
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe F19W mutation reduces the binding affinity of the transmembrane Aβ11–40 trimer to the membrane bilayer†
Thanh Thuy Tran *ab,
Feng Panc,
Linh Trande,
Christopher Rolandf and
Celeste Saguif
*ab,
Feng Panc,
Linh Trande,
Christopher Rolandf and
Celeste Saguif
aLaboratory of Theoretical and Computational Biophysics, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: tranthanhthuy@tdtu.edu.vn
bFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City, Vietnam
cDepartment of Statistics, Florida State University, Tallahassee, Florida, USA
dInstitute of Fundamental and Applied Sciences, Duy Tan University, Ho Chi Minh City, 700000, Vietnam
eFaculty of Natural Sciences, Duy Tan University, Da Nang City, 550000, Vietnam
fDepartment of Physics, North Carolina State University, Raleigh, North Carolina, USA
First published on 12th January 2021
Abstract
Alzheimer's disease is linked to the aggregation of the amyloid-β protein (Aβ) of 40 or 42 amino acids. Lipid membranes are known to modulate the rate and mechanisms of the Aβ aggregation. Point mutations in Aβ can alter these rates and mechanisms. In particular, experiments show that F19 mutations influence the aggregation rate, but maintain the fibril structures. Here, we used molecular dynamics simulations to examine the effect of the F19W mutation in the 3Aβ11–40 trimer immersed in DPPC lipid bilayers submerged in aqueous solution. Substituting Phe by its closest (non-polar) aromatic amino acid Trp has a dramatic reduction in binding affinity to the phospholipid membrane (measured with respect to the solvated protein) compared to the wild type: the binding free energy of the protein–DPPC lipid bilayer increases by 40–50 kcal mol−1 over the wild-type. This is accompanied by conformational changes and loss of salt bridges, as well as a more complex free energy surface, all indicative of a more flexible and less stable mutated trimer. These results suggest that the impact of mutations can be assessed, at least partially, by evaluating the interaction of the mutated peptides with the lipid membranes.
Introduction
According to the World Alzheimer Report 2016, there were 46.8 million patients with Alzheimer's disease (AD) in 2015, and the number of global cases was predicted to reach 131.5 million by 2050.1 AD is a neurodegenerative disease, which is pathologically characterized by amyloid plaques resulting from the aggregation of extracellular amyloid-β (Aβ) peptide, and by neurofibrillary tangles made by the accumulation of intracellular tau protein in the hippocampus and cerebral cortex.2–5 AD progressively affects normal brain functions such as memory, judgement, and cognition, and results in the failure of crucial cellular processes.6 Amyloid plaques consist of the extracellular accumulation of the Aβ40 and Aβ42 peptides derived from the transmembrane amyloid precursor protein (APP), which is located in the lipid-rich microdomains (lipid rafts) of endosome and the plasma membrane,7,8 after cleavage by β- and γ-secretases.9 The Aβ42 peptide is known to be more insoluble and with a higher probability of polymerization than the Aβ40 peptide. The latter is considered the primary constituent in cerebral amyloid angiopathy and is generally more abundant in plaques.10 The accumulation of soluble Aβ oligomers can cause neurovirulence and impair the synaptic transition.11,12Experimental and computational investigations in the Aβ peptide grew over the past years due to Aβ's dual nature: high intrinsic disorder and high aggregation propensity.5,13–17 Indeed, the diversity and flexibility of Aβ bring many challenges in its structural characterization by experiments, especially because the interaction between Aβ and the phospholipids in the cell membrane plays a crucial role in the aggregation mechanisms. Using a single electron method to study the interaction between the Aβ40 peptide and anionic lipid membranes, Ding et al. reported that trimers and tetramers may be the smallest Aβ40 oligomers in the lipid bilayers, and could lead to the initial neurotoxicity.18 Later, Jana et al. demonstrated that membrane-bound tetramer and trimer Aβ40 oligomeric species are associated with toxicity in cultured neurons.19 Several U-shape fibril models of Aβ40 that form in-register parallel β-sheets have been experimentally reported.20–23 In general, residues 1–10 in these models are disordered. Even though the conformations of Aβ40 depend on the peptide sequence and lengths,24 the contribution of the 1–10 residues is negligible. Additional experimental evidence suggests that the truncated Aβ11–40 peptide can capture the oligomerization/fibrillation behavior just as well as the full-length Aβ40 peptide.25 A scheme of the Aβ11–40 peptide is shown in Fig. 1, where the two hydrophobic patches (red) L17–A21 (central hydrophobic core, CHC) and A30–V40 (C-terminus) are separated by a hydrophilic (blue) loop region (E22–G29). The N-terminus (E11–K16) is also very hydrophilic, and plays an important role in metal ion interactions together with the residues H13 and H14.5
 | ||
| Fig. 1 Sequence of the Aβ11–40. Hydrophilic and hydrophobic regions are shown in blue and red, respectively. | ||
In order to complement experimental endeavors, Molecular Dynamics (MD) simulations have been employed successfully, such as in the search of potential inhibitors26,27 against the aggregation of wild-type Aβ oligomers.14–17 In particular, numerical studies employing replica exchange MD (REMD) have provided insight into the truncated Aβ11–40 peptide and its corresponding trimer (3Aβ11–40) in its wild type and mutant forms28–31 in solution. In addition to its soluble conformations, the insertion of the Aβ oligomers in membranes has been investigated via MD.32–35 For instance, for the transmembrane 4Aβ17–42 tetramer a helical structure35 has been described.
Mutations in the Aβ peptide modify its toxicity, assembly, and rate of fibril formation. Specifically, the mutations in the CHC and loop regions, including F19W,36 F20W,36 L17A/F19A,37 Flemish A21G,38 Dutch E22Q,39 Italian E22K,40 Arctic E22G,41,42 E22Δ,43 and Iowa D23N44 could affect the conformational changes in Aβ oligomers. Another example shown that the combination of mutation A2V in N-terminal and histidine tautomerism can affect the Aβ monomer structures and its aggregation process.45 Thus, numerous mutation studies have been carried out both experimentally and employing MD simulations. These studies and their main results are summarized in Table S1.† In particular, the influence of local physical interactions on the fibrillation kinetics and the structure and dynamics of Aβ40 has been characterized by experimental studies that include fluorescence, transmission electron microscopy, X-ray diffraction, and solid-state NMR spectroscopy.46 The hydrophobic contact between F19–L34 was modified by a series of mutations on the residues F19 and L34, including F19G, F19P, F19E, F19K, F19Y, F19W, L34E, and L34K. These mutants were studied to understand the effect of local interactions, including electrostatic interactions (F19E and F19K mutations); hydrophobic interactions (F19Y and F19W mutations); conformational flexibility (F19G and F19P mutations); and the salt-bridge interactions (L34E and L34K mutations). These local interactions were found to impact the fibrillation kinetics, the intermolecular hydrogen bonds and the dynamics of the Aβ40, without changing the general fibril structure. The results also demonstrated that the non-local F19–L34 contact plays an important role in early-oligomers of Aβ40. The F19W mutation showed slower fibrillation kinetics than the wild-type. While both F19Y and F19W mutations replace the Phe ring by another aromatic ring, the tryptophan mimicked Phe better. A subsequent study investigated how the F19K mutation altering the F19–L34 contact affects the fibril structure and the toxicity of the Aβ40.47 This mutation was found to alter the local structure of the fibril and to abolish cytotoxicity. In addition, computational studies have characterized the A21G mutation in Aβ40 and Aβ42,48 and also in transmembrane 3Aβ11–40.33
It has been shown that the “susceptibility of neuronal cells to different types of misfolded oligomeric assemblies is directly related to the extent of binding of such oligomers to the cellular membrane”.49 These experiments included relatively complex physiological scenarios that included Ca2+ influx and cellular damage and opened new lines of questioning, for instance, how mutations affect the binding to the membrane and which minimal models can capture changes in binding. In this work we set out to find a simple model that can show mutations affecting the binding affinity of the aggregates. For this, we noticed that the impact of F19 mutations on the oligomers' structure of Aβ40 in membrane lipid bilayers has not been characterized. Also, if one wants to characterize a sort of “threshold” for binding differences in the mutated oligomer, it is better to choose a “subtle mutation” that is close to the original amino acid and does not change its polar/non-polar nature. For the case of Phe, the closest non-polar one is Trp, as it lacks the –OH group of Tyr and therefore mimicks Phe better. We notice that MD simulations have been used to study the conformations of the 3Aβ11–40 trimer in both solution28 and dipalmitoylphosphatidylcholine (DPPC) lipid environment.34 In addition, the F19W mutant of the 11–40 truncated Aβ trimer (F19W 3Aβ11–40) in aqueous solution was recently characterized via MD simulations.31 In the present study, the (F19W 3Aβ11–40) trimer with an initial conformation obtained from the Aβ fibril was inserted into a DPPC lipid bilayer, solvated and then simulated using REMD techniques. The metastable structures of the transmembrane F19W 3Aβ11–40 were deduced using a combination of free energy surface and clustering methods. Our results provide detailed structural conformations of the transmembrane F19W 3Aβ11–40 and how they differ from the wildtype transmembrane 3Aβ11–40 obtained in previous studies.34 The binding free energy of the mutated oligomer clearly shows that the even the subtle F19W mutation greatly destabilizes the 3Aβ11–40 trimer with respect to its wild type counterpart.
Computational methods
Temperature-REMD simulations
The conformation of the transmembrane 3Aβ11–40 inserted in the DPPC membrane bilayers50 was taken from a previous study34 in which the crystal structure of the 3Aβ11–40 was obtained from a fibril-like structure.20 PYMOL tools51 were then used to create the mutated F19W version for the 3Aβ11–40. Finally, the F19W 3Aβ11–40 was inserted in the DPPC lipid bilayer. The mutant trimer was then represented using the united atom GROMOS 53a6 force field.52 While there are other IDP-specific force fields53,54 that may be used to simulate intrinsically disordered proteins (IDP) in solution, we used the united atom GROMOS 53a6 force field because it is known to be quite good for amyloid beta transmembrane proteins.55,56 In addition, to save computational time and compare with the 3Aβ11–40 wild-type results,34 we used united atom GROMOS 53a6 force field.The system was solvated using the simple point charge (SPC) water model.57 The solvated system was neutralized with three Na+ ions. The initial conformation of the transmembrane F19W 3Aβ11–40 is presented in Fig. 2, in which Na+ ions are represented by three black balls and the mutant points were highlighted. The entire solvated transmembrane F19W 3Aβ11–40 system consists of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 987 atoms, including the F19W Aβ oligomer, 3293 water molecules, 125 DPPC molecules and three Na+ atoms.
987 atoms, including the F19W Aβ oligomer, 3293 water molecules, 125 DPPC molecules and three Na+ atoms.
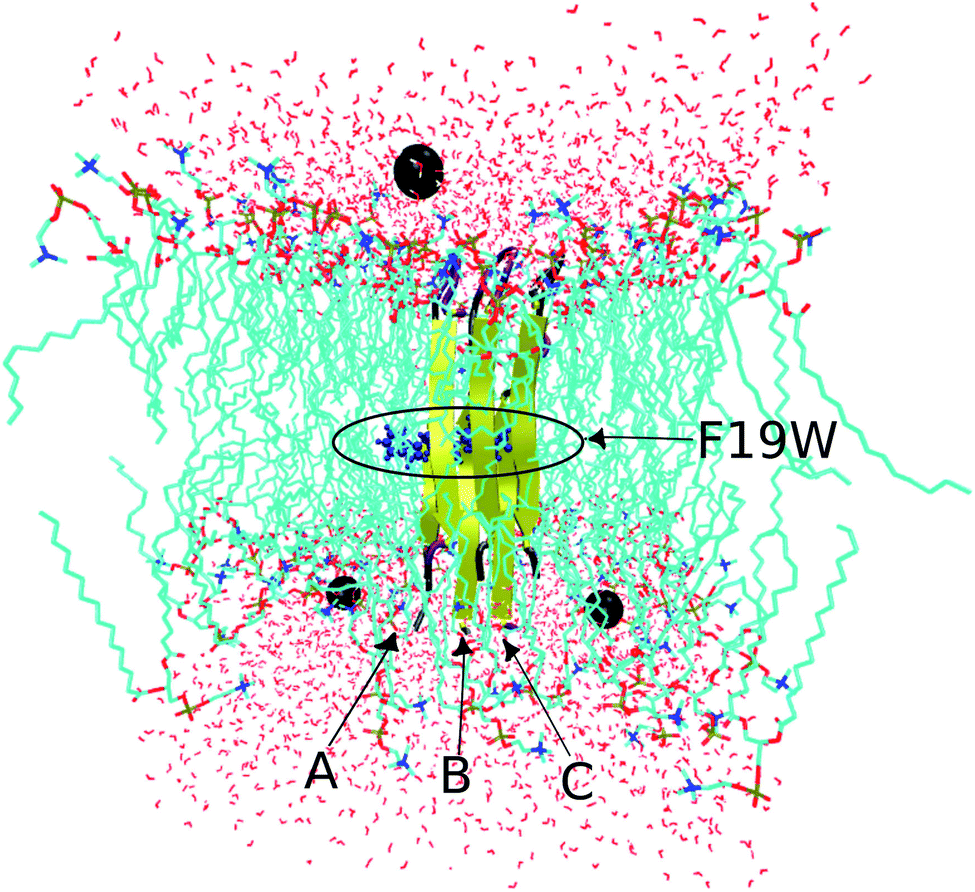 | ||
| Fig. 2 Initial conformation of the transmembrane F19W 3Aβ11–40 trimer with highlighted mutations. Water molecules are represented in red, DPPC molecules are shown in cyan, and neutralizing Na+ ions are shown as black balls. | ||
The initial structures for F19W 3Aβ11–40 were based on the previously deduced fibril-like structure. This choice is justified because it would take more than several microseconds per replica to simulating the aggregation of the Aβ from monomers. In this work, the transmembrane F19W 3Aβ11–40 was simulated using the T-REMD method with 32 replicas with temperatures varying from 321 to 423 K in the isothermal–isobaric (NPT) ensemble. The GROMACS version 5.1.3 was used with a periodic boundary condition (PBC) box with dimensions of 6.028 × 6.052 × 7.134 nm3 and a time step of 2 fs using a leap-frog algorithm.58 The electrostatic interactions were calculated using the particle mesh Ewald method with a 0.9 nm cut-off.59 The van der Waals interactions also had a 0.9 nm cut-off. The nonbonded pair lists were updated every 10 fs. The velocity-rescaling thermostat60 was used to control temperature, and the Parrinello–Rahman barostat61 was used to control pressure. All bonds were constrained by the LINear Constraint Solver (LINCS)62 with an order of 4. The individual temperatures were generated using a Web server.63 Exchanges between neighboring replicas were attempted every 1 ps, leading to mean acceptance ratios ranging from 18 to 25% (Fig. S2†). It was confirmed that all replicas have an efficient exchange rate over the whole temperature range, as is illustrated by the two examples shown in Fig. S3† which plots the temperature indices for the first replica (which has the lowest initial temperature) and last replica (which has the highest initial temperature), respectively. Each replica ran for 400 ns, resulting in a total of 12800 ns of MD simulations. Data was recorded every 10 ps. The results were analyzed for the last 150 ns of REMD simulations. The first 250 ns of the simulations were removed to avoid any starting bias. During the simulation, the membrane DPPC lipid bilayer was stable (Fig. S4†), in agreement with the non-mutated transmembrane 3Aβ11–40.34
Secondary structure analysis
The secondary structure parameters of the transmembrane F19W 3Aβ11–40, including coil, beta, turn, and helix contents, were predicted using DSSP tool.64Free energy surface (FES)
The free energy surface of the mutant trimer was constructed using the “gmx sham” tools65 of GROMACS with root-mean-square deviation (RMSD) and radius of gyration (Rg) serving as reaction coordinates.Free energy perturbation (FEP) method
The binding free energy between the mutant F19W 3Aβ11–40 and DPPC bilayer was predicted using the FEP method34,66 as described in the ESI.†Collision cross section (CCS)
The Ion Mobility Projection Approximation Calculation Tool (IMPACT) with the trajectory method was employed to compute the mutant trimer CCS.67Solvent-accessible surface area (SASA)
We calculated the total solvent-accessible surface area (SASA) for the transmembrane F19W 3Aβ11–40 with the double cubic lattice method68 as implemented in GROMACS.Computational analysis tools
The clustering method was carried out with a Cα RMSD cut-off of 0.3 nm.65,69 A non-bonded contact between heavy atoms of different residues was counted when their distance was smaller than 0.45 nm. A polar contact between two charged groups was counted when the distance between two specific atoms was equal or less than 0.46 nm. The intermolecular contact between heavy atoms of the F19W 3Aβ11–40 with the phosphate groups of the DPPC lipid bilayer was calculated by evaluating the minimum distance of the corresponding atoms with a cut-off of 0.45 nm. The contacts between side-chains (SCs) of the neighboring chains inside the trimer peptide were also counted if the distances between the SC were smaller than 0.45 nm. The lipid order parameters were computed using the formula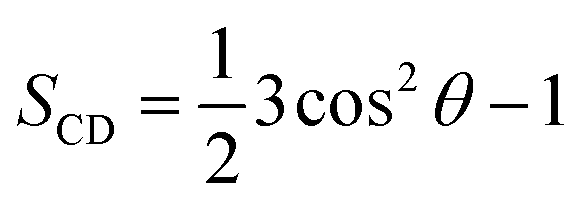 where C is carbon, D is deuterium, and θ is the angle between the molecular axis provided by the Ci−1 − Ci+1 vector and the bilayer normal and the results were averaged over the membrane during the simulation interval.
where C is carbon, D is deuterium, and θ is the angle between the molecular axis provided by the Ci−1 − Ci+1 vector and the bilayer normal and the results were averaged over the membrane during the simulation interval.
Results and discussion
Sampling convergence of the REMD simulations of the F19W 3Aβ11–40
REMD convergence at 324 K, above the phase transition 315 K temperature of the membrane DPPC lipid bilayer, was assessed by eight metrics calculated over time intervals of 250–350 ns and 250–400 ns. These included the percentage of secondary structures (beta, coil, turn and alpha contents), the radius of gyrations (Rg), the RMSD, the total solvent-accessible surface area, and the salt bridge D23–N27 of chain A of F19W 3Aβ11–40.Fig. 3 shows that the system had reached equilibrium at 324 K after 250 ns, with all metrics remaining consistent over the two time windows. Overall, the β content value varies in the range of 24–53% with the mean value of 44.36 ± 3.74% (Fig. 3a), while the random coil content spans the range of 27–59% with the average value of 41.7 ± 3.8% (Fig. 3b). The mean value of turn and helix contents are 1.18 ± 1.18% (Fig. 3c) and ∼0% (Fig. 3d), respectively. The mean Rg value of F19W 3Aβ11–40 is 1.47 ± 0.02 nm (Fig. 3e), which is larger than that in the wild type 3Aβ11–40 (1.42 ± 0.02 nm).34 The majority (71%) of the F19W 3Aβ11–40 population has a Rg higher than 1.45 nm (Fig. 3e), while the wild type 3Aβ11–40 has a Rg smaller than 1.45 nm.34 The mean RMSD value of the F19W 3Aβ11–40 is 0.53 ± 0.05 nm (Fig. 3f), larger than that of the wild 3Aβ11–40 (0.47 ± 0.07 nm).34 The distribution of total solvent accessible surface area of the F19W 3Aβ11–40 is rather broad, with the average value of 70.43 ± 3 nm2 (Fig. 3g) significantly higher than that of wild 3Aβ11–40 (64.73 ± 3.07 nm2). The distribution of the salt-bridge D23–N27 of chain A of the F19W 3Aβ11–40 is also broad, with the mean value of 1.04 ± 0.14 nm (Fig. 3h). The F19W 3β11–40 does not have a well-defined population with D23–N27 polar contacts, as was found in the wild 3Aβ11–40 (see below). As D23–N27 polar contacts play a crucial role in stabilizing the structures of the Aβ peptides and their fragments,34,70 the difference indicates that the F19W 3Aβ11–40 forms more extended structures that are less stable than those associated with wild 3Aβ11–40. Similar behavior of salt-bridge D23–N27 is also found in chain B and chain C (see Fig. S5 in the ESI†).
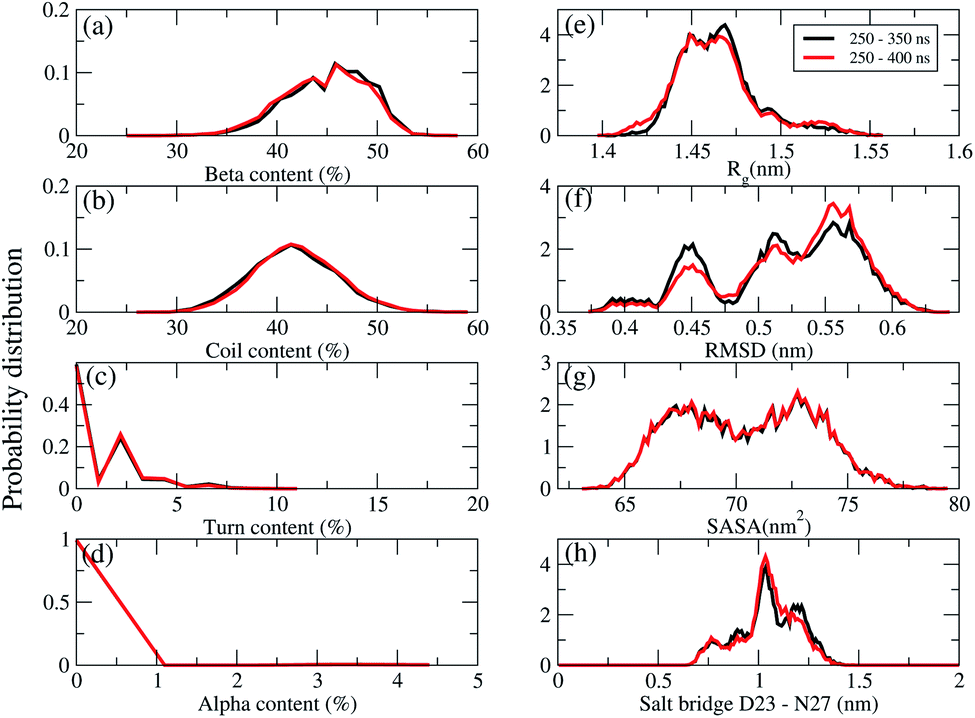 | ||
| Fig. 3 REMD simulations convergence at 324 K. Probability distributions (unnormalized) of the secondary structures (a) beta, (b) coil, (c) turn, (d) alpha contents, (e) the radius gyration (Rg), (f) the RMSD, (g) the total solvent-accessible surface area, (h) the salt bridge D23–N27 of chain A of the transmembrane F19W 3Aβ11–40. The results were calculated for two time windows 250–350 ns (black curves) and 250–400 ns (red curves). | ||
Distribution of secondary structures of the transmembrane F19W 3Aβ11–40 per residue
The averages of the random coil, beta, turn and α-helix structures are presented in Fig. 4. During our simulations, on average, the α-helix was rarely observed, comprising only ∼0.03% over the REMD simulations, which decreased in comparison with the wild type transmembrane 3Aβ11–40 ∼ 0.2% over the simulations. This data confirms that the α-helix is an intermediate step in the Aβ aggregation process.24,71,72 The turn population also decreased to ∼1.18% (it was ∼3% in the wild type transmembrane). The random coil conformation decreased from 57% in the wild type to 41.7% in the mutant type. The error bars of coil conformation for residues 22–29 is large due to the different behavior of the three chains (see Fig. S6†). In contrast, the β-content was dominant and increased from 40% in the wild type to 44.36% in the mutant type. This result is in good agreement with the solvated F19W 3Aβ11–40.31 Again, the error bars of the β-content is large for residues 20, 30–32 due to the fact that the population of β-content is quite different for each of the three chains (see Fig. S6†).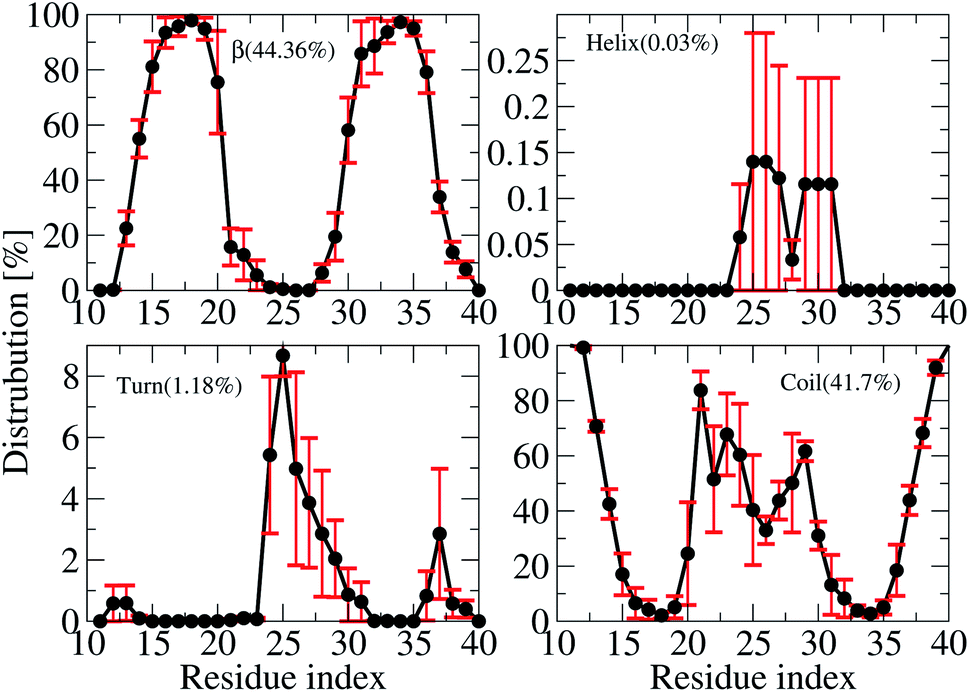 | ||
| Fig. 4 Secondary structure distributions per residue averaged for all three chains of the transmembrane F19W 3Aβ11–40. For clarity, the distribution of each secondary structure is presented on a different scale. The average values are given in the parenthesis. | ||
We also investigated the secondary structure along the sequence of each chain of the transmembrane F19W 3Aβ11–40 obtained in the last 150 ns of REMD simulations at 324 K (Fig. S6†). All chains were divided into five main sequences, in which sequences 14–20 and 30–36 exhibit mostly β-structures, while sequences 11–13, 21–29, and 37–40 exhibit mostly random coil structures. Overall, the two β-structure domains are separated by the three random coil regions. Turns are observed in the region dominant by coils, at residues 12–13, 24–32 and 36–37. The majority of turns were found at residues 24–26. The negligible amount of helical population was mostly found around residues 25–27 of chain B and 29–31 of chain C. In comparison with the wild-type truncated trimer,34 there are some slight shifts in the β-domains and random coils. In the wild type, β sheets were found in sequences 14–19 and 31–37, while random coils were observed at sequences 11–13, 20–30 and 38–40.
Interactions of the F19W Aβ11–40 chains with the other chains and with the lipid bilayer
To quantify the interactions of the peptides within the mutant truncated trimer, we constructed the backbone–backbone (BB–BB) and side-chain–side-chain (SC–SC) inter contact maps (Fig. 5) over the equilibrated snapshots. Looking at the inter-peptide SC–SC contact maps between neighboring chains of the transmembrane F19W 3Aβ11–40, we found diverse interactions between chain A–chain B (Fig. 5a) and chain B–chain C (Fig. 5b). In both Fig. 5a and b, these contacts can be divided into various regions: the interactions between CHC–CHC, C-terminal–C-terminal, C-terminal–CHC, loop–loop, N-terminal–CHC, N-terminal–C-terminal, C-terminal–loop, loop–CHC regions. The details of the contact probabilities between these chain pairs are given in Table S2.†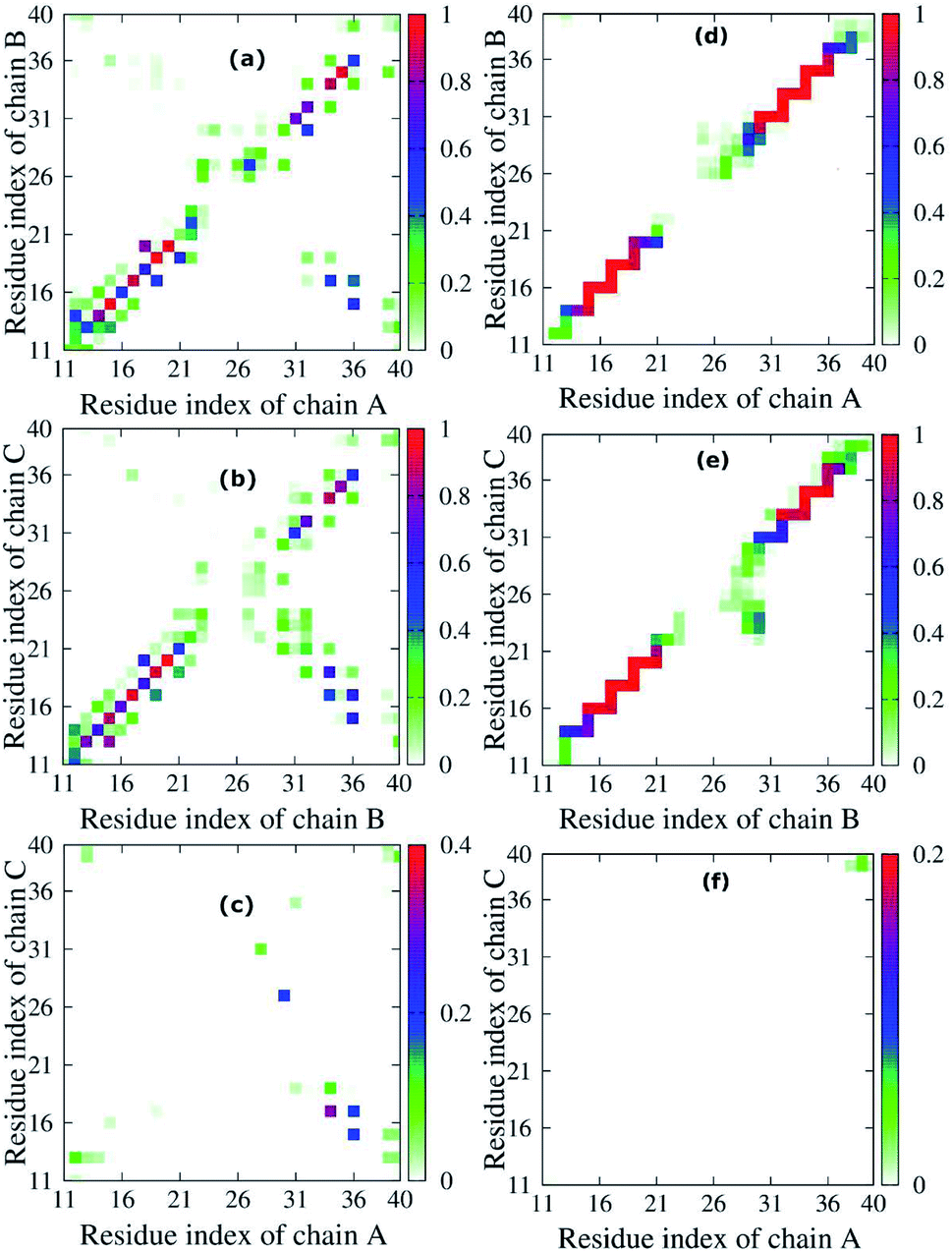 | ||
| Fig. 5 Interpeptide side-chain–side-chain, SC–SC, (left (a) Chain A - Chain B, (b) Chain B - Chain C, (c) Chain A - Chain C) and backbone–backbone, BB–BB, (right (d) Chain A -Chain B, (e) Chain B - Chain C, (f) Chain A - Chain C) contacts of the transmembrane F19W 3Aβ11–40. For clarity, the contact maps of neighbor pair – chains in the protein are presented on a different scale. The color in the figure indicates the probability of the contact between neighbor chains in the peptide. For example, in panel (a), the color varies from white to green, blue and red, indicating that the probability of the contact between chain A and chain B varies from 0% to 25%, 50% and 100%, respectively. | ||
By defining an 80% threshold, we found that the dominant interactions between both pairs chain A–chain B and chain B–chain C involves CHC–CHC, C-terminal–C-terminal and a part of N-terminal–N-terminal regions. The most populated residue-pair contacts are shown in Table S2.†
Other interaction regions have probability in the range of 10–80%. Importantly, the interpeptide SC–SC contact maps reveal many interactions between side-chains of residues L17 and W19 and side-chains of residues I32, L34 and V36 in both chain pairs: L34B–W19C (64.78%), V36B–L17C (61.65%), L34A–L17B (46.44%), etc. (Table S2†). These C-terminal–CHC interactions in early oligomers have been recently reported by experiments.21,23,46,73,74 The long-range contacts between CHC–CHC occur between V18–F20, A21–W19, W19–L17 in both chain pairs with populations between 35.83% to 79.65%. In addition, the C-terminal–C-terminal contacts show many long-range interactions between residues I32, L34, V36, V39 with residues A30, I32, L34, V39 and M35 in both chain pairs with populations varying from 10.71% to 50.71%. The strongest interactions are I32A–A30B (50.71%), L34A–I32B (26.90%) and V36A–L34B (21.12%), which have been studied by both experiments21,23,73 and computations.5,14 For C-terminal–N-terminal contacts, the interaction between V36–Q15 indicates that even polar side chains can be tolerated to a certain degree in the hydrophobic region. Finally, the N-terminal–N-terminal contacts cannot be ignored, with the strongest interactions occurring in Q15–H13 and V12–H14 between different chains. The details of the interactions for different residue pairs are shown in Table S1.† The C-terminal–loop, loop–loop and N-terminal–CHC contacts show interactions with much lower population (<30%) (Table S2†).
Unlike the diverse contacts between chain A–chain B and chain B–chain C, the chain A–chain C contact map (Fig. 5c) is rather sparse, indicating the lack of strong interactions. The hydrophobic interactions with highest probability appeared in the C-terminal–CHC regions between L34 and L17 (31.64%). The contacts in C-terminal–loop and C-terminal–N-terminal give interactions with probabilities around 20%. The contact probabilities of N-terminal–N-terminal, N-terminal–CHC and loop–C-terminal vary from 3.08% to 11.17%.
Fig. 5d–f shows the inter-peptide BB–BB contacts between adjacent chains of the transmembrane F19W 3Aβ11–40. The BB–BB contacts between chain A–chain B and chain B–chain C occur in the N-terminal–N-terminal, CHC–CHC, loop–loop and C-terminal–C-terminal regions. Details of contact probabilities between these chain pairs are given in Table S3.† Fig. 5d shows that the dominant BB–BB interactions are located in residues 15–19 of chain A with residues 14–20 of chain B and residues 30–36 of both chains, with probabilities in the range of 80% to 100% (Table S3†). Fig. 5e shows the dominant (probabilities of 80–100%) inter-peptide BB–BB interactions between chain B–chain C involve residues 16–21, 33–36 of both chains, and Q15B–H16C and V36B–G37C (Table S3†). Weaker, long-range interactions appear in the C-terminal–loop and loop–loop contacts, with probability ranging from 10% to 80%. The weakest interactions (probability lower than 10%) were all found in the loop regions. In Fig. 5f, the contact map is rather sparse, indicating that the contacts between chain A–chain C are negligible.
Overall, both SC–SC and BB–BB contact maps of chain pairs show diverse and strong interactions between chain A–chain B and chain B–chain C, while interactions between chain A–chain C are negligible. This implies that chain B stays in the middle between chain A and chain C during the simulation, contacting both A and C chains, which are thus separated from each other. In addition, the highest probabilities in both inter-peptide SC–SC and BB–BB contact maps appear in the parallel interactions between CHC–CHC, C-terminal–C-terminal and a small part of N-terminal–N-terminal of chain A–chain B and chain B–chain C which mainly correspond to the β sheets (sequences 14–20 and 30–36). This also support the fact that the trimer forms parallel β sheets, in good agreement with previous solid state nuclear magnetic resonance (ss-NMR), electron microscopy (EM) and electron paramagnetic resonance (EP) experimental studies on the structures of Aβ1–40 fibrils.23,74 The weakest interactions occur in the random coils (residues 11–13, 21–29, and 37–40) and helical contents regions (residues 25–27 of chain B and 29–31 of chain C).
The D23–N27, D23–K28 polar contacts have been shown in some previous studies21,22,34,70,75 to considerably contribute to stabilizing a loop that facilitates Aβ folding in solution. The distributions of the intra-molecular polar contacts of the transmembrane mutant F19W 3Aβ11–40 peptide are shown in Fig. 6, where upper and lower panels show results for D23–N27 and D23–K28, respectively. While chain A and B do not form D23–N27 polar contacts (Fig. 6a), chain C does with very low population (2.02%). The D23–K28 polar contact cannot be observed in chains A and C (Fig. 6b), and it rarely occurs in chain B (population 0.21%). Polar contacts are rarely observed in the chains of the F19W 3Aβ11–40 trimer because both K28 and N27 form contacts with the phosphate atoms of the DPPC lipid bilayers (see Fig. 7 below). This is in agreement with computational studies about the effect of lipid bilayers on the conformational changing of the Aβ40 monomer.76,77 Unlike the mutant, the wild-type truncated trimer has these polar contacts in all three chains, with very high population in chain C for D23–K28 contacts and in chains A and B34 for D23–N27 polar contacts. This suggests that the transmembrane wild-type trimer is more stable than the mutant, and that the F19W mutation would destabilize the folded trimer. In addition, the breakdown of essential salt-bridges can also lead to the lag of fibrillation. Sciarretta et al. studied the fibrillation rate of the Aβ1–40 and Aβ1–40Lactam (D23/K28).78 They proved that although Aβ1–40Lactam (D23/K28) forms fibrils similar to those formed by Aβ1–40, the fibrillogenesis rate increased to 1000-fold by suppressing the lag period. They highlighted that in Aβ1–40Lactam (D23/K28), the Lactam linkage resulted a bend-like structure in the peptide.
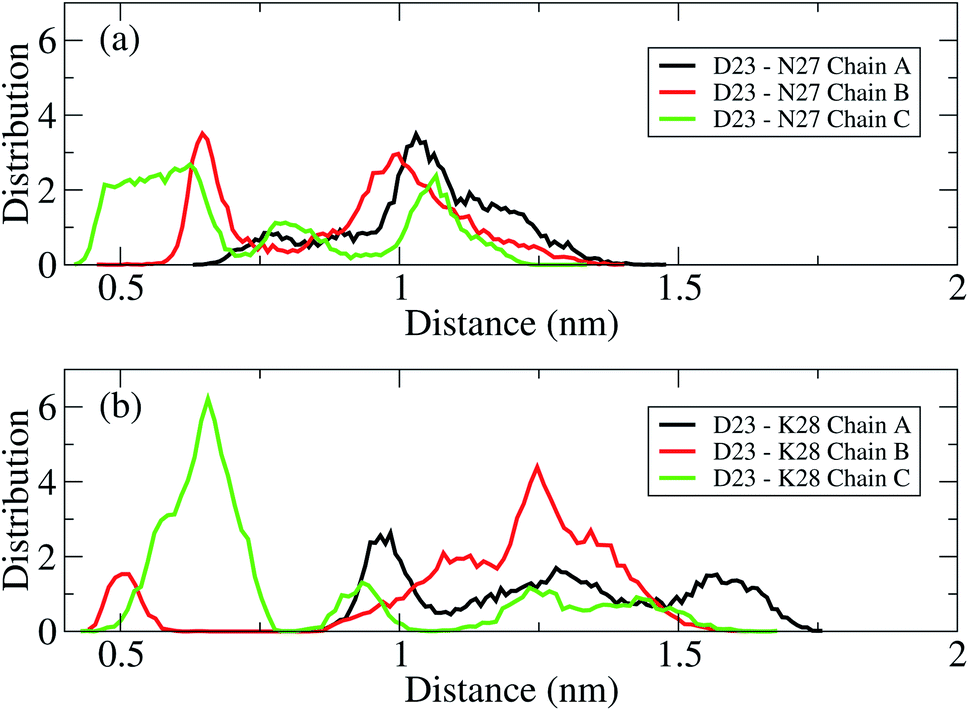 | ||
| Fig. 6 Distance distributions between the charge groups of D23 and N27 (a) and D23 and K28 (b) in chain A (black), chain B (red), and chain C (green) of the transmembrane mutant F19W 3Aβ11–40. The polar contacts are counted when their distance is within a 0.46 nm cutoff. | ||
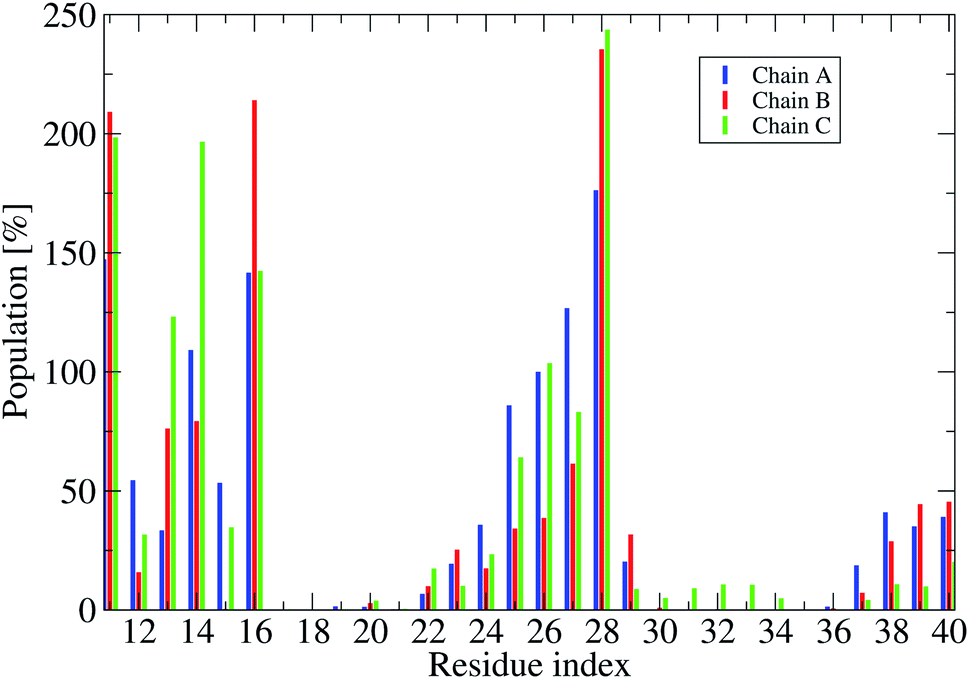 | ||
| Fig. 7 Population of intermolecular contacts between phosphate atoms of DPPC lipid bilayers and heavy atoms of the truncated F19W 3Aβ11–40. The population per residue is the sum of all populations of phosphate atoms contacts with one residue of the trimer (thus an individual value could be over 100%.). | ||
To understand how the protein interacts with the membrane lipid bilayers, we calculated the probabilities of intermolecular contacts between phosphate atoms of DPPC lipid bilayers and heavy atoms of the truncated trimer F19W (Fig. 7 and S8†), and the contact map between phosphate atoms of DPPC bilayers and each residue (Fig. S7†).
Fig. 7 shows that in all three chain residues E11, K16 and K28 have contacts populations higher than 100%. This means that after the F19W mutation, residues E11, K16 and K28 in all chains contact the membrane in all the conformations. This is in agreement with previous studies showing that K16 and K28 form the most regular contacts with lipid phosphate head groups.76,77 In comparison to the wild-type 3Aβ11–40,34 there are some remarkable changes in the membrane contacts of several regions of the mutant. In the F19W 3Aβ11–40 trimer, residues 22–23 in the random coil in the loop region of the chain (Fig. S6†) do contact the lipid bilayers, which does not happen in the wild-type 3Aβ11–40 trimer. In addition, there are fewer contacts in residues 17–21 and 35–36 in the mutant, while residues E11 and K28 increase their contact with the lipid bilayers, thus decreasing the D23–K28 polar contacts. Similarly, the increase of contacts between the N27 and D23 residues and the membrane results in the disappearance of the D23–N27 salt-bridge. In short, F19W mutation leads to more protein–membrane contacts and precludes the formation of crucial salt-bridges, which may decrease the aggregation rate.
Free energy surface and representative structures of the transmembrane F19W 3Aβ11–40 trimer
To characterize the conformations of the transmembrane F19W 3Aβ11–40 trimer, we constructed the free energy surface (FES) as a function of RMSD and radius gyration Rg and then used clustering methods65 to identify the metastable states. The FES is shown in Fig. 8, with the RMSD values in the range of 0.37–0.66 nm, and Rg values between 1.39–1.56 nm. The FES reveals twelve minima, denoted as S1–S12 with the representative structures shown in Fig. 9. The twelve states have populations varying from 17.29% to 0.51%, with their total population accounting for 67% of the system's fluctuation. All the twelve states form U-shaped conformations with three parallel β-strands in CHC and with the C-terminal regions separated by random coils in the loop region. Strong hydrophobic interpeptide contacts in these β-strands regions create a β-core of the trimer aligning parallelly to the lipid bilayer. In contrast, the random coil regions located at the end of the β-core strongly interact with phosphate head groups of DPPC lipid bilayers. This result captures the experimental structures of Aβ25–35 in membranes, in which the hydrophobic β-sheets are inserted into membranes while hydrophilic regions interact with the membrane surface.79 The result is also consistent with computational studies of the Aβ10–40 peptide.80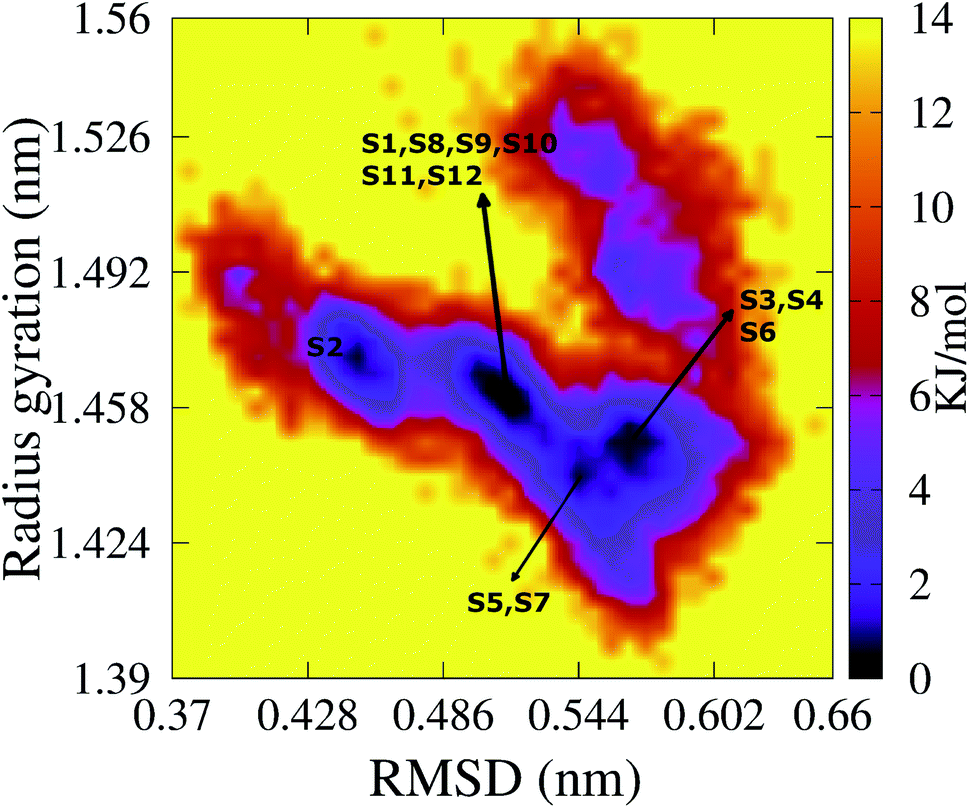 | ||
| Fig. 8 The FES of the transmembrane F19W 3Aβ11–40 as a function of RMSD and radius gyration Rg. Twelve minima are noted from S1 to S12 with those representative structures shown in Fig. 9. For clarity, several minima very closed to each other are shown in only one line with the names of the minima are noted. | ||
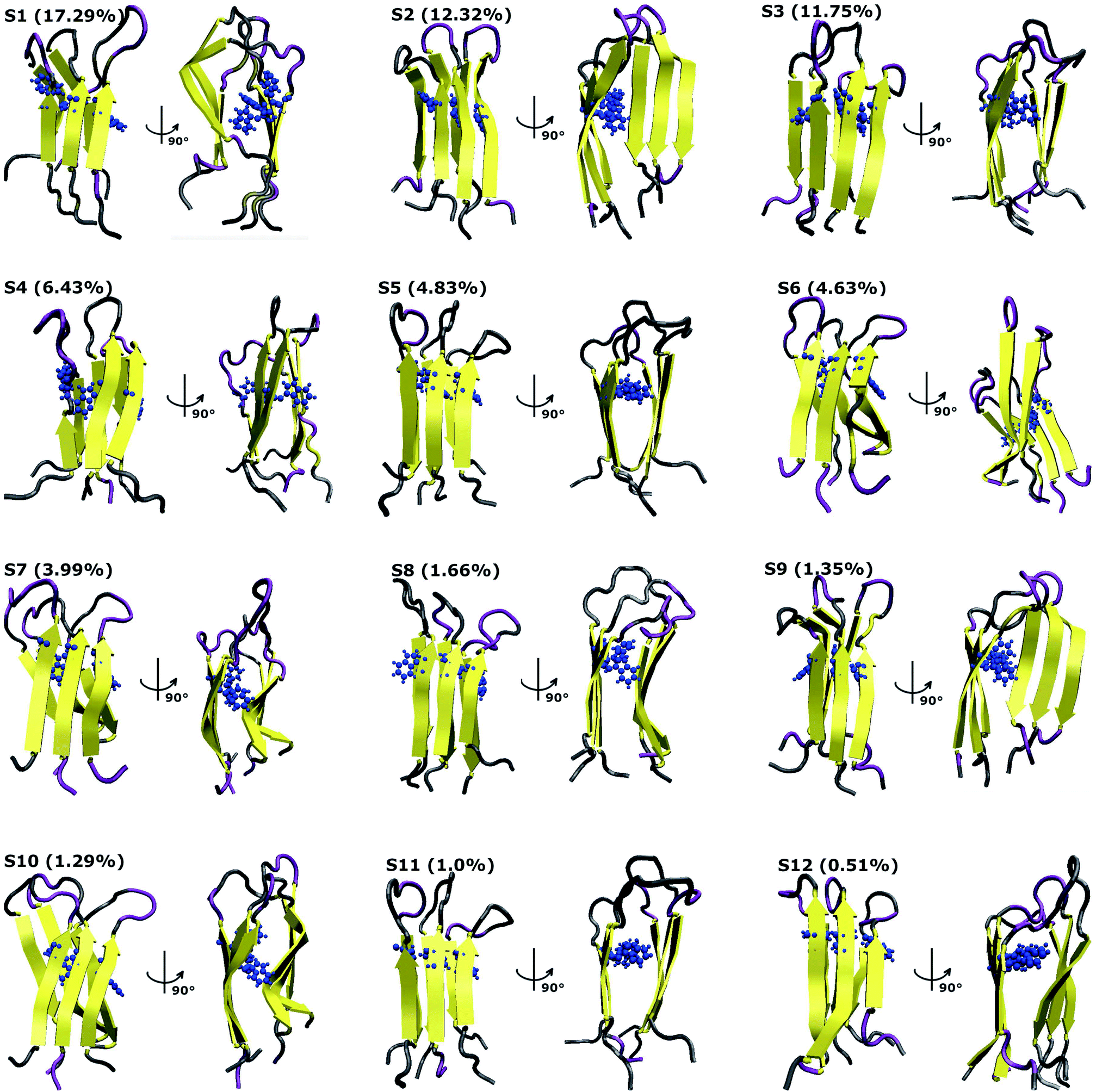 | ||
| Fig. 9 Representative structures of the twelve minima shown in Fig. 8. The population of each state is calculated using FES and clustering methods and is given in parentheses. Here, the residues β-contents are represented in yellow, those of mutant F19W are shown in blue, and coil conformations are shown as grey and purple colors, respectively. | ||
Based on all the conformations of each state, properties of the twelve states were computed and are presented in Table 1, which includes populations, the RMSD, the Rg, the secondary structures, the collision cross sections and solvent-accessible surface areas. Dominant secondary structures are β-strands and random coils. On average, the populations are 45% β-strand, 39.58% random coil, 0.42% turn and 0% helix.
| Minima | P (%) | Rg (nm) | RMSD (nm) | Coil (%) | β% | Turn (%) | Helix (%) | CCS (Å2) | SASA (nm2) |
|---|---|---|---|---|---|---|---|---|---|
| a Shown are the state population P in %, the Rg (nm), the RMSD (nm), the secondary structure terms in %, the collision cross sections in Å2 and the solvent-accessible surface areas in nm2.b Average reported in ref. 34.c Average reported in ref. 31. | |||||||||
| S1 | 17.29 | 1.464 | 0.516 | 39 | 47 | 1 | 0 | 1416 | 72.46 |
| S2 | 12.32 | 1.470 | 0.439 | 37 | 49 | 0 | 0 | 1381 | 67.49 |
| S3 | 11.75 | 1.454 | 0.581 | 45 | 40 | 0 | 0 | 1434 | 72.791 |
| S4 | 6.43 | 1.447 | 0.569 | 41 | 40 | 2 | 0 | 1376 | 69.24 |
| S5 | 4.83 | 1.448 | 0.533 | 41 | 43 | 0 | 0 | 1394 | 70.63 |
| S6 | 4.63 | 1.435 | 0.561 | 37 | 46 | 0 | 0 | 1345 | 65.34 |
| S7 | 3.99 | 1.435 | 0.551 | 38 | 45 | 0 | 0 | 1352 | 65.37 |
| S8 | 1.66 | 1.461 | 0.506 | 39 | 46 | 2 | 0 | 1410 | 68.79 |
| S9 | 1.35 | 1.469 | 0.484 | 39 | 49 | 0 | 0 | 1379 | 69.36 |
| S10 | 1.29 | 1.460 | 0.517 | 39 | 48 | 0 | 0 | 1392 | 68.78 |
| S11 | 1.00 | 1.451 | 0.528 | 46 | 41 | 0 | 0 | 1415 | 73.96 |
| S12 | 0.51 | 1.460 | 0.530 | 34 | 46 | 0 | 0 | 1379 | 67.70 |
| Average | 1.455 | 0.526 | 39.58 | 45 | 0.4 | 0 | 1389.4 | 69.33 | |
| Average of wild-typeb | 1.424 | 0.566 | 54 | 44 | 2 | 0 | 1340 | 64.18 | |
| Average of solvated F19Wc | 1.43 | 0.90 | 51 | 39 | 8 | 2 | 1347 | 63.30 | |
The first four states, S1–S4, account for 47.29% of the ensemble. The detailed topological characterization of those states are shown in Table S4,† including the positions for β-strands and coils, the orientation of the two β-strands and the inter-peptide contacts that stabilized the two β-domains. S1 with a population of 17.29% is characterized by parallel β-strands spanning residues 15–20, 28–36 in chain A, residues 15–19, 28–36 in chain B and residues 15–19, 28–35 in chain C. The two β-strands in each chain form two antiparallel β-sheets, resulting in two antiparallel, three-stranded β-sheets for the trimer. Random coils are present at residues 11–14, 37–40 in all three chains, and loop regions at residues 20–27 in chain A and 21–27 in chains B and C. Propensities for β-strands, coils, turns and helices are 47%, 39%, 1% and 0%, respectively. The state is stabilized by the inter-peptide contacts CHC–CHC, C-ter–C-ter, CHC–C-ter and N-ter–N-ter between chains A–B and chains B–C. The collision cross section is 1416 Å2, while the solvent-accessible surface area is 72.46 nm2. Each state in S2, S3, and S4 has also two three-stranded β-sheets spanning different residues in the chains, separated by 3 coils domains, and packed either perpendicularly (state S2), or in antiparallel (state S3 and S4) fashion (Table S4†). S1 and S2 have similar β populations, and these values are higher than those obtained in S3, S4 (Table 1). In addition, S1 and S3 have similar values of CCS and SASA, which are higher than those found in S2 and S4 (Table 1). Only S1 and S4 have turn contents with very low populations.
Among these twelve states, S2 and S9 have the highest β population (49%). States S11 and S3 rank highest in random coil (46%, 45%). Turn populations are very low in all cases. State S11 is most exposed to water, with a solvent accessible surface area of 73.96 nm2, while the states with least access to water are S6 and S7, with SASA 65.34 nm2 and 65.37 nm2. Each state among S1, S3, S4, S5, S8, S11 and S12 has two antiparallel β-sheets facing to each other, while the two β-sheets are located in two perpendicular planes for states S2, S6, S7, S9 and S10. The two β-sheets form the most in-register antiparallel in states S5, S8, S11, and they are the most perpendicular in states S2, S7, S10.
Average values over for the twelve states of the transmembrane F19W 3Aβ11–40 trimer are 45% β sheet, 39.58% coil, 0.42% turn, 0% helix, 1389 Å2 for CCS, and 69.33 nm2 for SASA. In comparison, those numbers in the truncated wild-type trimer are 44%, 54%, 2%, 0%, 1340 Å2 and 64.18 nm2 for β, coil, turn, helix contents, CCS and SASA, respectively. The only significant difference between the two is the decrease of the coil content, and increases for both the CCS and the SASA values for the mutant trimer with respect to the wild type.
Finally, the free energy values for these twelve states S1–S12 are −13.70, −12.56, −11.40, −13.13, −12.56, −12.56, −11.98, −13.7, −10.27, −13.7, −12.56 and −11.41 kJ mol−1, respectively. The global minimum, −13.70 kJ mol−1 is comparable to the wild type minimum in DPPC lipid bilayers (about ∼0.2 kJ mol−1 higher34) and it is about ∼0.9 kJ mol−1 lower than those found in solvated F19W 3Aβ11–40.31 The membrane truncated F19W trimer is more flexible than the wild-type trimer, due to the higher number of minima with smaller free energy barriers. This is consistent with the disappearance of essential polar contacts. Three states S1, S8 and S10 have the same lowest free energy value, −13.7 kJ mol−1, however, the populations are 17.29%, 1.66% and 1.29%, correspondingly. It indicates that the global representative structure of the system is S1. The total population of three states found in the global minimum is 20.24% of the ensemble. Meanwhile, four states were found in the global minimum of the wild-type truncated trimer (3Aβ11–40), with populations of 29%, 21%, 13% and 9%,34 resulting the total population 72% of the ensemble. From that point of view, the latter has a higher flexibility than the wild-type 3Aβ11–40.
Binding free energy of the F19W 3Aβ11–40 trimer to the DPPC lipid bilayer
To quantify the interactions between the mutant trimer and the lipid membrane, we calculated the binding free energy of the truncated mutant trimer and the DPPC using the double-annihilation binding free energy method. In this method, the peptide is annihilated by both the solvated and transmembrane systems. As the structure S1 has the highest population in the total ensemble, it was adopted as the initial structure for FEP computations.66 The binding free energy (ΔGbind) was estimated by the difference in the annihilation energy between the transmembrane protein and that of the corresponding solvated protein. ΔGbind consists of two terms, the Coulomb interaction energy ΔGCou and the van der Waals interaction energy ΔGvdW: ΔGbind = ΔGCou + ΔGvdW.For F19W 3Aβ11–40, the calculated values for ΔGCou and ΔGvdW are 132.65 ± 8.24 and −155.91 ± 2.70 kcal mol−1, respectively, resulting in a ΔGbind value of −23.26 ± 7.39 kcal mol−1. For the wild type, the ΔGCou and ΔGvdW values are 114 ± 18 and −184 ± 3 kcal mol−1, respectively,34 with a total ΔGbind value of −70 ± 18 kcal mol−1. Thus, the ΔGbind of F19W 3Aβ11–40 is significantly higher than that obtained for the wild type trimer using the same method, indicating that the mutant has less binding affinity to the membrane than the wild type. The significant difference in ΔGbind arises from the difference of collective Coulomb and van der Waals binding energies of the two proteins with DPPC bilayer. In comparison with the trimer wild type, both ΔGCou and ΔGvdW increase for the mutant type. In particular, the increase in ΔGCou is consistent with the increasing interactions between the phosphate groups of DPPC and the charged residues E11, E22 and D23 discussed previously (Fig. 7 and S8†).
Comparison with other studies
The collision cross section (CCS) is an important parameter for describing proteins in both experimental and computational methods. In experiments, CCS can be estimated by ion mobility mass spectrometry (IM-MS),81,82 while computationally, it can been calculated by the IMPACT method.67,83 CCS values of the representative structures based on IMPACT with the trajectory method are shown in Table 1. The CCS values of the twelve states range between 1345 and 1434 Å2, with an average of 1389.4 Å2. Although the experimental CCS value of the transmembrane F19W trimer is unavailable, the size of the trimer Aβ40 in solution was determined by IM-MS studies using distinct samples leading to mean collision-cross sections (CCSs) of 1265 and 1481 Å2.84,85 Our CCS values are in good agreement with these experimental results. Recently, using REMD simulations to study the stability of the Aβ11–40 trimer with antiparallel and parallel β-sheet organizations in the DPPC lipid bilayer, Ngo et al. shown that the CCS values of the F20W 3Aβ11–40 vary between 1351 and 1506 Å2,86 with an average of 1417.7 Å2. Our CCS values are smaller than that obtained from Ngo's paper due to the different point mutations between the two studies (F19W in our case, and F20W in Ngo's paper). Also, the starting structures in Ngo's paper are both U-shaped conformations (parallel β-sheets) and β-hairpin (antiparallel β-sheets), while the initial structure in our study is U-shaped.CD experiments with different sample preparations have shown that the β content of the Aβ trimer in solution is around 50% (ref. 24) or 40.8%.87 Our simulations gives 44.36%, in between the two CD-derived values. Also, during our simulations, α helices were rarely observed (∼0.03% over the simulations). In a previous study based on REMD simulations with the AMBER96 force field to study trimer Aβ10–35, Jang et al. reported the propensity of β-strands was ∼50% with negligible α helices.71 More recently, using the four-bead coarse-grained discrete molecular dynamics simulations to study the Aβ oligomers,88 Urbanc et al. found that the β-strand populations of 17% and 19%, and turn populations of 44% and 43% for the wild-type Aβ1–40 and Aβ1–42 trimers, respectively. Besides, based on REMD simulations to study the stability of the F20W Aβ11–40 trimer transmembrane with antiparallel and parallel β-sheet organizations, Ngo et al. reported that the β-contents is in the range of 44% to 60%, with a mean value of 49.71%, the coil-contents population of 37% to 58%, with an average value of 48%, and turn 2%.86 Our results, consistent with the first and third REMD studies and experimental results, do not support this: in particular, the β-strand propensities of residues 14–20 and 30–36 have an average value of 85%, while the largest β-strand content never exceeds 30% in the ref. 88. In addition, our computational studies found many non-local contacts between W19 and L34, and an increase in β population consistent with Huster and Hoffmann's experimental studies46,47 that were focused on the structure of the mature fibrils. The increase of β population also consistent with Ngo's results in F20W 3Aβ11–40 transmembrane studies.86 Our investigation provides a better understanding the structure of oligomers after the F19W mutation, specifically related to the conformational changes of the transmembrane F19W 3Aβ11–40 in DPPC lipid bilayers. In particular, the mutated residue inserted itself into the fibril core, in agreement with previous Thioflavin and tryptophan fluorescence and transmission electron microscopy experimental studies.36 Additionally, we found that F19W mutation destabilized the structure of 3Aβ11–40 in the membrane. It is consistent with Ngo's previous studies shown that F19P mutation destabilized the structure of 3Aβ11–40 in the DPPC lipid bilayer.86
We also found U-shaped conformations with two three-stranded β-sheets in the CHC and C-terminal regions that oriented in (i) the antiparallel form captured in many ss-NMR studies for Aβ oligomers varying between 4 and 33 chains5 – these forms may act as nucleation sites for antiparallel β-sheets fibrils as observed in Aβ1–40,23 and Aβ16–22;15 (ii) perpendicular orientation, consistent with the observations in coarse-grained15–17 and all-atom simulations14,89 of amyloid-peptides. In addition, the two β regions are completely inserted in the membrane, stabilized by the hydrophobic inter-peptide contacts, and separated by three random coils that interact with the phosphate head groups on the surface of the membrane. These findings are consistent with experimental studies of Aβ25–35 in membranes79 and with computational studies of Aβ10–40 in membranes.80 Finally, because the amyloid landscape is highly heterogeneous and sensitive to the experimental conditions, we cannot neglect that the β-hairpin conformations may also exist.86
Conclusions
In AD, the Aβ peptide is involved in neuronal toxicity via interactions with the cell membrane. Lipid membranes are known to modulate the rate and mechanisms of Aβ self-assembly by having the lipid molecules interact specifically with the growing fibrils, and thus accelerate the fibril growth rate.90 In addition, the vulnerability of cells to the effects of oligomeric aggregates is directly associated to the oligomer binding affinity to the cell membrane.49 Given these findings, we presented a minimal oligomer model, the 3Aβ11–40 trimer, with a “subtle mutation” F19W, where the Trp residue preserves the aromatic, non-polar character of the Phe residue, in order to investigate whether such minimal mutation can alter the binding affinity and the conformations of the mutated trimer compared to the wild type trimer. In order to achieve this, we ran extensive all-atom REMD simulations of the mutated F19W 3Aβ11–40 trimer both in solution and transmembrane DPPC lipid bilayers and computed the all-atom free energy landscape in terms of two order parameters (radius of gyration and RMSD). We then computed the F19W trimer binding free energies to the lipid bilayer.We found that the mutation brought about some non-negligible conformational changes with respect to the wild type. In particular, the average populations of alpha and turn motifs slightly decreased, but they were almost negligible in the wild type to start with. The random coil population decreased by 15.3%, from 57% in the wild type to 41.7% in the mutant. In contrast, the dominant β content increased by 4.4%, from 40% in wild type to 44.4% in the mutant type. Both the radius of gyration and the RMSD slightly increased, while the SASA increased from 64.73 nm2 in 3Aβ11–40 to 70.43 nm2 in the transmembrane F19W 3Aβ11–40. The latter suggests that the F19W 3Aβ11–40 trimer may aggregate more slowly than the wild type, which would be consistent with experiments46 showing that the F19W mutation slows down the fibrillation kinetics. The essential salt-bridges of 3Aβ11–40 disappeared in F19W 3Aβ11–40, indicating that the F19W mutation could destabilize the truncated trimer within the membrane. The interactions between the phosphate atoms of the DPPC lipid bilayers and the heavy atoms of the F19W 3Aβ11–40 trimer differ along the sequences and the residues of the trimer, and the total amount of contacts between the protein and membrane increases.
More important differences were found in the free energy surface in terms of the Rg and RMSD order parameters. This surface displayed twelve minima that account for 67% of the ensemble; by comparison, the wild-type free energy surface only displayed five that accounted for 100% of total conformations.34 The free energy values of the twelve states vary from −13.70 to −11.41 kJ mol−1, and the global minimum free energy is about ∼0.2 kJ mol−1 higher than those found in the minima of wild-type 3Aβ11–40. Although the free energy difference is small, the mutant is more flexible, due to the population of global minimum in F19W 3Aβ11–40 is much lower (20.24%) than that in wild-type truncated trimer (72%).34 The representative states are consistent with many other simulations' results and may act as nucleation sites for the fibrillation process. Finally, rather dramatic differences were found in the F19W 3Aβ11–40 binding free energy to the DPPC bilayer, computed using the FEP method. Our results indicate that this binding free energy is ∼40–50 kcal mol−1 higher than that in the wild-type 3Aβ11–40 trimer.
Altogether, our studies provide insight into the effect of mutation F19W on transmembrane 3Aβ11–40. The disappearance of crucial salt-bridges, the increase of the interactions between the peptides and the membrane as well as the greater structural diversity with higher free energy values indicate that the mutant is more flexible than the wild type, while the binding free energy indicates that F19W 3Aβ11–40 is considerably less stable in the lipid environment than its wild-type counterpart. These results suggest that the impact of mutations can be assessed, at least partially, by evaluating the interactions of both the wild-type and the mutated oligomers with the lipid membranes.
Author contributions
T. T. Tran run simulations. T. T. Tran and F. Pan analyze the simulations' data. T. T. Tran, F. Pan, L. Tran, C. Roland and C. Sagui write the original draft of the manuscript. T. T. Tran, F. Pan, C. Roland and C. Sagui review and edit the final pre-publication stages.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Son Tung Ngo for valuable discussion. This research is supported by Department for Management of Science and Technology Development (DEMASTED), Ton Duc Thang University, Ho Chi Minh City, Vietnam for T. T. Tran.Notes and references
- M. Prince, A. Comas-Herrera, M. Knapp, M. Guerchet and M. Karagiannidou, World Alzheimer Report 2016: Improving healthcare for people living with dementia: coverage, quality and costs now and in the future, Alzheimer's Disease International (ADI), London, 2016 Search PubMed.
- L. W. Chu, Hong Kong Med. J., 2012, 18, 228–237 CAS.
- A. Soldano and B. A. Hassan, Curr. Opin. Neurobiol., 2014, 27, 61–67 CrossRef CAS.
- R. Ossenkoppele, Y. A. Pijnenburg, D. C. Perry, B. I. Cohn-Sheehy, N. M. Scheltens, J. W. Vogel, J. H. Kramer, A. E. van der Vlies, R. La Joie, H. J. Rosen, W. M. van der Flier, L. T. Grinberg, A. J. Rozemuller, E. J. Huang, B. N. van Berckel, B. L. Miller, F. Barkhof, W. J. Jagust, P. Scheltens, W. W. Seeley and G. D. Rabinovici, Brain, 2015, 138, 2732–2749 CrossRef.
- J. Nasica-Labouze, P. H. Nguyen, F. Sterpone, O. Berthoumieu, N.-V. Buchete, S. Coté, A. D. Simone, A. J. Doig, P. Faller, A. Garcia, A. Laio, M. S. Li, S. Melchionna, N. Mousseau, Y. Mu, A. Paravastu, S. Pasquali, D. J. Rosenman, B. Strodel, B. Tarus, J. H. Viles, T. Zhang, C. Wang and P. Derreumaux, Chem. Rev., 2015, 115, 3518–3563 CrossRef CAS.
- K. F. Winklhofer, J. Tatzelt and C. Haass, EMBO J., 2008, 27, 336–349 CrossRef CAS.
- R. Ehehalt, P. Keller, C. Haass, C. Thiele and K. Simons, EMBO J., 2003, 160, 113–123 CAS.
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741–766 CrossRef CAS.
- R. Francis, G. McGrath, J. Zhang, D. A. Ruddy, M. Sym, J. Apfeld, M. Nicoll, M. Maxwell, B. Hai, M. C. Ellis, A. L. Parks, W. Xu, J. Li, M. Gurney, R. L. Myers, C. S. Himes, R. Hiebsch, C. Ruble, J. S. Nye and D. Curtis, Dev. Cell, 2002, 3, 85–97 CrossRef CAS.
- C. Bi, S. Bi and B. Li, Aging Dis., 2019, 10, 383–403 CrossRef.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS.
- L. Tran, Curr. Pharm. Des., 2018, 24, 3341–3346 CrossRef CAS.
- L. Nagel-Steger, M. C. Owen and B. Strodel, ChemBioChem, 2016, 17, 657–676 CrossRef CAS.
- B. Tarus, T. T. Tran, J. Nasica-Labouze, F. Sterpone, P. H. Nguyen and P. Derreumaux, J. Phys. Chem. B, 2015, 119, 10478–10487 CrossRef CAS.
- T. T. Tran, P. H. Nguyen and P. Derreumaux, J. Chem. Phys., 2016, 144, 205103 CrossRef.
- M. Chiricotto, T. T. Tran, P. H. Nguyen, S. Melchionna, F. Sterpone and P. Derreumaux, Isr. J. Chem., 2017, 57, 564–573 CrossRef CAS.
- F. Sterpone, S. Doutreligne, T. T. Tran, M. Baaden, S. Melchionna, P. H. Nguyen and P. Derreumaux, Biochem. Biophys. Res. Commun., 2018, 498, 296–304 CrossRef CAS.
- H. Ding, J. Schauerte, D. Steel and A. Gafni, Biophys. J., 2012, 103, 1500–1509 CrossRef CAS.
- M. K. Jana, R. Cappai, C. L. L. Pham and G. D. Ciccotosto, J. Neurochem., 2016, 136, 594–608 CrossRef CAS.
- I. Bertini, L. Gonnelli, C. Luchinat, J. Mao and A. Nesi, J. Am. Chem. Soc., 2011, 133, 16013–16022 CrossRef CAS.
- A. T. Petkova, W. M. Yau and R. Tycko, Biochemistry, 2006, 45, 498–512 CrossRef CAS.
- A. T. Petkova, et al., Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS.
- R. Tycko and R. B. Wickner, Acc. Chem. Res., 2013, 46, 1487–1496 CrossRef CAS.
- M. D. Kirkitadze, M. M. Condron and D. B. Teplow, J. Mol. Biol., 2001, 312, 1103–1119 CrossRef CAS.
- G. Bitan, S. S. Vollers and D. B. Teplow, J. Biol. Chem., 2003, 278, 34882–34889 CrossRef CAS.
- S. T. Ngo, D. T. Truong, N. M. Tam and M. T. Nguyen, J. Mol. Graphics Modell., 2017, 76, 1–10 CrossRef CAS.
- S. T. Ngo, X.-C. Luu, N. T. Nguyen, V. V. Vu and H. T. T. Phung, PLoS One, 2018, 13, e0204026 CrossRef.
- S. T. Ngo, H. M. Hung, D. T. Truong and M. T. Nguyen, Phys. Chem. Chem. Phys., 2017, 19, 1909–1919 RSC.
- S. T. Ngo, H. M. Hung, N. D. Hong and N. T. Tung, J. Mol. Graphics Modell., 2018, 83, 122–128 CrossRef CAS.
- S. T. Ngo, H. T. Thu Phung, K. B. Vu and V. V. Vu, RSC Adv., 2018, 8, 41705–41712 RSC.
- S. T. Ngo, X.-C. Luu, M. T. Nguyen, C. N. Le and V. V. Vu, RSC Adv., 2017, 7, 42379–42386 RSC.
- S. T. Ngo, Commun. Phys., 2018, 28, 265–276 CrossRef.
- S. T. Ngo, M. T. Nguyen, N. T. Nguyen and V. V. Vu, J. Phys. Chem. B, 2017, 121, 8464–8474 CrossRef.
- S. T. Ngo, H. M. Hung, K. N. Tran and M. T. Nguyen, RSC Adv., 2017, 7, 7346–7357 RSC.
- S. T. Ngo, P. Derreumaux and V. V. Vu, J. Phys. Chem. B, 2019, 123, 2645–2653 CrossRef CAS.
- R. T. McDonough, G. Paranjape, F. Gallazzi and M. R. Nichols, Arch. Biochem. Biophys., 2011, 514, 27–32 CrossRef CAS.
- R. K. Saini, S. Shuaib, D. Goyal and B. Goyal, J. Cell. Biochem., 2018, 119, 8949–8961 CrossRef CAS.
- L. Hendriks, C. M. van Duijn, P. Cras, M. Cruts, W. Van Hul, F. van Harskamp, A. Warren, M. G. McInnis, S. E. Antonarakis and J. J. Martin, et al., Nat. Genet., 1992, 1, 218–221 CrossRef CAS.
- C. Van Broeckhoven, J. Haan, E. Bakker, J. A. Hardy, W. Van Hul, A. Wehnert, M. Vegter-Van der Vlis and R. A. Roos, Science, 1990, 248, 1120–1122 CrossRef CAS.
- G. Rossi, G. Macchi, M. Porro, G. Giaccone, M. Bugiani, E. Scarpini, G. Scarlato, G. E. Molini, F. Sasanelli, O. Bugiani and F. Tagliavini, Neurology, 1998, 50, 688–692 CrossRef CAS.
- A. R. Lam, D. B. Teplow, H. E. Stanley and B. Urbanc, J. Am. Chem. Soc., 2008, 130, 17413–17422 CrossRef CAS.
- R. K. Saini, D. Goyal and B. Goyal, ACS Omega, 2020, 5, 23219–23228 CrossRef CAS.
- T. Tomiyama, T. Nagata, H. Shimada, R. Teraoka, A. Fukushima, H. Kanemitsu, H. Takuma, R. Kuwano, M. Imagawa, S. Ataka, Y. Wada, E. Yoshioka, T. Nishizaki, Y. Watanabe and H. Mori, Ann. Neurol., 2008, 63, 377–387 CrossRef CAS.
- T. J. Grabowski, H. S. Cho, J. P. Vonsattel, G. W. Rebeck and S. M. Greenberg, Ann. Neurol., 2001, 49, 697–705 CrossRef CAS.
- H. Li, Y. Nam, A. Salimi and J. Y. Lee, J. Chem. Inf. Model., 2020, 60, 3587–3592 CrossRef CAS.
- J. Adler, H. A. Scheidt, M. Kruger, L. Thomas and D. Huster, Phys. Chem. Chem. Phys., 2014, 16, 7461–7471 RSC.
- F. Hoffmann, J. Adler, B. Chandra, K. R. Mote, G. Bekçioglu-Neff, D. Sebastiani and D. Huster, J. Phys. Chem. Lett., 2017, 8, 4740–4745 CrossRef CAS.
- A. Huet and P. Derreumaux, Biophys. J., 2006, 91, 3829–3840 CrossRef CAS.
- E. Evangelisti, R. Cascella, M. Becatti, G. Marrazza, C. M. Dobson, F. Chiti, M. Stefani and C. Cecchi, Sci. Rep., 2016, 6, 32721 CrossRef CAS.
- J. F. Nagle, Biophys. J., 1993, 64, 1476–1481 CrossRef CAS.
- The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC Search PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS.
- H. Liu, D. Song, Y. Zhang, S. Yang, R. Luo and H.-F. Chen, Phys. Chem. Chem. Phys., 2019, 21, 21918–21931 RSC.
- M. U. Rahman, A. U. Rehman, H. Liu and H.-F. Chen, J. Chem. Inf. Model., 2020, 60, 4912–4923 CrossRef CAS.
- C. H. Davis and M. L. Berkowit, Biophys. J., 2009, 96, 785–797 CrossRef CAS.
- A. Brown and D. Bevan, Biophys. J., 2016, 111, 937–949 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, Interaction Models for Water in Relation to Protein Hydration. Intermolecular Forces: Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry Held in Jerusalem, Israel, April 13–16, 1981, Springer Dordrecht, Netherlands, 1981, pp. 331–342 Search PubMed.
- W. F. Van Gunsteren and H. J. C. Berendsen, Mol. Simul., 1988, 1, 173–185 CrossRef.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- M. Parrinello and A. Rahman, Phys. Rev. Lett., 1980, 45, 1196 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. Johannes Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- A. Patriksson and D. A. van der Spoel, Phys. Chem. Chem. Phys., 2008, 10, 2073–2077 RSC.
- W. G. Touw, C. Baakman, J. Black, T. A. te Beek, E. Krieger, R. P. Joosten and G. Vriend, Nucleic Acids Res., 2015, 43, D364–D368 CrossRef CAS.
- E. Papaleo, P. Mereghetti, P. Fantucci, R. Grandori and L. De Gioia, J. Mol. Graphics Modell., 2009, 27, 89–99 CrossRef.
- R. W. Zwanzig, J. Chem. Phys., 1954, 22, 1420–1426 CrossRef CAS.
- E. G. Marklund, M. T. Degiacomi, C. V. Robinson, A. J. Baldwin and J. L. Benesch, Structure, 2015, 23, 791–799 CrossRef CAS.
- F. Eisenhaber, P. Lijnzaad, P. Argos, C. Sander and M. Scharf, J. Comput. Chem., 1995, 16, 273–284 CrossRef CAS.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Peptide Folding: When Simulation Meets Experiment, Wiley-VCH Verlag GMBH, 1999, vol. 38, pp. 236–240 Search PubMed.
- A. Baumketner, S. L. Bernstein, T. Wyttenbach, N. D. Lazo, D. B. Teplow, M. T. Bowers and J.-E. Shea, Protein Sci., 2006, 15, 1239–1247 CrossRef CAS.
- S. Jang and S. Shin, J. Phys. Chem. B, 2006, 110, 1955–1958 CrossRef CAS.
- Y. Fezoui and D. B. Teplow, J. Biol. Chem., 2002, 277, 36948–36954 CrossRef CAS.
- A. K. Das, A. Rawat, D. Bhowmik, R. Pandit, D. Huster and S. Maiti, ACS Chem. Neurosci., 2015, 6, 1290–1295 CrossRef CAS.
- A. Potapov, W.-M. Yau, R. Ghirlando, K. R. Thurber and R. Tycko, J. Am. Chem. Soc., 2015, 137, 8294–8307 CrossRef CAS.
- T. Luhrs, et al., Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef CAS.
- J. A. Lemkul and D. R. Bevan, Arch. Biochem. Biophys., 2008, 470, 54–63 CrossRef CAS.
- J. A. Lemkul and D. R. Bevan, Protein Sci., 2011, 20, 1530–1545 CrossRef CAS.
- K. L. Sciarretta, D. J. Gordon, A. T. Petkova, R. Tycko and S. C. Meredith, Biochemistry, 2005, 44, 6003–6014 CrossRef CAS.
- E. Terzi, G. Holzemann and J. Seelig, Biochemistry, 1994, 33, 7434–7441 CrossRef CAS.
- C. Lockhart and D. Klimov, J. Phys. Chem. B, 2014, 118, 2638–2648 CrossRef CAS.
- S. Lazzaro, N. Ogrinc, L. Lamont, G. Vechhio, G. Pappalardo and R. M. A. Heeren, Anal. Bioanal. Chem., 2019, 411, 6353–6363 CrossRef CAS.
- G. Li, K. DeLaney and L. Li, Nat. Commun., 2019, 10, 5038 CrossRef.
- Y. Sun, S. Vahidi, M. A. Sowole and L. Konermann, J. Am. Soc. Mass Spectrom., 2016, 27, 31–40 CrossRef CAS.
- M. Kłoniecki, A. Jabłonowska, J. Poznański, J. Langridge, C. Hughes, I. Campuzano, K. Giles and M. Dadlez, J. Mol. Biol., 2011, 407, 110–124 CrossRef.
- E. Sitkiewicz, J. Olędzki, J. Poznański and M. Dadlez, PLoS One, 2014, 9, e100200 CrossRef.
- S. T. Ngo, P. H. Nguyen and P. Derreumaux, J. Phys. Chem. B, 2020, 124, 617–626 CrossRef CAS.
- K. Ono, M. M. Condron and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14745–14750 CrossRef CAS.
- B. Urbanc, M. Betnel, L. Cruz, G. Bitan and D. B. Teplow, J. Am. Chem. Soc., 2010, 132, 4266–4280 CrossRef CAS.
- A. Morriss-Andrews and J.-E. Shea, Annu. Rev. Phys. Chem., 2015, 66, 643–666 CrossRef CAS.
- D. J. Lindberg, E. Wesén, J. Björkeroth, S. Rocha and E. K. Esbjörner, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1921–1929 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table for the introduction summarizing mutation studies, Free Energy Perturbation method (FEP), Free Energy Surface (FES), Collision Cross Section (CCS), temperature simulations, exchange rates between neighboring replicas, temperature indices of replicas with the lowest and the highest initial temperatures, the stability of the lipid bilayers, convergence of REMD simulations for chain B and chain C (distance of D23–N27), the secondary structure distribution per residue of the transmembrane F19W 3Aβ11–40, the contacts map between phosphate atom of lipid bilayers with each residues of each chain of the F19W 3Aβ11–40, the probabilities of SC–SC, BB–BB inter-peptide contacts between neighbor pair chains of the truncated mutant trimer, and details structural characterization of the first four metastable states of the mutant truncated trimer. See DOI: 10.1039/d0ra08837d |
| This journal is © The Royal Society of Chemistry 2021 |