, U.G.C. Centre of Advance Study-II, Guru Nanak Dev University, Amritsar, Punjab, India. E-mail: sschimni@yahoo.com; sschimni.chem@gndu.ac.in
bPost-Graduate Department of Chemistry, Khalsa College Amritsar, Punjab, India. E-mail: jasneet208@gmail.com
First published on 8th January 2021
Abstract
Arenesulfonyl indoles, bearing a good leaving group, are effective precursors for vinylogous imine intermediates which are generated in situ under basic conditions. This intermediate can readily react with other nucleophilic reagents to obtain C-3 substituted indole derivatives. In the last few years, a plethora of exciting synthetic applications of this substrate have been exploited. The stability of arylsulfonyl-containing substrates, mild reaction conditions, and the large variety of nucleophiles involved in these procedures are the key to their success in organic synthesis.
 Banni Preet Kaur | Banni Preet Kaur was born in 1993 at Amritsar, India. She did her B. Sc. Chemistry (Hons. Sch.) in 2013 and M. Sc. Chemistry (Hons Sch.) in 2015 from Department of Chemistry, Guru Nanak Dev University, Amritsar. Since 2016 she is working for her doctoral degree with Prof. Swapandeep Singh Chimni. Currently, she is interested in the development of new enantioselective carbon–carbon bond formation reactions and enantioselective organocascade reactions employing hydrogen-bonding organocatalysts. |
 Jasneet Kaur | Jasneet Kaur was born in 1989 in Vain Poin village of Tarn Taran, Punjab, India. She obtained her B.Sc. (Hons. Sch.) in Chemistry and M.Sc. (Hons. Sch.) in Chemistry from Guru Nanak Dev University, Amritsar. She completed her Ph.D. under the supervision of Prof. Swapandeep Singh Chimni from Guru Nanak Dev University, Amritsar, India, in 2017. After that she worked as a Research Associate with same research group until July 2018. Since August 2018, she is working as an Assistant Professor in the Department of Chemistry at Khalsa College, Amritsar. So far she has published 19 research papers including review articles and one book chapter. Her research interests include synthesis of new chiral bifunctional organocatalysts and their applications for enantioselective carbon–carbon and carbon–heteroatom bond formations and domino reactions. |
 Swapandeep Singh Chimni | Swapandeep Singh Chimni was born in 1962 at Amritsar, India. He received his M.Sc. (Hons. Sch.) in Chemistry in 1985 and Ph.D. in 1991 from Guru Nanak Dev University, Amritsar. After two years as a lecturer at Regional Engineering College (now NIT) Jalandhar, he joined the Department of Chemistry, Guru Nanak Dev University as Lecturer in 1992. He is presently working as a Professor in the same department. He has a research experience of 30 years and published over 120 publications. He works in the area of synthetic organic chemistry with emphasis on asymmetric organocatalysis, biocatalysis and phase transfer catalysis as well as green chemistry. |
1. Introduction
The asymmetric synthesis of complex heterocyclic frameworks has always fascinated synthetic as well as medicinal chemists, looking at their wide occurrence in alkaloids, dyes, pharmaceuticals and agrochemicals.1–4 Indole, among them, is considered as a privileged motif, owing to its occurrence in numerous molecules showing promising bio-activities such as anti-histamine,5 anti-convulsant,6 anti-microbial,7 anti-tubercular,8 anti-inflammatory,9 anti-diabetic,10 anti-hypertensive,11 anti-cancer12,13 etc. (Fig. 1). The vast spectrum of bioactivity of indole derivatives can be attributed to the functionalization at the C-3 position of indole. For a long period, the C-3 functionalization of indoles has been carried out using Friedel–Crafts reaction,14 however the last few decades have witnessed the emergence of new synthetic approaches for C-3 derivatization of indoles. In recent times, indolyl nitroalkenes have been exploited as Michael acceptors with Michael donors to functionalize indole at C-3 position.15,16 Another relatively new methodology involves the presence of leaving group at the benzylic C-3 position of indole, which includes gramines, 3-(1′-hydroxyalkyl)-indoles and arenesulfonyl indoles. These substrates undergo elimination under acidic or basic conditions to generate reactive alkylideneindolenine, a vinylogous imine intermediate, which upon nucleophilic addition leads to the C-3 functionalized indole adducts (Scheme 1). Gramine is an indole containing alkaloid, found in plants and possess various pharmacological properties.17 They have been actively used for the synthesis of bioactive indole derivatives and their reactions have been reviewed in 2004 by Semenov and Granik.18 In 2009, Petrini reviewed the applications of arenesulfonyl indoles along with gramines and 3-(1-hydroxyalkyl)-indoles highlighting the importance of alkylideneindolenine, the vinylogous imine intermediate, to synthesize 3-substituted indole derivatives.19 Among them, arenesulfonyl indole is a relatively less explored precursor which contains arenesulfonyl as leaving group at the C-3 benzylic position. | ||
| Fig. 1 Examples of bioactive molecules containing indole in core structure. | ||
 | ||
| Scheme 1 Synthesis of 3-substituted indoles via reactive alkylideneindolenine intermediates. | ||
Arenesulfonyl indoles have a great potential to act as an electrophile since the arenesulfinic group acts as an efficient leaving group to generate the alkylideneindolenine intermediate 2.20 Since their serendipitous synthesis in 2006, arenesulfonyl indoles have acted as viable alternatives for synthesizing C-3 substituted indole derivatives.
The present review focuses primarily on the role of arenesulfonyl indole as a precursor for synthesizing chiral as well as racemic C-3 functionalized indole derivatives. The first section discusses different methods to synthesize arenesulfonyl indoles, followed by its synthetic applications in the synthesis of C-3 functionalized indole derivatives.
2. Synthesis
The arenesulfonyl indole 11 was synthesized serendipitously by Petrini and co-workers21 in 2006, in an attempt to prepare N-ethoxycarbonylaminoalkylindoles, (7) by reaction of α-amidoalkylaryl sulfones (4) with indoles (6) using montmorillonite K-10 as the acid promotor under solvent-free conditions (Scheme 2). The adduct undergoes elimination of ethyl carbamate to generate vinylogous iminium ion 9, which can either undergo the Friedel–Crafts addition of indole to give bis-indole product 10 or add ArSO2H to provide the thermodynamically favourable arenesulfonyl indole 11. | ||
| Scheme 2 First report of synthesis of arenesulfonyl indoles. | ||
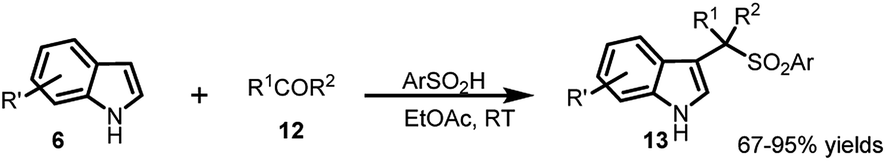
The following year, Petrini and co-workers22 reported a simplified procedure for the synthesis of 13 in 67–95% yields by three-component coupling of indole (6) with carbonyl derivative 12 and arylsulfinic acid at room temperature (Scheme 3).
 | ||
| Scheme 3 Three component coupling to synthesize arenesulfonyl indoles. | ||
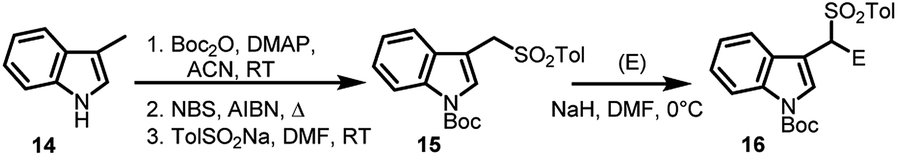
Later, Petrini's group,23 in 2008, proposed a new method of synthesizing arenesulfonyl indoles (16) making use of N-Boc-3-(1-tosylmethyl)indole (15), starting from 3-methyl indole (14). The tosyl group assists deprotonation at α-position facilitating the formation of anion which subsequently attacks on the electrophilic reagents, resulting in N-protected arenesulfonyl indoles (Scheme 4). Using this method, different alkyl and aryl groups can be incorporated at the 1′-position.
 | ||
| Scheme 4 Synthesis of arenesulfonyl indoles starting from 3-methylindole. | ||
3. Reactions of arenesulfonyl indoles
Arenesulfonyl indoles, since their first report in 2006, has undergone a vast variety of reactions including Michael reaction, Friedel–Crafts reaction, dearomatization, reductive desulfonylation, arylation/alkylation leading to the C-3 functionalization of indoles. Since, arenesulfonyl indoles react with different reagents via alkylideneindolenine intermediate 2, so the classification of the reaction types is based on the reaction of the vinylogous imine intermediate 2.3.1 Michael addition reaction
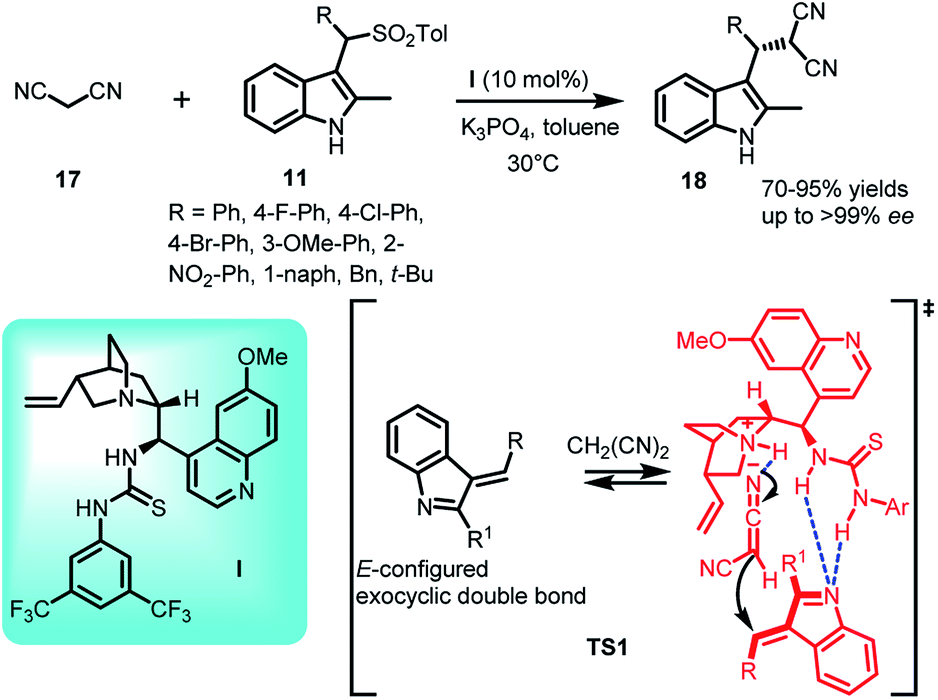
As one of the most powerful and useful carbon–carbon bond forming reactions, this conjugate addition reaction enables access to a variety of complex synthetic frameworks.24,25 The conjugate addition of resonance stabilized carbanions is both atom and step economic, and hence quite versatile for the construction of complex structures, which are expected to have potential biological significance.The carbanions of active methylene compounds are proven to be efficient Michael donors, since they are stabilized by the electron withdrawing groups present in conjugation. Its significance further increases when the addition product acts as a precursor to obtain biologically relevant molecules.26,27 In this context, various research groups are working on exploiting different active methylene compounds with arenesulfonyl indoles to synthesize C-3 functionalized indole derivatives. Jing et al.28 reported the use of malononitrile (17) as nucleophile in carrying out the conjugate addition to arenesulfonyl indoles 11 catalyzed by quinidine derived thiourea I. The resulting C-3 functionalized indole derivatives (S)-18 were obtained in 70–95% yields and excellent enantiomeric excess up to >99% (Scheme 5). The steric effect posed by methyl group at C-2 position is crucial for obtaining E/Z ratio, which further determined the enantioselectivity. In the proposed transition state TS1, the thiourea moiety interacted with the nitrogen atom of E-configured imine intermediate 2 through two hydrogen bonds whereas the carbanion which gets activated by the N-atom of quinuclidine ring to generate the ternary complex through hydrogen bond, consequently attacked the imine intermediate to generate 18.
 | ||
| Scheme 5 Quinidine thiourea catalysed Michael addition of malononitrile to arenesulfonyl indoles. | ||
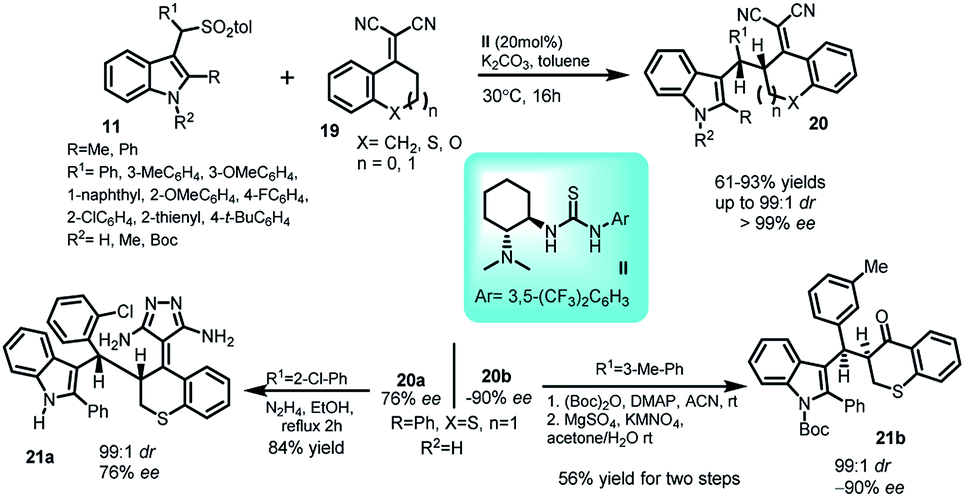
α,α-Dicyanoolefins derived from tetralone (19) have proven to be excellent vinylogous nucleophiles in various asymmetric reactions. In this regard, a doubly vinylogous Michael reaction of dicyanoolefins (19) with vinylogous imine intermediate 2 generated in situ from 3-arenesulfonyl indoles 11 have been reported by Zhu and co-workers.29 The reaction catalysed by Takemoto's catalyst II provided access to 3-substituted indole derivatives 20 in 61–93% yields with enantiomeric excess >99% and up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr (Scheme 6). The product 20a was transformed to corresponding pyrazolo derivative 21a in 84% yield through its reaction with hydrazine hydrate in ethanol. Additionally, another transformation was carried out, in which the ent-20b was converted to α-alkylated product 21b via a known oxidative cleavage procedure. In both the cases, the ee as well as dr remained intact.
:1 dr (Scheme 6). The product 20a was transformed to corresponding pyrazolo derivative 21a in 84% yield through its reaction with hydrazine hydrate in ethanol. Additionally, another transformation was carried out, in which the ent-20b was converted to α-alkylated product 21b via a known oxidative cleavage procedure. In both the cases, the ee as well as dr remained intact.
 | ||
| Scheme 6 Michael addition of α,α-dicyano olefins to arenesulfonyl indoles. | ||
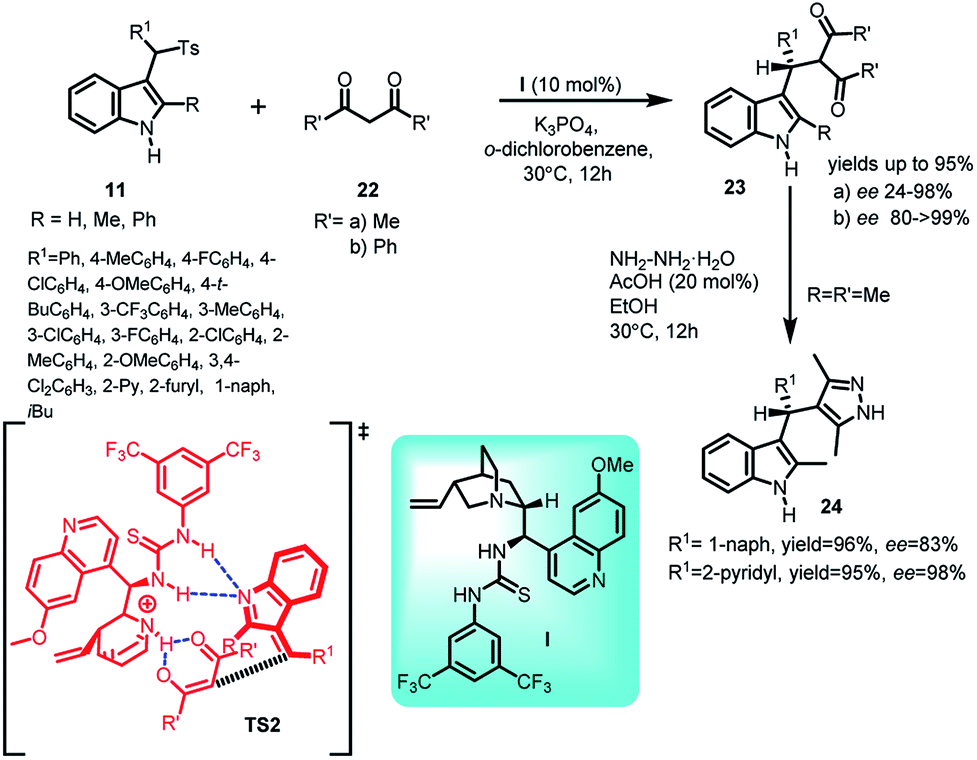
Zuo and co-workers30 documented quinidine thiourea I catalysed enantioselective Michael addition of 1,3-diketones 22 to alkylidineindolenine intermediate 2 generated from arenesulfonyl indoles 11 under basic conditions to afford a series of optically active C3-alkyl-substituted indole derivatives (S)-23 in 40–95% yields and enantiomeric excess up to >99% (Scheme 7). The author found that better enantioselectivity was achieved with dibenzoylmethane 22b as compared to acetylacetone 22a based on the steric factors. The final adduct-23 was transformed into 3-sec-alkyl indole derivative 24 involving pyrazole skeleton by carrying out its reaction with hydrazine hydrate in 95–96% yields without any loss of enantiomeric excess. A transition state TS2 was proposed by the author depicting the role of thiourea moiety as Bronsted acid and aliphatic tertiary amine as Bronsted base, simultaneously activating the E-alkylideneindolenine intermediate and 1,3-diketone respectively through hydrogen bonding.
 | ||
| Scheme 7 Enantioselective Michael addition of 1,3-diketones to arenesulfonyl indoles. | ||
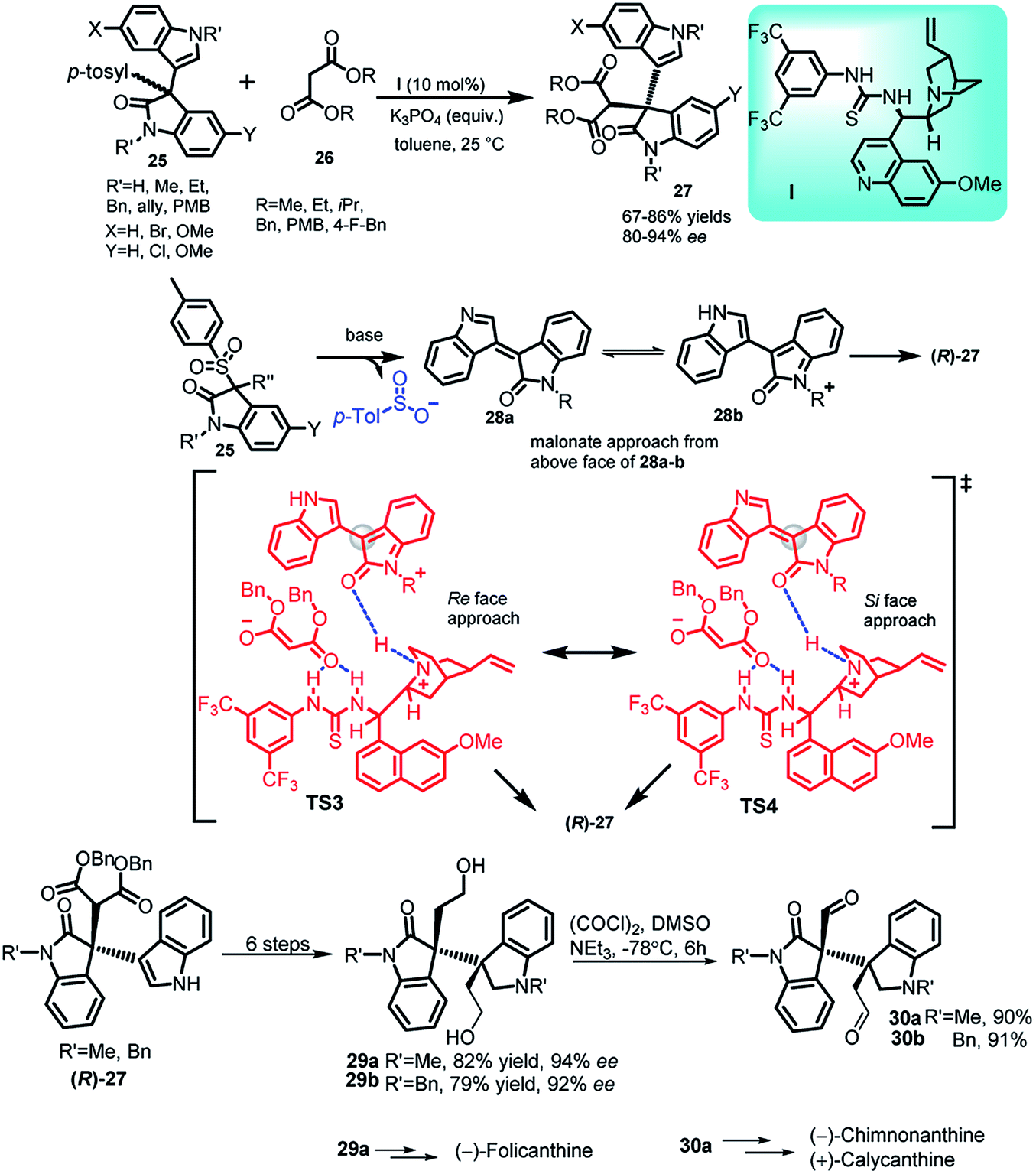
In recent years, the 3,3-disubstituted indole derivatives containing an all carbon quaternary stereocenter have experienced significant advances, just like 3-substituted indole molecules. E.g. gliocladin C, which contains a 3-substituted-3-indolyloxindole, is a marine alkaloid.31 Bisai and co-workers32 reported the thiourea I catalysed addition of malonates 26 to 3-sulfonyl-3′-indolyl-2-oxindoles (25) to obtain C-3 substituted indole derivatives 27 containing an all carbon quaternary stereogenic centre in 67–86% yields with 84–92% ee (Scheme 8). The author also mentioned the role of stronger π–π interactions behind the enhanced enantioselectivity. In the proposed transition state TS3 and TS4, the planar intermediate 28a and 28b gets activated by the quinuclidine moiety through H-bonding, whereas the malonate gets activated by the thiourea moiety via construction of two H-bonds. Intermediate 28b was stabilized by electron rich indole present at 3-position and attack from the above face led to the formation of (R)-27. The final adduct (R)-27 could be transformed in 6 steps to obtain C2-symmetric diol 29, which can further be transformed to 30 via Swern oxidation. Both these adducts 29a and 30a have been earlier reported to act as intermediates for the synthesis of (−)-folicanthine and (−)-chimonanthine, (+)-calycanthine, respectively.
 | ||
| Scheme 8 Thiourea catalysed addition of malonates to 3-sulfonyl-3′-indolyl-2-oxindoles. | ||
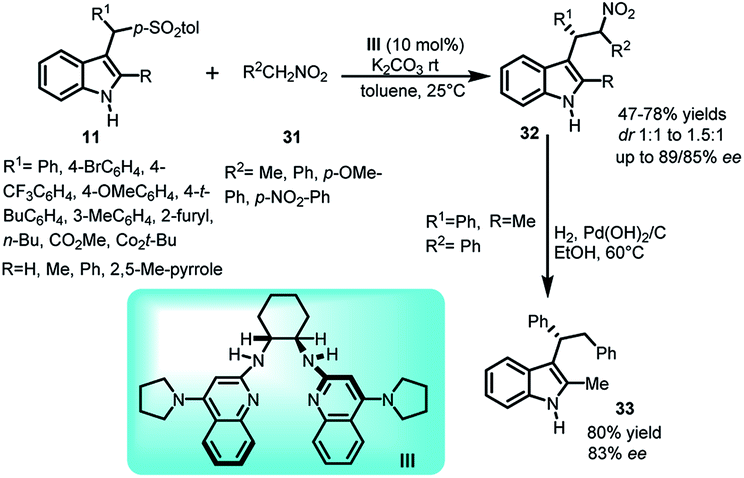
Nitroalkanes are highly valuable nucleophiles and their ease of transformation to other functional groups further enhances their worth. The reaction of nitroalkanes 31 with arenesulfonyl indoles 11 was carried out by Johnston's group33 in 2010 using pyrrolidine based bis-amidine catalyst (PBAM) III to generate chiral indole derivatives 32 bearing a sec-alkyl group at C-3 position in 47–78% yields and ee up to 89% for the major diastereomer while 85% for the minor diastereomer (Scheme 9). The adduct 32 was further denitrated to obtain 33 in 80% yield while ee gets slightly lower from 84% to 83%.
 | ||
| Scheme 9 PBAM catalysed addition of nitroalkanes to arenesulfonyl indoles. | ||
Fochi's group34 disclosed solvent-free asymmetric addition of nitroalkanes 34 to alkylideneindolenines 2 generated in situ from arenesulfonyl indoles 11 catalyzed by dihydroquinine urea catalyst IV to obtain (1S,2S)-35 in 65–99% yields and excellent ee up to 99% with dr up to 60:40 (Scheme 10). The synthetic use of this methodology was illustrated by the synthesis of tryptamine derivative, followed by its tosylation to obtain 36a and 36b in 82–98% yields with 91–92% ee. The author proposed the transition state TS5 in which soft enolization of nitroalkane 34 generated a nitronate coordinated by multiple H-bonds. It further attacked the Si-face of the prochiral (E)-alkylideneindolenine intermediate to generate 35. Moreover, low diastereoselectivity of the reaction is attributed to the inability of catalyst to control the face selectivity of prochiral intermediate.
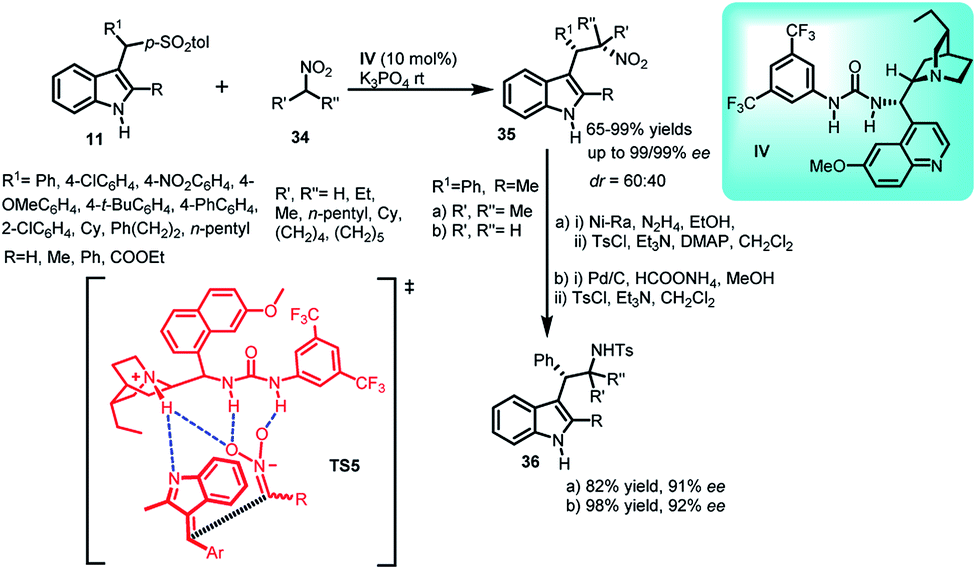 | ||
| Scheme 10 Quinine derived urea catalysed addition of nitroalkanes to arenesulfonyl indoles. | ||
In another study, the conjugate addition of nitroalkanes 34 with oxindolylideneindolenines (25) was reported, catalysed by quinidine-based urea catalyst V to construct a library of 3,3′-disubstituted indole derivatives 37 in 76–98% yields and 78–99% ee (Scheme 11).35 This protocol was further utilized for the formal total synthesis of (+)-gliocladin C, an active alkaloid exhibiting cytotoxicity against murine P388 lymphocytic leukemia cells. After undergoing a series of transformations, 38 was obtained, a known intermediate for the synthesis of gliocladin C.36
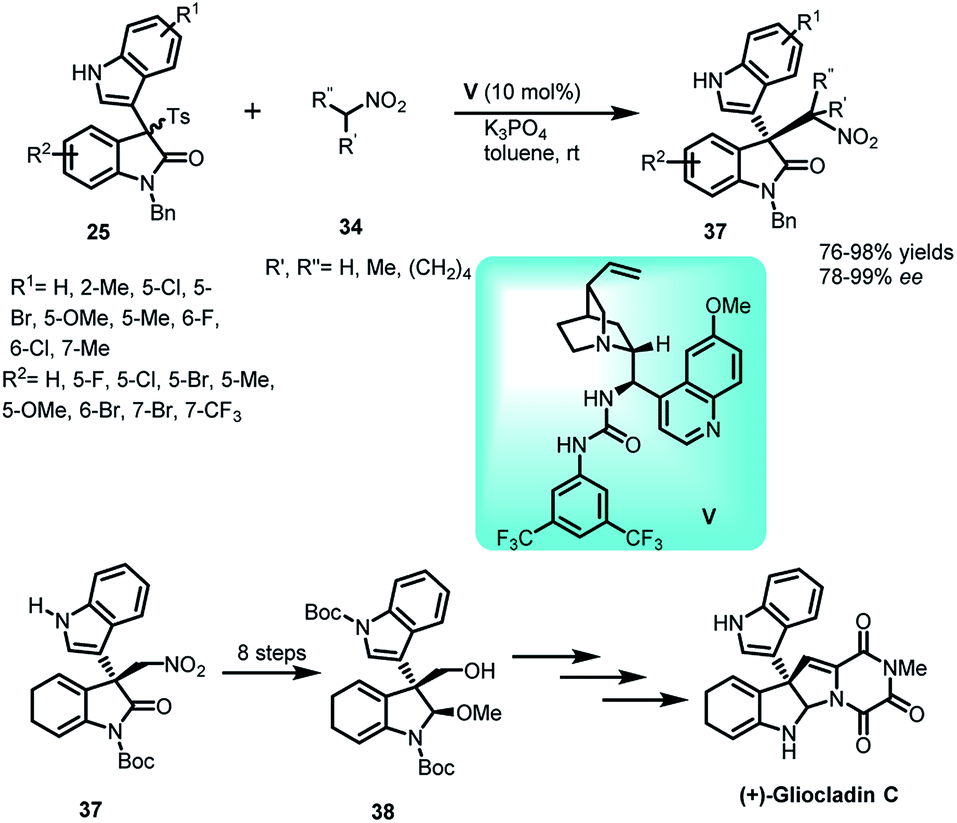 | ||
| Scheme 11 Quinidine derived urea catalysed addition of nitroalkanes to oxindolylideneindolenines. | ||
Petrini and co-workers37 reported the attack of easily enolizable methylene compounds, including nitroalkanes, malononitrile and dialkyl malonates with arenesulfonyl indoles 11 using potassium fluoride supported on basic alumina. The resultant 3-(2-nitroalkyl)indole derivatives 39 were obtained in 70–89% yields with nitroalkanes 34 as the nucleophile (Scheme 12). The significance of KF on alumina was examined using other methylene compounds 40 bearing electron withdrawing groups to obtain the adduct 41 in 63–95% yields. Inferior results were obtained while using sodium hydride in THF to carry out the same reaction. The author highlighted the importance of this reaction by proposing the synthesis of corresponding tryptamine analogue 42 via reduction of nitro group, followed by its conversion to β-carboline alkaloids 43 by means of Pictet–Spengler reaction. Additionally, they also proposed two different routes to access tryptophan derivatives. Mixed malonic acid esters 41a can easily undergo chemoselective cleavage of one ester group and the resulting adduct 44 undergoes Curtius rearrangement leading to desired tryptophan analogue 45. Moreover, hydrolysis and carboxylation of 41b can provide a direct route to access 45.
 | ||
| Scheme 12 KF/basic alumina promoted addition of methylene compounds to sulfonyl indoles. | ||
In another report by Petrini's group,38 potassium fluoride supported on basic alumina was utilized for reaction of arenesulfonyl indoles 11 with protected β-nitro ketones 46 to furnish 3-(2-nitroalkyl)indole derivatives 47, which underwent a sequence of cascade processes to finally furnish 1,4-unsymmetrical disubstituted carbazoles 48 in 44–68% yields (Scheme 13).
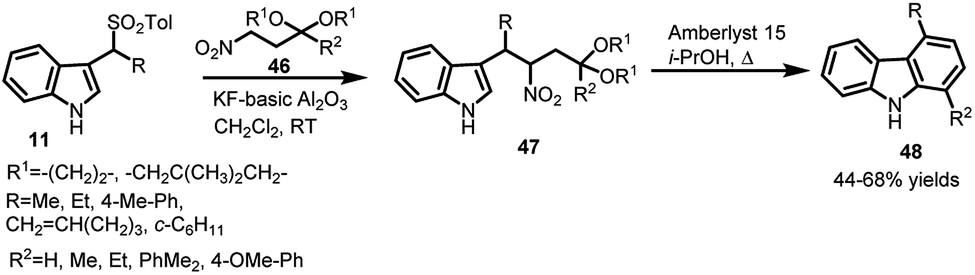 | ||
| Scheme 13 Michael addition of protected β-nitro ketones to arenesulfonyl indoles. | ||
The asymmetric Michael addition of oxazolones (49) to arenesulfonyl indoles 11 catalysed by cinchonine derived thiourea catalyst VI under mild conditions was reported by Jing and co-workers39 in 2012 to yield syn selective C-3 alkyl substituted indole derivative (10R,11S)-50 containing adjacent quaternary and tertiary stereocenters in 45–88% yields and high dr as well as ee up to 90:10 and 98% respectively (Scheme 14). The synthetic utility of the protocol was studied by transforming the Michael adduct-50 to syn-selective α,β-disubstituted tryptophan derivative 51 in 88% yield by ring opening of the oxazolone subunit with sodium methoxide while increasing ee from 96% to 98%. In the proposed transition state TS6, existence of π–π interaction between the phenyl group of the catalyst and that of the oxazolone ring was found to be the reason behind low stereoselectivity when R group is phenyl, because increased steric hindrance led to decreased π–π interaction of the ‘closed’ conformation in the transition state.
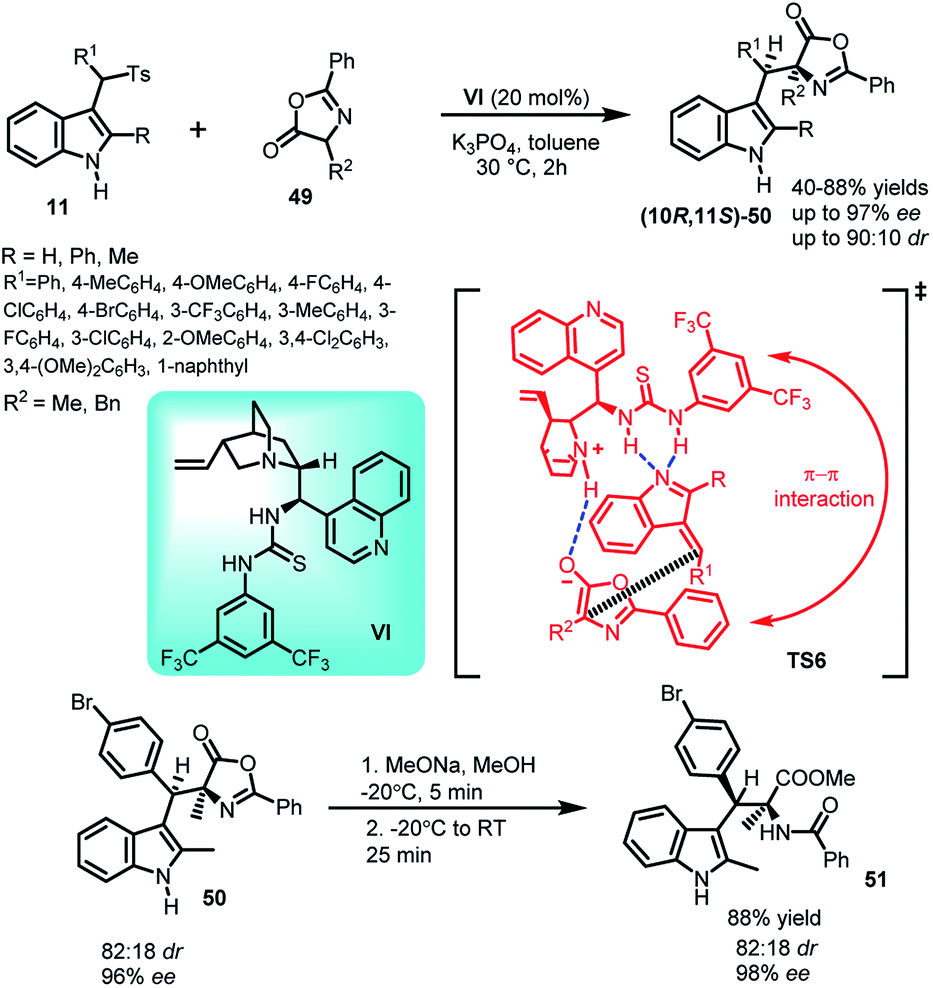 | ||
| Scheme 14 Asymmetric Michael addition of oxazolones to arenesulfonyl indoles. | ||
Benzofuran-2(3H)-one and derivatives are known to be present in a variety of valuable natural products.40–42 In this context, the synthesis of C-3 alkyl-substituted benzofuran-2(3H)-one derivatives (7S,8R)-53 containing indole skeleton with two adjacent stereocenters was reported via Michael addition of benzofuran-2(3H)-ones 52 to alkylidineindolenine intermediate 2 generated in situ by deprotonation of arenesulfonyl indole 11 under basic conditions. The final adduct 53 was obtained in 66–97% yields with ee and dr up to 94% and 93:7, respectively (Scheme 15).43 Based on the absolute configuration, the author proposed a transition state TS7, in which the catalyst VII simultaneously serves the purpose of activating both the benzofuran-2(3H)-one unit as well as arenesulfonyl indole through non-covalent catalysis. The thiourea moiety acts as Bronsted acid and interacts with the nitrogen atom of the vinylogous intermediate through double hydrogen bonding. The tertiary amine of the catalyst deprotonates benzofuranone unit and activates it through one hydrogen bond.
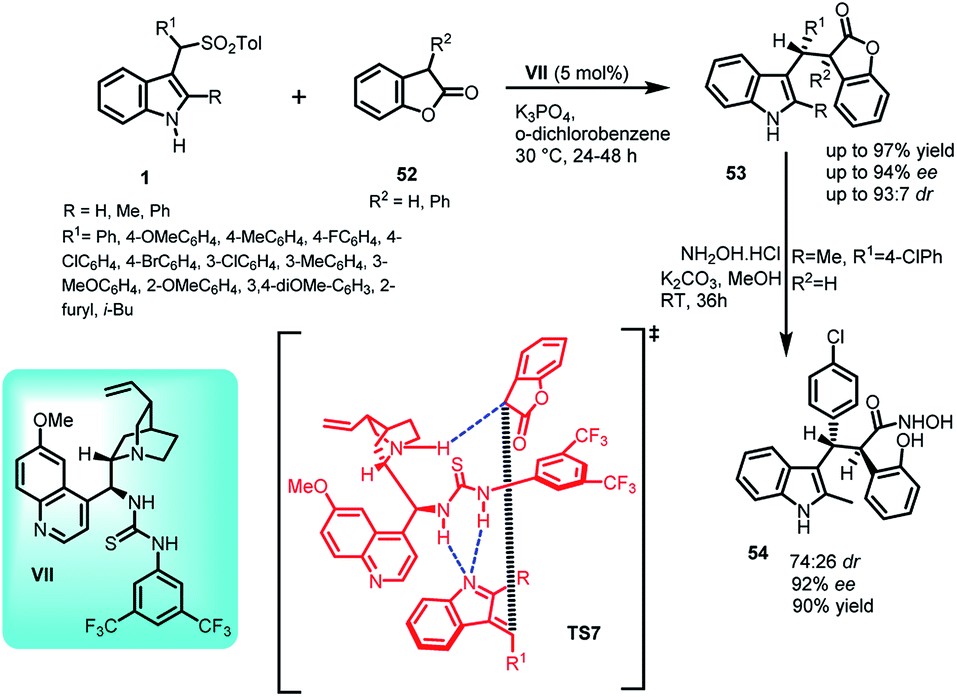 | ||
| Scheme 15 Quinine thiourea catalysed Michael addition of benzofuranone to arenesulfonyl indoles. | ||
Further, the adduct 53 was readily converted to hydroxamic acid derivative 54 by ring opening of benzofuranone using excess of hydroxyl amine hydrochloride in 90% yield with ee rising from 90% to 92%.
The asymmetric sulfidation of indole with thiol was carried out using arenesulfonyl indole 11 and tritylthiol (55) using quinidine derived thiourea catalyst I at 0 °C in water-chloroform solvent system to generate chiral C-3 indole derivatives containing C–S bond (Scheme 16).44 The imine intermediate generated by sodium carbonate was attacked by thiol to generate (S)-56 in 57–99% yields with enantiomeric excess up to 98%. The synthetic utility of the product was studied by transforming the resulting vulcanized indole derivative into corresponding indoline derivative using sodium cyanoborohydride to get 57 in 60% yield and 91% ee, which was further converted to free thiol 58 in 93% yield and 91% ee by eliminating the trityl group using trifluoroacetic acid and triethyl silane.
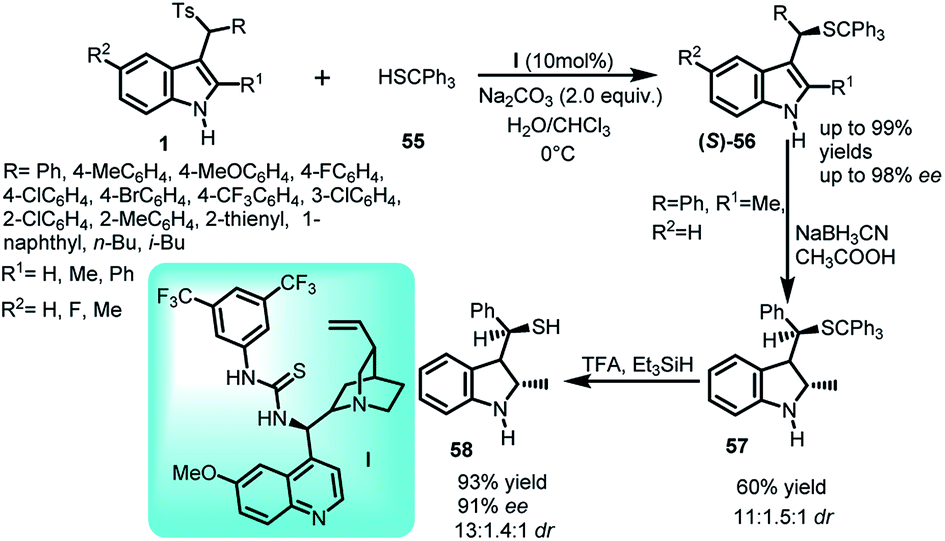 | ||
| Scheme 16 Sulpha-Michael addition of triphenylmethanethiol to arenesulfonyl indoles. | ||
Chiral Ni(II) complexes of glycine Schiff bases 59 have been used as Michael donors with arenesulfonyl indoles 11 by Wang and co-workers to synthesize syn-configured-β-substituted tryptophan derivatives (2S,3R)-60 in a highly enantio-as well as diastereoselective manner (Scheme 17).45 The resultant adduct-60 was obtained in 53–91% yields, syn:anti up to 94:6 and de up to >99% using DBU as the base. Further, they were disassembled using 6 N HCl in MeOH to afford the free amino-acid (2S,3R)-2-amino-3-(1H-indol-3-yl)-3-phenyl-propanoic acid 61 in 96% yield. The chiral ligand (S)-o-[N-(N-benzylprolyl)amino]benzophenone (S)-62 could be easily recovered and re-used without affecting the enantioselectivity.
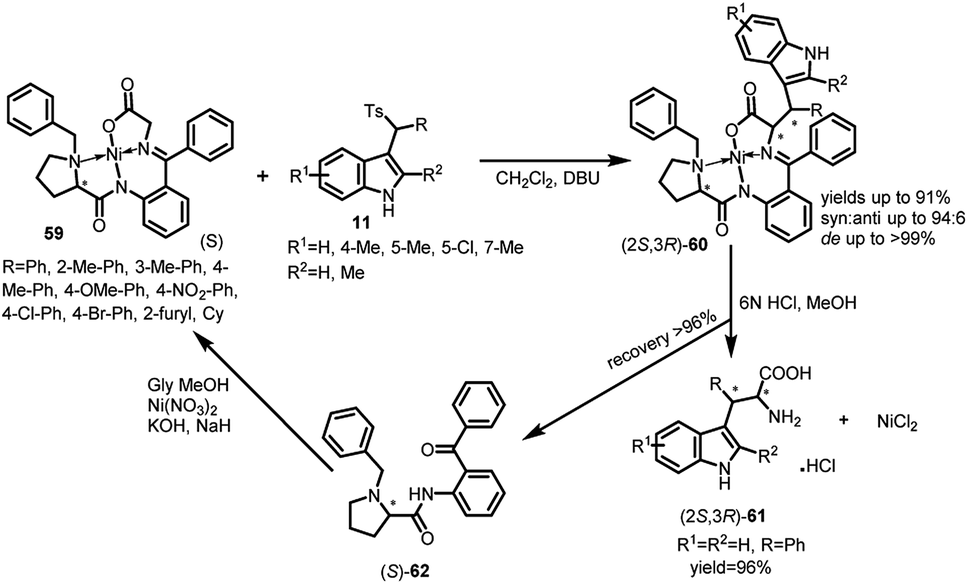 | ||
| Scheme 17 Michael addition of glycine derivatives to arenesulfonyl indoles. | ||
3.2 Friedel–Crafts reactions
Synthetic methodologies involving asymmetric indole frameworks containing phenolic –OH group in the substructure are found to be scarce,46 although they are present in various natural as well as synthetic bio-active compounds.47 Yu et al.48 utilized arenesulfonylalkyl indoles 11 as precursors to synthesize optically active indole derivatives containing phenolic hydroxyl group by carrying out the Friedel–Crafts reaction of 2-naphthols (63) using quinidine derived thiourea I as the chiral catalyst (Scheme 18). The imine intermediate was generated in situ by using potassium phosphate as the base at 30 °C to afford (R)-64 in yields up to 96% and enantiomeric excess up to 98%. A plausible transition state TS8 was proposed by the author, wherein the tertiary amine of the quinuclidine ring interacted with the phenolic hydrogen through hydrogen bonding to activate the nucleophile. On the other hand, double hydrogen bonding occurred between the –NH of thiourea moiety and nitrogen of the imine intermediate to activate the electrophile. The absolute configuration of the product was determined to be (R) on the basis of X-ray crystallographic analysis. | ||
| Scheme 18 Friedel–Crafts reaction of arenesulfonyl indoles with 2-naphthol. | ||
In 2016, Chang and co-workers reported the organocatalytic enantioselective Friedel–Crafts reaction of sesamol and electron rich phenols 65 to alkylideneindolenine intermediate 2 generated in situ from the deprotonation of arenesulfonyl indoles 11 under basic conditions.49 Using 20 mol% of quinine derived urea catalyst VIII, the Friedel–Crafts adduct (S)-66 containing a tertiary stereocenter was obtained in 54–99% yields and 54–90% ee (Scheme 19). Furthermore, by carrying out some control experiments wherein the reaction was carried out without base or catalyst, the author confirmed the requirement of both base and urea catalyst for obtaining optimized yield and enantioselectivity. Based on the results of the experiments performed and absolute stereochemistry, transition state TS9 was proposed, in which the base deprotonated arenesulfonyl indole for the in situ generation of alkylideneindolenine intermediate. This vinylogous imine intermediate was activated through double hydrogen bonding with the –NH groups of the urea moiety, whereas the protonated nitrogen of the quinuclidine ring activated sesamol through hydrogen bonding, resulting in the direct attack of sesamol to the Si face of the intermediate.
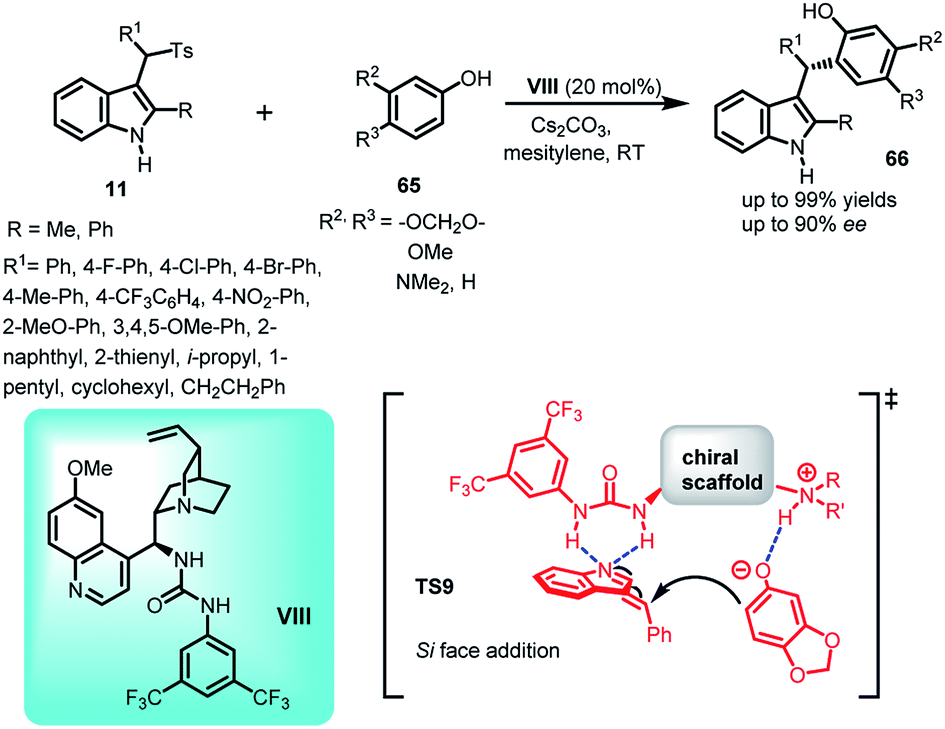 | ||
| Scheme 19 Friedel–Crafts reaction of arenesulfonyl indoles with electron rich phenols. | ||
Xing et al.50 reported the synthesis of 3,3-disubstituted oxindole derivatives 67 containing chiral quaternary carbon by carrying out the Friedel–Crafts reaction of 2-naphthol (63) with oxindolylideneindolenines (25) catalyzed by quinine derived thiourea IX as the organocatalyst (Scheme 20). The final adduct-67 was obtained in 39–99% yields with ee up to 95%. The results were improvised using co-solvent system to obtain 67 in 91–99% yields and ee up to 98%. The author proposed a transition state TS10 in which the base converted (1) to the corresponding vinylogous imine intermediate, followed by its complexation with the catalyst through H-bonding. The final addition of 2-naphthol resulted in the formation of adduct-67 with the regeneration of catalyst. The absolute configuration of the final adduct-67 was assigned as (R) by calculating and comparing the experimental ECD spectra with the simulated ECD spectra, which were in agreement with each other.
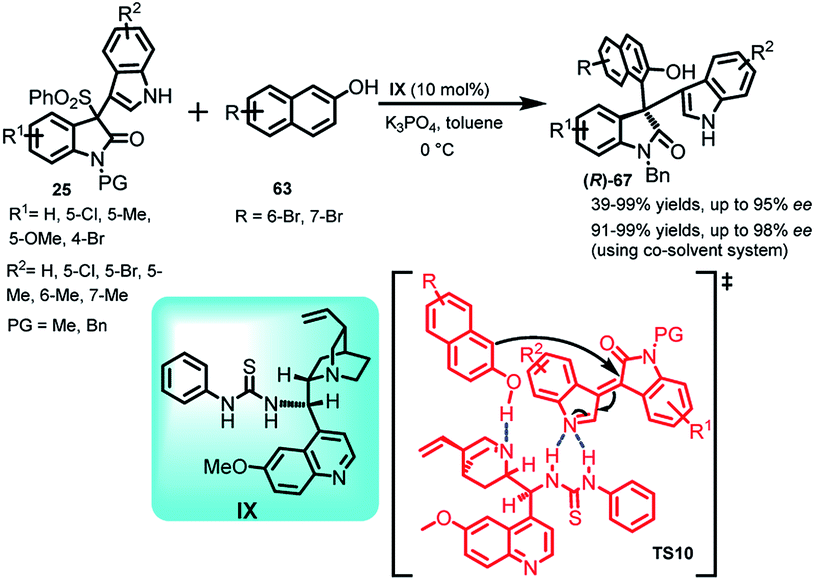 | ||
| Scheme 20 Friedel–Crafts reaction of oxindolylideneindolenines with 2-naphthol. | ||
In another study, toluene–water biphasic system was utilized to carry out the conjugate addition of electron rich phenols and active methylene compounds to arenesulfonyl indoles having quaternary C-3 position catalyzed by chiral organocatalysts.51 The Friedel–Crafts addition of 1-naphthol (68) to imine intermediate 2 by deprotonation of oxindolylideneindolenines 25 in the presence of cesium carbonate was catalyzed by 10 mol% of quinine derived thiourea VI to obtain the resultant 3,3-disubstituted indole derivatives (S)-69 in 52–99% yields and excellent ee up to 98% (Scheme 21). Using more diluted solutions of solvent system (toluene and water = 1:1) led to better yields and enantiomeric excess. The suppression of racemic reaction mediated by inorganic base due to ‘spatial separation’ created by water is expected to be the driving factor for this reaction. Further, the author also reported the attack of dibenzoyl malonate 70 to (25) catalyzed by VIII to obtain (S)-71 in 45–79% yields and 43% to >99% ee. Other active methylene compounds viz. acetyl acetone 72a, malononitrile 72b were also reacted with 25 using Xa and Xb as catalyst to obtain 73a in 58–87% yields, with up to 80% ee and 73b in 74–87% yields with up to 61% ee respectively. Several control experiments illustrated the requirement of both water as solvent and physical state to be liquid–liquid. The transition state TS11 was proposed, in which VII deprotonated 25 to generate 28a in the organic phase, while itself entered aqueous phase, and got neutralized by the inorganic base. The regenerated catalyst simultaneously activated both the deprotonated phenyl group and vinylogous intermediate through hydrogen bonding. Attack of the nucleophile from the Re face led to the formation of (S)-69.
 | ||
| Scheme 21 Addition of phenols and active methylene compounds to oxindolylideneindolenines. | ||
Recently, Chang52 et al. disclosed a solvent dependent enantiodivergent synthesis of both enantiomers of 69 by the Friedel–Crafts reaction of 1-naphthols 68 with arenesulfonyl indoles (25) using a single quinine derived bifunctional organocatalyst VIII (Scheme 22). The resultant (S)-69 was obtained using toluene as the solvent in 71–99% yields and up to 99% ee, while the (R)-enantiomer was obtained using 1,2-dichloroethane in 67–99% yields and up to 99% ee. The stereodiscrimination was explained by the author on the basis of DFT studies. The Eyring plot suggested the role of enthalpy–entropy in controlling the stereoselectivity. On its basis, different transition states TS12 and TS13 for both the enantiomers had been proposed, where Si-face addition took place in case of toluene, resulting in S-enantiomer and Re-face addition in case of 1,2-dichloroethane provided the R-enantiomer.
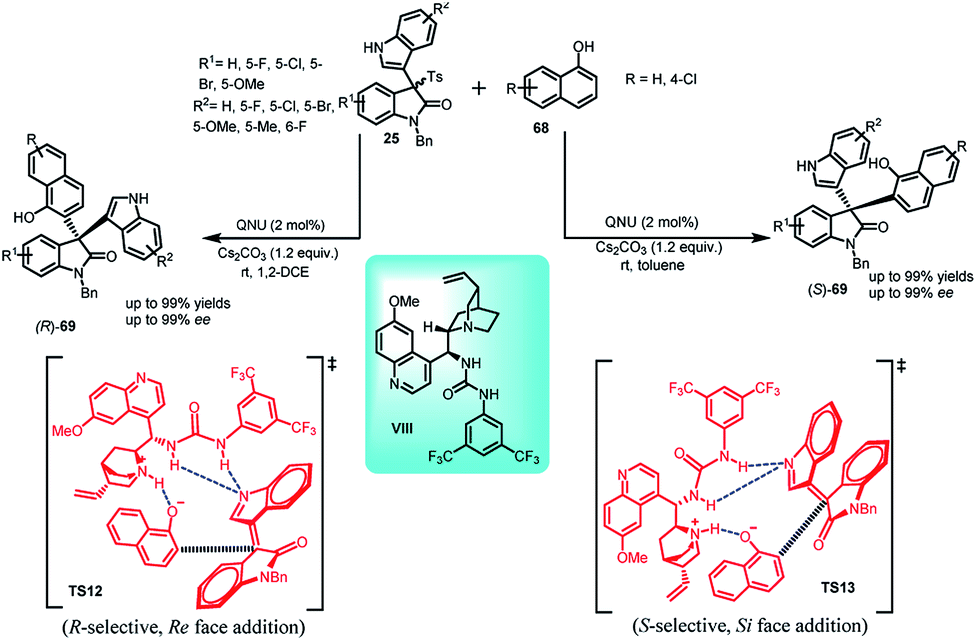 | ||
| Scheme 22 Enantiodivergent Friedel–Crafts reaction of arenesulfonyl indoles with 1-naphthol. | ||
3.3 Stetter-type Umpolung reaction
Umpolung reactions catalyzed by N-heterocyclic carbenes (NHCs) are an important route to synthesize various target molecules.53 In this context, arenesulfonyl indoles 11 have been successfully utilized by You and co-workers to carry out the N-heterocyclic carbene XIa catalysed Stetter-type Umpolung reaction with aldehydes 74 (Scheme 23).54 The thiazolium salt used as NHC precursor undergo deprotonation in the presence of base to generate carbene, which reacts with the aldehyde to give Breslow intermediate. Attack of this intermediate on the in situ generated imine electrophile affords the adduct 75 in up to 99% yields. The enantioselective version of this reaction was also attempted using XIb as the chiral catalyst to obtain the (S)-75 with 14–36% conversion and 90–97% ee.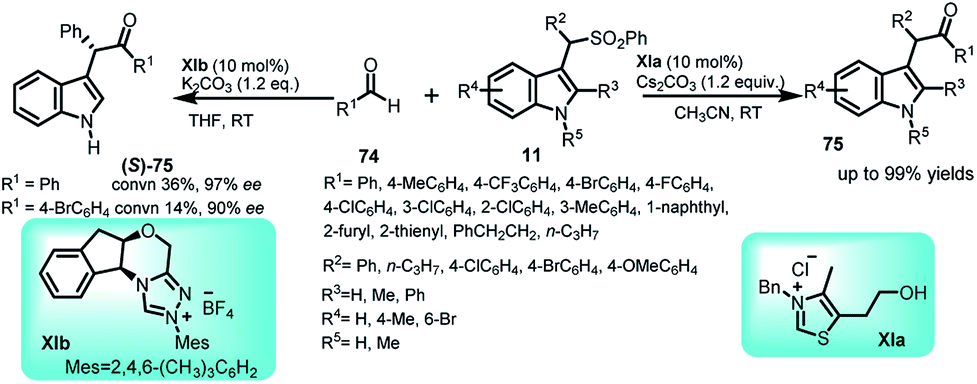 | ||
| Scheme 23 NHC-catalysed Stetter-type reaction of arenesulfonyl indole with aldehydes. | ||
3.4 Dearomatization reaction
Luo et al. utilized arenesulfonyl indoles 11 for carrying out the dearomatization reaction to synthesize spiro-cyclopropane derivatives. The vinylogous imine intermediate generated under mild basic conditions was attacked upon by sulfur ylides 76 to construct spiro-cyclopropane derivatives 77 in 55–88% yields with excellent dr up to 20:1 (Scheme 24).55 Additionally, an attempt to construct enantioselective spiro-cyclopropane indole derivative 77a in 29% yield and 64% ee was made using chiral sulfonium salt 78 with arenesulfonyl indole 11. The adduct 77a was further rearomatized in the presence of acid to obtain 2,3-disubstituted indole derivatives. Trifluoroacetic acid acts both as acid and nucleophile to obtain 79 in 65% yield and excellent dr. With water as nucleophile in the presence of p-TSA, the resultant adduct 80 was obtained in 89% yield and excellent dr up to >20:1.
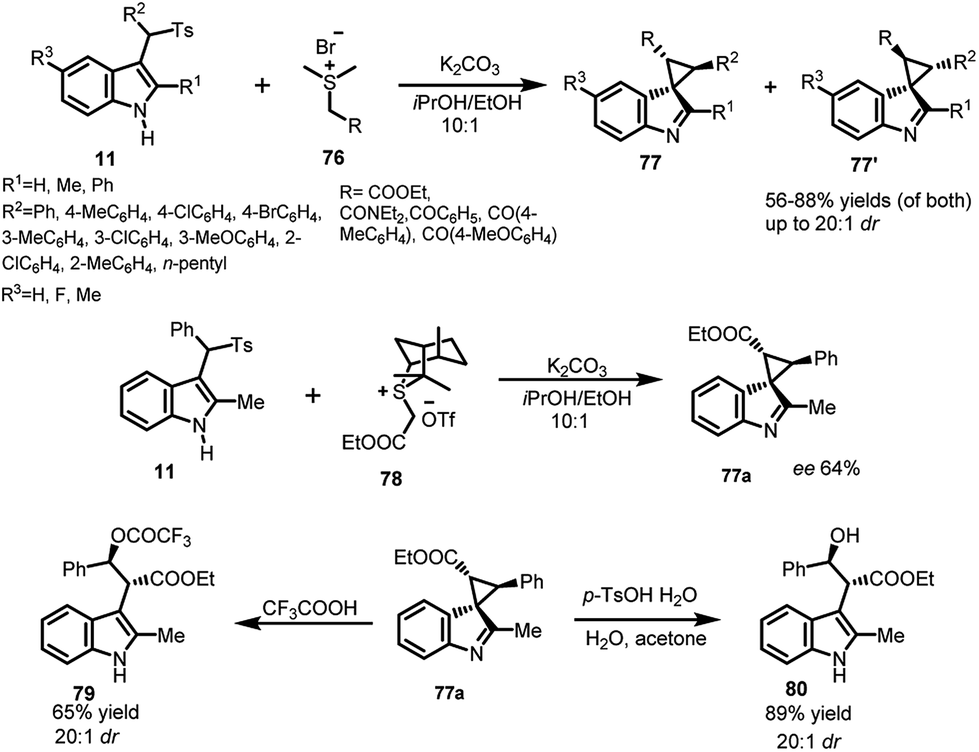 | ||
| Scheme 24 Dearomatization reaction of arenesulfonyl indole with sulfur ylides. | ||
3.5 Reductive desulfonylation reaction
The aptitude of tosyl group to act as a good leaving motif has been utilized to obtain 3-alkylated indoles starting from arenesulfonyl indoles. Petrini and co-workers21 documented the synthesis of arenesulfonyl indoles using montmorillonite K-10 at 55 °C. The final adduct 11 was obtained in 46–90% yields which was further desulfonylated using different reductive methods to furnish 3-alkyl indoles 81 in 56–86% yields (Scheme 25). | ||
| Scheme 25 Reductive alkylation of arenesulfonyl indoles. | ||
Use of flow chemical conditions for carrying out the reductive desulfonylation of arenesulfonyl indoles using polymer-supported sodium borohydride has been carried out by Petrini and co-workers56 to synthesize 3-alkyl indoles in 57–85% yields.
Similarly, indazoles, structurally analogous to indoles, hold a good position in biological and pharmaceutical advances for their anti-microbial, anti-inflammatory activities.57,58 However, due to reduced electron density, they suffer from lower reactivity towards Friedel–Crafts reaction and thus, functionalization at the C-3 position through this route is quite improbable. In this regard, indazoles (82) were converted to their corresponding arenesulfonyl indazoles (83) in the presence of aldehyde and p-toluenesulfinic acid in up to 82% yields.59 These adducts were then desulfonylated in the presence of sodium amalgam in protic solvents to obtain 84 in 77–94% yields (Scheme 26). The synthesis of 84 is otherwise not feasible using organometallic reagents or using sp3-carbon electrophiles on indazole.
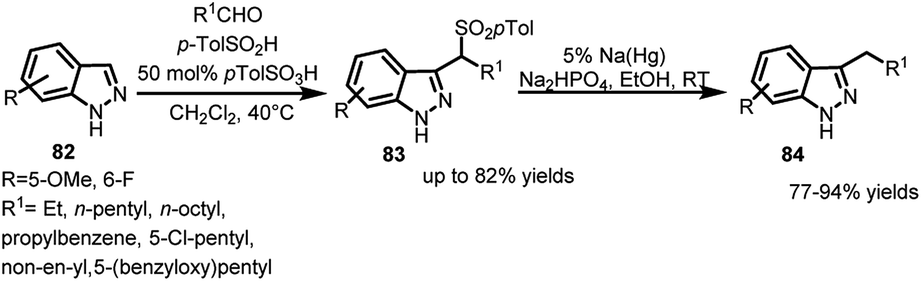 | ||
| Scheme 26 Desulfonylation of arenesulfonyl indazoles by sodium amalgam. | ||
Carrying out the reductive cleavage of ArSO2 group by Petrini and co-workers21 using LiAlH4 to obtain linear indoles was followed by using Grignard reagent 85 to obtain branched 3-alkyl indoles. The reaction between arylsulfonyl indoles 11 and Grignard reagent 85 was carried out at −35 °C in THF to provide access to 3-substituted indoles 86 in 75–94% yields (Scheme 27).
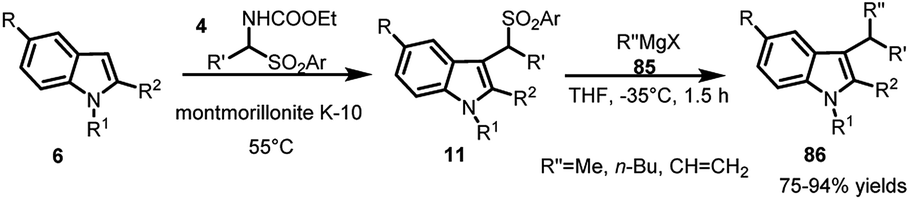 | ||
| Scheme 27 Reaction of arenesulfonyl indoles with Grignard reagent. | ||
An enantioselective version of the reaction has recently been reported by Harutyunyan's group60 carrying out the asymmetric addition of Grignard reagents (alkyl magnesium bromides) to vinylogous imine intermediate generated in situ from arenesulfonyl indoles. The reaction carried out using copper(I) salts and phosphoramidite ligand provided 3-sec alkyl indoles in high yields up to 97% with er up to 97.5:2.5.
Synthesis of bisindolylmethanes61 has been reported in a similar fashion via reaction of arenesulfonyl indoles with indolyl magnesium bromide in THF at 20 °C. Indolyl magnesium bromide was generated by reaction of methyl magnesium bromide with substituted indoles, which further adds on to the vinylogous imine intermediate to provide bisindolylmethanes in moderate to high yields up to 90%.
3.6 Arylation/alkylation/allylation reaction
Cao and co-workers disclosed the Rh-catalysed addition of arylboronic acids 87 to vinylogous imine intermediate 2 generated in situ from arenesulfonyl indoles 11 to obtain C-3 sec-alkyl-substituted indoles 88 in 52–99% yields with a wide range of substrates (Scheme 28).62 Using rhodium complex along with chiral ligand L, the asymmetric version of this reaction was carried out to furnish the adduct 88 in 21–29% yields and enantiomeric excess up to 84%.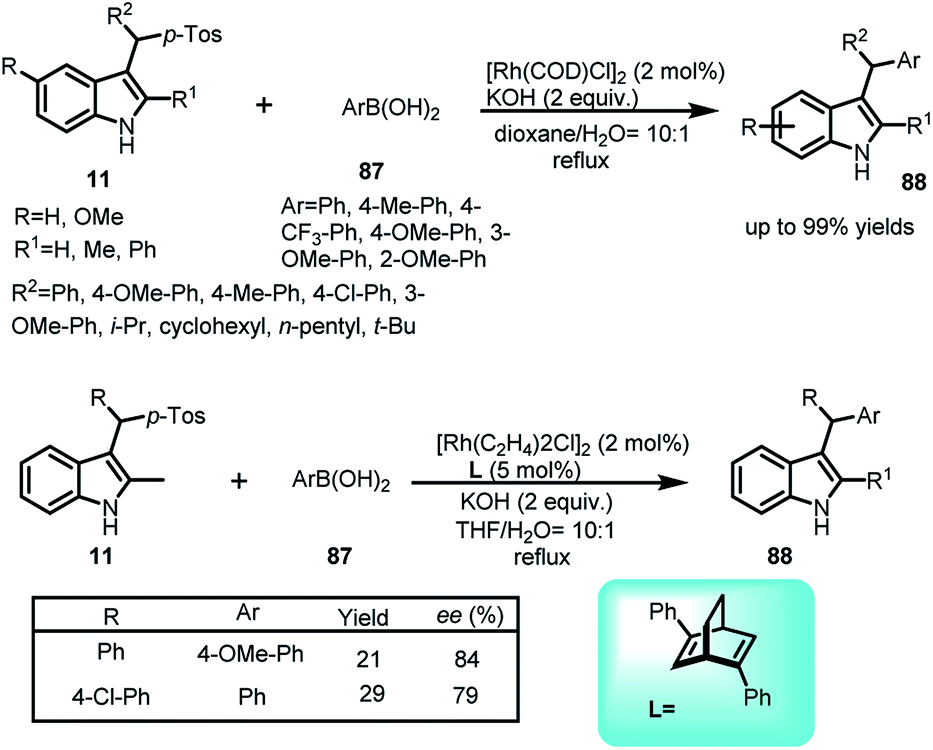 | ||
| Scheme 28 Rhodium catalysed addition of arylboronic acids to arenesulfonyl indoles. | ||
Same reaction was reported by Jiang and co-workers63 using palladium catalyst with tri-1-naphthylphosphine as ligand to obtain 3-sec-alkyl-indole derivatives 88 in 32–90% yields (Scheme 29).
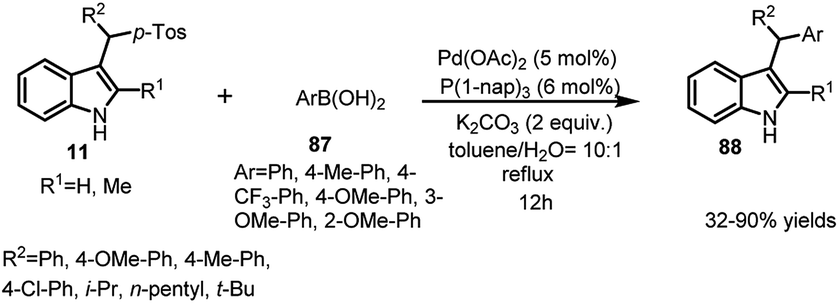 | ||
| Scheme 29 Addition of arylboronic acids to arenesulfonyl indoles catalyzed by palladium acetate. | ||
Jiang64 utilized arenesulfonyl indoles 11 to functionalize the otherwise unreactive α-position of cyclic and acyclic ethers 89, present in various biologically active compounds, medicines and natural products, using di-tert-butyl peroxide (DTBP). The in situ generated vinylogous imine intermediate gets attacked by α-position of ethers to result in the formation of 90 in 45–80% yields, with dr up to 67:33 (Scheme 30). The expected radical mechanism was confirmed by carrying out the reaction in the presence of TEMPO, after which a plausible mechanism was proposed. Homolysis of DTBP at high temperature delivered tert-butoxyl radical, which abstracted a proton from α-position of ether to generate alkoxy radical. The alkoxy radical attacked the imine intermediate (generated in situ from 11 under mild basic conditions) to generate product radical, which finally abstracted proton to give the final adduct.
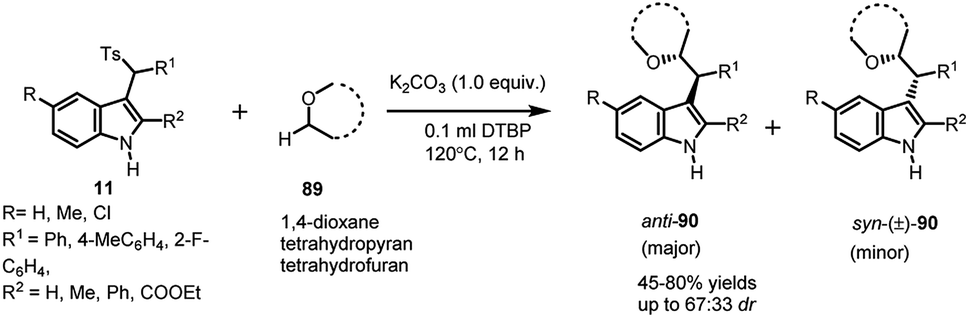 | ||
| Scheme 30 DTBP promoted α-alkylation of ethers with arylsulfnyl indoles. | ||
Petrini's group65 documented the use of Lewis acids for the elimination of ArSO2 group to convert the arenesulfonyl indoles or indazoles into their corresponding vinylogous iminium ions. These ions are quite stable and electrophilic to be able to react with faint nucleophiles such as allyltin reagents or enol ethers. The reaction of (91) with allyltributyl tin (92) in the presence of ethyl aluminium dichloride furnished the corresponding allylic adduct 93 in 46–86% yields (Scheme 31). In order to achieve 3-substituted indoles and indazoles with functionalized side chains, enol ethers and silyl ketene acetals have also been used.
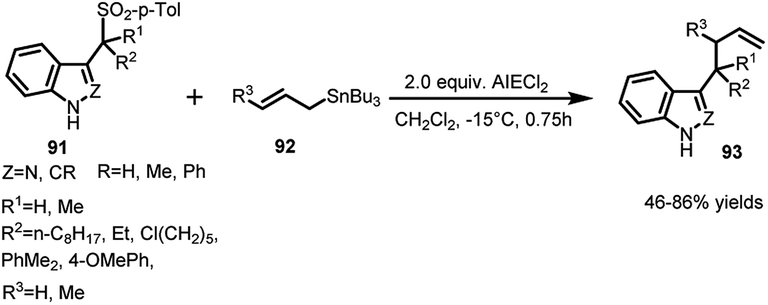 | ||
| Scheme 31 AlEtCl2 assisted allylation of sulfonyl indoles and indazoles. | ||
4. Addition-rearrangement domino reaction
Du66 et al., in 2016, developed a racemic Cs2CO3 promoted Michael addition of sulfur ylides 94 to imine intermediate generated from arenesulfonyl indoles 11 followed by [2,3]-sigmatropic rearrangement domino reaction to obtain 3-substituted indoles 95 and 95′ bearing a sulfide moiety in good to high yields up to 89% (Scheme 32). According to the mechanism proposed by the author, the ylide 96 attacks on the vinylogous imine intermediate 2, generated in situ from one equivalent of Cs2CO3. The Michael adduct further undergoes dehydrogenation from the methyl adjacent to sulfur to provide 97. Finally, 98 undergoes [2,3]-sigmatropic rearrangement to give access to the final adduct 95. The resulting adduct 95 was further reduced to obtain 99 in 62% yield by treatment with LiAlH4 in THF. Further, 95 was transformed to pyranoindole 100 in 4 steps with overall 27% yield.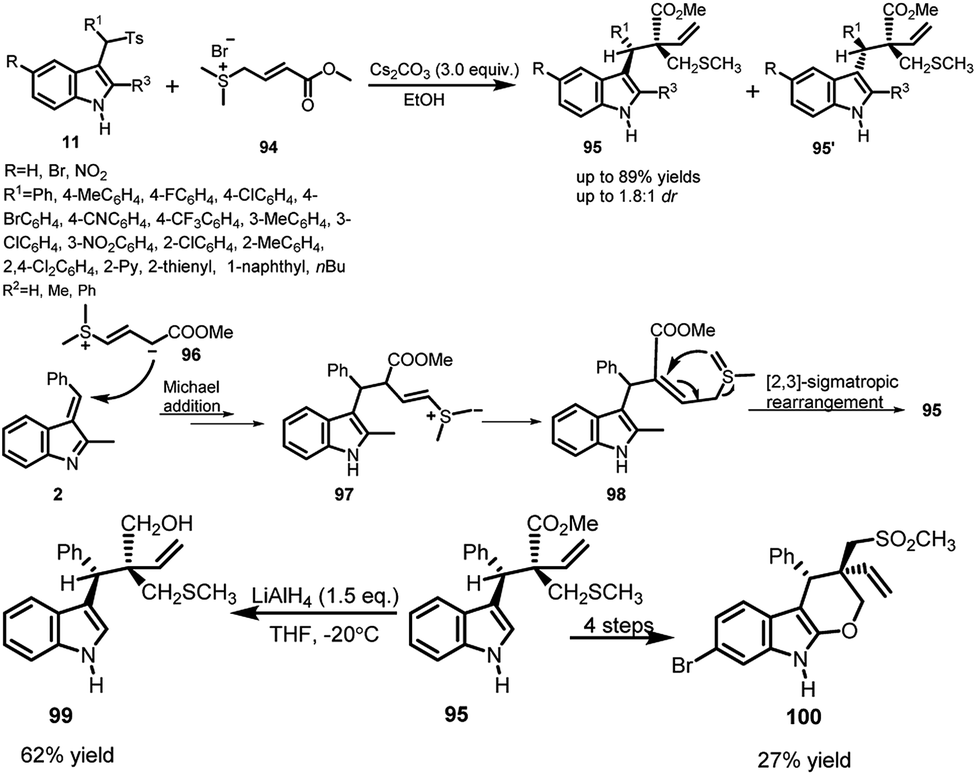 | ||
| Scheme 32 Cs2CO3 promoted [2,3]-sigmatropic rearrangement reaction between arenesulfonyl indoles and sulfur ylides. | ||
5. Miscellaneous reactions
Monofluoromethylation of indole was documented by Matsuzaki et al.67 by carrying out the addition of 1-fluoro-1,1-bis(phenylsulfonyl)methane (FBSM) (101) with arenesulfonyl indoles 11 using chiral ammonium salts derived from cinchona alkaloids XIII at −10 °C (Scheme 33). The resultant (R)-102 was obtained in 54–99% yields and enantiomeric excess up to 97%. Further, reduction of phenylsulfonyl group of 102 was carried out under Mg/MeOH to furnish the monofluoromethylated adduct 103 in 61% yield and 89% ee with 1% loss in enantiopurity. The utility of the reaction was further extended to asymmetric one-pot reaction starting from 2-substituted indoles to obtain 102 in 62–85% yields and 80–83% ee.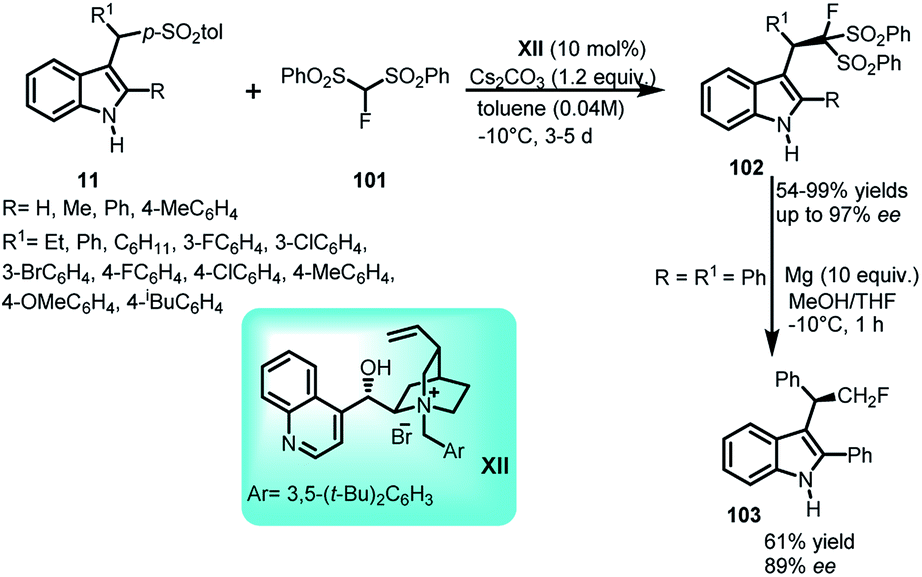 | ||
| Scheme 33 Asymmetric addition of FBSM to arenesulfonyl indoles. | ||
Arenesulfonyl indoles 11 as precursor for procuring indole core have been utilized by Shaikh and co-workers68 in 2008 to carry out the formal α-alkylation of various aldehydes 104 with indolyl derivatives (Scheme 34). Catalyzed by L-proline XIII, this reaction is an example of aminocatalysis where amine binds with substrate to form enamine complex, which then attacks on the stable imine intermediate complex generated by KF/alumina to give 105 in 63–92% yields, and ee up to 92% with dr up to 12:1. The absolute configuration of the corresponding tosylated alcohol obtained by simple aldehyde reduction of 105 was found to be 2S,3R on the basis of X-ray crystallography.
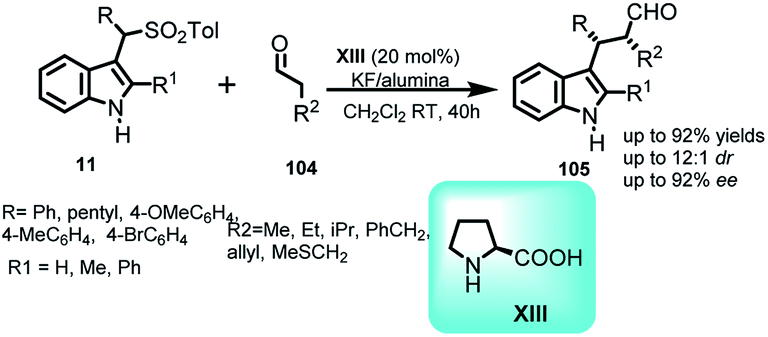 | ||
| Scheme 34 Proline catalyzed α-alkylation of aldehydes with vinylogous imine intermediates derived from arenesulfonyl indoles. | ||
Hou and co-workers69 carried out asymmetric reaction of glycine derivatives 106 with arenesulfonyl indole derivatives 11 at room temperature. Using cesium carbonate as base, this reaction was catalyzed by AgCl and commercially available chiral phosphoramidite ligand XIV to obtain a series of C-3 functionalized indole derivatives 107 in 58–95% yields with anti:syn ratio up to 98:2 and ee of the anti-isomer up to 98% (Scheme 35). The resultant adduct (2S,3S)-107 was conveniently transformed to β-phenyl tryptophan 108 with overall yield of 95% via a two-step route. The enantiomeric excess remained intact throughout the process.
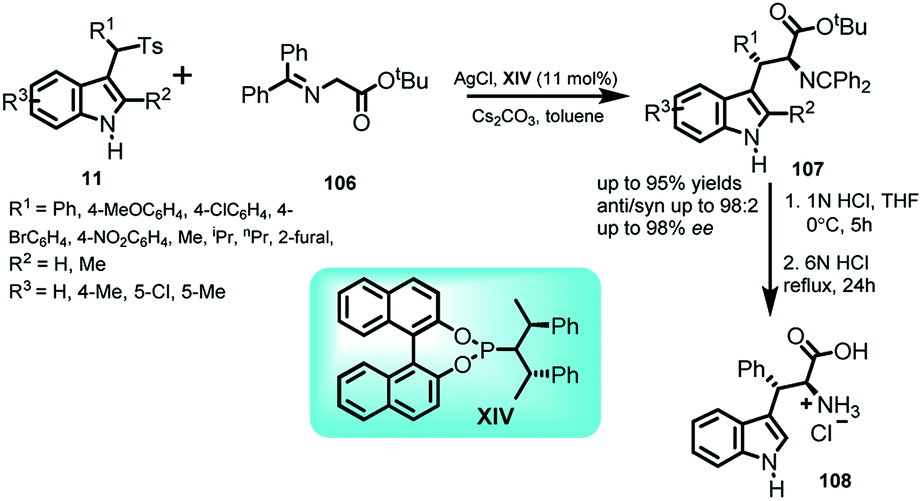 | ||
| Scheme 35 Addition of glycine derivatives to arenesulfonyl indole derivatives. | ||
The reaction of arenesulfonyl indoles 11 with Reformatsky reagents 109 (2-bromo carbonyl derivatives) in zinc metal with THF was reported by Petrini and co-workers22 to procure 3-indolyl propanoate esters 110 in 66–90% yields (Scheme 36). With C-2 position being occupied by ester group, the reactivity of arenesulfonyl indoles was sluggish and hence zinc–copper couple in dichloromethane was used to obtain 110 in 60–61% yields. The diester 110 underwent Dieckmann condensation with sodium hydride in THF under reflux conditions to further get converted to tricyclic β-ketoester 111 in 61% yield. Compounds of this type are pivotal intermediates in synthesizing tetracyclic lactam compounds.70
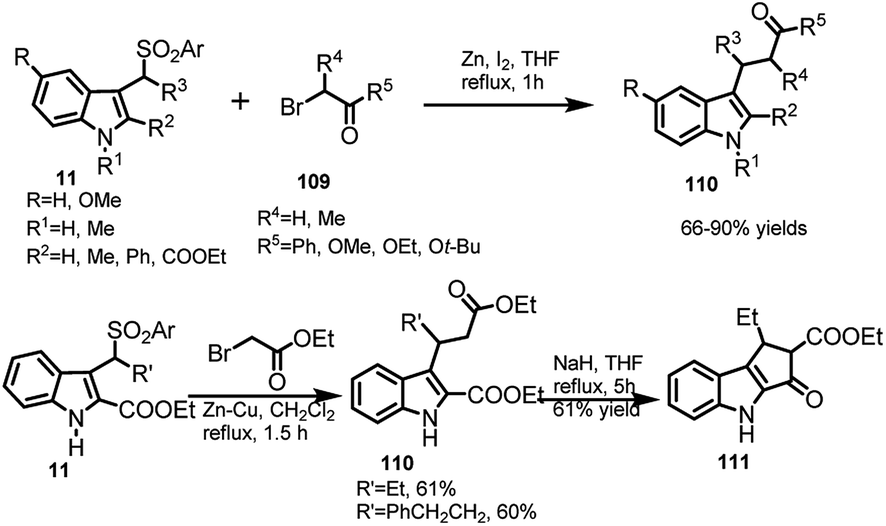 | ||
| Scheme 36 Reaction of 3-(1-arenesulfonylalkyl) indoles with Reformatsky reagents. | ||
3-(1-Tosylalkyl)indoles have been employed by Gong and co-workers71 with oxindoles to obtain 3,3′-disubstituted oxindoles. The asymmetric substitution reaction of 113 with oxindoles 112 catalyzed by quinine derived thiourea catalyst VII leads to the formation of (2S,3S)-114 in 91–97% yields, with dr up to 2.5:1 and ee up to 98% for both diastereomers (Scheme 37). The resultant adduct-114 was further transformed via seven steps to obtain (+)-trigolute B (115), containing spirooxindole core, in overall 18% yield.
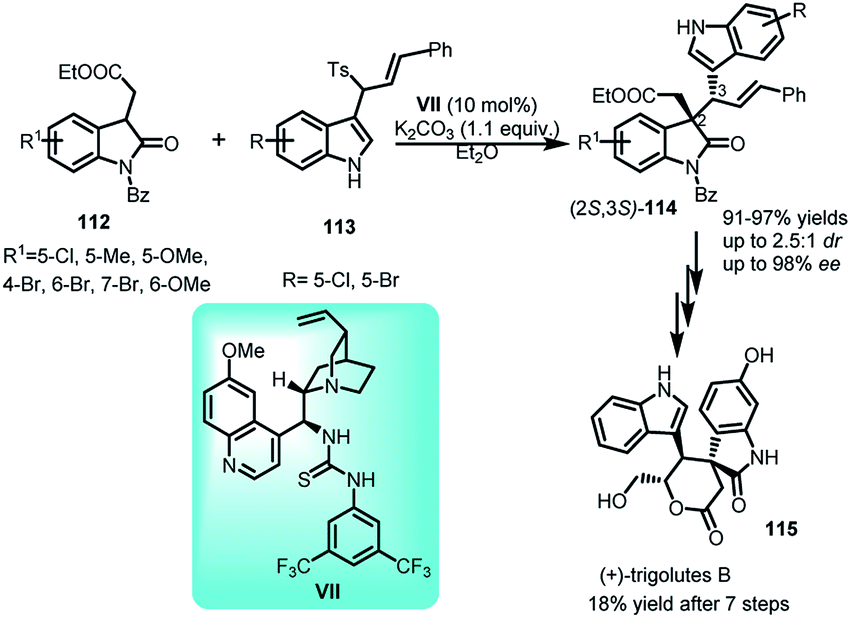 | ||
| Scheme 37 Quinine thiourea catalysed reaction of 3-(1-tosylalkyl)indoles with oxindoles. | ||
The absolute configuration of the major and minor diastereomer was found to be 2S,3S and 2S,3R respectively on the basis of X-ray crystallography. Based on this, a plausible mechanism was proposed (Fig. 2) in which the two isomers of imine intermediate (E) and (Z) isomer gets activated by bifunctional catalyst through double hydrogen bonding to give TS14 and TS15. The (Z)-isomer with lower steric repulsions between benzene ring and R group is thermodynamically more stable and hence the corresponding transition state TS14 provides the major diastereomer. The less energy gap between TS14 and TS15 attributes to the lower dr value.
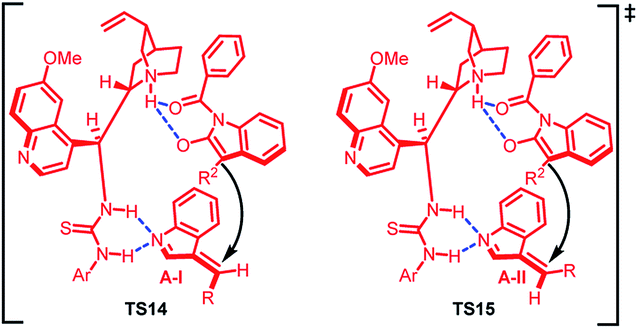 | ||
| Fig. 2 Proposed transition states. | ||
Emphasizing on the importance of indole-diketopiperazine bridge from structural aspect in many complex alkaloids of fungal origin possessing interesting biological activity, Olenyuk and co-workers72 documented the conjugate addition of diketopiperazines 116 to arenesulfonyl indoles 11 in KF/alumina using quinine derived organocatalyst (DHQD)2PHAL XV as the Lewis base to obtain indole derivatives containing indole-diketopiperazine 117 in 46–90% yields with dr up to 5:1 (Scheme 38).
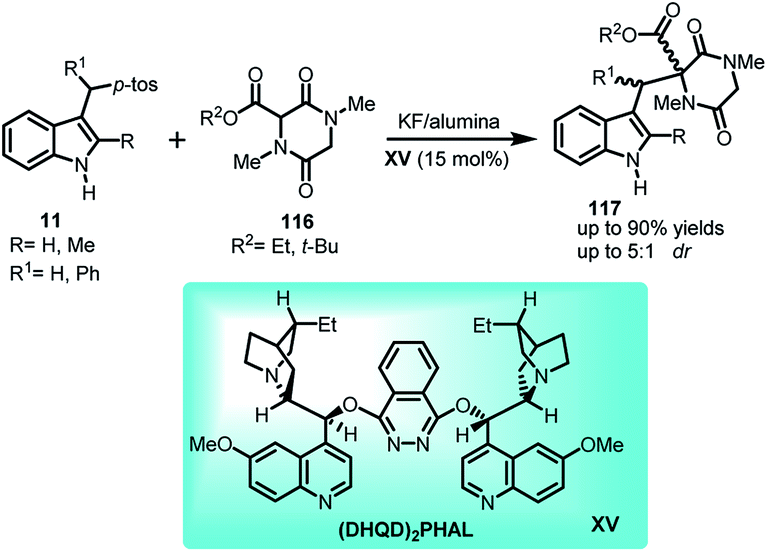 | ||
| Scheme 38 (DHQD)2PHAL catalysed conjugate addition of diketopiperazines to arenesulfonyl indoles. | ||
Petrini and co-workers23 documented the synthesis of arenesulfonyl indoles bearing diverse electrophiles at α-position by carrying out the reaction of 3-(tosylmethyl)indole (15) with different electrophilic reagents 118 in the presence of sodium hydride in DMF at 0 °C. The resultant sulfonyl indoles 16 were readily involved in the base promoted elimination of arenesulfinic acid followed by nucleophilic addition of active methylene compounds to provide 3-substituted indole derivatives 119 in 63–84% yields (Scheme 39).
 | ||
| Scheme 39 Synthesis of C-3 functionalized indole derivatives from 3-(1-tosylmethyl)indoles. | ||
Similarly, 3-[2-(1-alkyltriazol-4-yl)-1-tosylethyl]indoles (122) (containing 1,4-disubstituted triazole) conveniently underwent attack of methylene compounds facilitating the elimination of tosyl group under basic conditions to procure C-3 functionalized indole derivatives.73 3-(1-Tosylmethyl)indoles (15) were utilized as starting materials to synthesize 3-[2-(1-alkyltriazol-4-yl)-1-tosylethyl]indoles (122) in 70–81% yields via copper catalysed click cycloaddition of N-(tert-butoxycarbonyl)-3-(1-tosyl-3-butynyl)-1H-indole (120) with various aromatic and aliphatic azides 121. The resultant adduct-122 was then deprotected using TFA in dichloromethane, followed by sodium hydride assisted attack of diethyl malonate and malononitrile at the carbon bearing tosyl group leading to functionalized indole adducts 123 in 73–76% yields (Scheme 40).
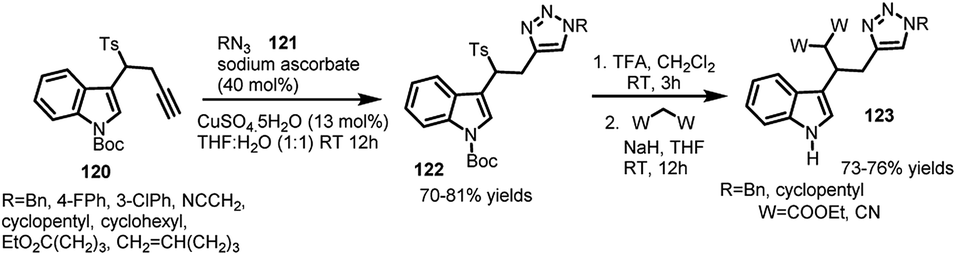 | ||
| Scheme 40 Cu catalysed click cycloaddition of 3-(1-tosyl-3-butynyl)-1H-indole. | ||
6. Summary and outlook
Arenesulfonyl indoles have proven to be excellent precursors and a viable alternative to Friedel–Crafts reaction for the synthesizing C-3 substituted indole derivatives. The propensity of arenesulfinic group to undergo elimination under mild basic conditions and obtain vinylogous imine intermediate opens new synthetic opportunities for the suitable functionalization of indoles at C-3 position by reaction with various nucleophilic reagents. This review confers the wide scope of reactions undergone by arenesulfonyl indoles since its serendipitous synthesis in 2006 by Petrini till 2020 such as Michael addition, Friedel–Crafts reaction, arylation and a few more. Different catalytic methodologies have been utilized during this tenure especially cinchona-based urea/thiourea catalysts, with a few examples with metal catalysis. Despite this, there is still abundant scope for this molecule as an electrophile with variety of nucleophiles which have not been explored yet to furnish functionalized indole derivatives with tertiary as well as quaternary C-3 center. Incorporation of different heterocycles at the 1′-position of arenesulfonyl indoles leads to new substrates which can further act as a crucial electrophile to provide indole derivatives containing quaternary stereocenter.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the research project (02(0359)/19/EMR-II) sanctioned to SSC by the CSIR, India. BPK is thankful to Council of Scientific and Industrial Research (CSIR), India for SRF fellowship. Financial support from the Department of Science and Technology (DST), India under FIST programme and UGC, India, under CAS-II and UPE programme is highly acknowledged.References
- M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Org. Biomol. Chem., 2019, 17, 3670–3708 RSC.
- P. Brandão and A. J. Burke, Tetrahedron, 2018, 74, 4927–4957 CrossRef.
- M. Kaur, M. Singh, N. Chadha and O. Silakari, Eur. J. Med. Chem., 2016, 123, 858–894 CrossRef CAS.
- J. Alemán and S. Cabrera, Chem. Soc. Rev., 2013, 42, 774–793 RSC; V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793 CrossRef CAS.
- R. J. Sundberg, The Chemistry of Indoles, Academic, New York, 1996 CAS; K. Swathi, A. Srinivas and M. Sarangapani, J. Chem. Pharm. Res., 2010, 2, 220–225 CAS.
- J. L. Stanton and M. H. Ackermann, J. Med. Chem., 1983, 26, 986–989 CrossRef CAS.
- R. A. Al-Qawasmeh, M. Huesca, V. Nedunuri, R. Peralta, J. Wright, Y. Lee and A. Young, Bioorg. Med. Chem. Lett., 2010, 20, 3518–3520 CrossRef CAS.
- G. A. Khan, J. A. War, A. Kumar, I. A. Sheikh, A. Saxena and R. Das, J. Taib Univ. Sci., 2017, 11, 910–921 CrossRef.
- C. A. Winter, E. A. Risley and G. W. Nuss, J. Pharmacol. Exp. Ther., 1963, 141, 369–376 CAS.
- M. Nazir, M. A. Abbasi, A.-U. Rehman, S. Z. Siddiqui, K. M. Khan, Kanwal, U. Salar, M. Shahid, M. Ashraf, M. A. Lodhi and F. A. Khan, Bioorg. Chem., 2018, 81, 253–263 CrossRef CAS.
- B. Venugopalan, P. D. Desai and N. J. DeSouza, J. Heterocycl. Chem., 1988, 25, 1633–1639 CrossRef CAS.
- J.-R. Weng, C.-H. Tsai, S. K. Kulp and C.-S. Chen, Cancer Lett., 2008, 262, 153–163 CrossRef CAS.
- S. A. Patil, R. Patil and D. D. Miller, Future Med. Chem., 2012, 4, 2085–2115 CrossRef CAS.
- I. P. Beletskaya and A. D. Averin, Curr. Organocatal., 2016, 3, 60–83 CrossRef CAS.
- J. Kaur, N. Islam, A. Kumar, V. K. Bhardwaj and S. S. Chimni, Tetrahedron, 2016, 72, 8042–8049 CrossRef CAS.
- J. Chen, Z.-C. Geng, N. Li, X.-F. Huang, F.-F. Pan and X.-W. Wang, J. Org. Chem., 2013, 78, 2362–2372 CrossRef CAS.
- B. G. Gower and E. Leete, J. Am. Chem. Soc., 1963, 85, 3683–3685 CrossRef CAS.
- B. B. Semenov and V. G. Granik, Pharm. Chem. J., 2004, 38, 287–310 CrossRef CAS.
- A. Palmieri, M. Petrini and R. R. Shaikh, Org. Biomol. Chem., 2010, 8, 1259–1270 RSC.
- F. Martinelli, A. Palmieri and M. Petrini, Arylsulfonyl Group: Activating Properties and Recent Synthetic Applications, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186(5), 1032–1045 CrossRef CAS.
- R. Ballini, A. Palmieri, M. Petrini and E. Torregiani, Org. Lett., 2006, 8, 4093–4096 CrossRef CAS.
- A. Palmieri and M. Petrini, J. Org. Chem., 2007, 72, 1863–1866 CrossRef CAS.
- A. Palmieri, M. Petrini and R. R. Shaikh, Synlett, 2008, 12, 1845–1851 Search PubMed.
- Reviews on Michael addition: N. Krause and A. H. Roder, Synthesis, 2001, 2, 171–196 CrossRef.
- D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058–11076 CrossRef CAS.
- R. M. de Figueiredo, A. Mazziotta, D. P. de Sant'Ana, C. Palumbo and T. Gasperi, Curr. Org. Chem., 2012, 16, 2231–2289 CrossRef.
- A. D. Mola, L. Palombi and A. Massa, Curr. Org. Chem., 2012, 16, 2302–2320 CrossRef.
- L. Jing, J. Wei, L. Zhou, Z. Huang, Z. Li, D. Wu, H. Xiang and X. Zhou, Chem.–Eur. J., 2010, 16, 10955–10958 CrossRef CAS.
- X.-L. Zhu, W.-J. He, L.-L. Yu, C.-W. Cai, Z.-L. Zuo, D.-B. Qin, Q.-Z. Liu and L.-H. Jing, Adv. Synth. Catal., 2012, 354, 2965–2970 CrossRef CAS.
- Z. Zuo, S. Zhang, R. Wang, W. He, S. Wu, X. Xie, D. Qin and L. Jing, Synthesis, 2013, 45, 2832–2842 CrossRef CAS.
- T. R. Hodges, N. M. Benjamin and S. F. Martin, Org. Lett., 2017, 19, 2254–2257 CrossRef CAS.
- K. N. Babu, A. Roy, M. Singh and A. Bisai, Org. Lett., 2018, 20, 6327–6331 CrossRef CAS.
- M. C. Dobish and J. N. Johnston, Org. Lett., 2010, 12, 5744–5747 CrossRef CAS.
- M. Fochi, L. Gramigna, A. Mazzanti, S. Duce, S. Fantini, A. Palmieri, M. Petrini and L. Bernardi, Adv. Synth. Catal., 2012, 354, 1373–1380 CrossRef CAS.
- J.-Z. Huang, X. Wu and L.-Z. Gong, Adv. Synth. Catal., 2013, 355, 2531–2537 CrossRef CAS.
- J. E. De Lorbe, S. Y. Jabri, S. M. Mennen, L. E. Overman and F.-L. Zhang, J. Am. Chem. Soc., 2011, 133, 6549–6552 CrossRef CAS.
- R. Ballini, A. Palmieri, M. Petrini and R. R. Shaikh, Adv. Synth. Catal., 2008, 350, 129–134 CrossRef CAS.
- R. Ballini, S. Gabrielli, A. Palmieri and M. Petrini, Adv. Synth. Catal., 2010, 352, 2459–2462 CrossRef CAS.
- C.-W. Cai, X.-L. Zhu, S. Wu, Z.-L. Zuo, L.-L. Yu, D.-B. Qin, Q.-Z. Liu and L. H. Jing, Eur. J. Org. Chem., 2013, 456–459 CrossRef CAS.
- S. A. Adediran, D. Cabaret, B. Drouillat, R. F. Pratt and M. Wakselman, Bioorg. Med. Chem., 2001, 9, 1175–1183 CrossRef CAS.
- X. Luo, L. Wang, L. Peng, J. Bai, L. Jia, F. Tian, X. Xu and L. Wang, Chin. J. Chem., 2012, 30, 2703–2706 CAS.
- A. A. Hussein, J. J. M. Meyer, M. L. Jimeno and B. J. Rodrίguez, J. Nat. Prod., 2007, 70, 293–295 CrossRef CAS.
- R. Wang, L. Jing and D. Qin, Tetrahedron Lett., 2015, 56, 2867–2870 CrossRef CAS.
- P. Chen, S.-M. Lu, W. Guo, Y. Liu and C. Li, Chem. Commun., 2016, 52, 96–99 RSC.
- J. Wang, S. Zhou, D. Lin, X. Ding, H. Jiang and H. Liu, Chem. Commun., 2011, 47, 8355–8357 RSC.
- D. Wilcke, E. Herdtweck and T. Bach, Synlett, 2011, 9, 1235–1238 Search PubMed.
- T. P. Pathak, K. M. Gligorich, B. E. Welm and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 7870–7871 CrossRef CAS.
- L. Yu, X. Xie, S. Wu, R. Wang, W. He, D. Qin, Q. Liu and L. Jing, Tetrahedron Lett., 2013, 54, 3675–3678 CrossRef CAS.
- C.-H. Chang and J.-L. Han, ChemistrySelect, 2016, 1, 5628–5632 CrossRef CAS.
- Z.-H. Xing, Y. Zhang, Y. Wang, X.-P. Xu and S.-J. Ji, Tetrahedron Lett., 2017, 58, 1094–1097 CrossRef CAS.
- J.-L. Han, Y.-T. Liao and C.-H. Chang, Eur. J. Org. Chem., 2019, 2019, 5815–5823 CrossRef CAS.
- C.-H. Chang, N. S. Kumar, Y.-T. Liao, H.-T. Chen and J.-L. Han, Adv. Synth. Catal., 2020, 362, 903–912 CrossRef CAS.
- Y. Que and H. He, Eur. J. Org. Chem., 2020, 5917–5925 Search PubMed; O. Hollóczki, Chem.–Eur. J., 2020, 26, 4885–4894 CrossRef CAS.
- Y. Li, F.-Q. Shi, Q.-L. He and S.-L. You, Org. Lett., 2009, 11, 3182–3185 CrossRef CAS.
- J. Luo, B. Wu, M.-W. Chen, G.-F. Jiang and Y.-G. Zhou, Org. Lett., 2014, 16, 2578–2581 CrossRef CAS.
- A. Chiurchiù, A. Palmieri and M. Petrini, Arkivoc, 2019, 4, 69–79 Search PubMed.
- X. Li, S. Chu, V. A. Feher, M. Khalili, Z. Nie, S. Margosiak, V. Nikulin, J. Levin, K. G. Sprankle, M. E. Tedder, R. Almassy, K. Appelt and K. M. Yager, J. Med. Chem., 2003, 46, 5663–5673 CrossRef CAS.
- X. Lin, J. Busch-Petersen, J. Deng, C. Edwards, Z. Zhang and J. K. Kerns, Synlett, 2008, 20, 3216–3220 CrossRef.
- S. Campetella, A. Palmieri and M. Petrini, Eur. J. Org. Chem., 2009, 2009, 3184–3188 CrossRef.
- L. Ge, M. Zurro and S. R. Harutyunyan, Chem.–Eur. J., 2020, 26, 16277–16280 CrossRef CAS.
- L. Yuan, A. Palmieri and M. Petrini, Adv. Synth. Catal., 2020, 362, 1509–1513 CrossRef CAS.
- L.-L. Cao, Z.-S. Ye, G.-F. Jiang and Y.-G. Zhou, Adv. Synth. Catal., 2011, 353, 3352–3356 CrossRef CAS.
- L.-L. Cao, X.-N. Li, F.-Y. Meng and G.-F. Jiang, Tetrahedron Lett., 2012, 53, 3873–3875 CrossRef CAS.
- Z. Gu, Y. Tang and G.-F. Jiang, J. Org. Chem., 2017, 82, 5441–5448 CrossRef CAS.
- L. Marsili, A. Palmieri and M. Petrini, Org. Biomol. Chem., 2010, 8, 706–712 RSC.
- Y. Du, A. Yu, Y. Zhang, J. Jia and X. Meng, Asian J. Org. Chem., 2016, 5, 1309–1313 CrossRef CAS.
- K. Matsuzaki, T. Furukawa, E. Tokunaga, T. Matsumoto, M. Shiro and N. Shibata, Org. Lett., 2013, 15, 3282–3285 CrossRef CAS.
- R. R. Shaikh, A. Mazzanti, M. Petrini, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2008, 47, 8707–8710 CrossRef CAS.
- B.-H. Zheng, C.-H. Ding, X.-L. Hou and L.-X. Dai, Org. Lett., 2010, 12, 1688–1691 CrossRef CAS.
- M.-L. Bennasar, T. Roca, R. Griera and J. Bosch, J. Org. Chem., 2001, 66, 7547–7551 CrossRef CAS.
- J.-Z. Huang, C.-L. Zhang, Y.-F. Zhu, L.-L. Li, D.-F. Chen, Z.-Y. Han and L.-Z. Gong, Chem.–Eur. J., 2015, 21, 8389–8393 CrossRef CAS.
- R. Dubey and B. Olenyuk, Tetrahedron Lett., 2010, 51, 609–612 CrossRef CAS.
- M. Petrini and R. R. Shaikh, Synthesis, 2009, 18, 3143–3149 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |