
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEco-friendly multivariate curve resolution-alternating least squares and chromatographic quantifications of some veterinary drug residues in pharmaceutical industrial wastewater†
Osama I. Abdel Sattara,
Hamed H. M. Abuseadaa,
Mohamed S. Emara a and
Mahmoud Rabee*b
a and
Mahmoud Rabee*b
aPharmaceutical Analytical Chemistry Dept., Faculty of Pharmacy, Al-Azhar University, 11751, Nasr City, Cairo, Egypt
bPharmaceutical Chemistry Department, Heliopolis University, Cairo-Belbeis Desert Rd, El-Salam, Cairo Governorate 11777, Egypt. E-mail: Mahmoud.rabee@hu.edu.eg; Tel: +20-1125490478
First published on 14th January 2021
Abstract
Three eco-friendly and cost-effective analytical methods were developed and optimized for quantitative analysis of some veterinary drug residues in production wastewater samples. The studied drugs were ivermectin, rafoxanide and sulfadimidine. A solid-phase extraction procedure was employed using Bond Elut C18 cartridges, prior to analysis. The first method was a chemometric approach called multivariate curve resolution – alternating least squares (MCR-ALS). A calibration model was developed and several figures of merit (RMSEP, SEP, bias, RE%) were calculated. The second method was a thin layer chromatography followed by densitometric measurements at 245 nm. The separation was performed using silica gel 60 F254 plates and ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile:toluene:ammonia (20:3:2:1, by volume) as a developing system. The third method was a high performance liquid chromatographic separation on HiQsil C18 HS column with UV detection at 245 nm. The mobile phase consisted of acetonitrile:methanol:water (60:25:15, by volume), with a flow rate of 1.5 mL min−1. The proposed methods were validated according to ICH guidelines. The described procedures were applied to quantify the studied drug residues in synthetic and real industrial wastewater samples. The proposed methods were statistically compared with the official and the reported methods, showing no significant difference with respect to accuracy and precision at P = 0.05.
:acetonitrile:toluene:ammonia (20:3:2:1, by volume) as a developing system. The third method was a high performance liquid chromatographic separation on HiQsil C18 HS column with UV detection at 245 nm. The mobile phase consisted of acetonitrile:methanol:water (60:25:15, by volume), with a flow rate of 1.5 mL min−1. The proposed methods were validated according to ICH guidelines. The described procedures were applied to quantify the studied drug residues in synthetic and real industrial wastewater samples. The proposed methods were statistically compared with the official and the reported methods, showing no significant difference with respect to accuracy and precision at P = 0.05.
1 Introduction
Analysis of pharmaceutical residues in the aquatic environment is considered to be an emergent research area. These pharmaceuticals (human or veterinary drugs) are frequently being released to the aquatic environment mainly through industrial processes, improper disposal, or metabolic excretions.1 Veterinary drug residues may also occur in aquatic systems due to manure application to soils.2 In the last few years, the amounts of veterinary drugs released into the environment were much higher due to the lack of control in relation to types and amounts of drugs in use, mainly when considering countries such as China or other Southeast Asia Nations.3Veterinary pharmaceuticals have been classified as emerging contaminants and established as parameters to be controlled in the environmental quality regulations.4 Anthelmintics and antibiotics are considered to be the most widely used veterinary pharmaceuticals. They are administered to a wide range of animals in agriculture and aquaculture, and comprise a large sector of the animal pharmaceutical industry.4 In this study, three of the most frequently used veterinary drugs were selected to be analyzed in industrial wastewater samples. Their names were ivermectin (IVM), rafoxanide (RFX), and sulfadimidine (SDD) = sulfamethazine.
IVM (Fig. S1†) is a macrocyclic lactone drug, chemically known as 22,23-dihydroavermectin B1a and 22,23-dihydroavermectin B1b. It is well known for its broad-spectrum anti-parasitic activity, therefore it has been widely used for controlling helminthes and ectoparasites in the veterinary field.5 Different methods were reported for IVM determination in various specimens, such as HPLC6,7 and LC-MS/MS.8,9
RFX (Fig. S1†) is a potent anthelmintic, chemically known as N-[3-chloro-4-(4-chloro-phenoxy)phenyl]-2-hydroxy-3,5-diiodobenzamide, widely used in veterinary medicine for the treatment of fascioliasis in sheep and cattle.10 Different techniques were reported for RFX determination, like spectrophotometry,11 HPLC,12 and TLC-densitometry.13
SDD (Fig. S1†) is 4-amino-N-(4,6-dimethyl-2-pyrimidine)benzene sulfonamide, is also called sulfamethazine (SMZ).14 It is a broad-spectrum antibacterial drug, belongs to the sulfonamide class. It is widely used in veterinary practice to treat livestock diseases such as gastrointestinal and respiratory tract infections.14 Various methods were reported for SDD determination, such as LC-MS/MS,15 HPLC.16,17
Pharmaceutical residues are expected to occur in concentrations ranging from ng L−1 to ppb levels, but due to their widespread usage, accumulations, and biological activities, they represent a significant problem.18 Recent review papers discussed various aspects of the ecotoxicology of pharmaceuticals in the environment.19,20
Therefore, novel, sensitive, and selective analytical methods are required for quantitative analysis of these residues in the different aquatic systems, especially industrial wastewater.21 The data obtained from these methods may serve to optimize the treatment processes in wastewater treatment plants (WWTPs), preventing the release of these undesired pollutants into the environment. Furthermore, the analytical data about the actual concentrations of pharmaceuticals in aquatic systems may help to provide clear environmental risk assessments.22
Due to their remarkable toxicities and biological activities, several analytical methods were proposed for quantitative analysis of antibiotics and veterinary drug residues in water samples. These methods mainly based on liquid chromatography-tandem mass spectrometry23,24 or gas chromatography–mass spectrometry.25,26 Despite the high sensitivity and selectivity of these methods, they require tediously and time-consuming procedures. As well as the requirement of expensive solvents and sophisticated instruments. Recently, several ambient MS-based techniques have been reported for the direct analysis of the pharmaceutical contaminants in different environmental systems, including desorption electrospray ionization,27 paper spray ionization,28 and filter cone spray ionization coupled to a portable MS system.29 These “ambient MS” methods aimed at overcoming the significant drawbacks of the “hyphenated MS” methods by reducing complexity, consumables load, and time/cost of analysis. High-performance liquid chromatography coupled with ultraviolet/fluorescence detection was also employed in residual analysis to a minor extent.30,31
Nowadays, chemometric/multivariate spectrophotometric methods were recognized to be innovative and promising techniques in this field.32 As they have the advantage of being simple and cost-effective.33 They also overcome the significant problems of matrix interferences and overlapped spectra which present in univariate or conventional spectrophotometric methods.34–36 Thin-layer chromatography coupled with densitometry has also the advantage of being a simple, rapid, and inexpensive method, compared to other chromatographic methods.37 Table 1 shows a direct comparison between the chemometric/multivariate spectrophotometric methods e.g. MCR-ALS and the other traditional chromatographic methods in terms of solvents consumption, consumables load, amount of energy used per sample, time of analysis per sample, and total costs to perform each method.
| MCR-ALS | TLC-densitometry | LC/GC | LC-MS/GC-MS | |
|---|---|---|---|---|
| a kW h = kilowatt per hour, is a unit of energy equal to 3600 kilojoules. | ||||
| Solvents consumption | Low | Low | Moderate to high | High |
| Consumables load | Low | Low | Moderate to high | High |
| Energy useda,54 | <0.1 kW h per sample | <0.1 kW h per sample | ≤1.5 kW h per sample | >1.5 kW h per sample |
| Time of analysis per sample | Seconds to minutes | Minutes | Minutes to hours | Minutes to hours |
| Total costs | Very low cost | Low cost | Low to moderate cost | High cost |
Most of the proposed methods require a sample preparation step prior to analysis, to isolate and concentrate the target analytes from the complex matrices.18 Classical extraction procedures, such as liquid–liquid extraction have the disadvantages of consuming large volumes of solvents and being extremely low in selectivity. Solid-phase extraction (SPE) and solid-phase micro-extraction (SPME) are used as effective alternatives.38
Analytical chemistry plays an important role in the sustainable development of the planet.39 This is true, not only for monitoring of pollutants in the environment but also for the development of more sustainable processes. Therefore, the analytical methodologies should be of high quality, such as being accurate, precise, and sensitive; and the method itself should be as environmentally sustainable as possible.39
The main purpose of this study was to develop validated, eco-friendly, and cost-effective (environmentally sustainable) analytical methods. The first one was a chemo-metric/multivariate spectrophotometric method called multivariate curve resolution – alternating least squares (MCR-ALS). The other methods are chromatographic methods (TLC-densitometry and HPLC). These accurate methods are suitable for simultaneous determination and routine quality inspection of the studied drugs in production wastewater. Extraction and pre-concentration of the target analytes from the synthetic and the real wastewater samples were carried out by using the solid-phase extraction (SPE) technique.
2 Experimental
2.1 Instruments
For spectrophotometric measurements, a UV-vis spectrophotometer model AE-S90-MD from A & E Lab (UK) with a 1 cm micro quartz cuvette (1.4 mL volume) was used. The range of wavelengths was 200–400 nm, with a bandwidth of 1 nm. Spectra were automatically acquired by UV-vis analyst 5.37 software and exported as Excel files. Microsoft Excel 2013 was used for plotting the acquired spectra. The MCR-ALS model was developed by using MATLAB 2012a version 7.14.0.739 with MCR-ALS 2.0 toolbox, in addition to the use of the Unscrambler X 10.4 chemometric software.TLC densitometry was performed using TLC aluminum plates (20 × 10 cm, 0.20 mm) pre-coated with silica gel 60 F254 (EMD Millipore, Sigma Aldrich). Samples were applied by CAMAG LINOMAT V (Muttenz, Switzerland) automatic applicator with 100 μL micro-syringe (Hamilton, Switzerland), and plates were scanned at 245 nm with TLC Scanner 3 operated with winCATS software (Camag, Switzerland).
HPLC system model 1100 (Agilent Technologies, USA), with a variable wavelength detector, a quaternary pump, an autosampler, and a chromatographic column of HiQsil C18 HS column (250 × 4.6 mm, 5 μm particle size) were used.
Solid-phase extraction (SPE) was performed using Bond Elut C18 cartridges (Agilent, USA). The cartridges were placed on the SPE apparatus which consists of a 12-port vacuum manifold with drying attachment.
2.2 Materials
2.3 Standard solutions
Stock solutions of IVM, RFX and SDD were prepared by dissolving 100 mg of each drug in 100 mL methanol to prepare a solution with a concentration of 1000 μg mL−1. Working standard solutions of each drug were freshly prepared by dilution from their stock solutions with methanol to obtain a concentration of 100 μg mL−1.2.4 Collection and storage of samples
Five wastewater samples were collected in amber glass bottles from different production areas of some pharmaceutical industries located at different sites in Egypt. Before extraction, samples were filtered immediately through 0.45 μm nylon membrane filters to eliminate the suspended matter. The filtered volume of each sample was approximately 200 mL. The samples were stored at 4 °C and protected from light to prevent any degradation or deterioration as previously recommended.402.5 Procedures
2.5.2.1 Theory. MCR-ALS represents a chemometric approach known as self-modeling mixture analysis. It aims to recover spectra and concentration profiles of pure components in an unresolved mixture using a minimal number of assumptions about the nature and composition of these mixtures.41 Initially, it assumes a bilinear relation between the data matrix (spectra of mixture samples), components pure spectra, and components concentrations. Second, it aims to decompose this bilinear data matrix into pure individual components, according to the following eqn (1):
| D = CST + E | (1) |
To achieve complete decomposition or resolution of data matrix, an iterative alternating least squares (ALS) procedure is employed.44 To initiate this ALS procedure, initial estimations of C and ST are required. Initial estimations of C and ST represent a vital step because various estimations by the proposed model may lead to diverse results. Various algorithms are employed for this purpose like evolving factor analysis (EFA)45 or the determination of purest variables.44 In this study, known pure spectra and concentrations of each target analyte (purest variables) were applied for initial estimations.
For optimizing the proposed MCR-ALS model several constraints can be applied like non-negativity, closure, unimodality and correlation constraints.46 Non-negativity constraint enforces concentrations and/or spectra to be greater than or equal zero. Correlation constraint allows simultaneous quantitative analysis of analytes in the presence of unknown interfering substances leading to enhance the selectivity of the proposed model. In this study, both non-negativity and correlation constraints were employed and the developed calibration model was then used for predicting concentrations of samples in both the validation and test sets. For more details about the technique, readers are referred to ref. 47 and 48.
2.5.2.2 Construction of calibration model and validation set. A three-factor five-level experimental design was constructed to develop the calibration model of IVM, RFX and SDD in the concentration range of 1–10 μg mL−1. A set of 20 calibration mixture solutions of the three target analytes were prepared. A validation set of further 15 mixture solutions containing different concentrations within the same concentration range were prepared. The calibration model and validation set design are represented in Table S1.† The UV spectra of all samples were scanned over the wavelength range of 200–400 nm with a 1 nm interval. Spectra were automatically acquired by UV-vis analyst 5.37 software and exported as Excel files. The further handling by the chemometric software to build up the MCR-ALS model was employed.
2.5.2.3 Evaluation of quantitative prediction capability of the proposed MCR-ALS model. To evaluate the quantitative prediction capability of the proposed MCR-ALS model, the model was employed for prediction of concentrations of the studied drugs in the mixture solutions of the validation set. The model performance was also evaluated by calculating several figures of merit, such as root mean square error of prediction (RMSEP), bias, standard error of prediction (SEP) and relative percentage error in the concentration predictions (RE%) according to the following eqn (2)–(5):43
-Root mean square error of prediction (RMSEP):
 | (2) |
-Bias, is the average value of the difference between predicted and measured values:
 | (3) |
-Standard error of prediction (SEP):
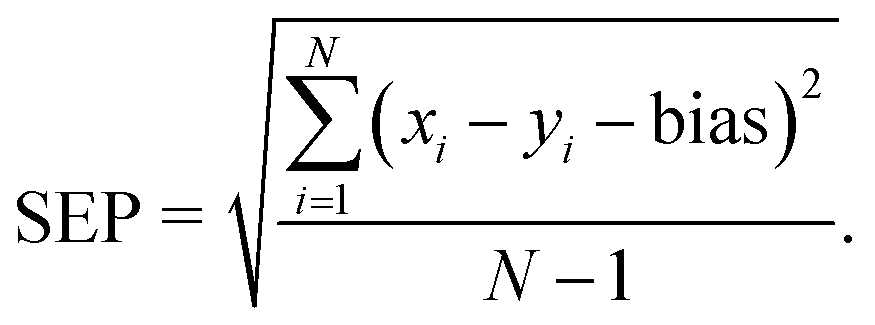 | (4) |
-Relative percentage error in the concentration predictions (RE%)
 | (5) |
Rotational ambiguity represents a problem that may occur during the development of the MCR-ALS model due to the presence of matrix interferences, especially in complex matrices like wastewater.49 In this study, SPE accompanied by MCR-ALS was employed to reduce the matrix interferences and eliminate the occurrence of such a problem.
2.5.3.1 Chromatographic conditions. Samples were applied as bands on TLC aluminum plates (20 × 10 cm, 0.20 mm) pre-coated with silica gel 60 F254 by Camag Linomat 5 automatic applicator using 100 μL Hamilton micro-syringe. The bandwidth was adjusted to be 6 mm. Each band was spaced 1 cm apart from each other and 1.5 cm from the bottom edge of the plate. The chromatographic chamber was pre-saturated with the mobile phase for 30 min. The plates were developed to a distance of approximately 8 cm by ascending technique using ethyl acetate
:acetonitrile:toluene:ammonia (33%) (20:3:2:1, by volume) as a mobile phase. The plates were dried in air at room temperature and scanned at 245 nm using Camag TLC scanner 3. This TLC scanner was operated in the absorbance mode with a deuterium lamp as a source of radiation. The slit dimension was kept at 3 mm × 0.45 mm and scanning speed was employed to be 20 mm s−1.
2.5.3.2 Construction of calibration curves. Aliquots (1–10 μL) from each working standard solution (100 μg mL−1) of IVM, RFX and SDD were accurately applied on TLC plates to obtain equivalent concentrations ranged from 0.1–1 μg per band for each drug. Calibration curves were constructed between peak areas of the studied drug at 245 nm versus their concentrations. The acquired regression equations were used to calculate the concentration of each drug throughout the whole study.
2.5.4.1 Chromatographic conditions. HPLC chromatographic separation was achieved using a HiQsil C18 HS column (250 × 4.6 mm, 5 μm particle size), using an isocratic elution of a mobile phase consisting of acetonitrile
:methanol:water (60:25:15, by volume), with a flow rate of 1.5 mL min−1. Analysis were employed at ambient temperature, and detection was carried out at 245 nm. The injection volume was 20 μL.
2.5.4.2 Construction of calibration curves. Aliquots from each working standard solution (100 μg mL−1) of IVM, RFX and SDD were accurately transferred into a series of 10 mL volumetric flasks. Each flask was completed with methanol to obtain a concentration range of 0.5–30 μg mL−1. These solutions were injected (20 μL) in triplicate into the HPLC system. The chromatographic conditions were employed, and the chromatograms were recorded. Calibration curves were constructed between peak areas of the studied drugs at 245 nm versus their concentrations. The acquired regression equations were used to calculate the concentration of each drug throughout the whole study.
In MCR-ALS method, the prepared samples were scanned over the wavelength range of 200–400 nm with a 1 nm interval. Spectra were exported and the MCR-ALS model was employed for the prediction of the concentrations of target analytes in the prepared samples. In TLC-densitometric method, the chromatographic conditions were adopted for each prepared water sample. Aliquots (10 μL) of each sample were applied on TLC plate as bands and scanned at 245 nm. In HPLC method, the chromatographic conditions were implemented for each prepared water sample. Aliquots (20 μL) of each sample were injected into the HPLC system, and the chromatograms were recorded. In each method, the concentration of each drug was calculated from the corresponding regression equations and conducted from an average of three experiments.
3 Results and discussion
The aim of this study was to develop validated, eco-friendly, and cost-effective chemometric and chromatographic methods for quantification and routine quality monitoring of residues of the most frequently used veterinary drugs; IVM, RFX, SDD in production wastewater. We overcame the problem of analysis of these drugs in a complex aquatic system like wastewater and reduced the interferences of various impurities by the proper selection of a successful extraction/pre-concentration procedure and by the efficient optimization of the proposed chemometric and chromatographic methods. These simple and rapid methods may serve to optimize the treatment processes in WWTPs, preventing the release of these undesired drug residues (pollutants) into the aquatic environment.3.1 MCR-ALS method
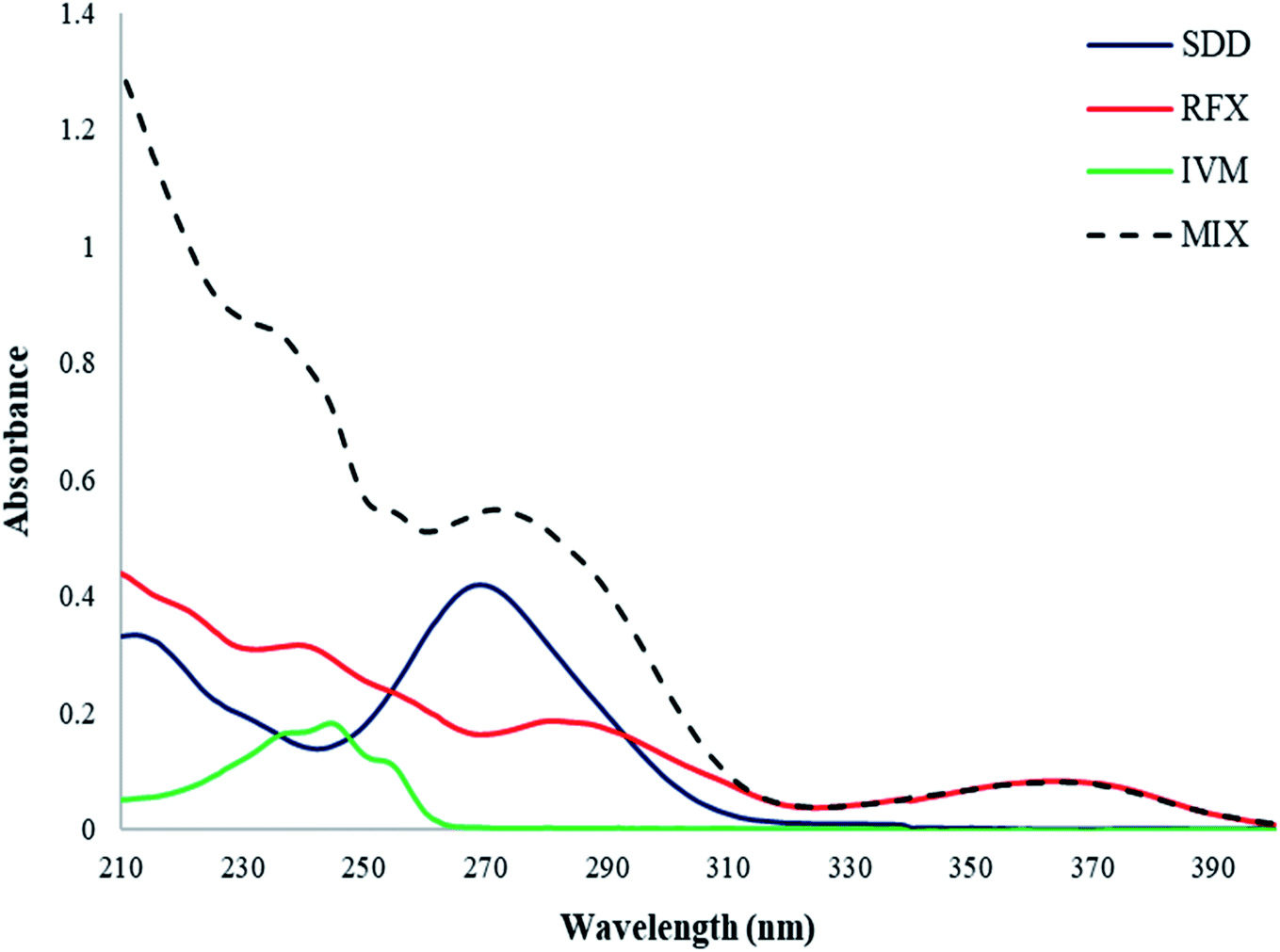 | ||
| Fig. 1 UV spectra of 5 μg mL−1 of IVM, RFX, and SDD and their mixture. | ||
The selection of the optimum wavelength range represents a vital factor that strongly affects the quality of multivariate analysis.50 In this work, the optimum wavelength range of the proposed MCR-ALS model was selected to be from 220 nm to 320 nm. Wavelengths below 220 nm were excluded due to the presence of remarkable noise. Furthermore, wavelengths above 320 nm were excluded due to poor absorption of both IVM and SDD at the measured concentration levels.
 | ||
| Fig. 2 Scatter plots of actual conc. vs. predicted conc. of the studied drugs for calibration and validation sets. | ||
Table 2 demonstrates the analytical figures of merit of the proposed MCR-ALS model. The results show a low relative percentage error in the concentration predictions (RE%) ≤0.8, low root mean square error of prediction (RMSEP) ≤0.064, low bias values ≤0.037, and low standard error of prediction (SEP) ≤0.063 for all target analytes. These low values indicated the high prediction capability and performance of the proposed method.
| Parameter | Calibration model | Validation set | ||||
|---|---|---|---|---|---|---|
| IVM | RFX | SDD | IVM | RFX | SDD | |
| a Root mean square error of prediction.b Standard error of prediction.c Relative percentage error in the concentration predictions. | ||||||
| RMSEPa | 0.053 | 0.039 | 0.061 | 0.043 | 0.064 | 0.060 |
| SEPb | 0.054 | 0.040 | 0.062 | 0.044 | 0.063 | 0.049 |
| Bias | 4.274 × 10−5 | 3.948 × 10−5 | −5.629 × 10−5 | 0.005 | 0.021 | 0.037 |
| RE%c | 0.801 | 0.302 | 0.033 | 0.015 | 0.329 | 0.592 |
3.2 TLC-densitometric method
The optimization of the chromatographic conditions for maximum separation was carried out by examining various developing systems with different ratios. The absolute separation of the studied drugs was accomplished by using ethyl acetate:acetonitrile:toluene:ammonia 33% (20:3:2:1, by volume) as mobile phase, which achieves excellent resolution, sharp and symmetrical peaks. The scanning wavelength was selected to be 245 nm as it is the wavelength of maximum absorption for both IVM and RFX. This selected wavelength also showed a good linearity curve for SDD. The retention factors (Rf) values were as following: RFX (0.28 ± 0.02), SDD (0.58 ± 0.04) and IVM (0.73 ± 0.03), as shown in Fig. 3(a).
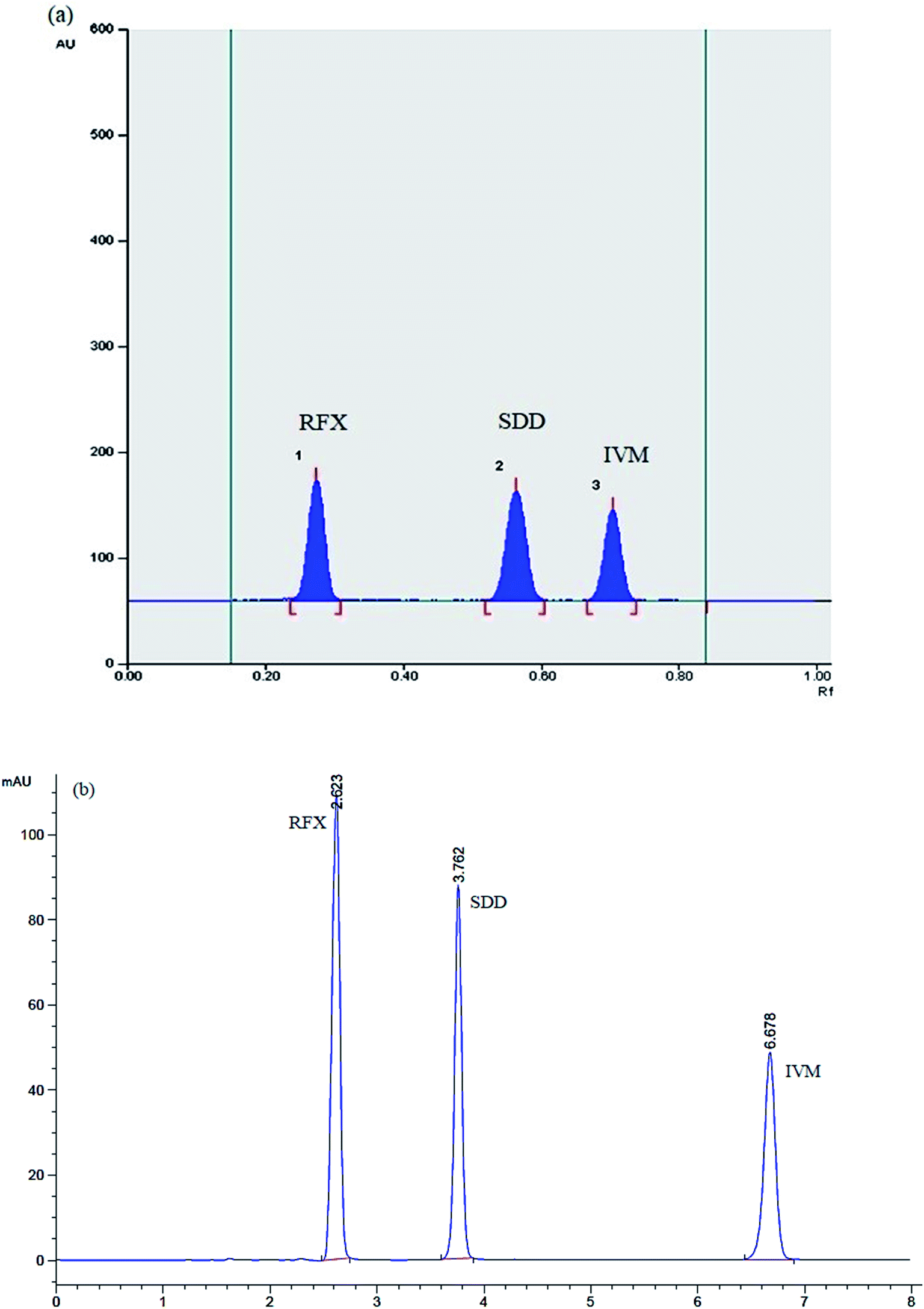 | ||
| Fig. 3 (a) TLC densitogram and (b) HPLC chromatogram showing Rf & tR of the studied drugs. | ||
3.3 HPLC method
The chromatographic conditions were optimized to get precisely the optimum separation pattern of the studied drugs. First, two types of stationary phases were tried (HiQsil C8 and HiQsil C18 HS column), but the latter showed a more suitable resolution. For the mobile phase, first; a simple mixture of acetonitrile and water (90:10) was tried and resulted in a poor separation between RFX and SDD and late appearance of the IVM peak with run time about 30 min. The addition of methanol in the composition of the mobile phase resulted in a satisfactory separation between RFX and SDD with a reduction of the run time to 20 min. The absolute separation was accomplished by using acetonitrile:methanol:water (60:25:15, by volume) as a mobile phase, which achieves excellent resolution, sharp and symmetrical peaks. The run time was 8 min, with a flow rate of 1.5 mL min−1, increasing the flow rate (>1.5 mL min−1) to get faster run time resulted in poor separation, asymmetric peaks, and an increase in column inlet pressure. The scanning wavelength was selected to be 245 nm for the same reasons in TLC-densitometric method. The retention times (tR) were found to be 2.6 ± 0.2, 3.7 ± 0.1, and 6.7 ± 0.3 min for RFX, SDD, and IVM, respectively, as shown in Fig. 3(b).
3.4 Method validation52
Method validation was carried out with all proposed methods as follows:In TLC-densitometric and HPLC methods, the calibration graphs for the studied drugs were constructed by plotting peak area at 245 nm versus the corresponding concentrations in μg per band & μg mL−1. The regression plots were found to be linear over the range of (0.1–1 μg per band; TLC), and (0.5–30 μg mL−1 for IVM, 0.5–25 μg mL−1 for RFX & SDD; HPLC), as shown in Fig. S2.†
Table 3 shows linearity, range, slopes, intercepts, and correlation coefficients (r2) for the proposed methods. The high value of coefficients of determination indicated good linearity of the calibration graphs.
| Parameters | MCR-ALS | TLC-densitometry | HPLC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IVM | RFX | SDD | IVM | RFX | SDD | IVM | RFX | SDD | |
| a MCR-ALS and HPLC-UV methods: in μg mL; TLC-densitometry method: in μg per band.b Repeatability (%RSD), average of three different concentrations repeated three times within the day.c Intermediate precision (%RSD), average of three different concentrations repeated three times in three different days. | |||||||||
| Linearitya | 1–10 | 0.1–1 | 0.5–30 | 0.5–25 | |||||
| Regression equation | Y = aX + b | ||||||||
| Slope (a) | 0.99998 | 1 | 1 | 2768 | 5407.1 | 3836.4 | 52.15 | 80.468 | 58.698 |
| Intercept (b) | 4.10 × 10−5 | 1.846 × 10−6 | −5.641 × 10−5 | 93.493 | −89.292 | 173.05 | −10.174 | 4.4505 | 2.3371 |
| Correlation coefficient (r2) | 0.9996 | 0.9997 | 0.9996 | 0.9999 | 0.9999 | 0.9999 | 0.9998 | 0.9999 | 0.9998 |
| LODa | 11 × 10−2 | 5 × 10−2 | 21 × 10−2 | 2 × 10−3 | 6 × 10−3 | 3 × 10−3 | 2.88 × 10−2 | 2.36 × 10−3 | 4.92 × 10−2 |
| LOQa | 34 × 10−2 | 15.3 × 10−2 | 64 × 10−2 | 8 × 10−3 | 19 × 10−3 | 10 × 10−3 | 8.74 × 10−2 | 7.16 × 10−3 | 1.49 × 10−1 |
| Accuracy mean ± S.D. | 100.66 ± 0.51 | 100.72 ± 0.75 | 100.37 ± 0.66 | 99.81 ± 0.64 | 99.89 ± 0.81 | 99.81 ± 0.51 | 99.88 ± 0.66 | 100.6 ± 0.31 | 100.35 ± 0.62 |
| Precision | |||||||||
| Repeatabilityb | 0.169 | 0.588 | 0.290 | 0.411 | 0.500 | 0.740 | 0.474 | 0.505 | 0.382 |
| Intermediate precisionc | 0.259 | 0.345 | 0.112 | 0.336 | 0.601 | 0.467 | 0.596 | 0.391 | 0.394 |
3.5 Application
| Method | IVM | RFX | SDD | |||
|---|---|---|---|---|---|---|
| Spiked levelsa | %R | Spiked levelsa | %R | Spiked levelsa | %R | |
| a MCR-ALS and HPLC methods: in μg mL; TLC-densitometry method: in μg per band.b Extraction efficiency (%). | ||||||
| MCR-ALS | 1 | 98.85 ± 0.37 | 1 | 97.56 ± 0.41 | 1 | 98.40 ± 0.28 |
| 3 | 97.85 ± 1.18 | 3 | 97.68 ± 0.99 | 3 | 98.11 ± 0.89 | |
| 7 | 98.40 ± 0.18 | 7 | 97.91 ± 0.11 | 7 | 98.82 ± 0.37 | |
| Meanb | 98.37 | Meanb | 97.72 | Meanb | 98.44 | |
| TLC-densitometry | 0.1 | 98.73 ± 0.94 | 0.1 | 98.40 ± 0.32 | 0.1 | 100.22 ± 0.54 |
| 0.3 | 99.59 ± 0.33 | 0.3 | 98.36 ± 0.33 | 0.3 | 100.47 ± 0.56 | |
| 0.7 | 99.67 ± 0.41 | 0.7 | 98.14 ± 0.13 | 0.7 | 100.24 ± 0.20 | |
| Meanb | 99.33 | Meanb | 98.30 | Meanb | 100.13 | |
| HPLC | 1 | 99.04 ± 0.12 | 1 | 99.17 ± 0.21 | 1 | 100.57 ± 0.18 |
| 3 | 99.25 ± 0.64 | 3 | 98.43 ± 0.32 | 3 | 101.21 ± 0.23 | |
| 7 | 99.23 ± 0.99 | 7 | 98.37 ± 0.27 | 7 | 100.41 ± 0.62 | |
| Meanb | 99.17 | Meanb | 98.66 | Meanb | 100.73 | |
| Samplesa | MCR-ALS | TLC-densitometry | HPLC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IVM | RFX | SDD | IVM | RFX | SDD | IVM | RFX | SDD | |
| a Samples are calculated in μg mL−1. | |||||||||
| W.W 1 | 6.04 | — | 6.16 | 6.00 | — | 6.11 | 6.09 | — | 6.08 |
| W.W 2 | 5.02 | — | 5.25 | 5.05 | — | 5.32 | 5.02 | — | 5.26 |
| W.W 3 | 7.36 | 5.18 | 5.64 | 7.42 | 5.133 | 5.46 | 7.45 | 5.13 | 5.53 |
| W.W 4 | 4.18 | 6.15 | — | 4.11 | 6.102 | — | 4.15 | 6.12 | — |
| W.W 5 | 5.65 | — | 6.19 | 5.69 | — | 6.33 | 5.70 | — | 6.28 |
 | ||
| Fig. 4 (a & c) TLC densitograms and (b & d) HPLC chromatograms of real and spiked real wastewater sample after SPE, respectively. | ||
3.6 Statistical analysis
Table 6 illustrates a statistical comparison between results obtained by applying the proposed methods and the official methods of IVM and SDD. It also shows a statistical comparison between the proposed methods and the reported method for RFX. The calculated t and F values were less than the theoretical ones indicating that there was no significant difference between the proposed, official and the reported methods which reflects high accuracy and precision of all proposed methods.| IVM | RFX | SDD | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MCR-ALS | TLC | HPLC | Officiala method | MCR-ALS | TLC | HPLC | Reported method13 | MCR-ALS | TLC | HPLC | Officiala method | |
| a Official BP methods were HPLC for IVM and titrimetric method for SDD.b Figures between parentheses represent the corresponding tabulated value of t and F value at P = 0.05. | ||||||||||||
| Mean | 100.66 | 99.81 | 99.88 | 100.54 | 100.72 | 99.89 | 100.6 | 100.21 | 100.37 | 99.81 | 100.35 | 100.23 |
| ±SD | 0.51 | 0.64 | 0.66 | 0.52 | 0.75 | 0.81 | 0.71 | 1.5 | 0.66 | 0.51 | 0.62 | 0.7 |
| %RSD | 0.169 | 0.588 | 0.290 | 0.247 | 0.411 | 0.500 | 0.740 | 0.385 | 0.474 | 0.505 | 0.382 | 0.524 |
| Variance | 0.26 | 0.40 | 0.43 | 0.27 | 0.56 | 0.65 | 0.50 | 2.25 | 0.43 | 0.26 | 0.38 | 0.49 |
| n | 15 | 5 | 8 | 5 | 15 | 5 | 7 | 5 | 15 | 5 | 7 | 5 |
| Student's t-test | 0.449(2.101)b | 1.979(2.306)b | 2.003(2.201)b | 0.730(2.101)b | 0.419(2.306)b | 0.539(2.228)b | 0.392(2.101)b | 1.084(2.306)b | 0.306(2.228)b | |||
| F value | 1.039(5.58)b | 1.514(6.39)b | 1.610(6.09)b | 4.012(5.58)b | 3.429(6.39)b | 4.463(6.16)b | 1.124(5.58)b | 1.883(6.39)b | 1.274(6.16)b | |||
3.7 Assessment of the proposed methods greenness
Greenness assessment of the proposed methods was accomplished using an assessment tool called Analytical Eco-Scale.54 This approach is based on penalty points which are assigned to different factors included in the analytical method and finally subtracted from a base of 100. The score will be more than 75 for excellent green analysis, more than 50 for acceptable green analysis, and less than 50 for inadequate green analysis. The reagent type and amount, the amount of energy of various electrical devices, the analytical waste treatment, and the occupational hazard are all given penalty points. For more information about calculating Eco-Scale scores, readers are directed to ref. 54. After assigning the penalty points for each analytical parameter, the proposed methods got an Eco-Scale score ranging from 79 to 92 out of 100, as shown in Table 7. The high Eco-Scale score (>75) indicated that all our proposed methods are excellent green methods, and the MCR-ALS method was the greenest one with a score of 92.| Hazard | MCR-ALS | TLC-densitometry | HPLC |
|---|---|---|---|
| a The penalty points were calculated according to ref. 54. | |||
| Reagents | |||
| Acetonitrile | — | 2 | 6 |
| Ammonia (33%) | — | 2 | — |
| Ethyl acetate | — | 4 | — |
| Methanol | 2 | — | 6 |
| Toluene | — | 2 | — |
| Water | — | — | 0 |
|
|||
| Instrument | |||
| Energy | 0 | 0 | 1 |
| Occupational hazard | 0 | 0 | 0 |
| Waste | 6 | 8 | 8 |
| Total penalty points (PPs)a | 8 | 18 | 21 |
| Analytical Eco-Scale total score | 100–8 = 92 | 100–18 = 82 | 100–21 = 79 |
4 Conclusion
SPE accompanied by chemometric and chromatographic methods were proposed for the simultaneous analysis of residues of the most frequently used veterinary drugs in pharmaceutical production wastewater. In the MCR-ALS method, the identification and quantification of the studied drugs were based on the decomposition of UV spectra of their mixture solutions into their pure spectra and concentration profiles. In the chromatographic methods, the identification and quantification of the studied drugs were based on Rf & tR values and peak areas. The SPE offers acceptable recoveries and the proposed methods proved to be sensitive, selective, eco-friendly, and low-cost alternatives to other sophisticated techniques. The MCR-ALS method proved to be the greenest one among the three proposed methods with low solvent/consumables load and low total time/cost to be performed. Although the 3-analyte system employed performed quite well with the proposed MCR-ALS model, the model will require further enhancements to be efficiently applied in the analysis of more complex and more representative wastewater samples. In future work, the enhancements may include trying different experimental designs, employment of different algorithms and constraints, or the usage of more advanced versions of the chemometric software.Conflicts of interest
The authors declare that there is no conflict of interest.References
- M. D. Hernando, M. Mezcua, A. R. Fernández-Alba and D. Barceló, Talanta, 2006, 69, 334–342 CrossRef CAS.
- A. B. A. Boxall, L. A. Fogg, P. A. Blackwell, P. Kay, E. J. Pemberton and A. Croxford, Veterinary Medicines in the Environment, 2004, vol. 180, pp. 1–91 Search PubMed.
- F. Kaczala and S. E. Blum, Curr. Anal. Chem., 2016, 12, 169–182 CrossRef CAS.
- A. J. M. Horvat, S. Babić, D. M. Pavlović, D. Ašperger, S. Pelko, M. Kaštelan-Macan, M. Petrović and A. D. Mance, TrAC, Trends Anal. Chem., 2012, 31, 61–84 CrossRef CAS.
- D. Molyneux and H. R. Taylor, Trends Parasitol., 2015, 31, 1 CrossRef.
- N. Abotaleb, T. Nasr, H. Ahmed and Z. Elsherif, J. Chem. Pharm. Res., 2017, 9, 135–140 CAS.
- K. Pietruk and P. Jedziniak, Sci. World J., 2013 DOI:10.1155/2013/362453.
- A. J. Ortiz, V. Cortez, A. Azzouz and J. R. Verdú, PLoS One, 2017, 12, 1–15 Search PubMed.
- Y. S. Chhonker, L. Ma, C. Edi and D. J. Murry, Bioanalysis, 2018, 10, 1841–1852 CrossRef CAS.
- S. C. Sweetman, Martindale: the complete drug reference, Pharmaceutical Press, London, 2009, vol. 3709 Search PubMed.
- M. Nassar, K. Attia, A. Mohamad, R. Said and A. Abdel-Monem, J. Adv. Pharm. Res., 2018, 2, 242–249 CrossRef.
- H. S. Yeung, W. H. Ching, S. S. L. Lai, W. O. Lee and Y. T. Wong, J. AOAC Int., 2010, 93, 1672–1677 CrossRef CAS.
- A. S. Saad, A. M. Hamdy, F. M. Salama and M. Abdelkawy, J. Chromatogr. Sci., 2016, 54, 1661–1669 CAS.
- B. Cancho Grande, M. S. García Falcón, M. Rodríguez Comesaña and J. Simal Gándara, J. Agric. Food Chem., 2001, 49, 3145–3150 CrossRef CAS.
- I. A. Unsal, M. Tasan, T. Gokcen and A. C. Goren, J. Chem. Metrol., 2018, 12, 70–78 CrossRef.
- E. Patyra, M. Przeniosło-Siwczynska and K. Kwiatek, Molecules, 2019, 24(3), 452 CrossRef.
- W. J. Pietroń, W. Cybulski, D. Krasucka, A. Mitura, K. Kos and M. Antczak, Bull. Vet. Inst. Pulawy, 2013, 57, 545–552 Search PubMed.
- W. W. Buchberger, Anal. Chim. Acta, 2007, 593, 129–139 CrossRef CAS.
- D. Ramírez-Morales, M. Masís-Mora, J. R. Montiel-Mora, J. C. Cambronero-Heinrichs, S. Briceño-Guevara, C. E. Rojas-Sánchez, M. Méndez-Rivera, V. Arias-Mora, R. Tormo-Budowski and L. Brenes-Alfaro, Sci. Total Environ., 2020, 746, 141200 CrossRef.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Green Chem., 2020, 22, 1458–1516 RSC.
- S. Babić, D. Ašperger, D. Mutavdžić, A. J. M. Horvat and M. Kaštelan-Macan, Talanta, 2006, 70, 732–738 CrossRef.
- S. Babić, D. Mutavdžić Pavlović, D. Ašperger, M. Periša, M. Zrnčić, A. J. M. Horvat and M. Kaštelan-Macan, Anal. Bioanal. Chem., 2010, 398, 1185–1194 CrossRef.
- C. Kim, H. D. Ryu, E. G. Chung and Y. Kim, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1084, 158–165 CrossRef CAS.
- I. Pugajeva, J. Rusko, I. Perkons, E. Lundanes and V. Bartkevics, J. Pharm. Biomed. Anal., 2017, 133, 64–74 CrossRef CAS.
- P. Lacina, L. Mravcová and M. Vávrová, J. Environ. Sci., 2013, 25, 204–212 CrossRef CAS.
- T. Kosjek, E. Heath and A. Krbavčič, Environ. Int., 2005, 31, 679–685 CrossRef CAS.
- I. S. Campbell, A. T. Ton and C. C. Mulligan, J. Am. Soc. Mass Spectrom., 2013, 5(2), 394–401 Search PubMed.
- M. F. Rodrigues, I. Pereira, R. L. Morais, G. S. Lobón, E. d. S. Gil and B. G. Vaz, J. Am. Soc. Mass Spectrom., 2020, 31(11), 2250–2257 CrossRef CAS.
- W. L. Fatigante, S. Mukta, Z. E. Lawton, A. M. Bruno, A. Traub, A. J. Gasa, A. R. Stelmack, C. R. Wilson-Frank and C. C. Mulligan, J. Am. Soc. Mass Spectrom., 2019, 31, 336–346 CrossRef.
- F. I. Khattab, H. Salem, S. M. Riad and H. T. Elbalkiny, J. Planar Chromatogr.--Mod. TLC, 2014, 27, 287–293 CrossRef CAS.
- K. He and L. Blaney, J. Hazard. Mater., 2015, 282, 96–105 CrossRef CAS.
- S. Mas, A. de Juan, R. Tauler, A. C. Olivieri and G. M. Escandar, Talanta, 2010, 80, 1052–1067 CrossRef CAS.
- S. E. Vignaduzzo, R. M. Maggio and A. C. Olivieri, J. Pharm. Biomed. Anal., 2020, 179, 112965 CrossRef CAS.
- H. Parastar and H. Shaye, RSC Adv., 2015, 5, 70017–70024 RSC.
- A. Mostafa and H. Shaaban, Acta Pharm., 2019, 69, 217–231 CAS.
- S. M. Riad, H. Salem, H. T. Elbalkiny and F. I. Khattab, Spectrochim. Acta, Part A, 2015, 140, 451–461 CrossRef CAS.
- H. El Balkiny, Anal. Chem. Lett., 2014, 4, 319–328 CrossRef CAS.
- B. Buszewski and M. Szultka, Crit. Rev. Anal. Chem., 2012, 42, 198–213 CrossRef CAS.
- C. Turner, Pure Appl. Chem., 2013, 85, 2217–2229 CAS.
- E. Turiel, G. Bordin and A. R. Rodríguez, J. Sep. Sci., 2005, 28, 257–267 CrossRef CAS.
- A. De Juan, J. Jaumot and R. Tauler, Anal. Methods, 2014, 6, 4964–4976 RSC.
- M. C. Bauza, G. A. Ibañez, R. Tauler and A. C. Olivieri, Anal. Chem., 2012, 84, 8697–8706 CrossRef CAS.
- H. Shaaban, A. Mostafa, B. Al-Zahrani, B. Al-Jasser and R. Al-Ghamdi, J. Anal. Methods Chem., 2020 DOI:10.1155/2020/1684172.
- T. Azzouz and R. Tauler, Talanta, 2008, 74, 1201–1210 CrossRef CAS.
- M. Maeder and A. D. Zuberbuehler, Anal. Chim. Acta, 1986, 181, 287–291 CrossRef CAS.
- H. Shaaban, A. Mostafa, Z. Almatar, R. Alsheef and S. Alrubh, J. Anal. Methods Chem., 2019 DOI:10.1155/2019/1863910.
- A. Olivieri and G. M. Escandar, Practical three-way calibration, Elsevier, 2014 Search PubMed.
- A. De Juan and R. Tauler, Crit. Rev. Anal. Chem., 2006, 36, 163–176 CrossRef CAS.
- A. N. Skvortsov, J. Chemom., 2014, 28, 727–739 CrossRef CAS.
- A. Mostafa, H. Shaaban, M. Almousa, M. Al Sheqawi and M. Almousa, J. AOAC Int., 2019, 102, 465–472 CrossRef CAS.
- R. G. Brereton, Analyst, 1997, 122, 1521–1529 RSC.
- ICH Q2 (R1), Validation of analytical procedures: text and methodology, 2005 Nov.
- United States Pharmacopeia, The National Formulary Usp, U.S.P.C.I, 2012 Search PubMed.
- A. Gałuszka, Z. M. Migaszewski, P. Konieczka and J. Namieśnik, TrAC, Trends Anal. Chem., 2012, 37, 61–72 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08850a |
| This journal is © The Royal Society of Chemistry 2021 |