
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-doped mixed Co, Ni-oxides with petal structure as effective catalysts for hydrogen and oxygen evolution by water splitting†
Hai Zhong‡
a,
Guofeng Cheng‡b,
Guangcai Mab,
Enhui Wuc,
Zhuo Zhangb,
Xuefeng She *a,
Shuqiang Jiaoa,
Jingsong Wanga and
Qingguo Xuea
*a,
Shuqiang Jiaoa,
Jingsong Wanga and
Qingguo Xuea
aState Key Laboratory of Advanced Metallurgy, University of Science and Technology, Beijing, 100083, China. E-mail: shexuefeng@ustb.edu.cn
bSchool of Physics and Physical Engineering, Qufu Normal University, Qufu, 273165, China
cPanzhihua International Research Institute of Vanadium and Titanium, Panzhihua University, Panzhihua, 617000, China
First published on 4th January 2021
Abstract
Developing electrocatalytic nanomaterials for green H2 energy is inseparable from the exploration of novel materials and internal mechanisms for catalytic enhancement. In this work, nano-petal N-doped bi-metal (Ni, Co) and bi-valence (+2, +3) (Ni1−xCox)2+Co23+O4 compounds have been in situ grown on the surface of Ni foam. The N3− atoms originate from the amino group in urea and doped in the compound during annealing. The as-synthesized N-doped (Ni1−xCox)2+Co23+O4 nano-petals demonstrate commendable hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) bi-functional catalytic efficiency and stability. Electrochemical measurements confirm that the nitrogen doping significantly improves the catalytic kinetics and the surface area. Density functional theory calculations reveal that the improved HER and OER kinetics is not only due to the synergistic effect of bi-metal and bi-valence, as well as the introduction of defects such as oxygen vacancies, but also it more depends on the shortened bond length between the nitrogen N3− atoms and the metal atoms, and the increased electron density of the metal atoms attached to the N3− atoms. In other words, the change of lattice parameters caused by nitrogen doping is more conducive to the catalytic enhancement than the synergistic effect brought by bi-metal. This study provides an experimental and theoretical reference for the design of bi-functional electrocatalytic nanomaterials.
1. Introduction
In response to the energy and environmental crisis, many countries are devoted to developing practical electrocatalytic materials for hydrogen and oxygen evolution by water splitting. As benchmarks, Pt is considered to be the most efficient catalyst for hydrogen evolution reaction (HER),1,2 while RuO2 and IrO2 have the leading positions for oxygen evolution reaction (OER).3,4 However, their rarity and high price greatly limit their industrialization and commercialization. Therefore, highly active and cost-effective catalysts are in urgent need to replace the precious metal catalysts. Recently, transition metal nickel (Ni) and cobalt (Co), and their compounds, such as (hydro)oxides, nitrides, and carbides, have attracted tremendous attention due to their intrinsic catalytic activity, environmental-friendliness and earth-abundance.5–10 Through intracrystalline or surface modification, Ni (or Co)-based catalysts can obtain comparable HER or OER performance with noble metal catalysts.For commercializing the catalysts, low-cost and bi-functionality in both HER and OER have long been the primary targets. Compared with the single-transition-metal-based catalysts, the bimetallic Ni–Co-based catalysts are more active to overcome the sluggish kinetics of OER with four electrons transfer, and thus possess commendable overpotentials for both HER and OER.11 For example, NiCo2O4/CoO/graphite composite can act as remarkable OER catalysis with an overpotential of 323 mV (vs. RHE) (recorded at 10 mA cm−2), which is superior to that of IrO2 (340 mV) and RuO2 (350 mV).12 Researchers attributed the highly improved electrocatalytic activity to the electron hopping between different valence states of Ni and Co, and also the redox couples of Co3+/Co2+ and Ni3+/Ni2+.13 As for alloys, Fu et al. revealed that Ni–Co alloy nanoparticles could even rival commercial Pt/C and RuO2 catalysts owing to the “rich valence state changes” between Ni and Co.14 Moreover, a large number of density functional theory studies have shown that the mixture of bi-metal and bi-valence can effectively increase the number of defects such as oxygen vacancy, thereby increasing the number of active sites as well as the density of electron cloud around them and consequently greatly reducing the free energy for both adsorption and desorption of intermediates.15
Beside bi-metallic oxides, in recent years, bi-metallic nitrides such as NiCo2N, NiMoN, FeNi3N, etc. have also attracted increasing attention due to their enhanced activity for water oxidation and reduction.16–18 Peng has proven that transition metal nitride has excellent electrocatalytic activity, stability and corrosion resistance, and it is a good substitute for noble metals in water splitting system.19 Yu Zhang et al. also demonstrated that the electronic structure of this metal nitride can be appropriately adjusted to promote the rapid transfer of electrons due to the inclusion of interstitial nitrogen atoms, thus greatly reducing the energy barrier to improve its catalytic activity.20 Considering the significant improvement of HER and OER kinetics via bimetallic and bivalent mixing, as well as nitrogen doping as mentioned above, it is very essential to combine the above three advantages in a bifunctional nano-catalytic system. Furthermore, it is of great significance to evaluate whether bimetal or nitrogen doping plays a much stronger role in improving catalytic efficiency.
In this work, large-scale and vertically aligned N-doped (Ni1−xCox)2+Co23+O4 nano-petals have been fabricated on the surface of Ni foam. The unique morphology like rose petals (Fig. 1a) enables the spacing between nano-petals to facilitate the chemical reactions and the evolution of hydrogen and oxygen. Electrochemical experimental results show that Ni–Co bimetal and bivalence (+2, +3), as well as nitrogen doping, can greatly improve HER and OER efficiencies; theoretical studies via density functional calculation reveal that these efficiencies improvement are due to the slight changes in atomic spacing, which greatly increases the number of defects and the electron densities. The effect of bimetal and nitrogen doping on catalytic performance enhancement has also been compared. The excellent HER and OER performances endowed by N-doped (Ni1−xCox)2+Co23+O4 nano-petals provide a technical and theoretical reference for the future design of bi-functional electrocatalytic materials.
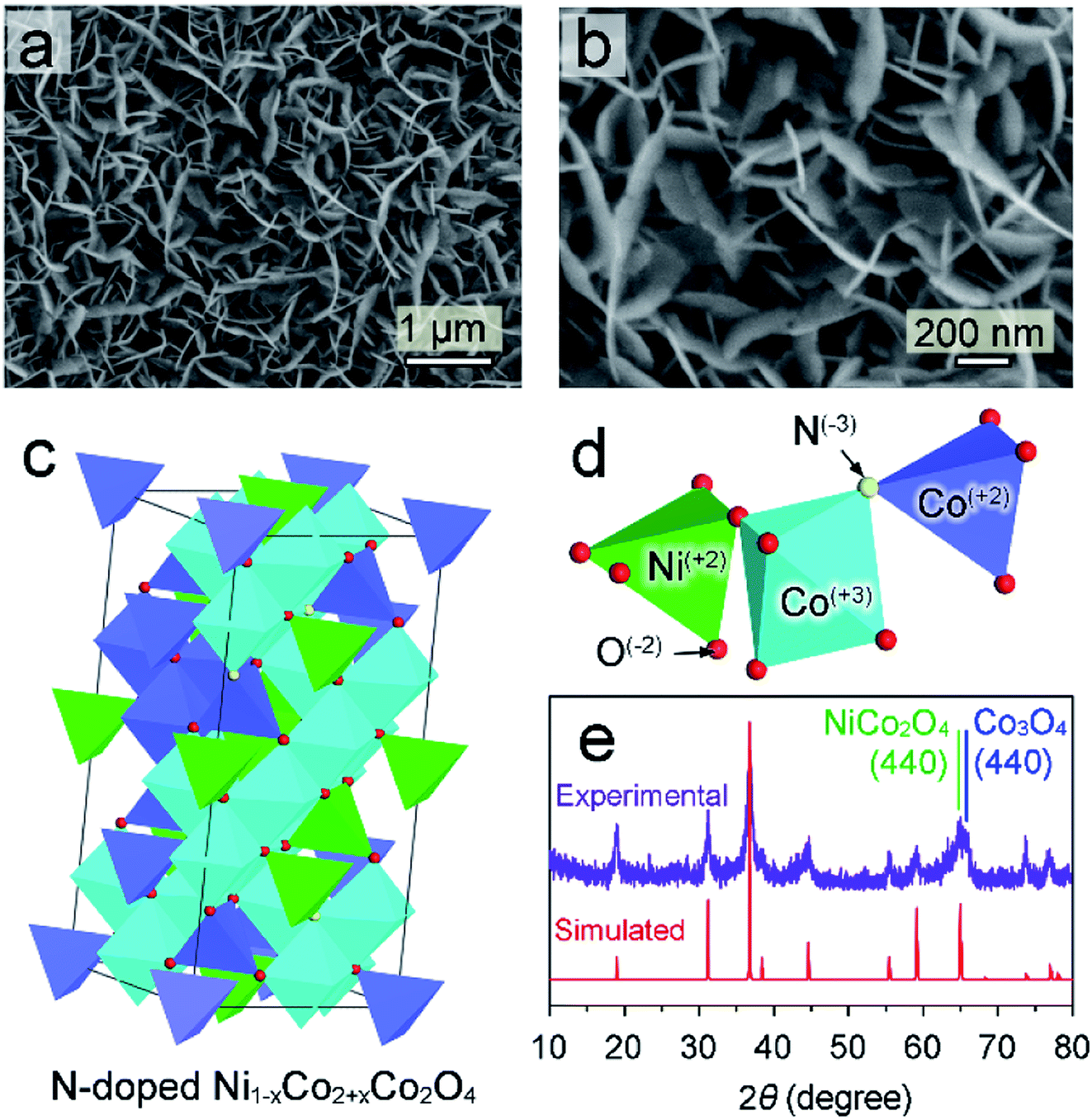 | ||
| Fig. 1 (a and b) Top-view SEM images of N–Ni1−xCo2+xO4 NPs; (c and d) crystal structure (c) and legend (d) of N-doped Ni1−xCo2+xO4; (e) experimental (upper row) and simulated (bottom row) XRD patterns of the N–Ni1−xCo2+xO4 NPs. | ||
2. Experimental and calculation section
2.1. Materials
Nickel nitrate hexahydrate [Ni(NO3)2·6H2O], cobalt nitrate hexahydrate [Co(NO3)2·6H2O], urea (CH4N2O), hexadecyl trimethyl ammonium bromide (CTAB, C19H42BrN), potassium hydroxide (KOH) were purchased from Aladdin. Ni foam was provided by JD.com. Prior to use, the Ni foam was cleaned by sonicating successively with acetone, ethanol and DI water for 10 min each.2.2. Preparation of N-doped (Ni1−xCox)2+Co23+O4 nano-petals
At first, 1 mmol Ni(NO3)2·6H2O and 3 mmol Co(NO3)2·6H2O were dissolved in 50 mL DI water, and 2 mmol urea and 1 mmol CTAB were dissolved in another 50 mL DI water by sufficient stir. Then, the above two solutions were mixed together. Next, pieces of Ni foam with 1 cm × 2 cm size were dipped into the glass bottle containing the mixed solution. After that, the bottle was put into air dry oven at 95 °C for 15 h. After 15 hours, the Ni foam was taken out from the bottle, followed by cleaning and drying. Finally, the Ni foam was placed in a tubular furnace and annealed at 300 °C in nitrogen atmosphere for 1 h.2.3. Characterization
SEM measurement was carried out on a XL 30S FEG scanning electron microscope at an acceleration voltage of 20 kV. XRD patterns were acquired using a D/MAX-2500/PC X-ray diffractometer equipped with a Cu Kα radiation source. The structures of the samples were characterized by a JEM-2200FS transmission electron microscopy (TEM) operated at 200 kV. XPS measurement was performed using a VG ESCALAB 250 X-ray photoelectron spectrometer with Mg as the excitation source.2.4. Electrochemical measurements
All of the electrochemical measurements were carried out using a CHI660E electrochemical workstation in a three electrodes system. As-grown samples, Pt wafer and saturated calomel electrode were used as the working, counter and reference electrodes, respectively. The iR correction was applied to all data. The experimental potentials were converted to RHE with the equation E(RHE) = E(calomel) + (0.219 + 0.0591 × pH)V.21 LSV measurements were performed in H2-saturated 1.0 M KOH solution at a scan rate of 5 mV s−1.2.5. Density functional theory calculation
All of the density functional theory (DFT) calculations were performed using the CASTEP program available in Materials Studio. The plane wave basis set cutoff energy was 400 eV for geometry optimization and energy calculation. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) function was employed to present exchange–correlation interactions. The energy convergence tolerance for the self-consistent field (SCF) was 2.0 × 10−6 eV per atom. All of the DFT calculations were carried out without spin restricted.3. Results and discussion
3.1. Characterization of Ni, Co-samples
N-doped (Ni1−xCox)2+Co23+O4 nano-petals is abbreviated as N–Ni1−xCo2+xO4 NPs in this work. The scanning electron microscope (SEM) image in Fig. 1a shows that large area N–Ni1−xCo2+xO4 NPs with uniform thickness and a certain radian are evenly distributed on the surface of Ni foam. Fig. 1b displays that the arc-shaped nano-petals could support and leave space for each other, which prevents the nano-petals from collapsing and utilizes the space to greatly facilitate the release of hydrogen or oxygen. (Ni1−xCox)2+Co23+O4 has very similar crystalline structure with NiCo2O4 and Co3O4, possessing spinel Fd3m geometries.22 Therefore, in the framework of N–Ni1−xCo2+xO4 NPs in Fig. 1c, the vertex-sharing CoOx or NiOx polyhedrons are cross-linked by the O atom to build a three-dimensional framework. Some Co2+ and a few of O2− are replaced by Ni2+ and N3−, respectively. Atoms with different valences are shown by different colors (Fig. 1d). The X-ray diffraction (XRD) pattern of the as-grown N–Ni1−xCo2+xO4 NPs is in good agreement with the DFT simulated pattern (Fig. 1e), justifying our theoretical crystal model. It should be noted that the (440) diffraction peak measured in the experiment is relatively wide, due to a slight deviation between the actual (440) peaks of NiCo2O4 and Co3O4.23,24A close-up side-view of the nano-petals in Fig. 2a shows a thickness about 16 nm. The original SEM image is exhibited in ESI† Part 1 (ESI, P1). It is a layered structure by the superposition of several ultra-thin nano-layers (Fig. 2b). Fig. 2c is a typical transmission electron microscope (TEM) image of an ultra-thin N–Ni1−xCo2+xO4 NP, which is obtained by ultrasonic peeling off from the nano-petals in DI water. Taken from the two red square regions in Fig. 2c and d demonstrates that the thickness of the ultra-thin NP is about 1–3 nm. The different lattice fringe images are shown in Fig. 2e by zooming in the blue region in Fig. 2c. Fig. 2f–h reveal the highly crystalline nature of the N–Ni1−xCo2+xO4 NP with lattice spacings of 0.243, 0.285, and 0.233 nm, corresponding to the (311), (220) and (222) planes, respectively.25,26 The spatial elemental analysis using electron energy loss spectroscopy (EELS) in Fig. 2i–l confirms that Co, Ni, O and N are distributed homogeneously throughout the selected flakes.
 | ||
| Fig. 2 (a) Close-up view of the N–Ni1−xCo2+xO4 NPs; (b) schematic diagram of an individual petal with multilayer structure; (c) TEM image of the N–Ni1−xCo2+xO4 NP; (d and e) enlarged views taken from the red (d) and blue (e) squares in (c); (f–h) HRTEM images taken from (e); (i–l) element mappings of Co(I), Ni (j), O (k) and N (l). | ||
XPS analysis was carried out to further investigate the chemical states of the N–Ni1−xCo2+xO4 NPs. Fig. 3a shows that the Ni 2p spectra consist of two groups of sub-peaks due to the spin–orbit coupling of the 2p1/2 and 2p3/2 states. For each group, there are three sub-peaks corresponding to Ni3+ and Ni2+ states, as well as a satellite peak. Similar to the Ni 2p, Fig. 3b reveals that the Co 2p spectra also include two sub-peaks due to the spin–orbit coupling of the 2p1/2 and 2p3/2 states.27 The N 1s spectrum only contains the N3−-metal chemical bond located at 398.2 eV (Fig. 3c), confirming that the N atoms only replace some of the O atoms.28 In Fig. 3d, the O 1s spectrum of the as-grown sample without annealing consists of two sub-peaks due to the O2--metal and –OH chemical bonds. With the annealing time increasing from 0.5 to 1 h, the sub-peak corresponding to –OH gradually fade away, while the peak position of O2−-metal bonds has a slight left-shift. This can be ascribed to that metal hydroxide is dehydrated and changed to oxide gradually during annealing, and nitrogen doping is produced during the thermal treatment.
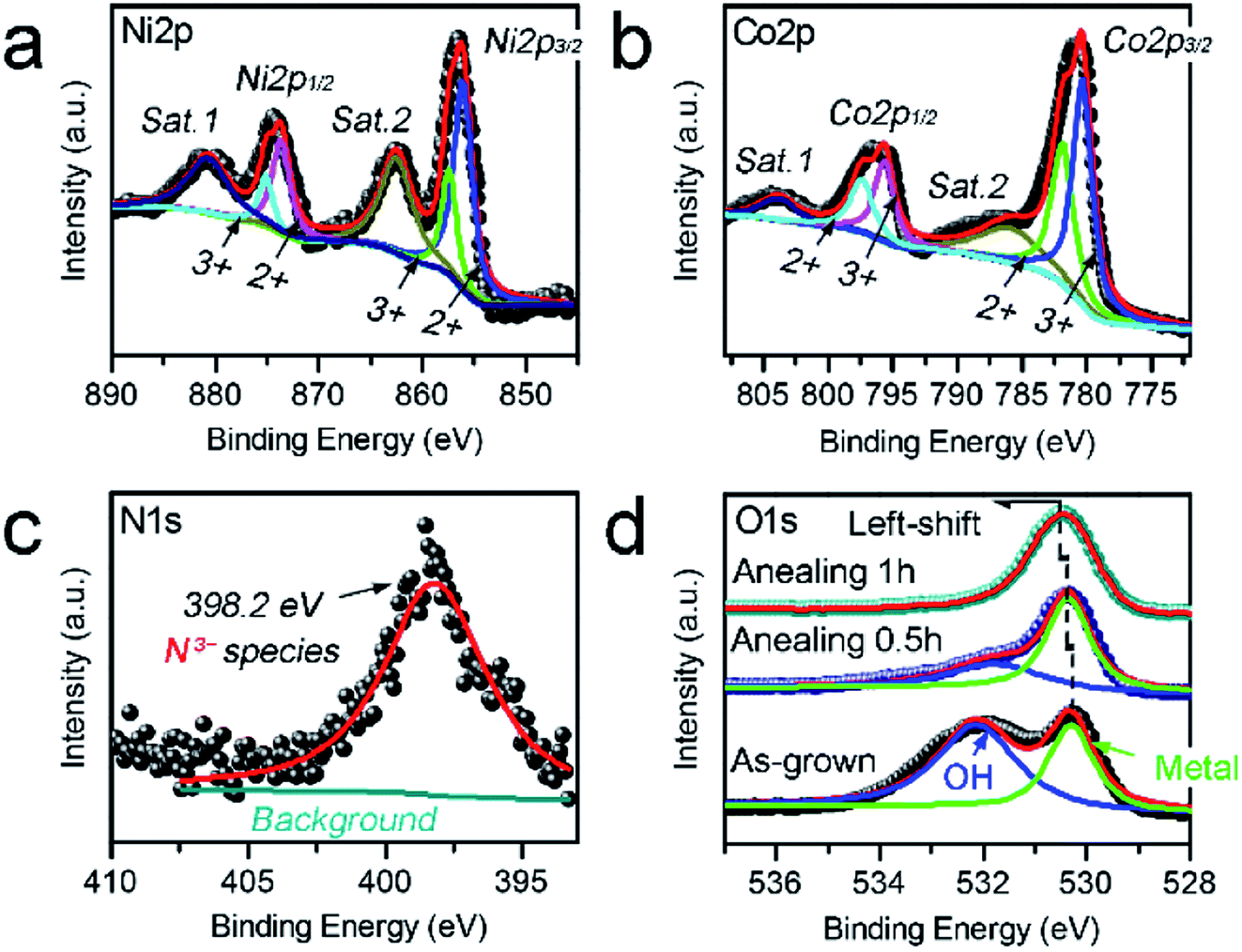 | ||
| Fig. 3 (a–d) Ni 2p (a), Co 2p (b), N 1s (c) and O 1 s (d) XPS spectra measured from the N–Ni1−xCo2+xO4 NPs. | ||
3.2. Influences of CTAB and urea over the morphology and N-doping of samples
During the fabrication process, the concentrations of CTAB and urea demonstrate significant influences over the morphology and N-doping of samples, respectively. It was confirmed by SEM observation (ESI, P2†) that, without adding CTAB, the morphology of product was one-dimensional nanowire; as the CTAB concentration increased to 5 mmol L−1, the product was a mixture composed by nanowires and nano-petals; with the CTAB concentration exceeded 10 mmol L−1, the product was formed by pure nano-petals. Therefore, during the sample construction process of this work, CTAB, as a surfactant, inhibits the growth of the nanostructure along the c-axis into a one-dimensional acicular structure, and promotes the formation of two-dimensional sheet structure of the N–Ni1−xCo2+xO4 NPs.Besides, with the aid of XPS, we found that the concentration of doped nitrogen (at%) could be increased with urea (ESI, P3†). As the concentration of urea in hydrothermal-solution increasing from 0 to 1.0, 2.0 and 3.0 mmol L−1, the N-doing concentration in samples increased from 1.36 to 2.20, 4.51 and 4.49%, respectively. Here, the N-doping of 1.36% may be induced by CTAB. This indicates that after the concentration of urea exceeds 3 mmol, the concentration of N-doping will not continue to increase.
3.3. Catalytic properties of Ni, Co-samples
 | ||
| Fig. 4 OER LSV curves (a) and corresponding Tafel plots (b) of the N–Ni1−xCo2+xO4 NPs with different N-doping concentrations. | ||
Derived from the logarithmic region of LSV curves in Fig. 4a, the Tafel slopes are employed to evaluate the HER kinetics difference of the four samples.29 Fig. 4b demonstrates that the sample with the N-doping concentration of 4.51% has the lowest Tafel slope than those of others, showing the most efficient OER kinetics of all.
 | ||
| Fig. 5 (a and b) HER (a) and OER (b) LSV curves of the N–Ni1−xCo2+xO4 NPs without annealing and annealed for 0.5, 1 and 1.5 h; (c) corresponding Tafel plots obtained from HER LSV curves; (d and e) CV curves of the N–Ni1−xCo2+xO4 NPs without annealing (d) and annealed for 1 h (e) measured in a potential range of 0.2–0.3 V vs. RHE under the scan rates from 60 to 140 mV s−1; (f) charging current density differences at 0.25 V vs. RHE (Δj = ja − jc) as a function of the scan rates; (g) HER and OER LSV curves of the N–Ni1−xCo2+xO4 NPs before and after 1000 cycles; (h) HER and OER stability measurements of the N–Ni1−xCo2+xO4 NPs. | ||
After HER analysis, the LSV curves for OER of all samples were measured (Fig. 5b). As expected, the sample with the annealing time of 1 h still has the lowest OER potential about 1.65 V vs. RHE at the current density of 10 mA cm−2. As revealed by Fig. 5a and b, the samples with different annealing times exhibit different HER and OER efficiencies. It is essential to explore the mechanism for the effect of catalytic enhancement by annealing. Taken HER efficiency as an example, the Tafel slopes and active surface areas of the above four samples are compared and shown in Fig. 5c–f. Fig. 5c confirms that the sample with the annealing time of 1 h has the lowest Tafel slope, demonstrating the most efficient HER kinetics. And then, the double layer capacitances (Cdl) of the four samples are tested to compare the active surface areas. Cdl is derived from the slope obtained by fitting the plot of Δj as a function of the scanning rate of potential (mV s−1). Fig. 5d and e collect the closed CV curves in the region of 0.2 to 0.3 V vs. RHE for the samples without annealing and annealed for 1 h, respectively. Taking the RHE potential of 0.25 V as reference, the Cdl slope of the sample without annealing is lower than that annealed for 1 h, indicating that the electrochemical effective area can be increased by annealing. The above results reveal that the superior HER and OER efficiencies of the N–Ni1−xCo2+xO4 NPs annealed for 1 h in alkaline media originates from the improved catalytic kinetics and increased active surface area.
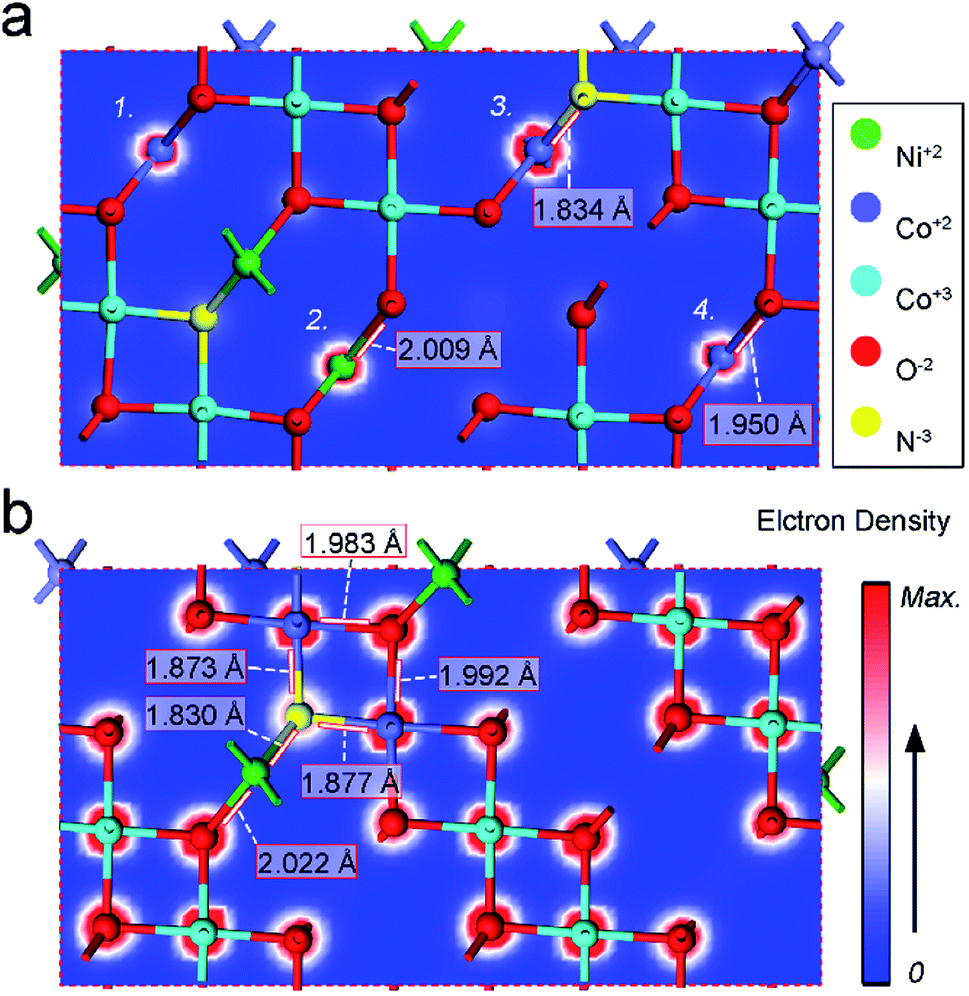 | ||
| Fig. 6 (a and b) Electron densities in different cross sections in N–Ni1−xCo2+xO4 NPs from DFT calculation. | ||
4. Conclusion
Large-area N–Ni1−xCo2+xO4 nano-petals have been in situ synthesized on the surface of Ni foam by hydrothermal and annealing strategies. The annealing introduced N3− atoms doping originates from the amino group in urea. The electrochemical measurements demonstrate that the N–Ni1−xCo2+xO4 NPs with 1 h annealing have a low overpotential and a creditable stability for both HER and OER. DFT calculation reveals that, compared with bimetallic and bivalent mixing, nitrogen doping contributes more significantly to the improvement of HER and OER performances, which can be ascribed to the N-doped induced creation of defects such as oxygen vacancies, increase of the electron density of the metal atoms connected to N3− atoms and the shortening of the bond length between N3− and metal atoms. The successful synthesis of high-performance bi-functional N–Ni1−xCo2+xO4 NPs opens a new avenue for rational design of effective electrocatalytic nanomaterials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.51874029).References
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Li, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS.
- C. Xu, P. K. Shen and Y. Liu, J. Power Sources, 2007, 164, 527–531 CrossRef CAS.
- K. S. Exner, J. Anton, T. Jacob and H. Over, ChemElectroChem, 2015, 2, 707–713 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and S. H. Yang, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS.
- C. Hitz and A. Lasia, J. Electroanal. Chem., 2001, 500, 213–222 CrossRef CAS.
- T. Zhang, M. Y. Wu, D. Y. Yan, J. Mao, H. Liu, W. B. Hu, X. W. Du, T. Ling and S. Z. Qiao, Nano Energy, 2018, 43, 103–109 CrossRef CAS.
- X. Zhang, R. Liu, Y. Zang, G. Liu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Chem. Commun., 2016, 52, 5949 Search PubMed.
- Y. Zhao, B. Sun, X. Huang, H. Liu, D. Su, K. Sun and G. Wang, J. Mater. Chem. A, 2015, 3, 5402–5408 RSC.
- K. L. Nardi, N. Yang, C. F. Dickens, A. L. Strickler and S. F. Bent, Adv. Energy Mater., 2015, 5, 1500412 CrossRef.
- J. W. D. Ng, M. García-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodicv and T. F. Jaramill, Nat. Energy, 2016, 1, 16053 CrossRef CAS.
- X. Yan, K. X. Li, L. Lyu, F. Song, J. He, D. Niu, L. Liu, X. Hu and X. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3208–3214 CrossRef CAS.
- N. Srinivasa, L. Shreenivasa, P. S. Adarakatti, J. R. N. Samuel, C. E. Banks and S. Ashoka, RSC Adv., 2019, 9, 24995–25002 RSC.
- J. Zhao, Y. He, Z. Chen, X. Zheng, X. Han, D. Rao, C. Zhong, W. Hu and Y. Deng, ACS Appl. Mater. Interfaces, 2018, 11, 4915–4921 CrossRef.
- Y. Fu, H. Y. Yu, C. Jiang, T. H. Zhang, R. Zhan, X. Li, J. F. Li, J. H. Tian and R. Yang, Adv. Funct. Mater., 2018, 28, 1705094 CrossRef.
- J. Diao, Y. Qiu, S. Liu, W. Wang, K. Chen, H. Li, W. Yuan, Y. Qu and X. Guo, Adv. Mater., 2019, 32, 1905679 CrossRef.
- R. Zou, M. Xu, S. A. He, X. Han, R. Lin, Z. Cui, G. He, D. J. L. Brett, Z. Guo, J. Hu and I. P. Parkin, J. Mater. Chem. A, 2018, 6, 19853–19862 RSC.
- B. Chang, J. Yang, Y. Shao, L. Zhang, W. Fan, B. Huang, Y. Wu and X. Hao, ChemSusChem, 2018, 11, 3198–3207 CrossRef CAS.
- L. Liu, F. Yan, K. Li, C. Zhu, Y. Xie, X. Zhang and Y. Chen, J. Mater. Chem. A, 2019, 7, 1083–1091 RSC.
- X. Peng, C. Pi, X. Zhang, S. Li, K. Huo and P. K. Chu, Sustainable Energy Fuels, 2019, 3, 366–381 RSC.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- D. Kim, J. Park, J. Lee, Z. Zhang and K. Yong, ChemSusChem, 2018, 11, 3618–3624 CrossRef CAS.
- L. Huang, D. Chen, Y. Ding, S. Feng, Z. L. Wang and M. Liu, Nano Lett., 2013, 13, 3135–3139 CrossRef CAS.
- F. Deng, L. Yu, G. Cheng, T. Lin, M. Sun, F. Ye and Y. Li, J. Power Sources, 2014, 251, 202–207 CrossRef CAS.
- P. Vikas, J. Pradeep, C. Manik and S. Shashwati, Soft Nanosci. Lett., 2012, 2, 1–7 CrossRef.
- J. Béjar, L. Álvarez-Contreras, J. Ledesma-García, N. Arjona and L. G. Arriagaa, J. Electroanal. Chem., 2019, 847, 113190 CrossRef.
- H. Xu, Z. X. Shi, Y. X. Tong and G. R. Li, Adv. Mater., 2018, 30, 1705442 CrossRef.
- Y. Guo, P. Yuan, J. Zhang, H. Xia, F. Cheng, M. Zhou, J. Li, Y. Qiao, S. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28, 1805641 CrossRef.
- M. Yu, Z. Wang, C. Hou, Z. Wang, C. Liang, C. Zhao, Y. Tong, X. Lu and S. Yang, Adv. Mater., 2017, 29, 1602868 CrossRef.
- B. Chen, Z. Zhang, S. Kim, S. Lee, J. Lee, W. Kim and K. Yong, ACS Appl. Mater. Interfaces, 2018, 10, 44518–44526 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08846c |
| ‡ Co-author. |
| This journal is © The Royal Society of Chemistry 2021 |