
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review on the structural dependent optical properties and energy transfer of Mn4+ and multiple ion-codoped complex oxide phosphors
Meng Gaoa,
Yuexiao Pan *a,
Yitian Jina and
Jun Lin*b
*a,
Yitian Jina and
Jun Lin*b
aKey Laboratory of Carbon Materials of Zhejiang Province, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, P. R. China. E-mail: yxpan@wzu.edu.cn; Fax: +86-577-88373017; Tel: +86-577-88373017
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: jlin@ciac.ac.cn; Fax: +86-431-85698041; Tel: +86-431-85262031
First published on 4th January 2021
Abstract
The tetravalent manganese Mn4+ ions with a 3d3 electron configuration as luminescence centers in solid-state inorganic compounds have been widely investigated because they emit bright light in the red to far-red region when they are excited by light with a wavelength in the UV to blue light region. Herein, we present an overview of the recent developments of Mn4+ and multiple ion such as Bi3+ and rare earth ion Dy3+, Nd3+, Yb3+, Er3+, Ho3+, and Tm3+ codoped complex oxide phosphors. Most of the specified host lattices of these complex oxide phosphors possess multiple metallic cations, which provide possible substitutions with different codopants and form various luminescence centers with diverse spectra. The luminescence of Mn4+ and multiple ion-codoped materials spans almost the whole visible light to near infrared (NIR) region. The crystal structures of complex oxide phosphors, the spectroscopic properties of Mn4+, and the energy transfer between Mn4+ and multiple ions are introduced and summarized in detail with regard to their practical applications. This review provides an insight into the optical properties of Mn4+ and the energy transfer process in multiple ion-codoped luminescence materials, which will be helpful in the development of novel excellent materials for applications in the lighting industry.
1. Introduction
The optical properties based on the structures of host lattices and the energy transfer between Mn4+ and multiple ion-codoped complex oxide phosphors described in this review make the identified luminescence materials promising for application in solar energy cells, white light-emitting diodes (WLEDs), indoor lighting for plant cultivation, and temperature sensors, as illustrated in Fig. 1.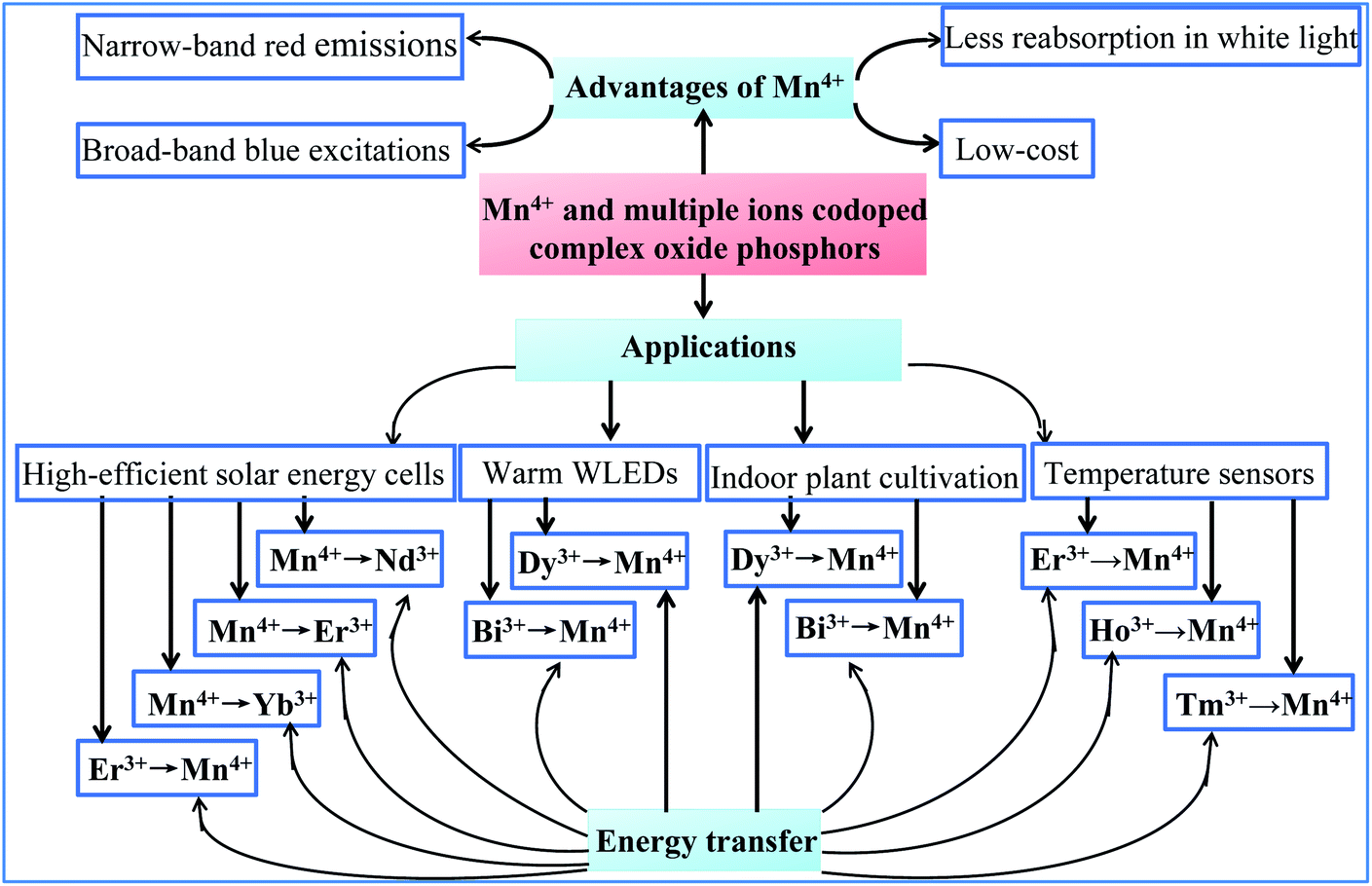 | ||
| Fig. 1 Optical properties, energy transfer, and potential application of Mn4+ and multiple ion-codoped complex oxide phosphors described in this review. | ||
Solar energy cells and WLEDs are considered as alternative approaches to relieve the energy crisis with the increasing global energy consumption. Solar energy cells using crystalline silicon solar cells have occupied majority of the solar cell market owing to their well-developed techniques and low cost; however, their conversion efficiency should be improved further for their wide commercial applications. It is well known that most of the energy of the solar spectrum is concentrated at wavelengths beyond 900 nm including UV-visible (UV-vis) and NIR light, which cannot be absorbed by the current crystalline silicon solar cells with high efficiency.1–4
Converting the energy of the solar spectrum at wavelengths beyond 900 nm into the range located 900–1100 nm, which matches the maximum spectral response of the absorption of crystalline silicon, is an important alternative approach to improve the energy conversion efficiency of crystalline silicon solar cells. Recently, much attention has been paid to developing Mn4+-doped phosphors because Mn4+ usually shows sharp line emissions in the red-infrared (IR) region due to its unique 3d3 electron configurations. It has been observed that Mn4+ shows red to far-red photoluminescence, which is assigned to the spin-forbidden 2Eg → 4A2g transition under the excitation of UV or blue light owing to its high effective positive charge and the influence of a strong local crystal-field.5–12 The reversible conversion of UV-vis into NIR, and NIR into visible light with dual-mode luminescence can be realized by codoping multiple ions such as Nd3+/Er3+/Yb3+ into Mn4+ ion-doped luminescence materials. The red emission of Mn4+ can be obtained when it is excited by 980 nm due to the energy transfer from Nd3+/Er3+/Yb3+ to Mn4+ ions.13 The NIR photoluminescence maxima at 1064, 1537, and 980 nm originating from Nd3+/Er3+/Yb3+ ions can be sensitized by Mn4+ with excitation in the UV-vis region (200–500 nm).13–16 The conversion of UV-vis light into NIR light at about 1064 nm through energy transfer from Mn4+ to multiple ions is desirable to improve the conversion efficiency of solar cells by coating the phosphor layer on the surface of a crystalline Si layer.
WLEDs have received extensive attention due to their high energy efficiency, long lifetime, and environmental friendliness. The WLEDs fabricated with blue semiconductor GaN chips and yellow phosphor Y3Al5O12:Ce3+ (YAG:Ce) can produce cold white light because the red component in their spectra is weak. To meet the requirement for indoor illumination, warm white light with a high color rendering index (CRI > 80) and a low correlated color temperature (CCT < 4000 K) is necessary.17,18 Accordingly, phosphors with strong absorption in the blue light region and intense emission in the red light region should be co-coated on blue semiconductor GaN chips to produce warm white light. Mn4+ ions located at octahedral crystallographic sites are favorable luminescent centers and promising for blue GaN-excited warm WLED applications because they have narrow-band red emissions, broad-band blue excitations, and no reabsorption in white light, while being free of expensive rare earth metals.5–12 Thus, much attention has been paid to developing red phosphors to provide alternatives to the commercial nitride phosphors. In particular, the interest in Mn4+-doped inorganic phosphors has increased because the Mn4+ luminescence center usually shows sharp line emissions in the red region with high color purity due to the sharp feature of its emission spectrum.5–12
Recently, indoor plant cultivation has attracted considerable attention because this advanced technology can exclude the unfavorable influence of the climate and natural damage. To meet the requirement in lighting for indoor plant cultivation, blue-violet light in wavelength range of 420–500 nm is indispensable for chlorophyll A and chlorophyll B, and red-far red light in wavelength range of 640–750 nm is indispensable for phytochrome PR and phytochrome PFR.19–24 The fabrication of red Mn4+-doped phosphors in blue LED chip results in a superior performance in lighting for indoor plant cultivation due to the blue light from LED chips and red light from Mn4+-doped luminescence materials excited by blue light. This type of light device is a promising light source for large scale industrial application because of the energy saving and long working time of LEDs, and low cost of Mn4+-doped luminescence materials. Bi3+ and Mn4+ codoped oxide phosphors, which emit dual blue and red light upon excitation by near UV (NUV) LEDs, are alternative candidates for application in the agricultural industry to improve the efficiency of photosynthesis. To maintain the electroneutrality of the compound, excess metal ion vacancies and O2− ions in the lattices of complex oxides may be formed for charge compensation.25–30
The upconverted NIR luminescence of Mn4+ was realized with the aid of the efficient energy transfer of Yb3+ → Ln3+ → Mn4+ in the specially prepared Yb3+/Ln3+/Mn4+ (Ln = Er, Ho, Tm) codoped YAlO3 and its energy transfer efficiency was systematically clarified by its steady-state and time-resolved upconverted emission spectra.31 The dual emission based on Mn4+ and multiple ion (such as Yb3+, Ln3+, and Mn4+) codoped phosphors is promising for accurate temperature sensors due to the fact that the thermal quenching mechanisms of Mn4+ and Ln3+ are different.32 Fig. 2 presents a summary of the energy transfer between Mn4+ and multiple ions, the emission wavelengths, and corresponding electronic transitions of both the donor and acceptor. The octahedral environment-coordinated Mn4+ ions emit red to far-red emissions in the region of 600 to 700 nm. Thus, tunable spectral emissions from the visible to NIR region can be realized by codoping Mn4+ and multiple ions.
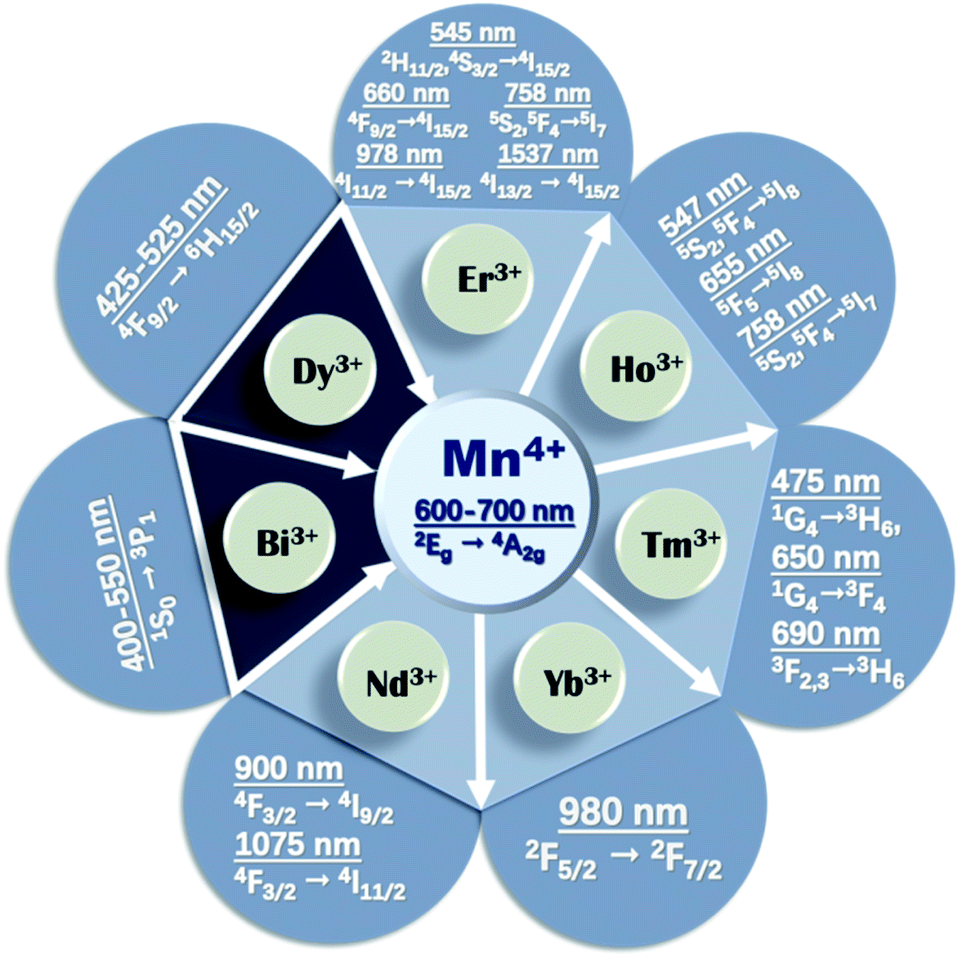 | ||
| Fig. 2 Summary of the energy transfer between Mn4+ and rare earth ions. | ||
In all the host lattice of complex oxides, as summarized in Table 1, Mn4+ ions perfectly substitute the sites in the centers of the octahedral environment coordinated with six oxygen atoms due to their similar radius and valence, such as Ga3+, Al3+, Ti4+, Ta5+, and Mg2+–Te6+ pairs, and Nb5+ ions. Multiple cation sites in the complex oxide host lattice provide the possibility for codoping Mn4+ ions and Bi3+ or trivalent rare earth ions. The optical characteristics of Mn4+ and other ions are strongly dependent on the structural symmetry of the host materials. This review aims to comprehensively present the structural-dependent optical properties based on the energy transfer between Mn4+ and multiple ions in codoped complex oxide phosphors for potential applications in high-efficient solar energy cells, warm WLEDs, indoor plant cultivation, and temperature sensors.
| Mn4+ doping octahedral centers | RE doping | Excitation of Mn4+ at 200–500 nm (maximum band) | Emission of Mn4+ at 650–800 nm (maximum peak) | Ref. | |
|---|---|---|---|---|---|
| Ca14Zn6Ga10O35 | Ga3+ | Ca2+ | 313 nm | 712 nm | 13, 46, 51 and 61–63 |
| Ca14Zn6Al10O35 | Al3+ | Ca2+ | 460 nm | 710 nm | 14, 15, 54, 65 and 66 |
| Ca3ZnAl4O10 | Al3+ | Ca2+ | 467 nm | 715 nm | 18 |
| Gd2ZnTiO6 | Ti4+ | Gd3+ | 365 nm | 704 nm | 33, 74 and 75 |
| La2LiTaO6 | TaO6 | La3+ | 495 nm | 709 nm | 34 |
| NaMgLaTeO6 | Mg2+ and Te6+ | La3+ | 365 nm | 705 nm | 16 |
| La2MgTiO6 | Ti4+ | La3+ | 355 nm | 710 nm | 35, 79 and 80 |
| Ba2LaNbO6 | Nb5+ | La3+ | 352 nm | 677 nm | 35 |
| CaAl12O19 | Al3+ | Ca2+ | 400 nm | 654 nm | 36, 116 and 117 |
| Mg2TiO4 | Ti4+ | Mg2+ | 475 nm | 657 nm | 37 |
| La2ZnTiO6 | Ti4+ | La3+ | 345 nm | 710 nm | 38 |
| MgAl2Si2O8 | Si4+ | Mg2+ | 258 nm | 710 nm | 39 and 145–147 |
| YAlO3 | Al3+ | Y3+ | 414 nm | 714 nm | 32 |
Mn4+ is isoelectronic with Cr3+, but the crystal field at the higher charged Mn4+ ions is stronger than that of Cr3+ and the vibronic emission 2Eg → 4A2g of Mn4+ is more intense than that of Cr3+. The Tanabe–Sugano energy diagram presents the energy splitting of the Mn4+ ion with an octahedral coordination dependent on the crystal field strength (Fig. 3a).5–12,38,40 The Stokes shift and the features of the photoluminescence emission and excitation (PL and PLE) spectra of Mn4+ ions are known to be tunable by changing the crystal-field of the host. The spectral position of Mn4+ ions can be easily tuned over a wide range from 620 nm to 723 nm by modifying the crystal field environment.8,41 Mn4+ ions are inclined to form Mn4+–Mn4+ pairs due to the O2− impurities in oxides, which significantly influence the excited state dynamics and reduce the luminescence efficiency of Mn4+.36,42
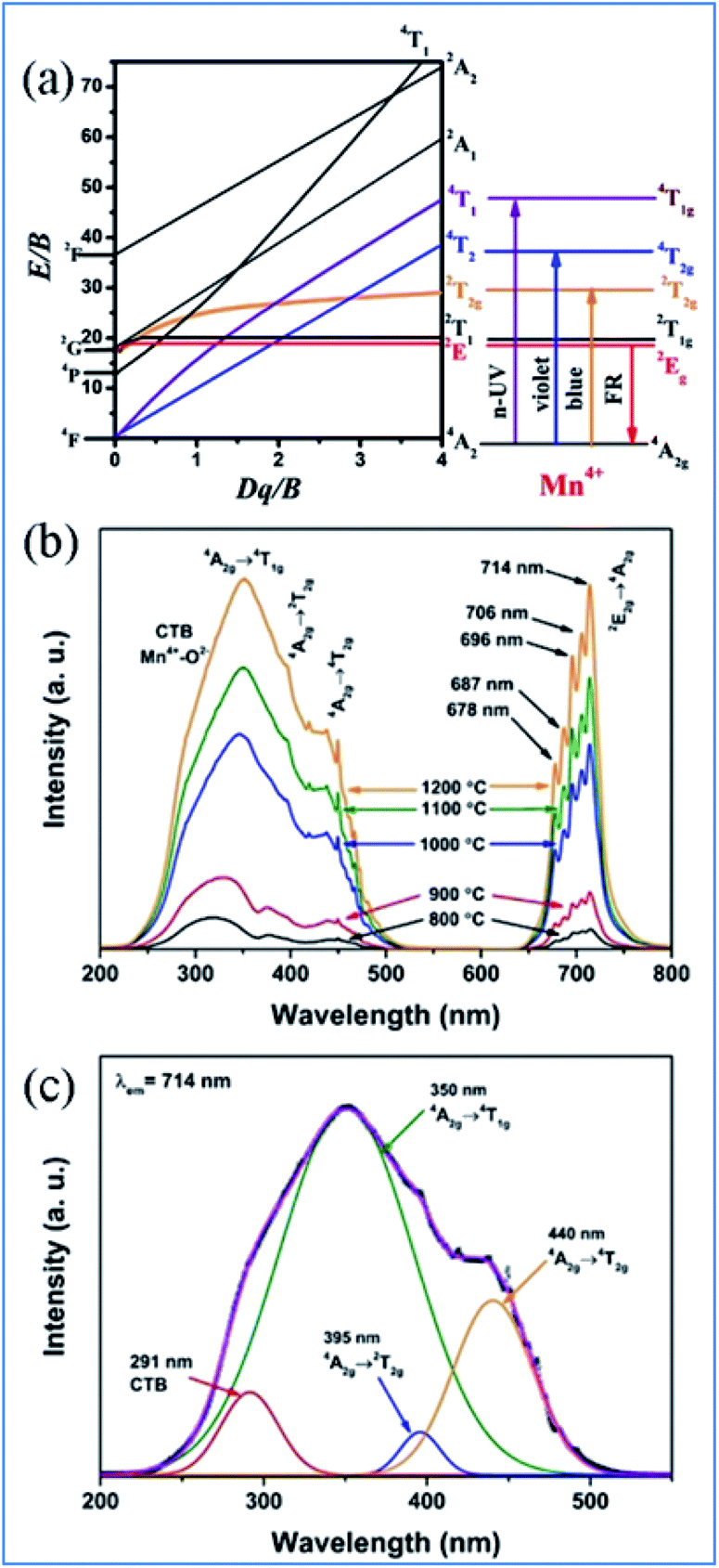 | ||
| Fig. 3 (a) Tanabe–Sugano diagram for Mn4+ dependent on the crystal field in complex oxides, (b) typical PLE and PL spectra, and (c) Gaussian curves of PLE spectra of Mn4+ in Ca14Zn6Ga10O35 phosphors. Reprinted with permission from ref. 39, Copyright 2017, The Royal Society of Chemistry. | ||
2. Luminescent properties of Mn4+ in complex oxide phosphors
Mn4+ ions generally occupy octahedral sites coordinated by eight oxygens in complex oxide phosphors, and the PLE and PL spectra of Mn4+ ions are located in the range of 200–500 nm and 600–700 nm, respectively. As shown in Fig. 3b and c, the excitation bands located at 350 and 440 nm in the PLE spectrum of Mn4+ in Ca14Zn6Ga10O35 (CZGO) are assigned to the spin-allowed transitions of Mn4+. The three Gaussian peaks at 313, 356, and 462 nm are attributed to the 4A2g → 4T1g, 4A2g → 2T2g, and 4A2g → 4T2g of Mn4+ transitions, respectively. The broad band at 291 nm in the PLE spectrum of Mn4+ is ascribed to both the charge transfer transitions of Mn4+ → O2− and 4A2g → 4T1g transitions of Mn4+ ions. Under excitation at 310 nm, the intense red emission is composed of some distinguishable sharp R lines and Stokes/anti-Stokes side-peaks located at 676, 684, 695, 704 and 713 nm due to the different vibrational modes for the 3d3 electrons when Mn4+ is in the [MnO6]8− octahedral complex, which correspond to the vibronic sidebands of the 2Eg → 4A2g transition of the Mn4+ ions.433. NIR emission of Nd3+, Yb3+, Er3+, Ho3+, and Tm3+ sensitized by Mn4+
3.1 Ca14Zn6Ga10O35 as host lattice for Mn4+ and multiple ion codoping
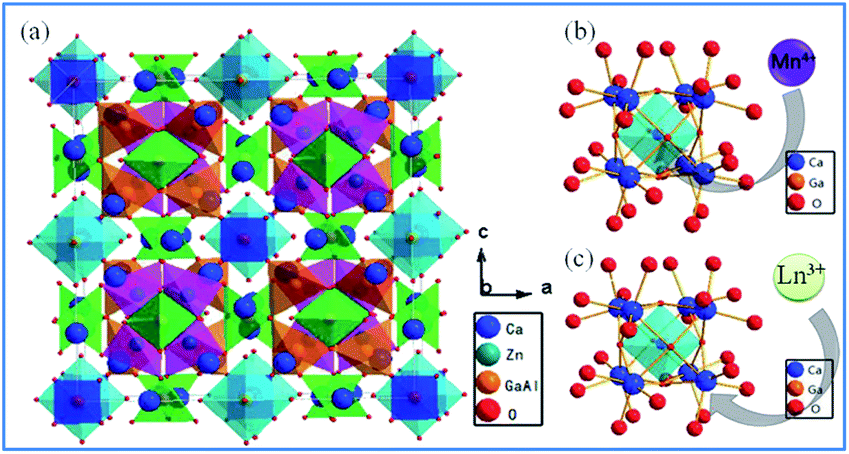 | ||
| Fig. 4 (a) Crystal structure of Ca14Zn6Ga10O35, (b) schematic diagram of Mn4+ ions occupying the octahedral lattice sites of Ga3+, and (c) Ln3+ ions occupying the Ca2+ ion sites in the Ca14Zn6Ga10O35 host. Reprinted with permission from ref. 44, Copyright 2017, The Royal Society of Chemistry. | ||
Based on the effective ionic radii of cations with different coordination numbers (CN),47 trivalent rare earth ions are expected to randomly occupy six- and seven-coordinated Ca2+ (CN = 6, r = 1.00 Å and CN = 7, r = 1.06 Å) sites, and Mn4+ (CN = 6, r = 0.53 Å) ions are preferentially accommodated at the Ga3+ (CN = 5, r = 0.62 Å) sites with an octahedral coordination in the crystal structure.41 Electroneutrality in the Mn4+ and multiple ion-codoped CAZO phosphors can be easily achieved due to some defects such as the formation of Ca2+ vacancies and excess O2− ligands for charge compensation.48
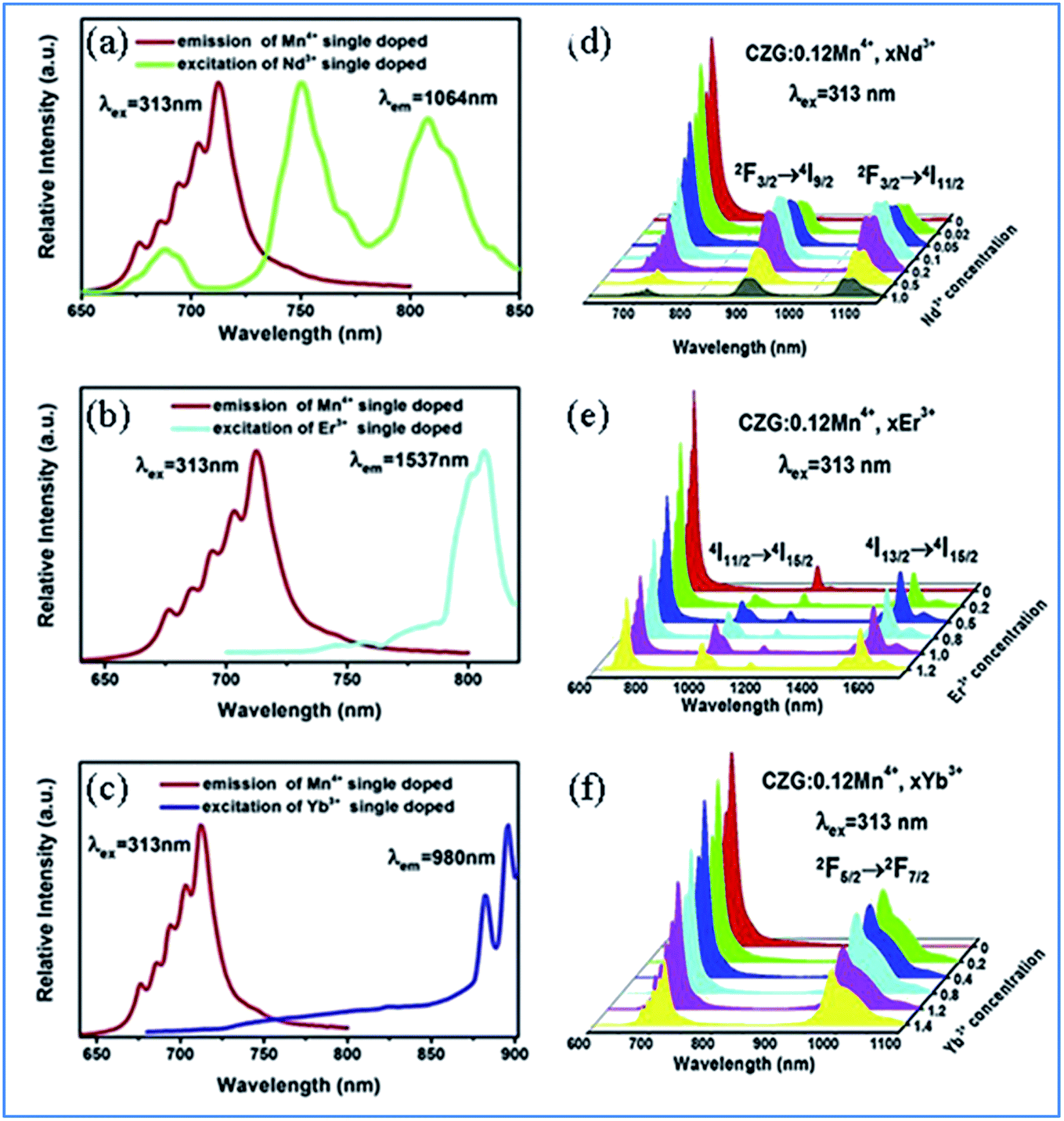 | ||
| Fig. 5 (a–c) Overlap between the PL spectrum of Ca14Zn6Ga10O35:Mn4+ and PLE spectra of Ca14Zn6Ga10O35:Ln3+ (Ln = Nd, Er, and Yb), respectively. (d–f) Changes in the PL spectra of Mn4+ and multiple ion Nd3+/Er3+/Yb3+ codoped Ca14Zn6Ga10O35 with a change in the concentration of Nd3+/Er3+/Yb3+, respectively. Reprinted with permission from ref. 49, Copyright 2019, Elsevier BV. | ||
Fig. 5e–g show the emission spectra of Mn4+ and multiple ions Nd3+/Er3+/Yb3+ codoped CZGO with different doping concentrations of Ln3+ ions, respectively. Upon excitation at 313 nm, the NIR emissions of Nd3+/Er3+/Yb3+ such as the emission peaks at 900 and 1075 nm are assigned to the 4F3/2 → 4I9/2 and 4F3/2 → 4I11/2 transitions of Nd3+, that at 978 and 1537 nm are ascribed to the 4I11/2 → 4I15/2 and 4I13/2 → 4I15/2 transitions of Er3+, and that at 980 nm is caused by the 2F5/2 → 2F7/2 transition of Yb3+. The emission intensity of Mn4+ monotonously decreases with an increase in the content of Ln3+, which indicates energy transfer occurs from Mn4+ to Nd3+/Er3+/Yb3+.
The energy transfer process from Mn4+ to multiple ions, Nd3+/Er3+/Yb3+, in CZGO is illustrated in Fig. 6a. Under excitation of NUV to visible light ranging from 250 to 550 nm, the Mn4+ ions are excited into their charge transfer or excited states of 4T1g and 4T2g, which then rapidly relax to the metastable state of 2Eg of the Mn4+ ions. The energy transfer occurs via Mn4+: 2Eg + Nd3+: 4I9/2 → Mn4+: 4A2g + Nd3+: 4F9/2, 4F7/2, 4S3/2 or Mn4+: 2Eg + Yb3+: 2F7/2 → Mn4+: 4A2g + Yb3+: 2F5/2. The NIR emissions at 896 and 1064, 1540, and 980 nm are generated by the radiative transitions of the Nd3+: 4F3/2, Er3+: 4I13/2 and Yb3+: 2F5/2 levels, respectively.52
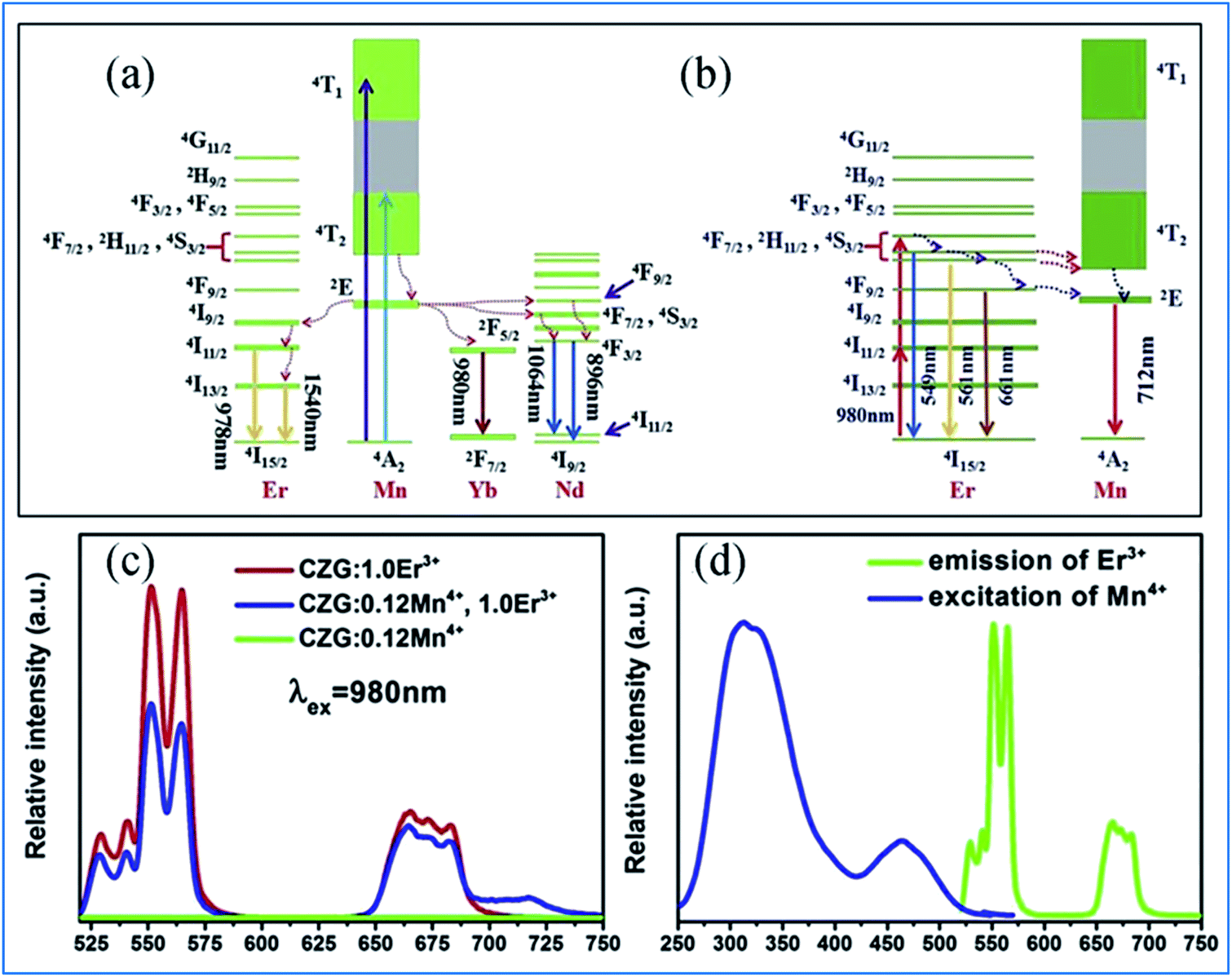 | ||
| Fig. 6 (a) Excitation/emission and energy transfer mechanism of Mn4+ to Ln3+ in Ca14Zn6Ga10O35:Mn4+,Ln3+ (Ln = Nd, Er, and Yb) phosphors. (b) Mechanism of the up-conversion of Er3+ and energy transfer from Er3+ to Mn4+. (c) Emission spectra of Mn4+ and Er3+ codoped Ca14Zn6Ga10O35 upon 980 nm excitation and (d) spectral overlap between the emission of Er3+ and excitation of Mn4+. Reprinted with permission from ref. 49, Copyright 2019, Elsevier BV. | ||
Under 980 nm light excitation, green upconverted emission peaks at 551 and 561 nm attributed to the 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions of Er3+ are produced, as shown in Fig. 6b. Therefore, the red emission centered at 712 nm of Mn4+ and Er3+ codoped CZGO phosphor produced by excitation at 980 nm is ascribed to the energy transfer from Er3+ to Mn4+.
The green and red emission centered at 551 (561) and 661 nm can be ascribed to the transitions of 2H11/2 → 4I15/2 (4S3/2 → 4I15/2) and 4F9/2 → 4I15/2 of Er3+, respectively, via the multiple non-radiative multiphonon relaxations from the 4F7/2 to H11/2, 4S3/2 and 4F9/2 levels.53 The deep red emission ascribed to the transition of 2Eg → 4A2g of Mn4+ is attributed to the energy transfer from Er3+ to Mn4+, as illustrated in the corresponding mechanism diagram in Fig. 6c. The spectral overlap observed between the emission spectrum of Er3+ and the excitation spectrum of Mn4+ makes the reversal energy transfer from Er3+ to Mn4+ possible, as presented in Fig. 6d.
3.2 Ca14Zn6Al10O35 as host lattice for Mn4+ and multiple ion codoping
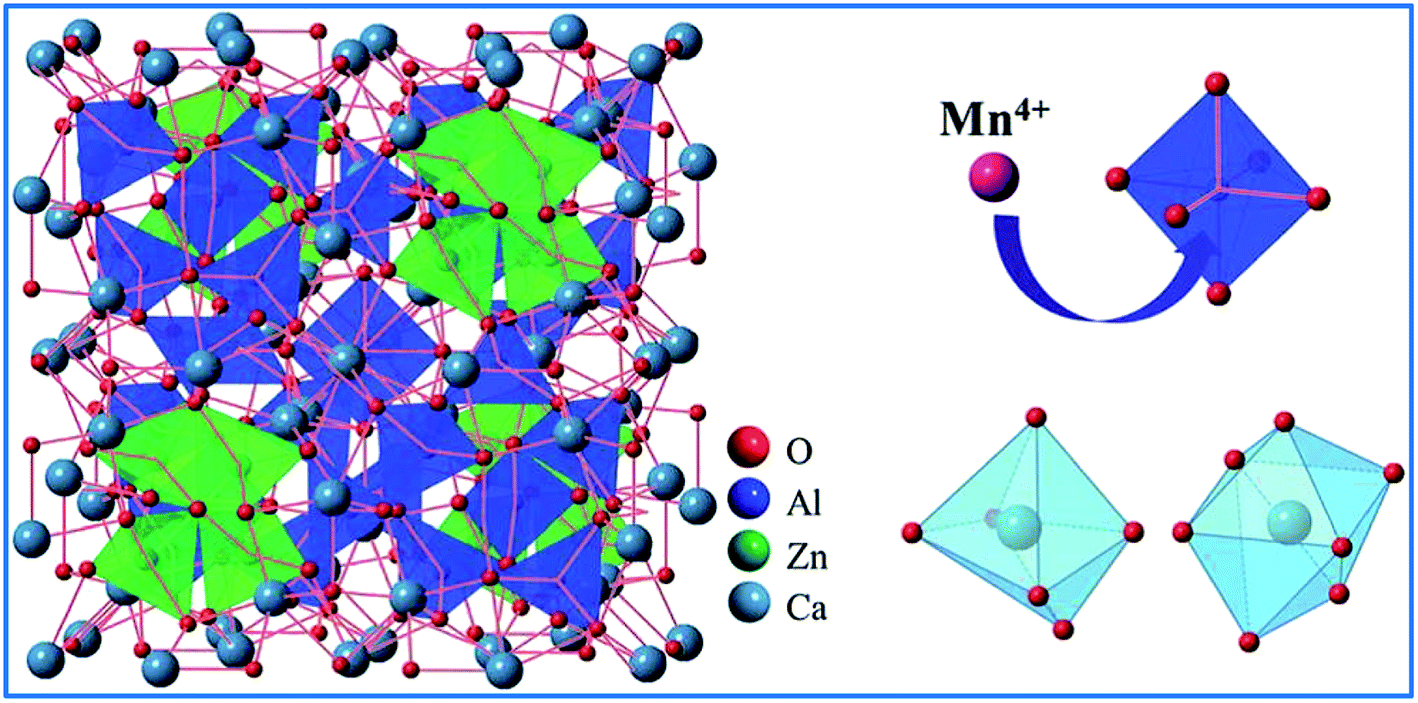 | ||
| Fig. 7 Schematic of the crystal structure of Ca14Zn6Al10O35. Reprinted with permission from ref. 14, Copyright 2016, The Royal Society of Chemistry. | ||
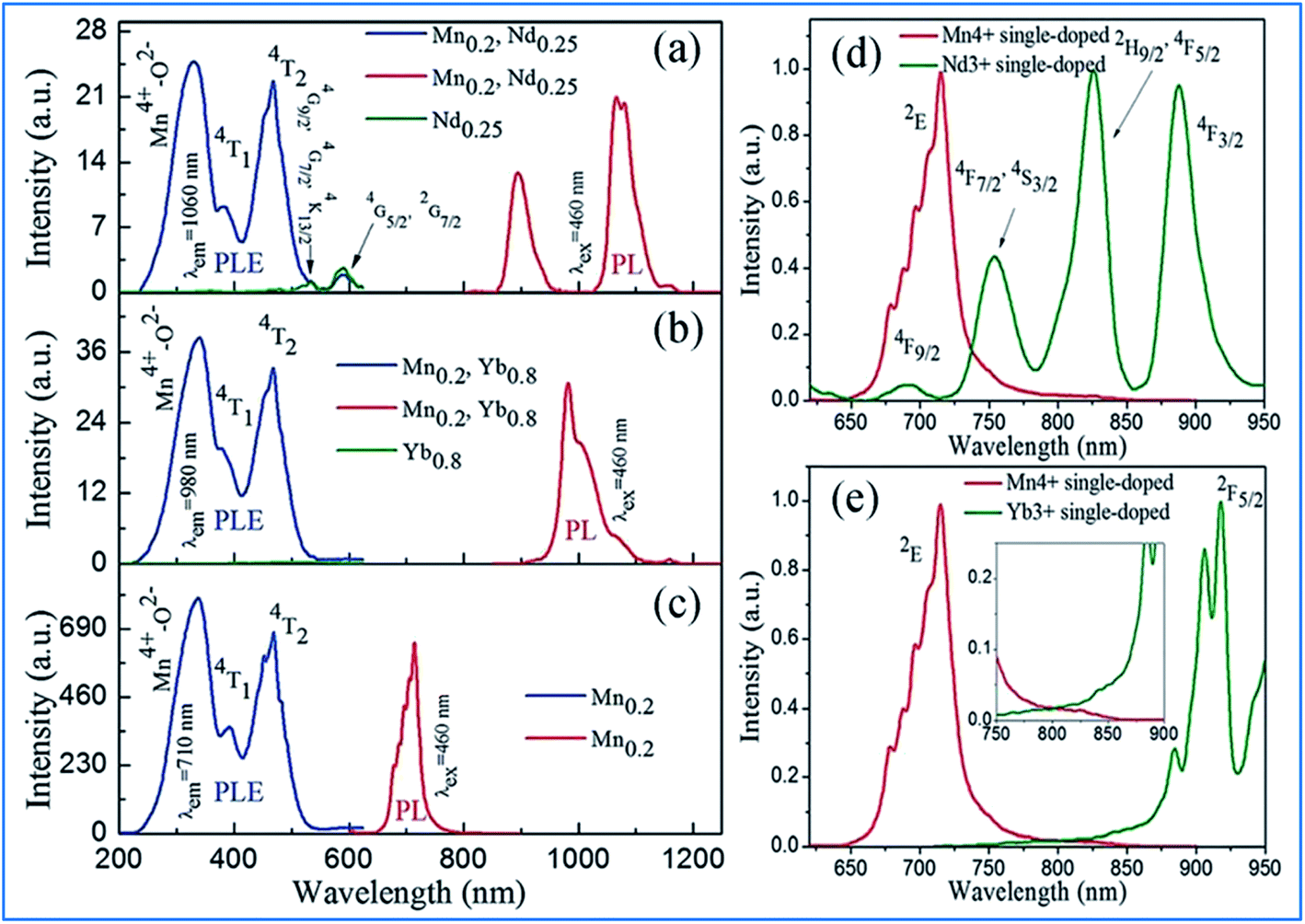 | ||
| Fig. 8 (Left) PLE spectra and/or (right) PL spectra for (a) Ca13.75Zn6Al9.8O35:Mn0.2,Nd0.25 and Ca13.75Zn6Al10O35:Nd0.25, (b) Ca13.2Zn6Al9.8O35:Mn0.2, Yb0.8 and Ca13.2Zn6Al10O35:Yb0.8, and (c) Ca14Zn6Al9.8O35:Mn0.2. Spectral overlap between the emission spectrum of Mn4+ and the excitation spectrum of (d) Nd3+ monitored at 1060 nm and (e) Yb3+ monitored at 980 nm. Reprinted with permission from ref. 14, Copyright 2016, The Royal Society of Chemistry. | ||
The energy transfer efficiency depends on the spectral matching of the excitation of the acceptor and emission spectra of the donor. As shown in Fig. 8d, good spectral overlap can be observed between the 2Eg emission of Mn4+ and the 4F9/2, 4F7/2, and 4S3/2 excitations of Nd3+. It can be seen from Fig. 8e that although there is a relatively large energy gap between the excited state 2Eg of Mn4+ and 2F5/2 of Yb3+, an efficient energy transfer from Mn4+ to Yb3+ can still occur in the Mn4+ and Yb3+ codoped samples with strong electron-phonon coupling.64 Therefore, the NIR luminescence of Yb3+ may be mainly generated by phonon-assisted energy transfer from Mn4+ to Yb3+. The excitation/emission and energy transfer pathways for the Mn4+ and codoped Nd3+/Yb3+ ion couples in CZAO are quite similar to that in the host lattice of CZGO.14,49
NIR emissions from Nd3+/Yb3+ have been observed in Mn4+ and Nd3+/Yb3+ codoped CZAO phosphors. The intensity of the NIR emissions of Nd3+/Yb3+ increases initially with an increase in the content of rare earth ions Nd3+/Yb3+, and then decreases gradually as a result of concentration quenching.62 The NIR luminescence intensity is enhanced by 338 times at 1060 nm for Ca13.75Zn6Al9.4O35:Mn0.6,Nd0.25 and 306 times at 980 nm for Ca13.2Zn6Al9.4O35:Mn0.6,Yb0.8, respectively, which is attributed to the efficient energy transfer from Mn4+ to the Nd3+/Yb3+ ions, respectively.65,66 Fig. 9a and b illustrate the excitation spectra of Mn4+ and emission spectra of Er3+ in Mn4+ and/or Er3+ codoped samples with various doping concentrations. The two broad and intense excitation bands (monitored at Mn4+ 710 nm emission) correspond to the spin-allowed transitions 4A2g → 4T1g and 4A2g → 4T2g of Mn4+ (Fig. 9a). The weak and discrete excitation peaks (monitored at Er3+ 1540 nm emission) are ascribed to the transitions from 4I15/2 to 4G11/2, 4F5/2, 4F7/2, 2H11/2, and 4S3/2 of Er3+. From the excitation spectrum of the Mn4+ and Er3+ codoped sample monitored at the 1540 nm emission of Er3+ in Fig. 9b, it can be seen that not only broad and intense excitation bands ascribed to Mn4+ ions but also the superimposed excitation peaks assigned to the 4I15/2 to 2H11/2 and 4S3/2 transitions of Er3+ appear, indicating the energy transfer from Mn4+ to Er3+.
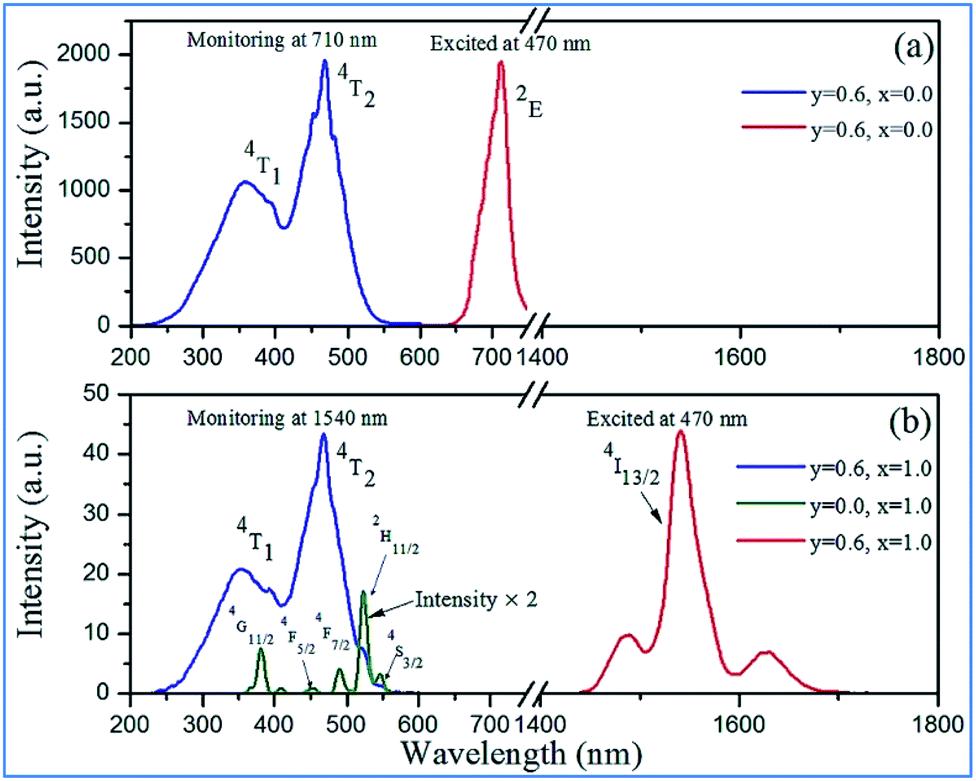 | ||
| Fig. 9 Excitation and emission spectra of Ca14−xZn6Al10−yO35:Mny,Erx (y = 0.0, 0.6; x = 0.0, 1.0) in (a) NIR and (b) IR regions. Reprinted with permission from ref. 4, Copyright 2016, The Royal Society of Chemistry. | ||
3.3 Complex hexoxides as host lattices for Mn4+ and multiple ion codoping
As shown in Fig. 10a,65 Gd2ZnTiO6 (GZT) crystallizes in a double-perovskite monoclinic structure with the space group P21/n, with the cell parameters of a = 5.3664(9) Å, b = 5.6631(9) Å, c = 7.6847(9) Å and β = 90.294(2)°. In the crystal structure of GZT, the Zn2+ and Ti4+ ion centers are at two slantwise octahedral sites surrounded by six oxygen atoms, and the Gd3+ ion occupies the decahedron site coordinated with twelve oxygen atoms. La2LiTaO6 is built up of alternating strands of LiO6 and slightly disordered TaO6 with La3+ located in the cavities of the interconnected network of octahedral sites, as shown in Fig. 10b.67–70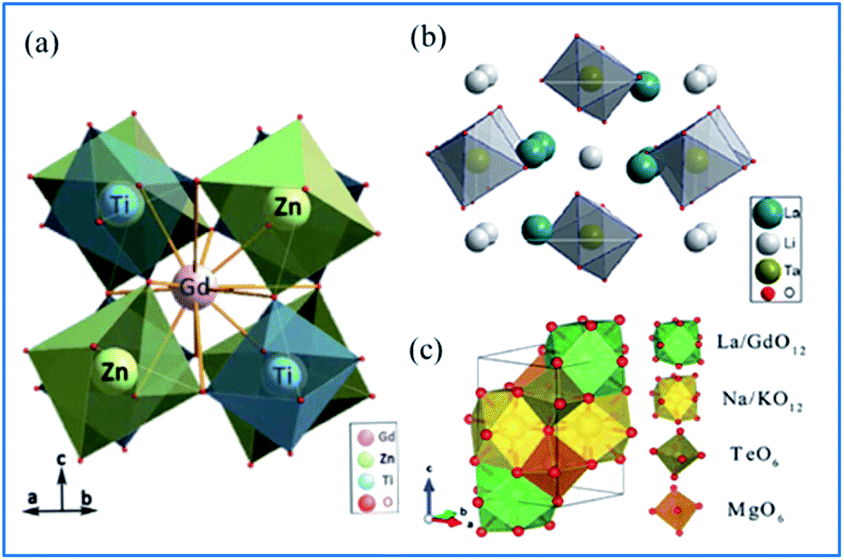 | ||
| Fig. 10 Crystal structure of (a) Gd2ZnTiO6. Reprinted with permission from ref. 65, Copyright 2014, The Chemical Society of Japan. (b) La2LiTaO6, Reprinted with permission from ref. 34, Copyright 2014, Springer Nature. (c) NaMgLaTeO6 Reprinted with permission from ref. 16, Copyright 2018, The Royal Society of Chemistry. | ||
According to the doping rule that with a similar radius and the same valence of the dopants and host cationic ions, Mn4+ ions perfectly enter the centers of the octahedral environment coordinated with six oxygen atoms and the trivalent rare earth ions can occupy the Gd3+ and/or La3+ sites in the host lattices of complex hexoxides, respectively. NaMgLaTeO6 crystallizes in a monoclinic system with the P121/m1(11) space group, as depicted in Fig. 10c.16,71,72 Both Mg2+ and Te6+ are located at the six-fold sites to form MgO6 and TeO6 octahedra with a shared oxygen atom, respectively. Moreover, the La/Gd and Na/K atoms are coordinated with twelve oxygen atoms to form polyhedral La/GdO12 and Na/KO12. These four types of polyhedra connect closely to construct the space framework of this crystal structure.73 The Mg2+ and Te6+ sites at the centers of the octahedra are expected to be substituted by Mn4+ ions and red luminescence centers of Mn4+ are formed. In the Mn4+ and Er3+ coped GZT sample, efficient energy transfer from Mn4+ to Er3+ was observed, and the mechanism is quite similar to that in Mn4+ and Er3+ codoped CZAO.14
It can be seen from Fig. 11a that the emission spectrum of Gd2ZnTiO6:yMn4+,0.02Er3+ (y = 0, 0.002) is excited at 335 nm, corresponding to the 4A2g → 4T1g of Mn4+, and in that of Gd2ZnTiO6:0.002Mn4+,2xEr3+ (x = 0, 0.005) are excited at 379 nm, corresponding to 4A2g → 4T1g of Mn4+ and 4I15/2 → 4G11/2 of Er3+.75 Only the characteristic emission peaks (2Eg) of Mn4+ can be observed and no characteristic visible emission peaks (2H11/2/4S3/2) of Er3+ for the GZT:0.002Mn4+,0.02Er3+ sample in the emission excited at 335 nm. Spectral overlap exists between the emission for Er3+ (2H11/2/4S3/2) and the absorption for Mn4+ (4A2g), which provides a possible energy transfer pathway from Mn4+ to Er3+.76 The emission intensity of 2Eg of Mn4+ upon the codoping of Er3+ in GZT is much stronger than that of Mn4+ single-doped GZT under the common excitation wavelength of 379 nm, which indicates that energy back transfer occurs from Er3+ (2H11/2/4S3/2) to Mn4+ (4A2) under the common excitation wavelength of 379 nm (see Fig. 11b).
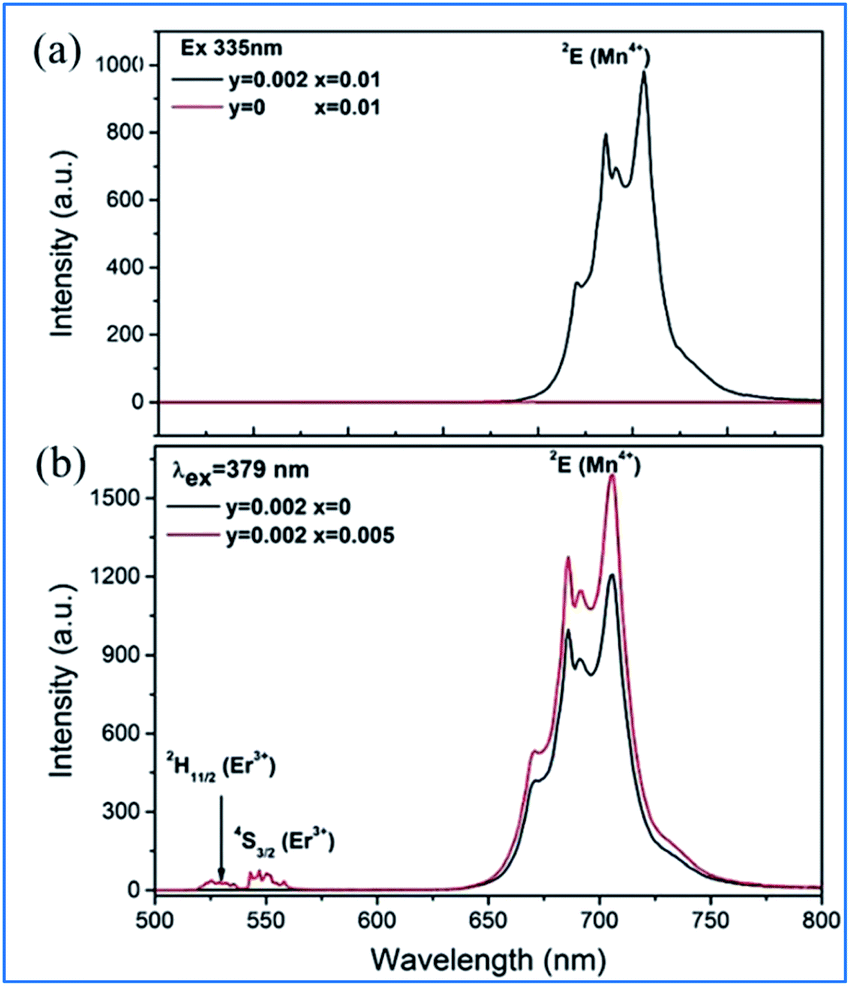 | ||
| Fig. 11 (a) Emission spectra of Gd2ZnTiO6:yMn4+,0.02Er3+ (y = 0, 0.002) excited at 335 nm, and (b) Gd2ZnTiO6:0.002Mn4+,2xEr3+ (x = 0, 0.005) excited at 379 nm. Reprinted with permission from ref. 74, Copyright 2017, The Royal Society of Chemistry. | ||
The IR emission at 1529 nm is ascribed to the 4F9/2 (4I9/2) → 4I13/2 transition of Er3+ through energy transfer from Mn4+ in the Mn4+ and Er3+ codoped GZT phosphor and the corresponding mechanism is illustrated in Fig. 12a. The Mn4+ ions are excited into their excite states under irradiation by short-wavelength light in the region of 250–550 nm, and then the energy transfer of 2E (Mn4+) → 4F9/2, 4I9/2 (Er3+) happens between the Mn4+ and Er3+ ions to populate the 4F9/2 and 4I9/2 levels of Er3+ followed by nonradiative relaxation to 4I13/2. Finally, IR emission at 1529 nm is produced by radiative transition from 4I13/2 to 4I15/2 of Er3+.
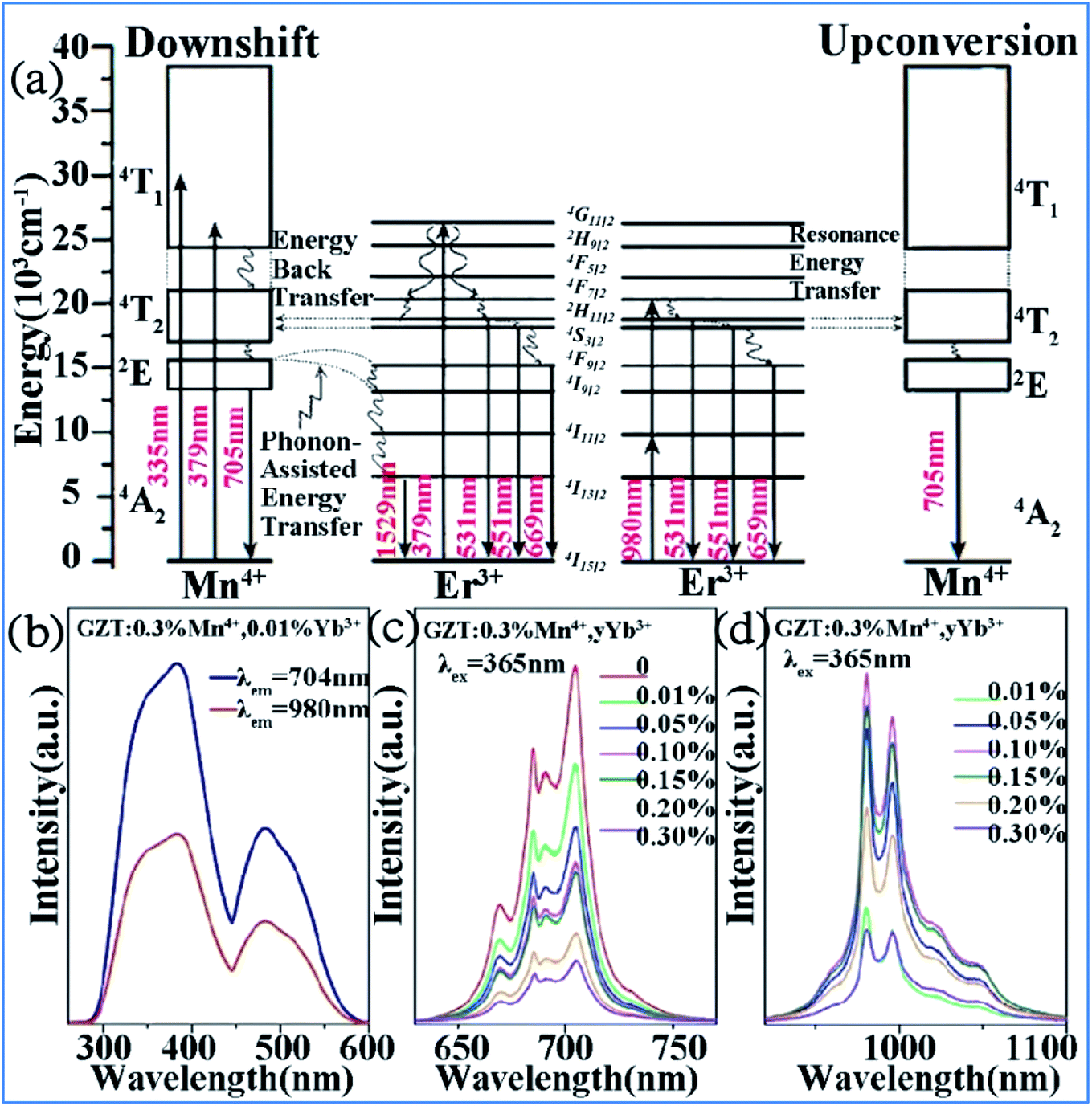 | ||
| Fig. 12 (a) Electron transitions and mutual sensitized energy transfer scheme between Mn4+ and Er3+ in the Gd2ZnTiO6 matrix. Reprinted with permission from ref. 74, Copyright 2017, The Royal Society of Chemistry. (b) PLE, (c) visible, and (d) NIR spectra of Gd2ZnTiO6:0.3%Mn4+,yYb3+. Reprinted with permission from ref. 33, Copyright 2018, Elsevier BV. | ||
Far-red (FR) and near-infrared (NIR) double-wavelength emissions have been observed in the Mn4+ and Yb3+ codoped GZT phosphor, which are expected to application in LEDs towards plant cultivation.77–79 The PLE and PL spectra of the Mn4+ and Yb3+ codoped samples are shown in Fig. 12b–d. The shapes and positions of both PLE spectra (Fig. 12b) monitored at emission 704 nm from the 2Eg → 4A2g transition of Mn4+ and that at 980 nm from the Yb3+ transition 2F5/2 → 2F7/2 are similar to that of Mn4+ singly doped GZT, which indicates that energy transfer between Mn4+ and Yb3+ occurs in the codoping systems. Under the excitation of 365 nm light, both FR emission from Mn4+ and NIR emission from Yb3+ are observed in Fig. 12c and d. The FR emission intensity of Mn4+ gradually decreases with an increase in the content of Yb3+, whereas the NIR emission intensity first increases and then decreases due to the concentration quenching effect, which further prove the occurrence of energy transfer from Mn4+ to Yb3+.
The similar energy transfer from Mn4+ to Yb3+ has been also observed in Mn4+ and Yb3+ codoped La2MgTiO6 samples. Broad excitation bands from 250 nm to 550 nm corresponding to the absorptions involving the 4A2g → 4T1g, and 4A2g → 4T2g transitions of Mn4+ monitored at 710 nm and Yb3+ ions monitored at 980 nm were observed in the PLE of La1.91MgTi0.998O6:Mn0.002,Yb0.09 sample, as shown in Fig. 13a.36,80,81 The excitation spectrum monitored at 980 nm of Yb3+ emission is similar to that monitored at 710 nm of the Mn4+ emission in the La1.91MgTi0.998O6:Mn0.002,Yb0.09 sample, which clearly proves that energy transfer from Mn4+ to Yb3+ takes place in the Mn4+ and Yb3+ codoped La2MgTiO6 samples when the Mn4+ ions are excited.
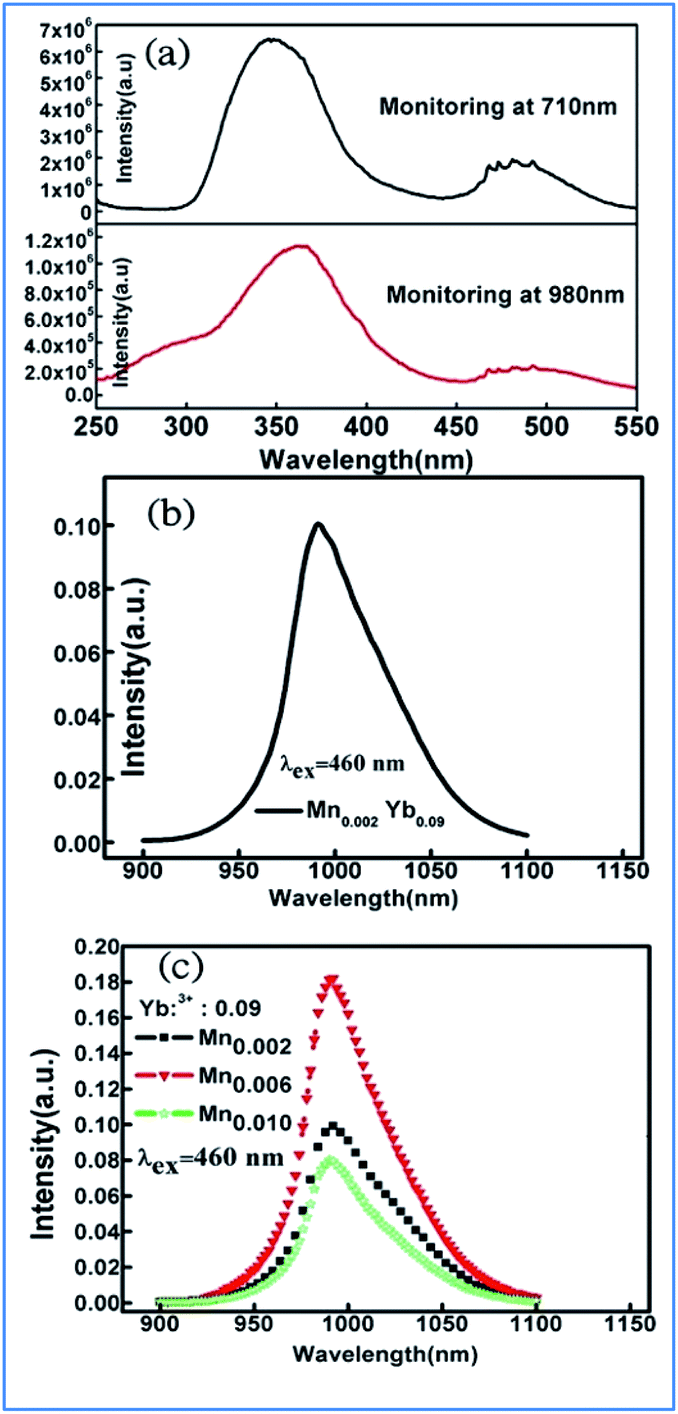 | ||
| Fig. 13 (a) Excitation spectra of Mn4+ monitored at 710 nm and Yb3+ monitored at 980 nm in La1.91MgTi0.998O6:Mn0.002,Yb0.09 sample, emission spectra of (b) La2−xMgTi1−yO6:Mny,Ybx, (c) La2−xMgTi1−yO6:Mny,Ybx samples pumped by 460 nm light. Reprinted with permission from ref. 35, Copyright 2018, Elsevier BV. | ||
Fig. 13b and c exhibit the emission spectra of the La2−xMgTi1−yO6:Mny,Ybx and La2−xMgTi1−yO6:Mny,Ybx samples pumped by 460 nm light. The NIR emission band with the highest peak at 990 nm is from the 2F5/2 → 2F7/2 transition of Yb3+ ions and its emission is strongly dependent the concentrations of Yb3+ ions.35,82,83 The integrated intensity of the NIR emission band centered at 990 nm increases initially with an increase in the concentration of Yb3+ ions.
Energy transfer from Mn4+ to Yb3+ occurs in the Mn4+ and Yb3+ codoped Ba2LaNbO6 (BLNO) samples, as illustrated in Fig. 14.35 The spectral shapes and positions of the excitation spectra monitored at 677 nm (Mn4+ emission) and 998 nm (Yb3+ emission) remain the same, but their intensities are different, which indicates that energy transfer from Mn4+ to Yb3+ occurs in the Mn4+ and Yb3+ codoped BLNO, as show in Fig. 14a and b. The emission centered at 998 nm is consistent with the infrared light needed for bacterial chlorophyll.22,84 The intensity of the Mn4+ emission at 677 nm decreases, while that of the Yb3+ emission at 998 nm increases due to the transfer of energy from Mn4+ to Yb3+.
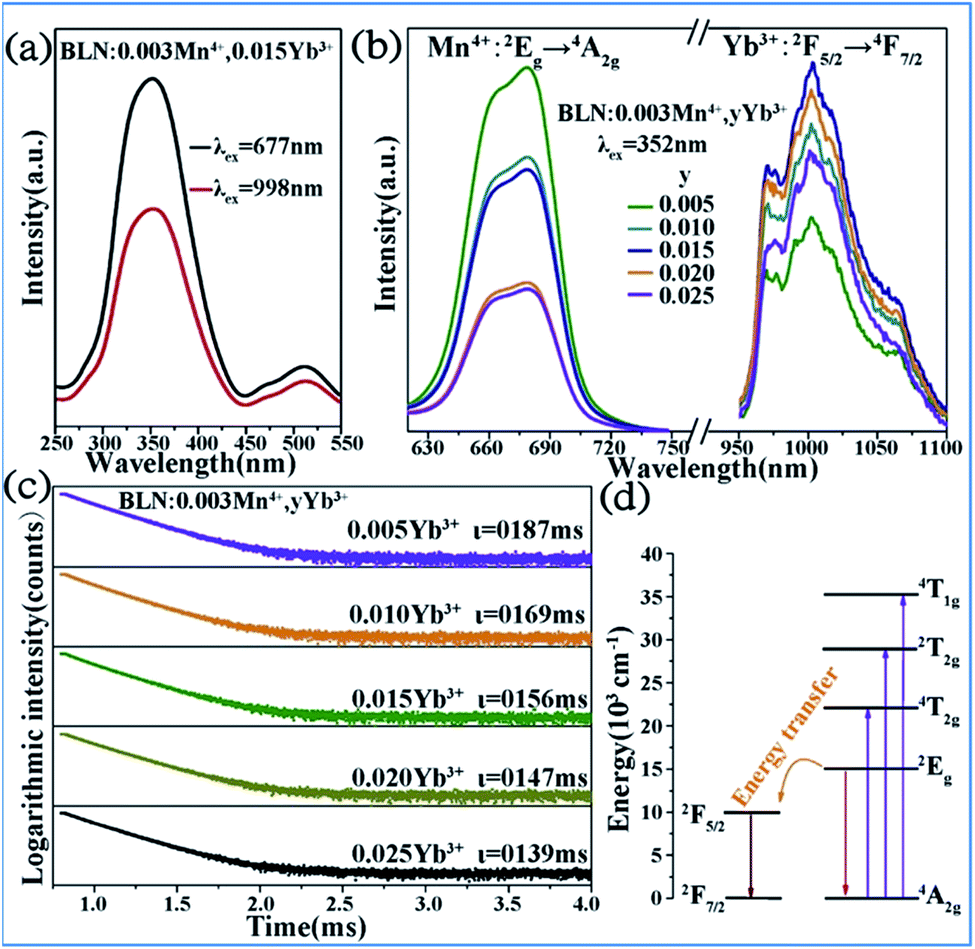 | ||
| Fig. 14 (a) PLE and (b) PL spectra of Ba2LaNbO6:Mn4+,yYb3+ phosphors. (c) Decay curves of Ba2LaNbO6:Mn4+,yYb3+. (d) Energy transfer schematic diagram of Mn4+ and Yb3+ codoped system. Reprinted with permission from ref. 35, Copyright 2019, Elsevier BV. | ||
Fig. 14c shows the decay lifetimes of BLNO:0.003Mn4+,yYb3+, which decrease with an increase in the Yb3+ concentration, thus proving the occurrence of energy transfer from Mn4+ to Yb3+ in the phosphor. According to the mechanism of energy transfer of Mn4+ and Yb3+ based on Fig. 14d,35,85,86 the Mn4+ ions are excited from the ground state (4A2g) to excited states (4T1g, 2T2g, and 4T2g) under UV light excitation, and then relax to the 2Eg state. The energy can be transferred from the 2Eg state of Mn4+ to the 2F5/2 level of Yb3+ through nonradiative transition, thereby producing the NIR emission observed at 998 nm.
The energy transfer from Mn4+ to Nd3+ occurs in the Mn4+ and Nd3+ codoped (Na,K)Mg(La,Gd)TeO6 samples, as illustrated in Fig. 15.16 Upon excitation at 365 nm UV, both emissions from Mn4+ and Nd3+ are observed, and the Mn4+ emission intensity and the corresponding decay time of Mn4+ at 705 nm decrease monotonously with an increase in Nd3+ concentration, which strongly confirms the efficient energy transfer from the Mn4+ to Nd3+ ions in these samples.87,88
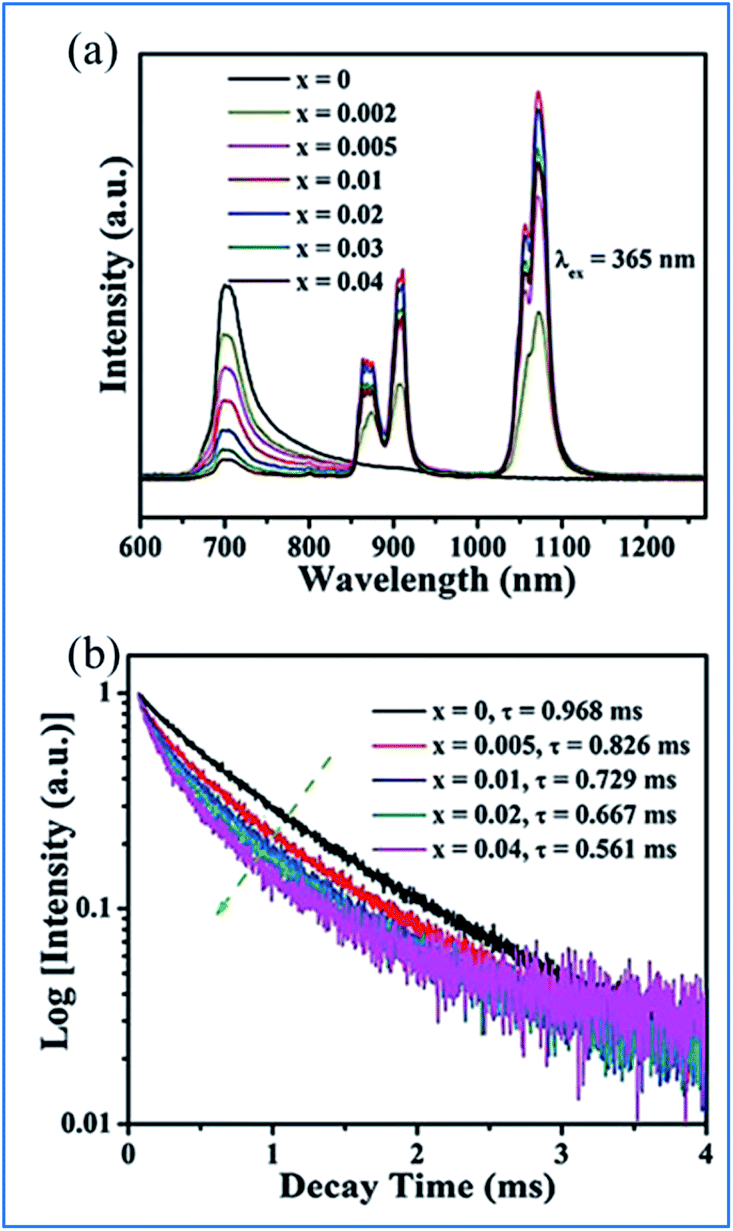 | ||
| Fig. 15 (a) PL emission spectra (λex = 365 nm) of NaMgLaTeO6:0.02Mn4+,xNd3+ and (b) corresponding decay curves for NaMgLaTeO6:0.02Mn4+,xNd3+ (λex = 365 nm, λem = 705 nm). Reprinted with permission from ref. 16, Copyright 2018, The Royal Society of Chemistry. | ||
The energy transfer processes of Mn4+ → Nd3+ → Yb3+ occurring in the Mn4+, Nd3+ and Yb3+ codoped NaMgLaTeO6 (NMLTO) samples are illustrated in Fig. 16.16 The emission spectra of NML:0.02Mn4+,0.30Yb3+ excited at 365 nm contains both the Mn4+ emission band at around 705 nm due to the Mn4+ 2Eg → 4A2g transition, and the Yb3+ emission band with a maximum at around 1003 nm attributed to the Yb3+ 2F5/2 → 2F7/2 transition. The excitation spectrum (200–900 nm) monitored at 1003 nm clearly contains the Mn4+ absorption band, suggesting energy transfer from Mn4+ to Yb3+ ions.89–91 In the Mn4+, Nd3+, and Yb3+ codoped NMLTO sample, the emission spectra of the obviously present bands from all three ions Mn4+, Nd3+, and Yb3+ in the range of 600–1300 nm upon 365 nm UV excitation.92,93 The emission intensity of Nd3+ decreases monotonously with an increase in Yb3+ concentration, which illustrates the possibility of energy transfer from the Nd3+ to Yb3+ ions as shown in Fig. 16a–c.
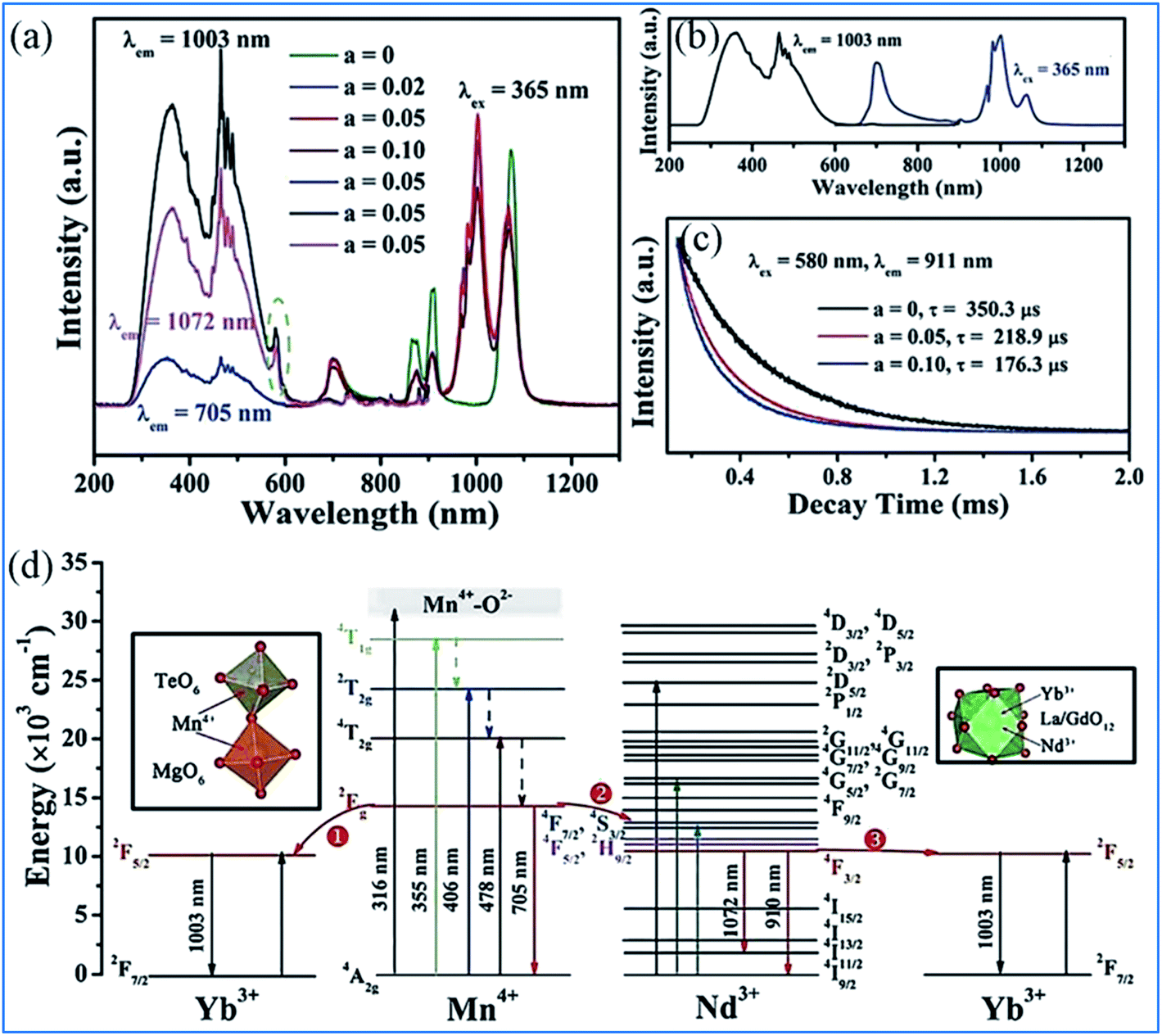 | ||
| Fig. 16 PL excitation and emission spectra of (a) NaMgLaTeO6:0.02Mn4+,0.30Yb3+, (b) NaMgLaTeO6:0.02Mn4+,0.01Nd3+,aYb3+, and (c) decay curves of NaMgLaTeO6:0.02Mn4+,0.01Nd3+,aYb3+ (λex = 580 nm, λem = 911 nm). (d) Partial coordination environment in the NaMgLaTeO6 structure and schematic energy-level diagram illustrating the possible energy transfer processes in the NaMgLaTeO6:Mn4+,Nd3+,Yb3+ materials. Reprinted with permission from ref. 16, Copyright 2018, The Royal Society of Chemistry. | ||
Fig. 16d shows an overview of the partial electronic energy level diagram of Mn4+, Nd3+, and Yb3+ in NMLTO and a schematic diagram illustrating the possible energy transfer processes occurring in Mn4+, Nd3+, and Yb3+ codoped NMLTO.16 The energy at the Mn4+ excited state 2Eg can be transferred to the Nd3+ levels 4F7/2 and 4S3/2 via the Forster resonant energy transfer process to produce the emissions at 910 and 1072 nm.90,94
The NIR emissions of Nd3+ at 910 and 1072 nm from the 4F7/2 and 4S3/2 levels, respectively, increases and the red emission of Mn4+ at 705 nm from the 2Eg excited state decreases with an increase in the concentration of Mn4+, which indicates the energy transfer from Mn4+ to Nd3+.68,95 Then the excited 4F7/2 and 4S3/2 energy levels of Nd3+ can relax nonradiatively to the 4F5/2 and 2H9/2 Nd3+ energy levels, and transfer the energy to the 2F5/2 Yb3+ excited state and enhance the Yb3+ emission.
As can be seen in Fig. 17, the excitation spectra of the Mn4+, Nd3+ and Yb3+ codoped NMLTO samples match well with the solar spectrum in the UV and visible regions, and the emission bands are located at the ideal 930–1100 nm region for excellent response for crystal silicon solar energy cells.68,96 Thus, the Mn4+, Nd3+ and Yb3+ codoped NMLTO sample has potential for the effective broadband spectral conversion of UV/visible light to the NIR band utilizing the energy transfer processes of Mn4+ → Nd3+ → Yb3+.
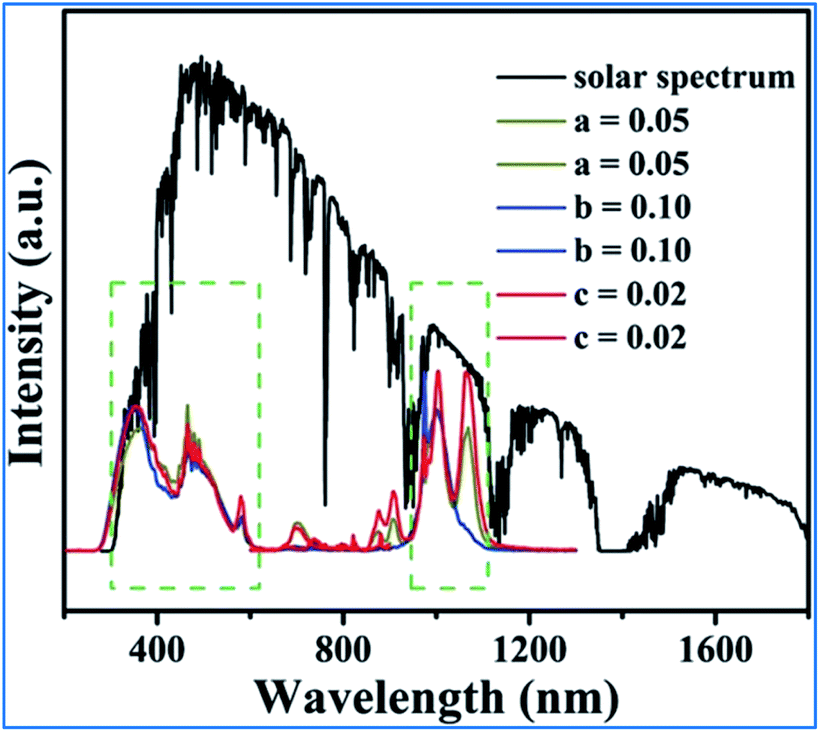 | ||
| Fig. 17 Solar spectrum and PL excitation and emission spectra of NaMgLaTeO6:0.02Mn4+,0.01Nd3+,aYb3+, NaMgGdTeO6:0.01Mn4+,0.02Nd3+,bYb3+ and KMgLaTeO6:0.006Mn4+,0.03Nd3+,cYb3+. Reprinted with permission from ref. 16, Copyright 2018, The Royal Society of Chemistry. | ||
4. Tunable multiple emissions via energy transfer in a single host lattice
4.1 Energy transfer between Dy3+ and Mn4+
As displayed in Fig. 18a, the PLE spectrum of the Ca13.88Al10Zn6O35:0.12Dy3+ phosphor monitored at 576 nm consists of a series of sharp peaks with the strongest absorption at 351 nm due to the 6H15/2 → 6P7/2 transition of Dy3+. Under excitation at 351 nm, the PL spectrum consists of two dominant peaks at around 482 nm (blue) and 576 nm (yellow), corresponding to the 4F9/2 → 6H15/2 and 4F9/2 → 6H13/2 transitions of Dy3+, respectively.97–99 As shown in Fig. 18b, significant spectral overlap was observed between the PLE of Mn4+ and PL of Dy3+, indicating that effective energy transfer from Dy3+ to Mn4+ is expected.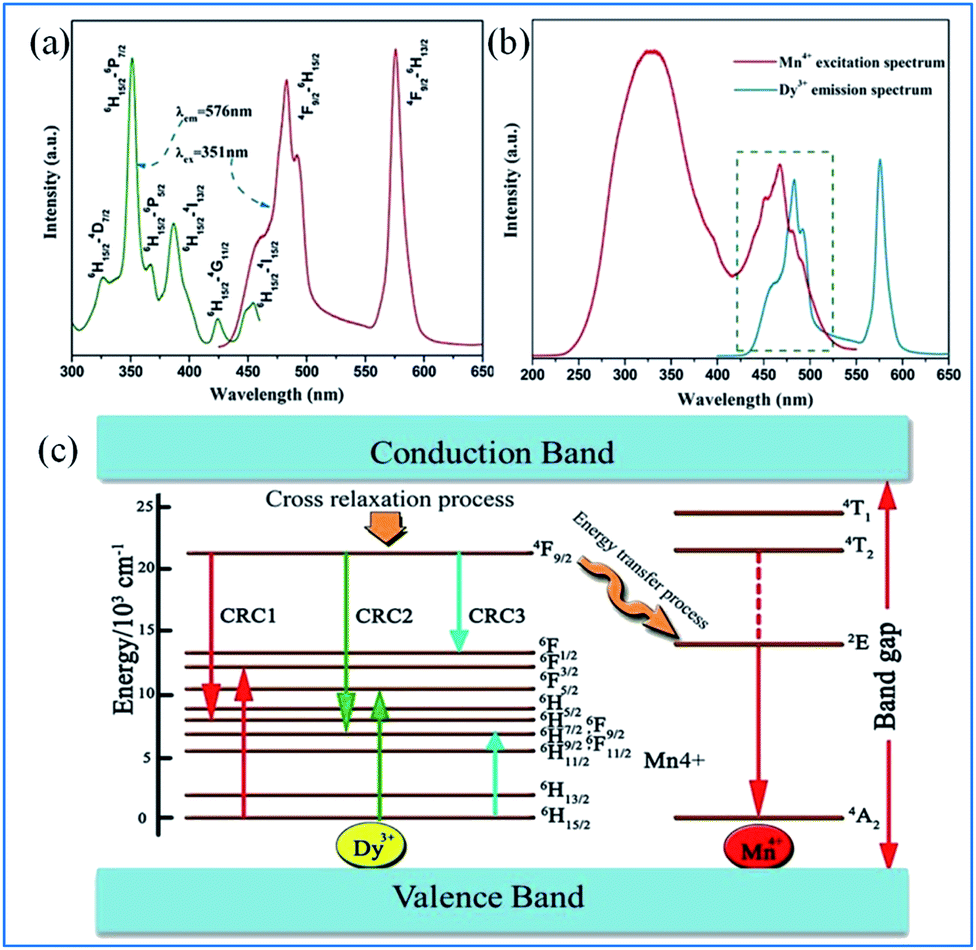 | ||
| Fig. 18 (a) PLE and PL spectra of Ca13.88Al10Zn6O35:0.12Dy3+, (b) spectral overlap between the PLE of Mn4+ and the PL of Dy3+, (c) schematic level diagram for the cross-relaxation process and energy transfer process from Dy3+ to Mn4+. Reprinted with permission from ref. 97, Copyright 2016, Kluwer Academic Publishers. | ||
The energy transfer process from Dy3+ to Mn4+ is elucidated according to the schematic energy level diagram in Fig. Fig. 18c. In the cross-relaxation processes, the Dy3+ ions at the 4F9/2 level can be de-excited to the 6F9/2/6H7/2, 6H9/2/6F11/2, or 6F1/2 level, while the ions at the 6H15/2 ground state will accept the energies excited simultaneously to the 6F3/2, 6F5/2, and 6H9/2/6F11/2 levels. Although the energy level 4F9/2 of Dy3+ (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 747 cm−1) is higher than the 2Eg energy level of Mn4+ (14025 cm−1), the energy transfer from the 4F9/2 level of Dy3+ to the 2E level of Mn4+ may be realized via the assistance of phonons.97,100–104
747 cm−1) is higher than the 2Eg energy level of Mn4+ (14025 cm−1), the energy transfer from the 4F9/2 level of Dy3+ to the 2E level of Mn4+ may be realized via the assistance of phonons.97,100–104
The PL spectra of Ca13.88Al10−yZn6O35:0.12Dy3+,yMn4+ (y = 0, 0.01, 0.05, 0.10, 0.15, 0.20, and 0.25) upon excitation at 351 nm and the change in the emission intensities of Dy3+ and Mn4+ with the concentration of Mn4+ are presented in Fig. 19.97 The emissions at 482 and 576 nm are due to the 4F9/2 → 6HJ/2 (J = 15, 13) transitions of Dy3+, and the red emission with a multi-peak structure in the wavelength range of 650 to 750 nm corresponds to the vibronic emission 2Eg → 4A2g of Mn4+. The emission intensity of Mn4+ increases, whereas that of Dy3+ is simultaneously found to decrease monotonically with an increase in concentration of Mn4+, indicating that the energy transfer from Dy3+ to Mn4+ is efficient.105–108
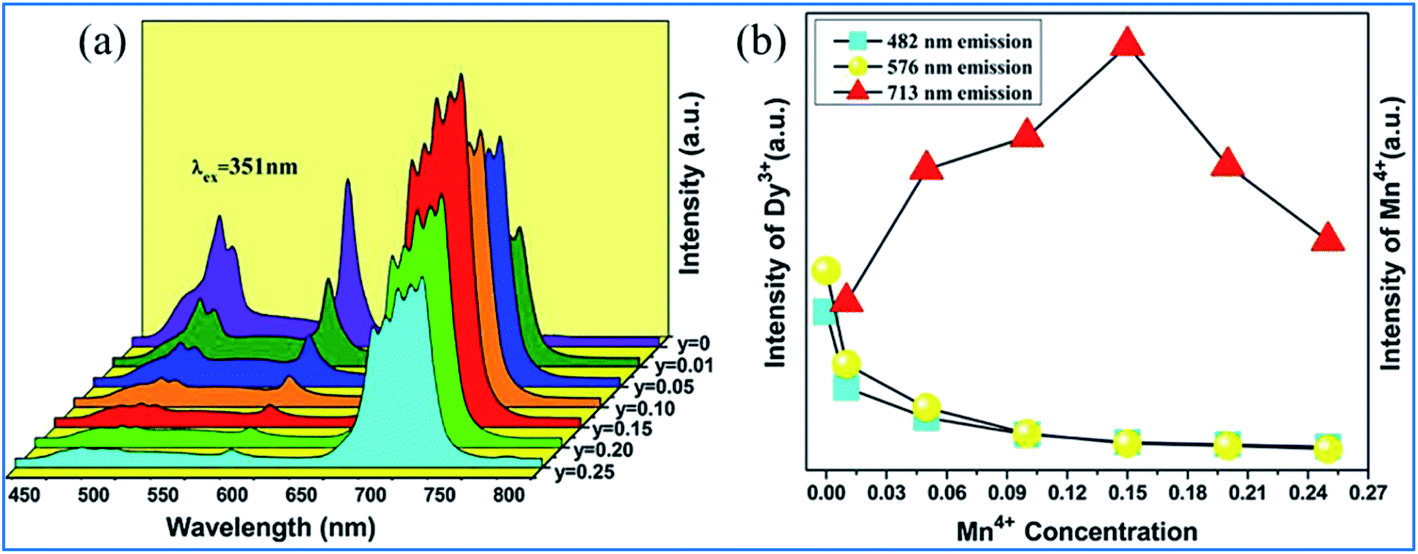 | ||
| Fig. 19 (a) PL spectra of Ca13.88Al10-yZn6O35:0.12Dy3+,yMn4+ (y = 0, 0.01, 0.05, 0.10, 0.15, 0.20, and 0.25) under the excitation at 351 nm and (b) emission intensities of Dy3+ and Mn4+ as a function of the concentration of Mn4+. Reprinted with permission from ref. 97, Copyright 2016, Kluwer Academic Publishers. | ||
4.2 Tunable dual emissions for Bi3+ and Mn4+ codoped phosphors
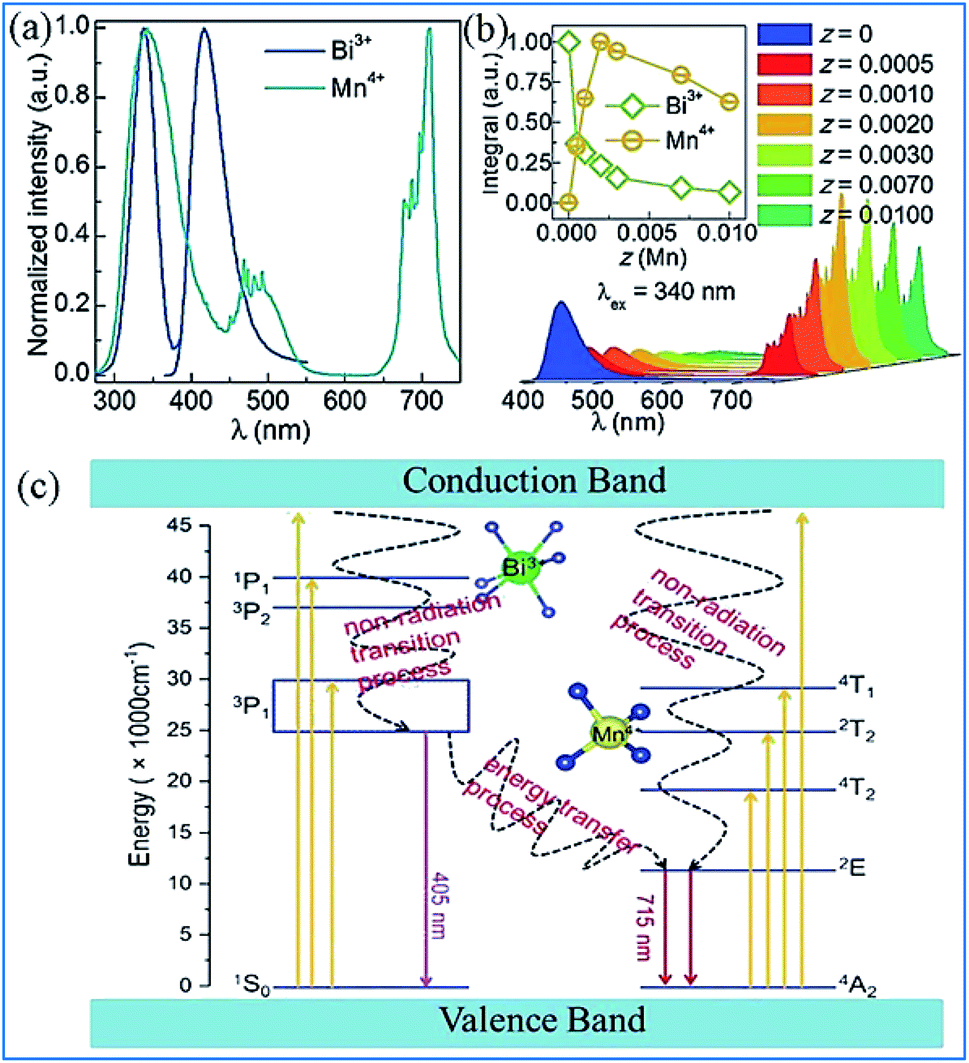
It was found that both the blue light from Bi3+ and red light from Mn4+ are produced in all the Bi3+ and Mn4+ codoped CZAO samples, as illustrated in Fig. 20a. The emission band from 400 nm to 550 nm with a maximum at 410 nm is ascribed to the 3P1 → 1S0 transition of the Bi3+ ions, while that from 650 nm to 750 nm is ascribed to the 2Eg → 4A2g emission of the Mn4+ ions.25,109 The intensity of the blue emission decreases and that of the red emission increases with an increase in the Mn4+ concentration, as shown in Fig. 20b, which indicates the occurrence of energy transfer from Bi3+ to Mn4+. The dual-emission color can be tuned by changing the Bi3+/Mn4+ ratio.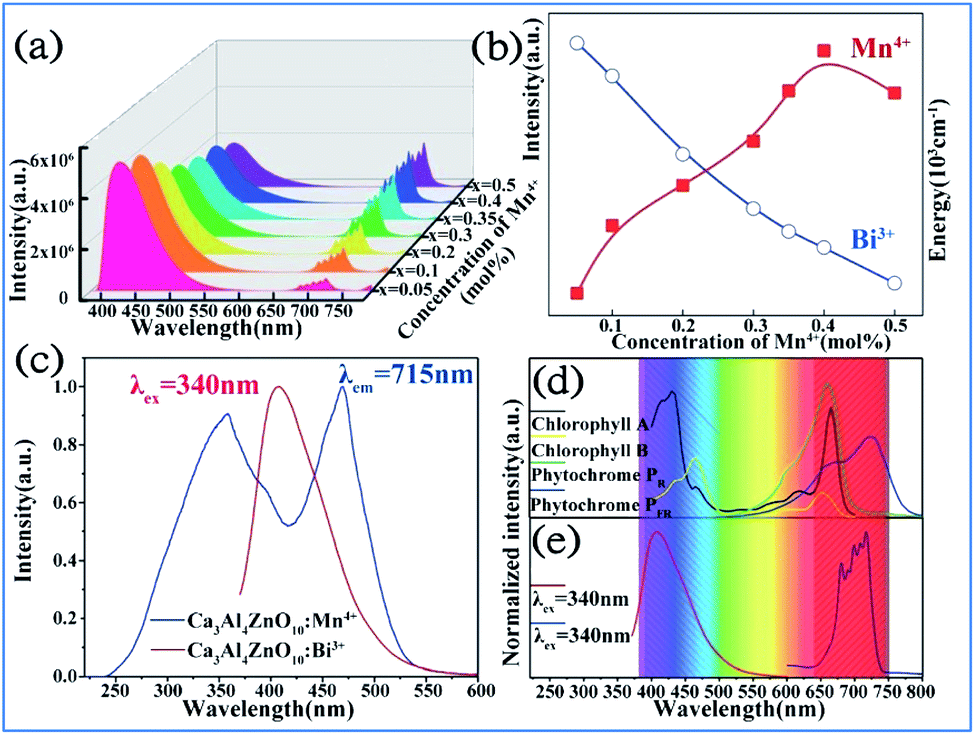 | ||
| Fig. 20 (a) Emission spectra (λex = 351 nm) of samples of Ca14Zn6Al10O35:0.5% Bi3+,x% Mn4+ (x = 0.05, 0.1, 0.2, 0.3, 0.35, 0.4 or 0.5) and (b) dependence of the luminescence intensities of the red emission from Mn4+ and blue emission from Bi3+ on the Mn4+ doping concentrations. Reprinted with permission from ref. 25, Copyright 2017, The Royal Society of Chemistry. (c) PLE spectrum of Ca3ZnAl4O10:0.008Mn4+ and PL spectrum of the Ca3ZnAl4O10:0.008Bi3+ phosphor. (d) Absorption spectra of chlorophyll A, chlorophyll B, and phytochromes PR and PPR, and (e) PL spectra of Bi3+ and Mn4+ in Ca3ZnAl4O10. Reprinted with permission from ref. 26, Copyright 2018, The Royal Society of Chemistry. | ||
A similar energy transfer from Bi3+ to Mn4+ was also observed in the Bi3+ and Mn4+ codoped CZAO phosphor due to the spectral overlap in the PLE of Mn4+ and PL of CZAO:0.008Bi3+, as shown in Fig. 20c. Under the same excitation source, Bi3+ and Mn4+ codoped CZAO phosphors show dual emissions, where the blue-violet emission is mainly from the 3P1 → 1S0 transition of Bi3+ and the far red emission is attributed to the 2Eg → 4A2g transition of Mn4+.26,110,111 As presented in Fig. 20d and e, the blue emission of Bi3+ matches the absorption spectra of chlorophyll A and chlorophyll B, while the red emission from Mn4+ matches the absorption spectra of phytochrome PR and phytochrome PFR, which indicate that the phosphor has potential for application in plant growth LED lighting.
The energy transfer process from Bi3+ to Mn4+ realized in Bi3+ and Mn4+ codoped La2MgTiO6 (LMTO) phosphors is illustrated in Fig. 21. The absorption bands from 275 to 375 nm in the PLE spectra for LMT:0.005Bi3+ in Fig. 21a are ascribed to the 1S0 → 1P1 and 1S0 → 3P1 transitions of Bi3+. A blue emission (375–500 nm) with a maximum at 417 nm of Bi3+ is detected, which is due to the 3P1 → 1S0 transitions. The strong red emission band from 650 to 750 nm with an emission peak at 710 nm is observed owing to the 2Eg → 4A2g transition of Mn4+. The spectral overlap between the emission spectrum of Bi3+ and the excitation spectra of Mn4+ provides strong evidence for the energy transfer between Bi3+ and Mn4+. The emission intensity of Bi3+ gradually decreases and that of Mn4+ presents a monotonous increase with an increase in the Mn4+ doping concentration, which indicates that energy transfer occurs in the Bi3+ and Mn4+ codoped LMTO phosphors, as shown in Fig. 21b.
 | ||
| Fig. 21 (a) PLE and PL spectra of La2MgTiO6:0.005Bi3+ (blue) and La2MgTiO6:0.002Mn4+ (cyan). (b) PL spectra of La2MgTi(1-z)O6:0.005Bi3+,zMn4+ (0 ≤ z ≤ 0.01). The inset shows the integrated intensity of the Bi3+ and Mn4+ emission as a function of the concentration of Mn4+. Reprinted with permission from ref. 112, Copyright 2018, The Royal Society of Chemistry. (c) Schematic illustration of the electronic transitions and energy transfer process in Ca3ZnAl4O10:Bi3+,Mn4+. Reprinted with permission from ref. 26, Copyright 2018, The Royal Society of Chemistry. | ||
The electronic transitions and the energy transfer process in the Bi3+ and Mn4+ codoped phosphors are illustrated the schematic energy level diagram shown in Fig. 21c.26 The Bi3+ ions are initially excited from the ground state 1S0 to the excited state 3P1, 3P2, and 1P1 or even the conduction bands under the irradiation of UV light. Then, the Bi3+ ions relax to the lowest excited state of 3P1 and return to the 1S0 ground state through radiative transition and yield blue emission. Simultaneously, the Bi3+ ions in the 3P1 state can also transfer their energy to the adjacent Mn4+ ions and promote the Mn4+ ions from the 4A2g ground state to the 4T2g, 2T2g, and 4T1g energy levels and relax to the 2Eg level through a nonradiative transition and then produce red emission when they return to the 4A2g ground state.113–115 The energy transfer occurring between Bi3+ and Mn4+ eventually lead to an enhancement in the far-red emission of Mn4+.
5. Red emitting phosphors for plant growth LED lights
5.1 Enhanced red emission of Mn4+ by codoping rare earth ions
The Dy3+ and Mn4+ codoped Ca14Ga10−mAlmZn6O35 (CGAZO:Dy3+,Mn4+) phosphor can exhibit strong far-red emission, which has potential application for plant growth LED lighting.44 As shown in Fig. 22a, the three absorption bands A (200–290 nm), B (290–420 nm), and C (420–550 nm) of the phosphors in the UV-vis absorption spectra can be attributed to the host lattice absorption, charge transfer transition of Mn4+-O2-, and spin-allowed transitions 4A2g → 4T1g and 4A2g → 4T2g of the Mn4+ ions, respectively.116 The absorption intensity of bands A and C decrease, but that of band B is enhanced with elevated Al3+ concentrations, which indicates that the absorption intensity of the phosphor powder is enhanced in the ultraviolet light range, but reduced slightly in the blue light range.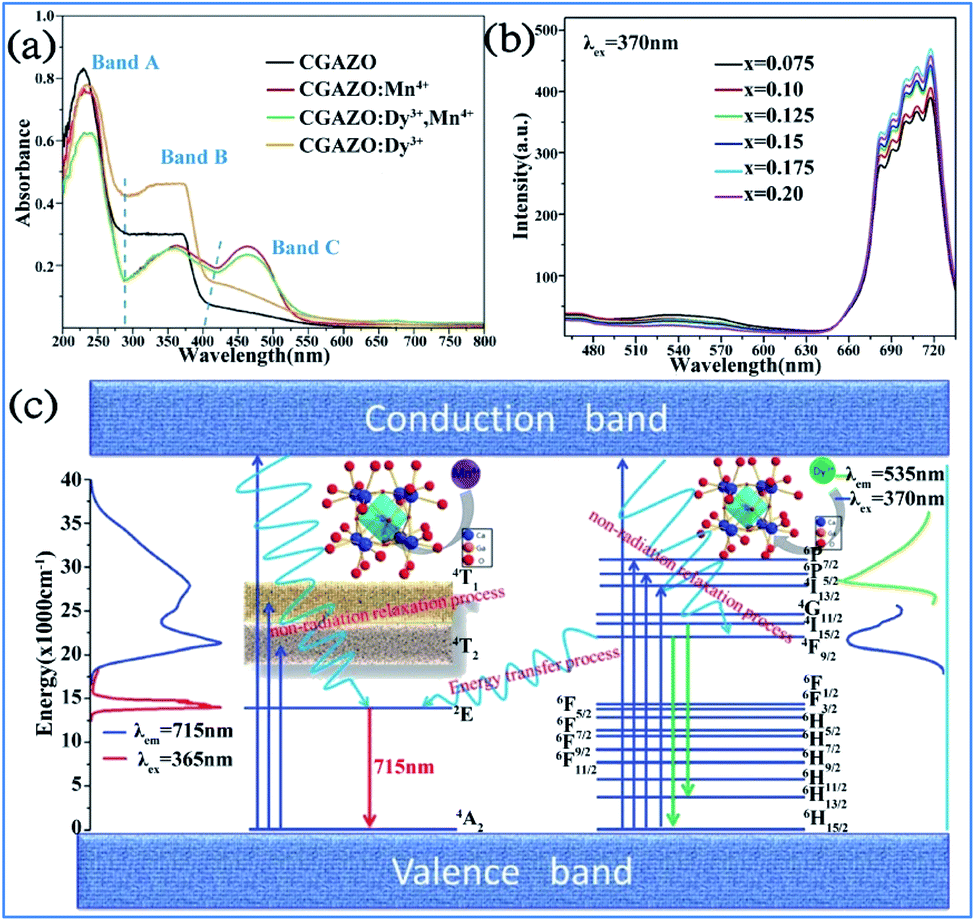 | ||
| Fig. 22 UV-vis absorption spectra of (a) Ca14Ga10−mAlmZn6O35 with different dopants. (b) PL spectra of Ca14Ga10−mAlmZn6O35:0.12Dy3+,xMn4+ phosphor. (c) Energy level, electron transitions and energy transfer schematic diagram of Dy3+, Mn4+ in Ca14Ga10−mAlmZn6O35 matrix. Reprinted with permission from ref. 44, Copyright 2017, The Royal Society of Chemistry. | ||
As shown in Fig. 22b, the PL intensity of the Mn4+ activator increased, whereas that of the Dy3+ sensitizer simultaneously decreased monotonically with an increase in the concentration of Mn4+ ions, which demonstrates that energy transfer from Dy3+ to Mn4+ occurred in the Dy3+ and Mn4+-co-activated CGAZO, as described using Fig. 22c. The Dy3+ ions are excited to their 6P7/2 or 6P5/2 or 4I13/2 excited states or conduction band under irradiation of near UV light and nonradiatively relax to their 4F9/2 state. The energy transfer process between the Dy3+ and Mn4+ ions occurs via 4F9/2 (Dy3+) → 2Eg (Mn4+) and the Mn4+ ions return from the lowest excited level 2Eg (Mn4+) to the 4A2g ground state (Mn4+) through a radiative transition, which produces the far-red light emission at 715 nm.
5.2 Enhanced red emission of Mn4+ by codoping Bi3+
The red emission of the Mn4+ ions in the phosphors based on the CaAl12O19,117,118 Mg2TiO4,37,119–121 and La2ATiO6 (ref. 112) (A = Mg, Zn) host lattices can be dramatically enhanced by the incorporation of Bi3+ codopant. The spectral profiles of the excitation and emission spectra of Mn4+ with or without codoping Bi3+ ions in these Mn4+ doped phosphors are quite similar. Therefore, it can be speculated that the synergetic effect of codoping Bi3+ plays a key role in the modification of the crystal structure and the luminescence efficiency of Mn4+. Thus, the strategy for enhancing the luminescence performance of Mn4+ plays a pivotal role in the development of highly efficient red-emitting phosphors.122–1256. Luminescent thermometers based on Mn4+ and multiple ion-doped materials
By employing the highly temperature-sensitive Mn4+ luminescence as the temperature detecting signal, while the temperature-insensitive rare earth ion (Eu3+, Tb3+ or Dy3+) emission was used as a reference signal, Mn4+ and multiple rare earth ion codoped phosphors exhibited an excellent temperature sensing performance with absolute and relative sensitivities as high as 0.114–0.441 K−1 and 2.32–4.81% K−1, respectively, which indicate their potential application in luminescent thermometers.126–128In Fig. 23a and b, the bright red luminescence of the Eu3+ and Mn4+ codoped YAG samples originated from both the transitions 5D0 → 7FJ of Eu3+ and 2Eg → 4A2g of Mn4+. With an increase in temperature, the luminescence of Mn4+ weakens quickly, whereas that of Eu3+ exhibits a slight decrease. As shown in Fig. 23c, the remarkable change in IMn/IEu with a variation in temperature measured on the cycling process of heating-cooling can almost be restored to the original states after the heating-cooling cycle.130–134 As confirmed in Fig. 23d, this temperature-dependent IMn/IEu is repeatable and reversible after several cycling experiments. Therefore, a highly sensitive temperature determination can be expected if the Mn4+ emission is employed as the detection signal of temperature, while the Eu3+ emission is used as the reference signal.
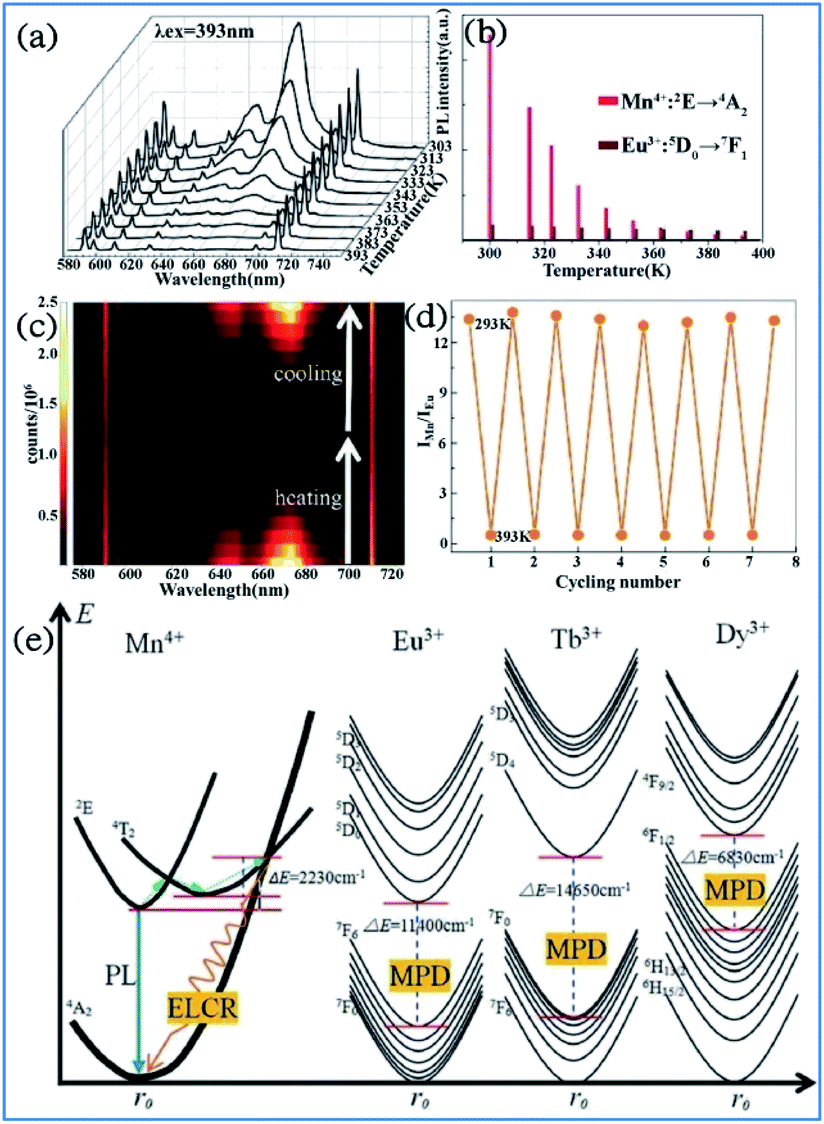 | ||
| Fig. 23 Temperature-dependent (a) PL spectra of a Mn4+/Eu3+:YAG sample recorded from 303 K to 393 K. (b) PL intensities for Mn4+ and Eu3+. (c) Emission mapping upon the cycling process of heating and cooling. (d) Temperature-induced switching of FIR between Mn4+ and Eu3+ (alternating between 293 K and 393 K). Reprinted with permission from ref. 122, Copyright 2016, The Royal Society of Chemistry. (e) Configurational coordinate diagrams of the Mn4+/Eu3+/Tb3+/Dy3+ emitting centers in the Y3Al5O12 host, showing the energy-level crossing relaxation (ELCR) quenching mechanism for the Mn4+ activator and the multi-phonon de-excitation (MPD) quenching mechanism for the Eu3+/Tb3+/Dy3+ centers. Reprinted with permission from ref. 129, Copyright 2016, The Royal Society of Chemistry. | ||
The photon generation and energy transfer between Mn4+ and rare earth ions in Mn3+, Mn4+, and Nd3+ codoped YAG nanocrystals can be illustrated by an energy level diagram, as presented in Fig. 23e. The Mn4+ ions are excited from the 4A2g ground state to the 4T2 excited state, followed by nonradiative multiphonon relaxation, leading to population of the 2Eg state, and then emit red emission at 670 nm, which is ascribed to the radiative electronic 2Eg → 4A2g transition of Mn4+. The appearance of an intersection point between the 4T2 parabola and the 4A2g parabola at ΔE (activation energy, in this case ΔE1 = 2506 cm−1) is due to the strong electron-phonon coupling. The value of ΔE1 is associated with the distortion of the Mn4+ energy states, which is strongly dependent on the crystal field. With an increase in the temperature, the population of higher vibrational states gradually increases up to the moment when the provided thermal energy is sufficiently high to overcome the intersection point (ΔE1), above which electrons from the 2Eg level are transferred through 4T2g to the 4A2g ground state via nonradiative multiphonon relaxation. In contrast, rare earth ions are expected to be less affected by luminescence temperature quenching because their energy diagram usually consist of numerous f energy states due to low electron-phonon coupling. Therefore, Mn4+ and rare earth ions codoped in a single host lattice can be applied in a luminescent thermometer.135–138
The structural coordinate diagram in Fig. 23e proposes a possible mechanism for elucidating the high temperature sensitivity of Mn4+ and rare earth ion (such as Eu3+/Tb3+ and Dy3+) codoped samples. The Mn4+ luminescence is easily thermally quenched through an energy-level crossing relaxation (ELCR) between the 4T2g excited state and the 4A2g ground state due to the role of strong electron–phonon coupling. The thermal quenching of rare earth ions is completely different to that of Mn4+ since there is no crossing point between the excited states and the ground state of rare earth ions because their 4f orbitals are shielded from the surroundings by the filled 5S2 and 5P6 orbitals,139,140 and consequently the multi-phonon deexcitation (MPD) mode is the dominant mechanism responsible for the thermal-quenching of rare earth ions. The thermal-quenching probability of Eu3+, Tb3+, and Dy3+ luminescence is quite low because the required phonon numbers to bridge the energy gaps of Eu3+, Tb3+ and Dy3+ are 16, 21 and 10, respectively.
The representative thermal evolution of the emission spectra of Y3Al5O12:Mn3+, Mn4+, Nd3+ nanocrystals presented in Fig. 24a indicates that the emission intensity of both the 2Eg → 4A2g emission band of Mn4+ and the 4F3/2 → 4I9/2 band of Nd3+ decreases with an increase in temperature. In contrast, the 5T2 → 5E′′ emission of Mn3+ exhibits different behavior. The upper lying 5T2 state of Mn3+ can be populated via phonon-assisted energy transfer with the phonon absorption. The probability of this process increases with temperature according to the Miyakawa–Dexter theory.31,141,142 On the other hand, Kuck et al. explained the increase in the Mn3+ emission at elevated temperatures in terms of the thermal population from the 3T1 state.143
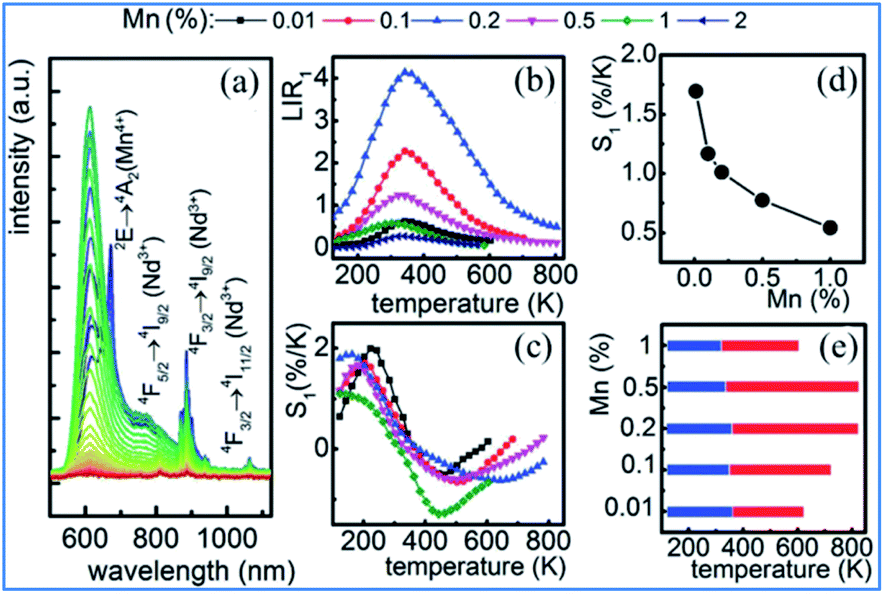 | ||
| Fig. 24 (a) Thermal evolution of 20 nm Y3Al5O12:0.1% Mn,1% Nd3+ nanocrystal emission spectra. (b) Impact of temperature on LIR for different Mn concentrations of Y3Al5O12:Mn3+, Mn4+, Nd3+ nanocrystals. (c) Thermal evolution of S1 for thermometers with different Mn concentrations. (d) Dependence of sensitivity on manganese concentration at T = 273 K. (e) UTR of thermometers with different Mn concentrations. Reprinted with permission from ref. 31, Copyright 2018, Pergamon Press Ltd. | ||
The thermal evolution of LIR1 for the series of 20 nm nanocrystals with different manganese concentrations is presented in Fig. 24b. In the low temperature range, LIR1 increases with temperature, reaching the maximum at T = 400 K. A further increase in temperature causes a reduction in the value of LIR1. In the low temperature range (below 350 K), the LIR2 value is thermally independent, which is related to the high thermal stability of the Mn4+ luminescence at low temperatures. This shows that the emission intensity of the 5T2 state of Mn3+ increases at low temperatures, while that of the 2Eg state of Mn4+ becomes stable.
The thermoluminescence glow curves of the all the Mn4+ and rare earth ion (La3+, Gd3+, Dy3+, and Ho3+) codoped MgAl2Si2O8 host phosphors recorded after β- and α-irradiation are shown in Fig. 25.39 All the phosphors exhibit one main peak at about 261 ± 3 °C for β-irradiation and many satellite peaks in the low temperature range up to 200 °C. Furthermore, the α-irradiated phosphors had one main peak at about 245–252 °C and the same satellite peaks. The addition of La3+, Gd3+, Dy3+, and Ho3+ dopants in the MgAl2Si2O8:Mn4+ phosphor did not cause any new TL peaks, but the peak intensities changed. In addition, the Dy3+ and Gd3+ co-doped phosphors had relatively high peak intensities compared with the other phosphors. The main peaks were shifted towards the lower temperature region when the phosphors were exposed to α-irradiation.144–147 The TL curves of the β- and α-irradiated phosphors exhibited substantial changes, which can be associated with the type of radiation. Therefore, the TL peak positions of MgAl2Si2O8:Mn4+ with codoping La3+, Gd3+, Dy3+, and Ho3+ activators did not change for α- and β-irradiation.
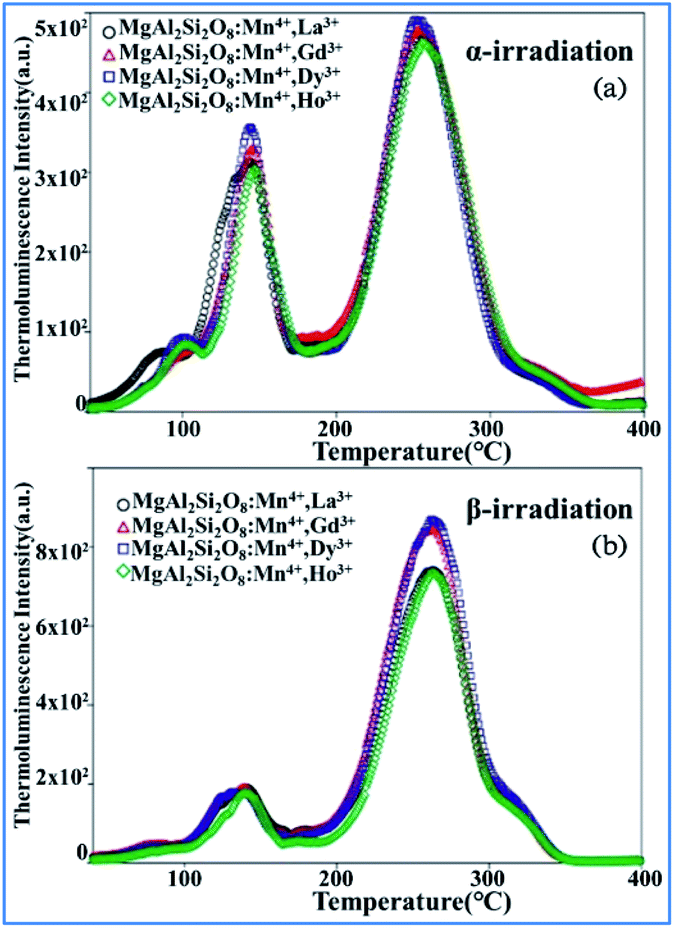 | ||
| Fig. 25 TL glow curves of all the phosphors recorded after 1 h of (a) α-irradiation and (b) 37.5 Gy β-irradiation. Reprinted with permission from ref. 39, Copyright 2018, John Wiley & Sons Inc. | ||
The upconversion (UC) luminescence of Mn4+ can be realized by energy transfer from Yb3+ to Er3+: 2H11/2/4S3/2, 4F9/2, Ho3+: 5S2/5F4, 5F5 and Tm3+: 1G4, and further to Mn4+: 2T2g and 2Eg exited states in Mn4+, Yb3+, and Er3+/Ho3+/Tm3+ codoped YAlO3.32 The different influence of temperature on the emission spectra and decay behaviors of Mn4+ and rare earth ions exhibits their possible application in optical thermometry. Fig. 26a shows down the converted PL and PLE spectra for Mn4+ single doped and Yb3+/Ln3+ (Ln = Er, Ho, Tm) codoped YAlO3. The PL spectrum of Mn4+ exhibits two emission bands centered at 694 and 714 nm, which are assigned to the spin-forbidden transition 2Eg → 4A2g of Mn4+. The PLE spectrum monitored at 714 nm consists of two strong excitation peaks centered at 340 and 484 nm, which are attributed to the spin-allowed 4A2g → 4T1g and 4A2g → 4T2g transitions of Mn4+, respectively.148–152
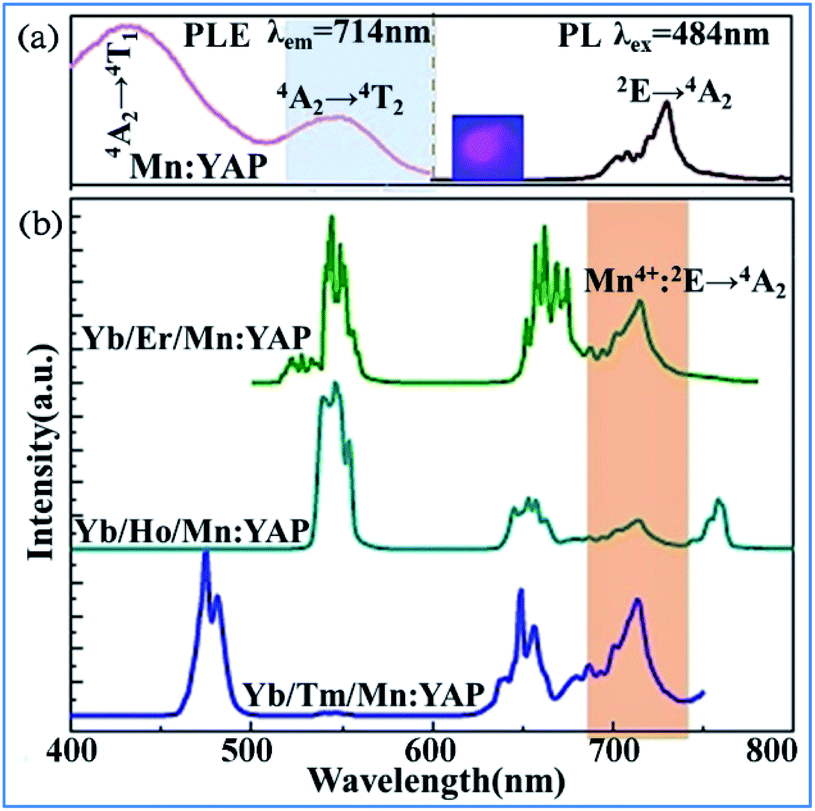 | ||
| Fig. 26 (a) PL (λex = 484 nm) and PLE (λem = 714 nm) spectra of single Mn4+-doped YAlO3 and (b) UC emission spectra of Yb3+/Ln3+/Mn4+ codoped YAlO3 (Ln = Er, Ho, Tm) under 980 nm laser excitation. Reprinted with permission from ref. 32 and 148, Copyright 2017, Elsevier BV. | ||
The obvious spectral overlap between the Ln3+ emission band and Mn4+ excitation band indicates the possible resonant energy transfer from Ln3+ to Mn4+. As evidenced in Fig. 26b, an extra NIR emission band at around 714 nm assigned to the 2Eg → 4A2g transition of Mn4+ is observed for all these Yb3+/Ln3+/Mn4+ tri-doped samples, confirming the existence of Ln3+ → Mn4+ (Ln = Er, Ho, Tm) energy transfer.
Similar behavior was observed in the Mn4+ and Tb3+ codoped Sr4Al14O25 nanocrystalline phosphor. The intense red emission associated with the 2E → 4A2 electronic transition of Mn4+ ions was drastically quenched, while the 5D4 → 7F5 emission of Tb3+ remained almost thermally independent above 100 °C. The combination of the thermally quenched luminescence from the Mn4+ ions to the almost temperature-independent emission from Tb3+ provided a sensitive luminescent thermometer (SR = 2.8%/°C at 150 °C) with strong emission color variability. Thus, the developed thermochromic luminescent nanomaterials based on codoped Mn4+ and Tb3+ possess the high application potential for thermal sensing and mapping.153
7. Challenges and perspectives
Mn4+ and multiple ion-codoped complex oxide phosphors have high stability, abundant starting materials, simple synthetic technology (solid state sintering), and tunable luminescence spectra covering the full visible light region from blue to red, and extending to the NIR region. The challenges and perspectives of the future work focusing on Mn4+ and multiple ion-codoped materials are proposed as follows:(1) Applying the developed Mn4+ and multiple ion-codoped phosphors for the fabrication of WLED devices, solar energy cells, etc.
(2) Enhancement of the luminescence efficiency of Mn4+ by optimizing the synthetic parameters including codoping some content of multiple ions.
(3) Discovery of novel host lattice materials with multiple crystals sites to accommodate various dopants and luminescence centers in a single host lattice.
(4) Improvement of the efficiency of energy transfer between Mn4+ and multiple ion-codoped phosphors to obtain tunable luminescence spectra.
8. Conclusions
This review summarized the recent research progress of Mn4+ and multiple ion such as Bi3+ and rare earth ions Dy3+/Nd3+/Yb3+/Er3+/Ho3+/Tm3+ codoped phosphors in the complex oxide host lattice, including their structural-dependent optical properties, energy transfer mechanism, and potential optical applications. Thus, these Mn4+-and multiple ion-codoped phosphors are potential candidates for application in the fields of solar energy cells, WLEDs, indoor plant cultivation, and temperature sensors. This review provides extensive insight for developing novel Mn4+-doped phosphors with desirable functional properties from an application point of view and helps to reveal the underlying energy transfer mechanism between Mn4+ and multiple ions.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We are grateful to the financial support from National Natural Science Foundation (NSF) of China (51572200) and NSF of Key Projects of NSF of Zhejiang Province (LZ20E020003), and Wenzhou Major Scientific and Technological Innovation Project (ZG2020025).References
- T. Trupke, M. A. Green and P. Würfel, J. Appl. Phys., 2002, 92, 1668–1674 CrossRef CAS.
- M. A. Green, Third generation photovoltaics: advanced solar energy conversion, Berlin, Springer, 2003 Search PubMed.
- X. Y. Huang, S. Y. Han, W. Huang and X. G. Liu, Chem. Soc. Rev., 2013, 42, 173–201 RSC.
- J. Chen, J. Liu, H. Yin, S. Jiang, H. Yao and X. Yu, J. Am. Ceram. Soc., 2016, 99, 141–145 CrossRef CAS.
- Q. Zhou, L. Dolgov, A. M. Srivastava, L. Zhou, Z. L. Wang, J. X. Shi, M. D. Dramićanin, M. G. Brik and M. M. Wu, J. Mater. Chem. C, 2018, 6, 2652–2671 RSC.
- Q. Wang, Z. Y. Yang, H. Y. Wang, Z. K. Chen, H. L. Yang, J. Yang and Z. L. Wang, Opt. Mater., 2018, 85, 96–99 CrossRef CAS.
- H. Ming, S. F. Liu, L. L. Liu, J. Q. Peng, J. X. Fu, F. Du and X. Y. Ye, ACS Appl. Mater. Interfaces, 2018, 10, 19783–19795 CrossRef CAS.
- D. Q. Chen, Y. Zhou and J. S. Zhong, RSC Adv., 2016, 6, 86285–86296 RSC.
- S. Adachi, J. Lumin., 2018, 202, 263–281 CrossRef CAS.
- H. Sijbom, R. Verstraete, J. Joos, J. Poelman and D. Philippe, Opt. Mater., 2017, 7, 3332–3362 CAS.
- Z. G. Xia and Q. L. Liu, Mater. Sci., 2016, 84, 59–117 CAS.
- P. P. Dang, D. J. Liu, G. G. Li, A. A. Al Kheraif and J. Lin, Adv. Opt. Mater., 2020, 8, 1901993 CrossRef CAS.
- Y. Lv, Y. H. Jin, T. Sun, J. L. Su, C. L. Wang, G. F. Ju, L. Chen and Y. H. Hu, Dyes Pigm., 2019, 161, 137–146 CrossRef CAS.
- X. J. Gao, W. B. Xia, T. J. Chen, X. L. Yang, X. L. Jin and S. G. Xiao, RSC Adv., 2016, 6, 7544–7552 RSC.
- X. J. Gao, W. Li, X. L. Yang, X. L. Jin and S. G. Xiao, J. Phys. Chem. C, 2015, 119, 28090–28098 CrossRef CAS.
- K. Li and R. V. Deun, J. Mater. Chem. C, 2018, 6, 7302–7310 RSC.
- W. J. Chung and Y. H. Nam, ECS J. Solid State Sci. Technol., 2020, 9, 016010 CrossRef.
- X. L. Shi, Y. L. Huang, Y. Jie, L. Shi, X. B. Qiao and H. J. Seo, J. Rare Earths, 2010, 28, 693–696 CrossRef CAS.
- J. X. Hu, T. H. Huang, Y. P. Zhang, B. Lu, H. Q. Ye, B. J. Chen, H. P. Xia and C. Y. Ji, Dalton Trans., 2019, 48, 2455–2466 RSC.
- Q. Sun, S. Y. Wang, B. Devakumar, L. L. Sun, J. Liang, X. Y. Huang and Y. C. Wu, J. Alloys Compd., 2019, 785, 1198–1205 CrossRef CAS.
- L. Jia, B. Devakumar, L. L. Sun, Q. Sun, S. Y. Wang, B. Li, D. Q. Chen and X. Y. Huang, Ceram. Int., 2019, 45, 4564–4569 CrossRef.
- Z. Z. Lu, Y. B. Meng, L. L. Wen, M. X. Huang, L. Y. Zhou, L. J. Liao and D. T. He, Dyes Pigm., 2019, 160, 395–402 CrossRef CAS.
- Z. W. Zhou, J. M. Zheng, R. Shi, N. M. Zhang, J. Y. Chen, R. Y. Zhang, H. Suo, E. M. Goldys and C. F. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 6177–6185 CrossRef CAS.
- X. Chen, M. Z. Chi, L. L. Xing, X. Xie, S. M. Liu, Y. R. Liang, M. T. Zheng, H. Hu, H. W. Dong, Y. L. Liu, S. P. Jiang and Y. Xiao, ACS Sustainable Chem. Eng., 2019, 7, 5845–5855 CrossRef CAS.
- L. Li, Y. X. Pan, Z. Chen, S. M. Huang and M. M. Wu, RSC Adv., 2017, 7, 14868–14875 RSC.
- Z. Zhou, Y. Zhong, M. Xia, N. Zhou, B. Lei, J. Wang and F. Wu, J. Mater. Chem. C, 2018, 6, 8914–8922 RSC.
- Y. Ding, N. Guo, X. Lv, H. T. Zhou, L. Wang, R. Z. Ouyang, Y. Q. Miao and B. Q. Shao, J. Am. Ceram. Soc., 2019, 102, 7436–7447 CrossRef CAS.
- D. Huang, P. Dang, Y. Wei, B. Bai, H. Lian, Q. Zeng and J. Lin, Mater. Res. Bull., 2020, 124, 110743 CrossRef CAS.
- D. Huang, P. Dang, H. Lian, Q. Zeng and J. Lin, Inorg. Chem., 2019, 58, 15507–15519 CrossRef CAS.
- Y. C. Chen, Q. Wang, Z. F. Mu, J. Q. Feng, D. Y. Zhu and F. G. Wu, Optik, 2019, 179, 1035–1041 CrossRef CAS.
- K. Trejgis and L. Marciniak, Chem. Phys. Lett., 2018, 20, 9574–9581 CAS.
- D. Q. Chen, W. Xu, S. Yuan, X. Y. Li and J. S. Zhong, J. Mater. Chem. C, 2017, 5, 9619–9628 RSC.
- J. M. Xiang, J. Y. Chen, N. M. Zhang, H. B. Yao and C. F. Guo, Dyes Pigm., 2018, 154, 257–262 CrossRef CAS.
- L. Wang, L. Yuan, Y. D. Xu, R. L. Zhou, B. Y. Qu, N. Ding, M. Shi, B. Zhang, Y. Q. Chen, Y. Jiang, D. Wang and J. Y. Shi, Appl. Phys. A, 2014, 117, 1777–1783 CrossRef CAS.
- W. Li, T. J. Chen, W. B. Xia, X. L. Yang and S. G. Xiao, J. Lumin., 2018, 194, 547–550 CrossRef CAS.
- J. Lu, Y. X. Pan, J. G. Wang, X. A. Chen, S. M. Huang and G. K. Liu, RSC Adv., 2013, 14, 4510–4513 RSC.
- Z. X. Qiu, T. T. Luo, J. L. Zhang, W. L. Zhou, L. P. Yu and S. X. Lian, J. Lumin., 2015, 158, 130–135 CrossRef CAS.
- J. H. Ou, X. L. Yang and S. G. Xiao, Mater. Res. Bull., 2020, 124, 110764 CrossRef CAS.
- E. Uzun, E. Öztürk and N. K. Ozpozan, Luminescence, 2018, 33, 1346–1357 CrossRef CAS.
- S. Adachiz, ECS J. Solid State Sci. Technol., 2020, 9, 026003 CrossRef.
- M. G. Brik, S. J. Camardello and A. M. Srivastava, ECS J. Solid State Sci. Technol., 2015, 4, R39–R43 CrossRef CAS.
- Y. X. Pan and G. K. Liu, Enhancement of phosphor efficiency via composition modification, Opt. Lett., 2008, 33, 1–3 CrossRef.
- A. M. Srivastava and W. W. Beers, J. Electrochem. Soc., 1996, 143, L203–L205 CrossRef CAS.
- Z. Zhou, M. Xia, Y. Zhong, S. J Gai, S. X. Huang, Y. Tian, X. Y. Lu and N. Zhou, J. Mater. Chem. C, 2017, 5, 8201–8210 RSC.
- I. D. Brown, The Chemical Bond in Inorganic Chemistry: The Bond Valence Model, IUCr Monographs on Crystallography, Oxford University Press, 2002 Search PubMed.
- B. Jiang, F. F. Chi, L. Zhao, X. T. Wei, Y. H. Chen and M. Yin, J. Lumin., 2019, 206, 234–239 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- X. D. Zhou, E. L. Wang, X. D. Lao, Y. M. Wang and X. K. Sun, Opt. Mater., 2020, 107, 110044 CrossRef CAS.
- Y. B. Wu, Y. Lv, K. B. Ruan and Z. Xie, Dalton Trans., 2018, 47, 15574–15582 RSC.
- S. J. Qiu, H. W. Wei, X. M. Wang, S. Zhang, M. M. Wang, Y. Y. Wang, L. Xu and H. Jiao, J. Lumin., 2020, 226, 117426 CrossRef CAS.
- C. Yang, Z. F. Zhang, G. C. Hu, R. Cao, X. J. Liang and W. D. Xiang, J. Alloys Compd., 2017, 694, 1201–1208 CrossRef CAS.
- F. Vetrone, J. C. Boyer, J. A. Capobianco, A. Speghini and M. Bettinelli, Appl. Phys. Lett., 2002, 80, 1752–1754 CrossRef CAS.
- M. R. N. Soares, T. Holz, F. Oliveira, F. M. Costaa and T. Monteiro, RSC Adv., 2015, 5, 20138–20147 RSC.
- T. Hasegawa, S. W. Kim, T. Abe, S. Kumagai, R. Yamanashi, K. Seki, K. Uematsu, K. Toda and M. Sato, Chem. Lett., 2016, 45, 1096–1098 CrossRef CAS.
- L. Li, Y. X. Pan, Y. Huang, S. M. Huang and M. M. Wu, J. Alloys Compd., 2017, 724, 735–743 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef.
- J. J. Zhou, Y. C. Teng, X. F. Liu, S. Ye, X. Q. Xu, Z. J. Ma and J. R. Qiu, Opt. Express, 2010, 18, 21663–21668 CrossRef CAS.
- V. Singh, R. P. S. Chakradhar, I. Ledoux-Rak, L. Badie, F. Pelle and S. Ivanova, J. Lumin., 2009, 129, 1375–1380 CrossRef CAS.
- J. Lv, Y. Huang, Y. Tao and H. J. Seo, J. Alloys Compd., 2010, 500, 134–137 CrossRef.
- K. Li and R. V. Deun, Dyes Pigm., 2019, 162, 990–997 CrossRef CAS.
- Z. F. Liao, H. F. Xu, W. R. Zhao, H. X. Yang, J. Y. Zhong, H. Zhang, Z. G. Nie and Z. K. Zhou, Chem. Eng. J., 2020, 395, 125060 CrossRef CAS.
- H. H. Liu, L. Yuan, X. F. Wu, X. Y. Hou, M. Tang, C. M. Hou, H. W. Chen and S. H. Feng, J. Mater. Chem. C, 2020, 8, 9615–9624 RSC.
- F. Q. Sun, R. R. Xie, L. Guan and C. Y. Zhang, J. Lumin., 2016, 180, 251–257 CrossRef CAS.
- T. Miyakawa and D. L. Dexter, Phys. Rev. B: Solid State, 1970, 1, 2961–2969 CrossRef.
- K. Seki, K. Uematsu, K. Toda and M. Sato, Chem. Lett., 2014, 43, 1213–1215 CrossRef CAS.
- W. Lv, W. Z. Lv, Q. Zhao, M. M. Jiao, B. Q. Shao and H. P. You, Inorg. Chem., 2014, 53, 11985–11990 CrossRef.
- Q. Sun, S. Y. Wang, B. Devakumar, L. L. Sun, J. Liang and X. Y. Huang, ACS Omega, 2019, 8, 13474–13480 CrossRef.
- K. Hayashi, H. Noguchi and M. Ishii, Mater. Res. Bull., 1986, 21, 401–406 CrossRef CAS.
- K. Hayashi, H. Noguchi and S. Fujiwara, Mater. Res. Bull., 1986, 21, 289–293 CrossRef CAS.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer, Berlin, 1994 Search PubMed.
- L. I. Kazakova, A. B. Dubovsky, G. V. Semenkovich and O. A. Ivanova, Radiat. Meas., 1995, 24, 359–360 CrossRef CAS.
- M. Nyman, M. A. Rodriguez, L. E. Shea-Rohwer, J. E. Martin and P. P. Provencio, J. Am. Chem. Soc., 2009, 131, 11652–11653 CrossRef CAS.
- K. Li, H. Z. Lian, R. V. Deun and M. G. Brik, Dyes Pigm., 2019, 162, 214–221 CrossRef CAS.
- J. S. Liao, Q. Wang, H. R. Wen, H. L. Yuan, S. J. Liu, J. X. Fu and B. Qiu, J. Mater. Chem. C, 2017, 5, 9098–9105 RSC.
- H. Chen, H. Lin, Q. M. Huang, F. Huang, J. Xu, B. Wang, Z. B. Lin, J. C. Zhou and Y. S. Wang, J. Mater. Chem. C, 2016, 4, 2374–2381 RSC.
- R. P. Cao, X. O. Yang, Y. M. Jiao, X. T. Wang, Q. L. Hu, T. Chen, C. X. Liao and Y. J. Li, J. Lumin., 2019, 209, 1–7 CrossRef.
- S. W. Le, J. M. Seo, M. K. Lee, J. H. Chun, P. Antonisamy, M. V. Arasu, T. Suzuki, N. A. Al-Dhabi and S. J. Kim, Ind. Crops Prod., 2014, 54, 320–326 CrossRef.
- G. G. Li, D. L. Geng, M. M. Shang, C. Peng, Z. Y. Cheng and J. Lin, J. Mater. Chem., 2011, 21, 13334–13344 RSC.
- X. Yin, J. Y. Yao, Y. M. Wang, C. C. Zhao and F. Q. Huang, J. Lumin., 2012, 132, 1701–1704 CrossRef CAS.
- M. L. Hu, C. X. Liao, L. B. Xia, W. X. You and Z. F. Li, J. Lumin., 2019, 211, 114–120 CrossRef CAS.
- L. Qin, D. L. Wei, S. L. Bi, C. L. Chen, J. Wang, H. H. Yin and H. J. Seo, Opt. Mater., 2019, 98, 109496–109503 CrossRef CAS.
- R. Watanabe, Y. Iso and T. Isobe, Ceram. Int., 2019, 2, 4009–4017 CAS.
- U. Rambabu and S. D. Han, Ceram. Int., 2013, 39, 1603–1612 CrossRef CAS.
- X. Y. Huang and H. Guo, Dyes Pigm., 2018, 152, 36–42 CrossRef CAS.
- L. Shi, J. X. Li, Y. J. Han, W. L. Li and Z. W. Zhang, J. Lumin., 2019, 208, 201–207 CrossRef CAS.
- A. J. Fu, A. X. Guan, D. Y. Yu, S. Y. Xia, F. F. Gao, X. S. Zhang, L. Y. Zhou, Y. H. Li and R. G. Li, Mater. Res. Bull., 2017, 88, 258–265 CrossRef CAS.
- J. Q. Hu, E. H. Song, S. Ye, B. Zhou and Q. Y. Zhang, J. Mater. Chem. C, 2017, 5, 3343–3351 RSC.
- A. B. Hassen, F. I. H. Rhouma, M. Daoudi, J. Dhahri, M. Zaidi and N. Abdelmoula, RSC Adv., 2019, 9, 19285–19296 RSC.
- H. H. Lin, T. Yu, G. X. Bai, M. K. Tsang, Q. Y. Zhang and J. H. Hao, J. Mater. Chem. C, 2016, 4, 3396–3402 RSC.
- M. Rathaiah, P. Haritha, A. D. Lozano-Gorrin, P. Babu, C. K. Jayasankar, U. R. Rodriguez-Mendoza, V. Lavin and V. Venkatramu, Chem. Phys., 2016, 18, 14720–14729 CAS.
- W. Lv, M. M. Jiao, B. Q. Shao, L. F. Zhao, Y. Feng and H. P. You, Dalton Trans., 2016, 45, 466–468 RSC.
- S. Ye, J. J. Zhou, S. T. Wang, R. X. Hu, D. P. Wang and J. R. Qiu, Opt. Express, 2013, 21, 4167–4173 CrossRef CAS.
- Y. J. Han, S. Wang, H. Liu, L. Shi, J. Y. Zhang, Z. N. Zhang, Z. Y. Mao, D. J. Wang, Z. F. Mu, Z. W. Zhang and Y. Zhao, J. Lumin., 2020, 220, 116968 CrossRef CAS.
- A. S. Laia, D. A. Hora, M. V. dos S. Rezende, Y. T. Xing, J. J. Rodrigues Jr, G. S. Maciel and M. A. R. C. Alencar, Chem. Eng. J., 2020, 399, 125742 CrossRef CAS.
- H. Nurhafizah and M. S. Rohani, Solid State Phenom., 2019, 290, 16–21 Search PubMed.
- C. Cao, M. Xue, X. J. Zhu, P. Y. Yang, W. Feng and F. Y. Li, ACS Appl. Mater. Interfaces, 2017, 9, 18540–18548 CrossRef CAS.
- J. H. Chen, W. R. Zhao, N. H. Wang, Y. J. Meng, S. P. Yi, J. He and X. Zhang, J. Mater. Sci., 2016, 51, 4201–4212 CrossRef CAS.
- P. You, G. Yin, X. Chen, B. Yue, Z. Huang, X. Liao and Y. Yao, Opt. Mater., 2011, 33, 1808–1812 CrossRef CAS.
- Q. Liu, Y. Liu, Z. Yang, Y. Han, X. Li and G. Fu, J. Alloys Compd., 2012, 515, 16–19 CrossRef CAS.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- G. Q. Zhang, M. S. Molokeev, Q. C. Ma, X. N. Yang, S. Q. Han, Q. Chen, B. N. Zhong and B. Ma, CrystEngComm, 2020, 22, 5809–5817 RSC.
- Y. R. Shi, Y. H. Wang and Z. G. Yang, J. Alloys Compd., 2011, 509, 3128–3131 CrossRef CAS.
- H. Zhong, X. Li, R. Shen, J. Zhang, J. Sun, H. Zhong, L. Cheng, Y. Tian and B. Chen, J. Alloys Compd., 2012, 517, 170–175 CrossRef CAS.
- Q. Du, G. Zhou, J. Zhou, X. Jia and H. Zhou, J. Alloys Compd., 2013, 552, 152–156 CrossRef CAS.
- Z. Yang, C. Hou, G. Duan, F. Yang, P. Liu, C. Wang, L. Liu and G. Dong, J. Alloys Compd., 2014, 604, 346–351 CrossRef CAS.
- D. L. Monika, H. Nagabhushana, R. H. Krishna, B. M. Nagabhushana, S. C. Sharma and T. J. Thomas, RSC Adv., 2014, 4, 38655–38662 RSC.
- X. Liu, W. Xiang, F. Chen, Z. Hu and W. Zhang, Mater. Res. Bull., 2013, 48, 281–285 CrossRef CAS.
- G. Q. Wang, X. H. Gong, Y. J. Chen, J. H. Huang, Y. F. Lin, Z. D. Luo and Y. D. Huang, Opt. Mater., 2014, 36, 1255–1259 CrossRef CAS.
- P. C. Ma, Y. H. Song, B. Yuan, Y. Sheng, C. Y. Xu, H. F. Zou and K. Y. Zheng, Ceram. Int., 2017, 43, 60–70 CrossRef CAS.
- P. C. Chen, F. W. Mo, A. X. Guan, R. F. Wang, G. F. Wang, S. Y. Xia and L. Y. Zhou, Appl. Radiat. Isot., 2016, 108, 148–153 CrossRef CAS.
- R. P. Cao, H. D. Xu, W. J. Luo, Z. Y. Luo, S. L. Guo, F. Xia and H. Ao, Mater. Res. Bull., 2016, 81, 27–32 CrossRef CAS.
- G. C. Xing, Y. X. Feng, M. Pan, Y. Wei, G. G. Li, P. P. Dang, S. S. Liang, M. S. Molokeev, Z. Y. Cheng and J. Lin, J. Mater. Chem. C, 2018, 6, 13136–13147 RSC.
- T. Orihashi, T. Nakamura and S. Adach, RSC Adv., 2016, 6, 66130–66139 RSC.
- Z. B. Xia, Q. Xue, K. F. Zhang, H. P. Zhang and T. He, J. Mater. Sci.: Mater. Electron., 2015, 26, 8078–8082 CrossRef CAS.
- R. F. Wang, J. Liu and Z. Zhang, J. Alloys Compd., 2016, 688, 332–336 CrossRef CAS.
- L. Kong, Y. Y. Liu, L. P. Dong, L. Zhang, L. Qiao, W. S. Wang and H. P. You, Dalton Trans., 2020, 49, 1947–1954 RSC.
- Y. J. Zhu, Z. X. Qiu, B. Y. Ai, Y. T. Lin, W. L. Zhou, J. L. Zhang, L. P. Yu, Q. H. Mi and S. X. Lian, J. Lumin., 2018, 201, 314–320 CrossRef CAS.
- B. Wang, H. Lin, J. Xu, H. Chen and Y. S. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 22905–22913 CrossRef CAS.
- R. Dittmann, D. Hahn and J. Stade, Luminescence of Crystals, Molecules, and Solutions, 1973, 518–523 CAS.
- Z. G. Xia, R. S. Liu, K. W. Huang and V. Drozd, J. Mater. Chem., 2012, 22, 15183–15189 RSC.
- J. Ueda, K. Kuroishi and S. Tanabe, Appl. Phys. Express, 2014, 7, 062201–062203 CrossRef.
- M. Zhao, Z. Xia, M. S. Molokeev, L. Ning and Q. Liu, Chem. Mater., 2017, 29, 6552–6559 CrossRef CAS.
- S. Z. Liao, X. Y. Ji, Y. F. Liu and J. L. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 39064–39073 CrossRef CAS.
- W. L. Wu, M. H. Fang, W. Zhou, T. Lesniewski, S. Mahlik, M. Grinberg, M. G. Brik, H. S. Sheu, B. M. Cheng, J. Wang and R. S. Liu, Chem. Mater., 2017, 29, 935–939 CrossRef CAS.
- S. Adachi, J. Lumin., 2018, 202, 263–281 CrossRef CAS.
- X. T. Rao, T. Song, J. K. Gao, Y. J. Cui, Y. Yang, C. D. Wu, B. L. Chen and G. D. Qian, J. Am. Chem. Soc., 2013, 135, 15559–15564 CrossRef CAS.
- D. Wawrzynczyk, A. Bednarkiewicz, M. Nyk, W. Strek and M. Samoc, Nanoscale, 2012, 4, 6959–6961 RSC.
- M. D. Dramićanin, Z. Antić, S. Ćulubrk, S. P. Ahrenkiel and J. M. Nedeljković, Nanotechnology, 2014, 25, 485501–485506 CrossRef.
- D. Q. Chen, S. Liu, Y. Zhou, Z. Y. Wan, P. Huang and Z. G. Ji, J. Mater. Chem. C, 2016, 4, 9044–9051 RSC.
- D. Q. Chen, W. Xu, Y. Zhou and Y. Chen, J. Alloys Compd., 2016, 676, 215–223 CrossRef CAS.
- A. F. Pereira, K. U. Kumar, W. F. Silva, W. Q. Santos, D. Jaque and C. Jacinto, Sens. Actuators, B, 2015, 213, 65–71 CrossRef CAS.
- S. Cúlubrk, V. Lojpur, S. P. Ahrenkiel, J. M. Nedeljkovic and M. D. Dramićanin, J. Lumin., 2016, 170, 395–400 CrossRef.
- E. C. Ximendes, U. Rocha, K. U. Kumar, C. Jacinto and D. Jaque, Appl. Phys. Lett., 2016, 108, 253103–253107 CrossRef.
- E. C. Ximendes, W. Q. Santos, U. Rocha, U. K. Kagola, F. SanzRodríguez, N. Fernández, A. daSilva Gouveia-Neto, D. Bravo, A. M. Domingo, B. del Rosal, G. D. S. Brites, L. D. Carlos, D. Jaque and C. Jacinto, Nano Lett., 2016, 16, 1695–1703 CrossRef CAS.
- D. Q. Chen, Y. Zhou, W. Xu, J. S. Zhong, Z. G. Ji and W. D. Xiang, J. Mater. Chem. C, 2016, 4, 1704–1712 RSC.
- K. S. Kumar, C. G. Lou, A. G. Manohari, H. H. Cao and D. Pribat, RSC Adv., 2017, 7, 24674–24678 RSC.
- Pushpendra, R. K. Kunchala, R. Kalia and B. S. Naidu, Ceram. Interfaces, 2020, 46, 18614–18622 CrossRef CAS.
- Y. P. Tai, X. Z. Li, X. G. Du, B. L. Pan and G. H. Yuan, RSC Adv., 2018, 8, 23268–23273 RSC.
- M. G. Nikolić, Z. Antić, S. Cúlubrk, J. M. Nedeljkovic and M. D. Dramićanin, Sens. Actuators, B, 2014, 201, 46–50 CrossRef.
- H. Nakamura, K. Shinozaki, T. Okumura, K. Nomura and T. Akai, RSC Adv., 2020, 10, 12535–12546 RSC.
- Pushpendra, R. K. Kunchala, R. Kalia and B. S. Naidu, RSC Adv., 2020, 10, 14525–14530 RSC.
- T. Miyakawa and D. L. Dexter, Phys. Rev. B: Solid State, 1970, 1, 2961–2969 CrossRef.
- S. Kück, S. Hartung, S. Hurling, K. Petermann and G. Huber, Spectrochim. Acta, Part A, 1998, 54, 1741–1749 CrossRef.
- E. Öztürk and N. O. Kalaycioglu, Solid State Phenom., 2015, 230, 217–220 Search PubMed.
- N. O. Kalaycioglu and E. Irir, J. Alloys Compd., 2011, 510, 6–10 CrossRef.
- E. Uzun, E. Öztürk, N. K. Ozpozan and E. Karacaoglu, J. Lumin., 2016, 173, 73–81 CrossRef CAS.
- E. Öztürk and N. O. Kalaycioglu, J. Therm. Anal. Calorim., 2014, 115, 573–577 CrossRef.
- M. Buryi, V. Laguta, M. Nikl, V. Gorbenko, T. Zorenko and Y. Zorenko, CrystEngComm, 2019, 21, 3313–3321 RSC.
- Pushpendra, R. K. Kunchala, S. N. Achary, A. K. Tyagi and B. S. Naidu, Cryst. Growth Des., 2019, 19, 3379–3388 CrossRef CAS.
- Y. Zhang, W. T. Gong, J. J. Yu, Z. Y. Cheng and G. L. Ning, RSC Adv., 2016, 6, 30886–30894 RSC.
- Pushpendra, R. K. Kunchala, S. N. Achary and B. S. Naidu, ACS Appl. Nano Mater., 2019, 2, 5527–5537 CrossRef CAS.
- W. Li, X. J. Gao, X. L. Yang, X. L. Jin and S. G. Xiao, J. Alloys Compd., 2016, 664, 181–187 CrossRef CAS.
- W. Piotrowski, K. Trejgis, K. Maciejewska, K. Ledwa, B. Fond and L. Marciniak, ACS Appl. Mater. Interfaces, 2020, 12, 44039–44048 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |