
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerfluoroalkylated alternating copolymer possessing solubility in fluorous liquids and imaging capabilities under high energy radiation†
Hyun-Taek Oha,
Seok-Heon Junga,
Kang-Hyun Kimb,
Yina Moon‡
c,
Do Hyeon Jeongc,
Yejin Kua,
Sangsul Lee d,
Byeong-Gyu Parkd,
Jiyoul Leec,
Chawon Kohe,
Tsunehiro Nishie,
Hyun-Woo Kime and
Jin-Kyun Lee*a
d,
Byeong-Gyu Parkd,
Jiyoul Leec,
Chawon Kohe,
Tsunehiro Nishie,
Hyun-Woo Kime and
Jin-Kyun Lee*a
aDepartment of Polymer Science and Engineering, Inha University, Incheon 22212, Republic of Korea. E-mail: jkl36@inha.ac.kr
bDepartment of Mechanical Engineering, POSTECH, Pohang 37673, Republic of Korea
cDepartment of Graphic Arts Information Engineering, Pukyong National University, Busan 48513, Republic of Korea
dPohang Accelerator Laboratory, POSTECH, Pohang 37673, Republic of Korea
eSamsung Electronics Co., Ltd., Semiconductor R&D Center, Gyeonggi-do 18448, Republic of Korea
First published on 5th January 2021
Abstract
A highly fluorinated alternating polymer, P(RFMi-St), possessing improved thermal properties and patterning capabilities over perfluoroalkyl polymethacrylates under high energy radiation was achieved with semi-perfluorododecyl maleimide (RFMi) and styrene (St). RFMi could be synthesised efficiently via a Mitsunobu reaction condition and copolymerised with St by free radical and reversible-deactivation radical polymerisation protocols. P(RFMi-St) showed a satisfactory glass-transition temperature (108 °C) and intermolecular cross-linking behaviour under electron-beam and commercially more important extreme UV (λ = 13.5 nm) irradiation. The exposed regions lost their solubility, resulting in the successful formation of mechanically non-deteriorated negative-tone images down to 50 nm. In addition, P(RFMi-St) could be solution-processed with chemically non-damaging fluorous liquids, which enabled the polymer to be applied effectively on top of an organic semiconductor layer as a dielectric material (dielectric constant 2.7) for the organic field-effect transistor fabrication.
Introduction
Highly fluorinated polymers have a large number of strong C–F bonds with low-lying energy levels that provide excellent stability against harsh oxidizing conditions.1–4 On the other hand, the corresponding depressed anti-bonding orbitals render them vulnerable under reducing environments; electrons injected into the fluorinated molecular framework result in the generation of radical anion species, which are subsequently decomposed to reactive radicals.5,6 Recently, the present authors have investigated the chemical behaviour of thin films consisting of perfluoroalkylated polymethacrylates when subjected to an accelerated electron-beam (e-beam). A decrease in solubility was observed in the irradiated regions, thus enabling polymeric nanostructures to be built up after washing in a fluorous solvent. This phenomenon was understood in terms of crosslinking reactions among the radical species generated on the perfluoroalkyl moieties.7Driven by these preliminary results and the benefits of polymers possessing chemical orthogonality,8–10 the authors began a larger study to develop the chemistry into meaningful applications. The starting point was to identify the limits of the current perfluoroalkylated polymethacrylate substrates, and to improve their thermal and mechanical properties. Initial characterizations indicated that the polymers had low glass-transition temperatures (Tg) and a melting behaviour.11,12 This semi-crystalline nature is not desirable for the construction of uniform and robust microstructures because the presence of grain boundaries of the crystalline domains and resulting anisotropic properties cause problems, such as non-uniform intermolecular cross-linking and dissolution behaviour of the exposed areas.13 Moreover, the mechanical strength of the polymer films was found to weaken under e-beam irradiation. This criticism motivated the search for cross-linkable polymers with solubility in fluorous solvents, enhanced Tg, and non-deteriorated mechanical strength under high energy radiation. The results are reported in detail in the present paper.
Results and discussion
In general, highly fluorinated polymers are classified into two categories, namely (i) the fluoropolymers such as polytetrafluoroethylene (PTFE), which are based on fluorinated backbones, and (ii) the vinyl polymers with perfluoroalkyl side chains. A variety of studies are recently focused on the latter category because structurally refined polymeric materials are conveniently accessed by, for example, reversible-deactivation radical polymerization protocols in an ordinary laboratory environment.14–17 The present authors were attracted by these benefits and selected the alternating vinyl copolymers as a promising polymer platform.It is well known that monomer combinations with a large difference in electron density in their vinyl functional groups, such as N-alkyl maleimide and styrene, form copolymer structures with an alternating monomer sequence.19 In addition, N-alkyl maleimide-styrene copolymers are known to exhibit high Tg and tunable solubility due to their rigid backbones and alkyl substituents, respectively.20 These intriguing properties motivated the authors to explore a copolymer composed of N-perfluoroalkyl maleimide and styrene, P(RFMi-St), as shown in Fig. 1. To access the target polymer, synthesis of a comonomer, semi-perfluorododecyl maleimide (RFMi, 3), was attempted by reacting the fluorinated dodecyl amine 2 with maleic anhydride (Fig. 1a). The resulting amic acid intermediate was subjected to imidisation at 110 °C in the presence of Ac2O, but RFMi (3) was recovered in only ca. 10% yield. Various conditions, including base catalysis, were employed, but no significant improvement was made. Another pathway based on the Mitsunobu reaction was examined with maleimide and semi-perfluorododecanol 5. This proved successful in providing RFMi (3) in 70% yield.21 Interestingly, however, the same conditions did not work equally well for a commercially-available decanol 6 with a shorter non-fluorinated alkyl unit.
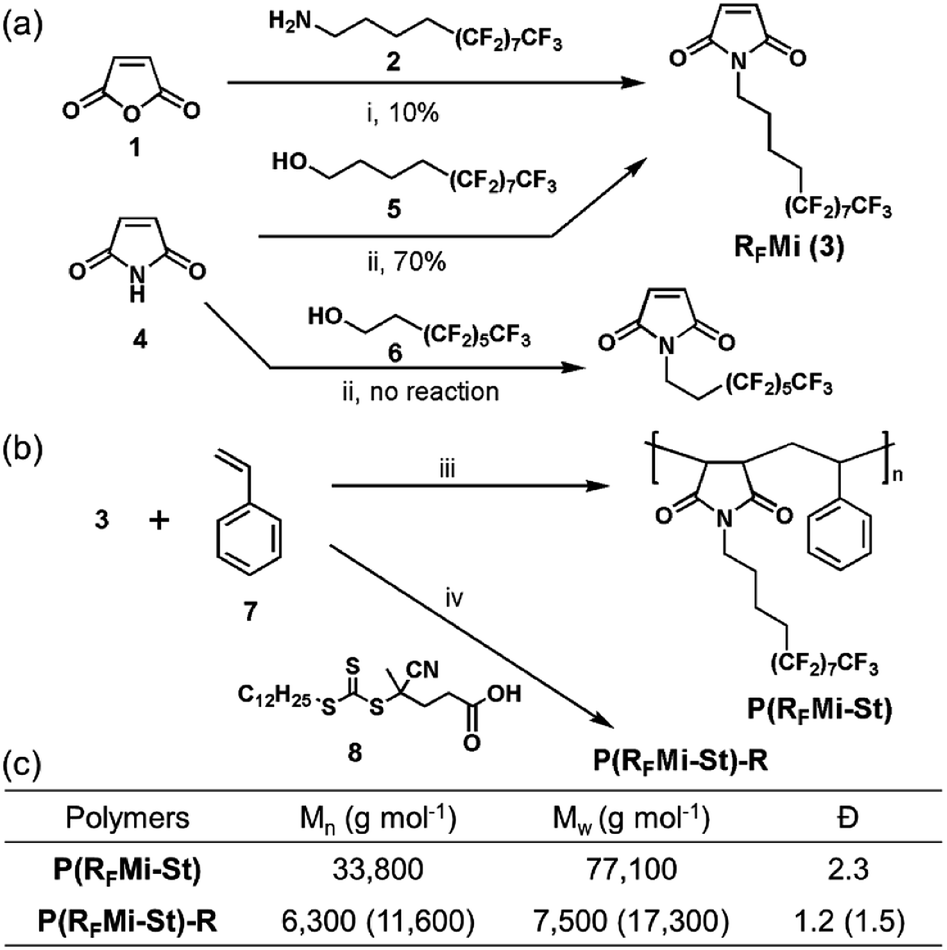 | ||
| Fig. 1 (a) Synthesis of RFMi (3). (b) Free radical copolymerisation of RFMi and styrene in the absence and presence of chain-transfer agent 8 to achieve P(RFMi-St) or P(RFMi-St)-R, respectively. (c) Molecular weights of the copolymers measured by size-exclusion chromatography with monodisperse PMMA as reference standard.18 Reagents and conditions: (i) CH2Cl2, acetone, 25 °C, 1 h, followed by Ac2O, 110 °C, 12 h; (ii) 5, PPh3, diisopropyl azodicarboxylate (DIAD), THF, −78 °C to rt, 12 h; (iii) 2,2-azobisisobutyronitrile (AIBN), benzotrifluoride, 80 °C, 12 h; (iv) 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoic acid (CDSTSP, 8), benzotrifluoride, 80 °C, 12 h. | ||
Free radical copolymerization with equimolar RFMi and styrene was then conducted in benzotrifluoride (Fig. 1b). The polymer recovered by precipitation into hexane was found to have Mn = 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 and a polydispersity index (PDI) of 2.3 (Fig. S3, ESI†). The molar monomer composition was found to be 1:1 by 1H NMR spectrometry (Fig. S4, ESI†), and was anticipated to involve alternating copolymers. Differential scanning calorimetry (DSC) indicated a high Tg of 108 °C for the P(RFMi-St) (Fig. 2a), and thermogravimetric analysis (TGA) indicated 5% weight loss at a temperature of ca. 350 °C (Fig. 2a). As previous studies have demonstrated that the Tg increases with increasing chain stiffness,22 this explains how the P(RFMi-St) can have a Tg comparable to that of the well-known commercially available fluoropolymer, Cytop™ (ca. 105 °C).23 Further, the P(RFMi-St) was found to dissolve in 1,1,1,2,3,3-hexafluoro-4-(1,1,2,3,3,3-hexafluoropropoxy)pentane (PF-7600) at a concentration above 40 wt%, thus providing suitable solutions for thin film casting (Fig. S5a and b, ESI†). The dissolution rate of the cast 300 nm-thick film in PF-7600 was measured using a quartz crystal microbalance (QCM) to indicate fast and complete dissolution in only a few seconds (Fig. S6, ESI†). In addition, the powder X-ray diffraction (XRD) pattern in Fig. S7† exhibits two broad halo patterns, rather than narrow diffraction peaks, thus indicating an amorphous solid structure for the P(RFMi-St).
800 and a polydispersity index (PDI) of 2.3 (Fig. S3, ESI†). The molar monomer composition was found to be 1:1 by 1H NMR spectrometry (Fig. S4, ESI†), and was anticipated to involve alternating copolymers. Differential scanning calorimetry (DSC) indicated a high Tg of 108 °C for the P(RFMi-St) (Fig. 2a), and thermogravimetric analysis (TGA) indicated 5% weight loss at a temperature of ca. 350 °C (Fig. 2a). As previous studies have demonstrated that the Tg increases with increasing chain stiffness,22 this explains how the P(RFMi-St) can have a Tg comparable to that of the well-known commercially available fluoropolymer, Cytop™ (ca. 105 °C).23 Further, the P(RFMi-St) was found to dissolve in 1,1,1,2,3,3-hexafluoro-4-(1,1,2,3,3,3-hexafluoropropoxy)pentane (PF-7600) at a concentration above 40 wt%, thus providing suitable solutions for thin film casting (Fig. S5a and b, ESI†). The dissolution rate of the cast 300 nm-thick film in PF-7600 was measured using a quartz crystal microbalance (QCM) to indicate fast and complete dissolution in only a few seconds (Fig. S6, ESI†). In addition, the powder X-ray diffraction (XRD) pattern in Fig. S7† exhibits two broad halo patterns, rather than narrow diffraction peaks, thus indicating an amorphous solid structure for the P(RFMi-St).
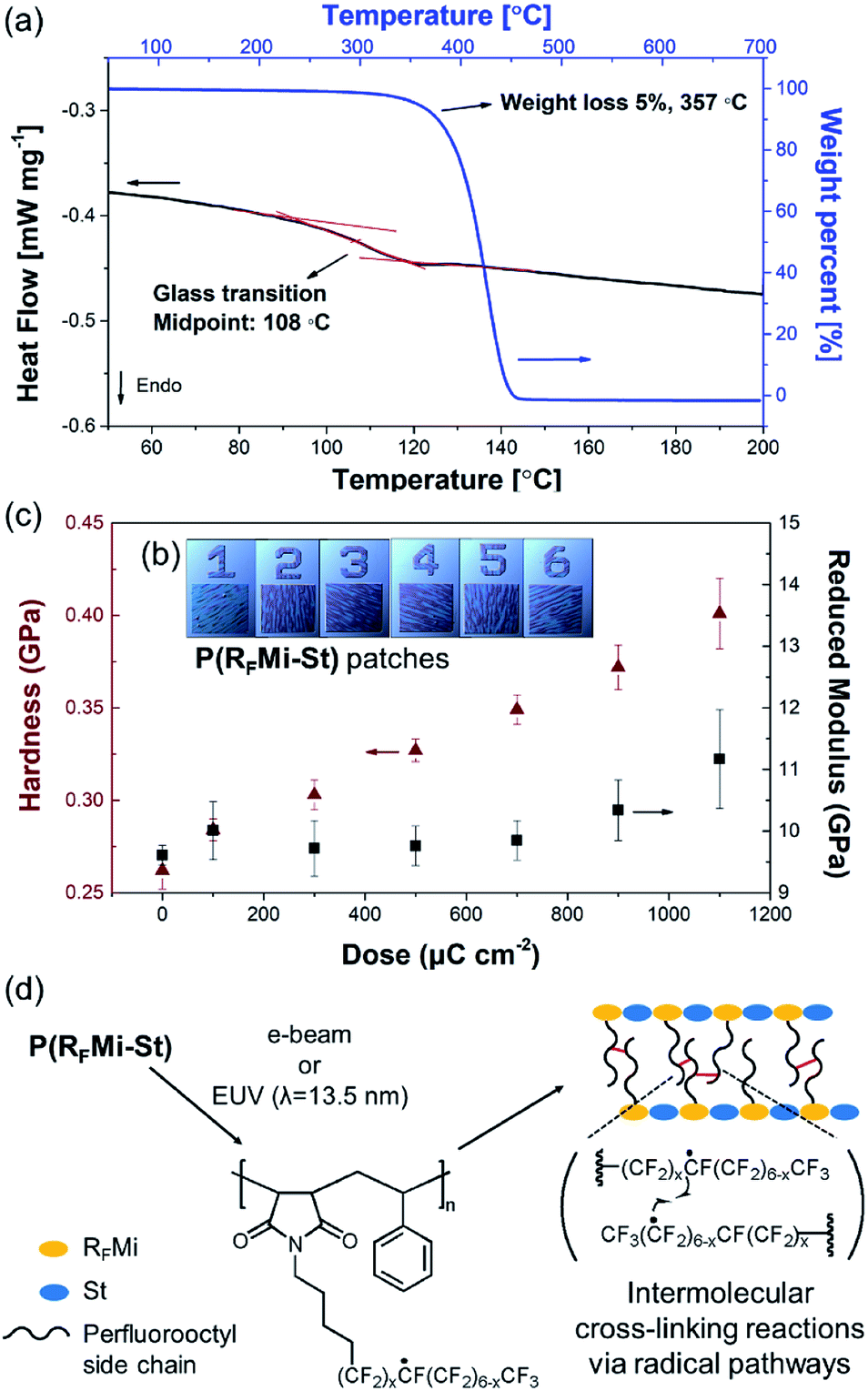 | ||
| Fig. 2 (a) DSC and TGA curves of the P(RFMi-St). (b) Photographic images of the 1 × 1 mm2 patches of P(RFMi-St) on an Si substate which were used for measuring the mechanical properties via nano-indentation. The e-beam exposure dose was increased from 100 μC cm−2 for patch 1 to 1100 μC cm−2 for patch 6 in increments of 200 μC cm−2. (c) Hardness and reduced Young's moduli of the patterned P(RFMi-St) patches shown in (b). (d) A proposed cross-linking reaction mechanism for P(RFMi-St) under a high energy radiation, including e-beam and extreme UV (λ = 13.5 nm). | ||
The chemical behaviour of the P(RFMi-St) under high energy radiation was then investigated. An 800 nm-think film was spin-cast on a Si wafer from its solution in PF-7600, and exposed to an e-beam with an energy of 500 μC cm−2. The irradiated regions of the film did not wash off when soaked in the casting solvent due to the decrease in solubility, thus producing a negative-tone patterned structure. This feature is also compared to the case of Cytop™ polymer. According to the paper reported by Park et al., solubility of Cytop™ films after e-beam irradiation increases to make positive-tone images. This behaviour is understood with polymer main-chain scission and resulting molecular weight reduction of Cytop™.24
A comparison of the FT-IR spectra of the P(RFMi-St) film recorded before and after e-beam exposure revealed no noticeable differences (Fig. S8†). The intensities of the stretching peaks corresponding to the perfluoroalkyl moieties did not decrease along the e-beam exposure. This suggests that β-scission reactions of those units, which are typically related to the free radicals generated in the molecular chains, did not occur significantly, resulting in little changes in the molecular structure.7 The mechanical properties of the e-beam exposed regions were also characterised. As shown in Fig. 2b, a range of patterned P(RFMi-St) specimens were prepared by varying the energy of the e-beam between 100 μC cm−2 and 1100 μC cm−2. When each patch was tested by nanoindentation, the hardness was found to increase steadily with increasing e-beam energy (Fig. 2c). This was attributed to the cross-linking reactions among the perfluorooctyl side chains (Fig. 2d).25 In addition, the Young's modulus of the remaining patches remained unchanged up to an exposure energy of ca. 900 μC cm−2 and increased thereafter (Fig. 2c). Thus, in contrast to the poly(perfluorodecyl methacrylate),7,26,27 the mechanical properties of the P(RFMi-St) did not deteriorate under e-beam irradiation, thereby demonstrating that one goal of the present study could be achieved through alternating copolymerization of N-perfluoroalkyl maleimide and styrene.
Improved thermal and mechanical properties, solubility in fluorous solvents, and intermolecular cross-linking capabilities under high-energy radiation are all valuable features of the P(RFMi-St), enabling its adoption for advanced applications. Hence, its potential applications as an imaging material for microfabrication and as a dielectric in organic electronic devices are examined herein. The process of e-beam or extreme UV (EUV) irradiation followed by solvent washing is shown schematically in Fig. 3a. As demonstrated by the micrograph in Fig. 3b, 100 nm-sized structures were cleanly achieved by e-beam irradiation at an energy of 600 μC cm−2. The aspect ratio of 1.5 observed in these structures may be attributed to the high Tg and improved mechanical strength of the P(RFMi-St). The construction of smaller structures (≤70 nm) was then attempted but proved unsuccessful (Fig. S9†). When the irradiated film was washed in PF-7600, it could be seen in the SEM micrograph that the material in between the line patterns was not completely washed off. There were many bridges connecting the lines which could be due to the insufficient dissolution of polymer chains. This result could be attributed to the large molecular weight and molecular weight distribution of P(RFMi-St) (Fig. 1c) which did not allow quick and uniform dissolution behaviour of polymer chains in such small and densely packed structures shown in Fig. S9.† Hence, to address this problem, P(RFMi-St)-R with Mn = 6300 and Ð = 1.2 was prepared via reversible addition–fragmentation chain-transfer (RAFT) polymerization (Fig. 1b). Under the same e-beam lithography conditions, structures as small as 70 nm were successfully formed in the P(RFMi-St)-R thin film (Fig. 3c).
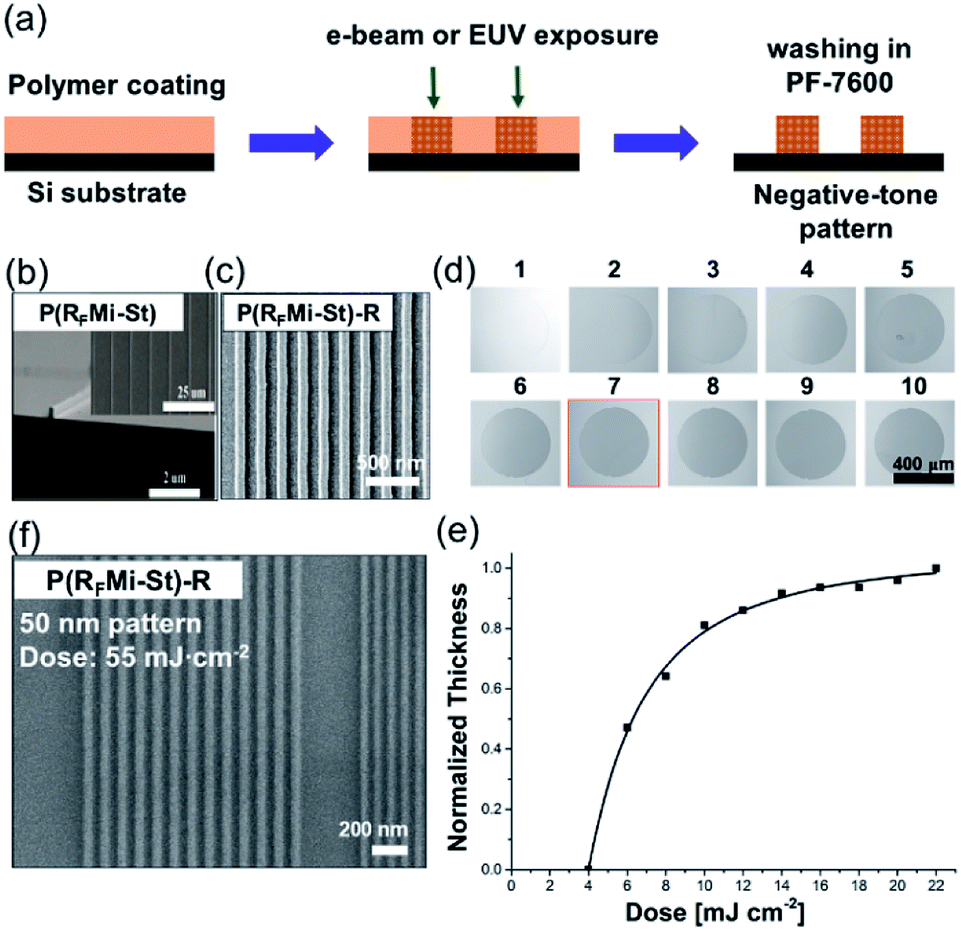 | ||
| Fig. 3 (a) A schematic diagram of e-beam and extreme UV (EUV) lithography. (b) and (c) SEM images of the line-and-space patterns after e-beam exposure of the thin films: (b) P(RFMi-St) (line width: 100 nm), and (c) P(RFMi-St)-R (line width: 70 nm). (d) Thin film patches left after EUV irradiation and washing in PF-7600. The exposure dose was increased from 4 mJ cm−2 for pattern 1 to 22 mJ cm−2 for pattern 10 in steps of 2 mJ cm−2. (e) The change in the remaining film thickess of the patches shown in (d). (f) SEM image of a P(RFMi-St)-R thin film (line width: <50 nm) after EUV exposure at a dose of 55.0 mJ cm−2 and washing in PF-7600. | ||
Recently, in semiconductor industry significant attention is focused on employing extreme UV (EUV, λ = 13.5 nm) as an innovative light source for the commercial production of high-resolution integrated circuit chips.28 Huge progress has been made in the manufacture of high power light source and precision exposure equipment, which strongly requires performance imaging materials working effectively and efficiently with that short wavelength light.29,30 EUV is classified as an ionising radiation, resulting in the secondary electron generation when it impinges on a materials surface.31 It is therefore necessary to consider the chemical reactions induced by high energy electrons along with those by the EUV photons in order to develop useful, sensitive imaging materials. Researchers in academia and industry proposed candidates belonging in a few categories; organic photoresists,32–34 small molecular organometallic compounds35,36 and transition metal oxide nanoclusters37–39 and nanoparticles.40,41 Organic photoresists, basically a refined version of conventional materials for 248 nm and 193 nm deep UV, work mainly in catalytic acidolysis processes with strong acid molecules. These materials can achieve pattern generation in a highly efficient manner but have disadvantages in controlling the acid diffusion phenomenon.42 For organometallic small molecules, metal oxide nanoclusters and nanoparticles, strategies of ligand dissociation and agglomeration upon irradiation are commonly employed. In particular, Sn-based nanoclusters are reported to show promising results although concerns about metal contamination are not yet completely cleared.43
In view of the change in solubility of P(RFMi-St) under e-beam irradiation, it is natural to wonder about the potential effects of EUV radiation. In the present work, 44 nm-thick P(RFMi-St)-R films (Mn = 11600 and Ð = 1.5) were exposed to a circular EUV beam with increasing energy of up to 22 mJ cm−2, followed by immersion in PF-7600. As shown in Fig. 3d, the exposed areas became insoluble and did not wash off. Moreover, the remaining thickness of the patches increased with increasing EUV energy of up to 10 mJ cm−2, levelling off thereafter (Fig. 3e). In view of the generally-accepted mechanism of imaging under EUV irradiation,44 this result could again be attributed to the intermolecular cross-linking reactions among the perfluoroalkyl moieties by the secondary or lower-energy electrons. When the P(RFMi-St)-R was tested under EUV lithographic conditions, 50 nm patterned structures were successfully resolved under an exposure energy of 55 mJ cm−2 (Fig. 3f).
As shown in Fig. 4a, the dielectric properties of the P(RFMi-St) were measured using a 310 nm-thick film sandwiched between two metal electrodes, indicating a capacitance of ca. 8 nF cm−2 at frequencies between 100 Hz and 1 MHz. This corresponds to a dielectric constant of 2.7, which is higher than that of the amorphous fluoropolymer, Cytop™ (i.e., 2.1).45 In addition, the current density vs. electric field (E-field) graph in Fig. 4b indicates a breakdown strength of 4.5 mV cm−1 for the P(RFMi-St), which is slightly smaller than that of Cytop™ (i.e. 5 mV cm−1) but high enough for a typical organic field-effect transistor (OFET) to withstand the driving voltage range. An OFET device with a top gate/bottom contact geometry was fabricated using the solution-cast P(RFMi-St) on top of a DPP-BTZ thin film (Fig. 4c).46,47 An examination of the transfer curve (ID − VG) in Fig. 4d indicates that the OFET operated stably. The hole and electron mobilities extracted from the linear region were ca. 0.4 cm2 V−1 s−1 and 0.02 cm2 V−1 s−1, respectively, for the P(RFMi-St) device, compared to ca. 0.3 and 0.008 cm2 V−1 s−1, respectively, for the Cytop™ device.
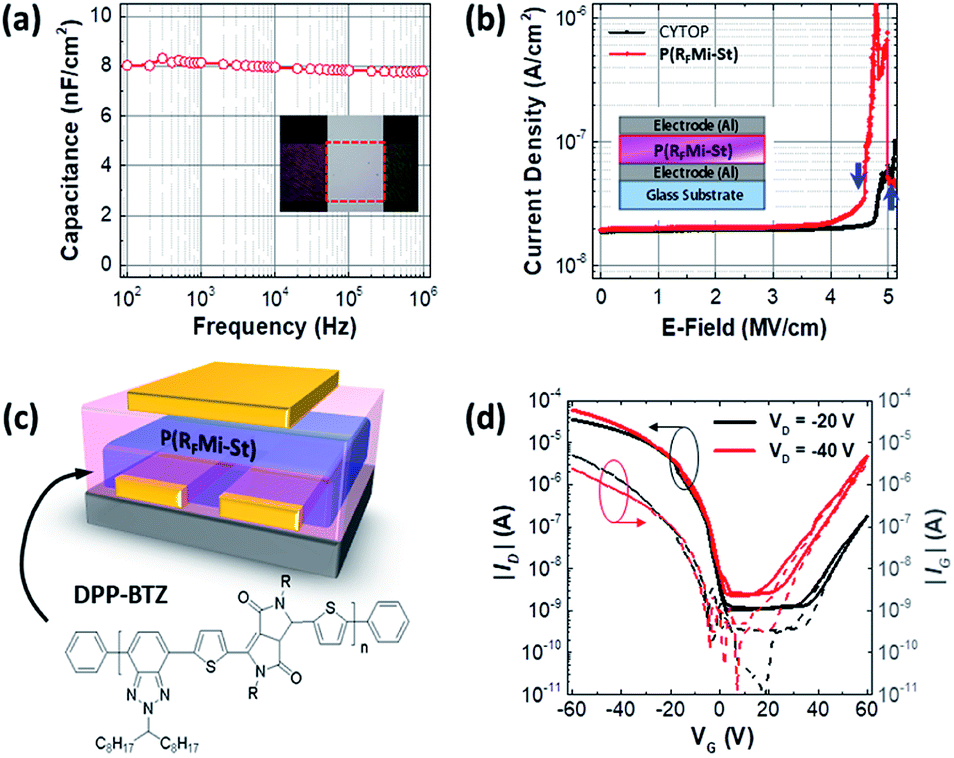 | ||
| Fig. 4 (a) Capacitance-frequency curve of the P(RFMi-St) film. The inset shows an image of the metal-insulator-metal (MIM) device. (b) Current density vs. electric field (E-field) graph for Cytop™ and P(RFMi-St) dielectric films. The arrows indicate the breakdown points and the inset shows a schematic of the MIM device. (c) The device configuration of the tested organic field-effect transistor (OFET) and the chemical structure of the semiconducting copolymer DPP-BTZ. (d) Transfer curves of the OFETs employing the P(RFMi-St) film as a dielectric layer in the linear and saturation regime (drain voltage VD = −20 V and −40 V). The solid and dashed lines show the drain current (ID) and gate leakage current (IG), respectively. | ||
Conclusions
A highly fluorinated alternating polymer, P(RFMi-St), possessing improved thermal properties and patterning capabilities under a high energy radiation was achieved with semi-perfluorododecyl maleimide (RFMi) and styrene (St). RFMi could be synthesised efficiently via a Mitsunobu condition and copolymerisation with St was performed by a free radical and reversible-deactivation radical polymerisation protocols. The recovered copolymer showed a satisfactory glass-transition temperature (108 °C) and intermolecular cross-linking behaviour under electron-beam and commercially more important EUV irradiation. The exposed regions lost their solubility, resulting in the successful construction of mechanically non-deteriorated negative-tone patterns down to 50 nm, which is the opposite to the case of amorphous fluoropolymer Cytop™. In addition, P(RFMi-St) revealed a dielectric constant of 2.7 and solution-processability in chemically non-damaging fluorous liquids, which enabled it to be applied on top of an organic semiconductor layer as a dielectric material for OFET fabrication. A more detailed study on P(RFMi-St) as a patterning material is going on currently, and the results will be reported in the near future.Experimental
Materials
Benzotrifluoride, diisopropyl azodicarboxylate, maleimide, and anhydrous THF were purchased from Sigma-Aldrich and used as received. 1,1,1,2,3,3-Hexafluoro-4-(1,1,2,3,3,3-hexafluoropropoxy)pentane (PF-7600) was bought from 3 M Korea and used without further purification. 2,2-Azobisisobutyronitrile (AIBN) was obtained from Junsei Chemical and used after recrystallization from a mixed solvent of MeOH and CHCl3. 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (CDSTSP),48 5,5,6,6,7,7,8,8,9,9,10,10,11,11,12,12,12-heptadecafluorododecan-1-ol,49 and 5,5,6,6,7,7,8,8,9,9,10,10,11,11,12,12,12-heptadecafluorododecan-1-amine50 were prepared according to the literature.Characterisation
1H NMR spectra were recorded on a Bruker Avance III (400 MHz) spectrometer at ambient temperature using the chemical shift of a residual protic solvent (CHCl3 at δ 7.24 ppm) as a reference. All chemical shifts were quoted in parts per million (ppm) relative to the internal reference, and the coupling constants, J, were reported in Hz. Signal multiplicity was indicated as follows: s (singlet), d (doublet) and t (triplet). Size exclusion chromatography (SEC) was carried out on a Younglin GPC system (YL9100) equipped with a refractive index detector and a series of two columns (PL gel mixed-C 5 μm and PL gel mixed-D 5 μm) by eluting 1,3-dichloro-1,1,2,2,3-pentafluoropropane (ASAHIKLIN™ AK-225G) at 35 °C. Monodisperse PMMA (molecular weight from 860 to 2200000 g mol−1, Shodex™, Showa-Denko) was used as a reference standard for the SEC experiments. Electron beam (e-beam) lithography was carried out on a NANOBEAM NB3 exposure tool operating at an acceleration voltage of 80 keV with a probe current of 1.0 nA. Developed patterns of the polymer films were observed by scanning electron microscopy (SEM, NanoBeam NB3, 80 keV) after sputter-coating of Pt. The nanoindentation tests were performed by a nano-indenter (Nanomechanics, USA) with a continuous stiffness measurement (CSM) technique. A Berkovich diamond tip was used in all the experiments.
Synthesis
:hexane = 1:4) and crystallized from iPrOH to give RFMi (3) as a white solid (2.90 g, 70%); (found: C, 33.8; H, 1.8; N, 2.5. C16H10NF17O2 requires C, 33.6; H, 1.8; N, 2.5%); IR (KBr) νmax: 1701, 1412, 1250, 1203, 1151, 845 cm−1; 1H NMR (400 MHz, CDCl3): δ = 6.71 (s, 2H), 3.56 (t, J = 7 Hz, 2H), 2.24–1.92 (m, 2H), 1.75–1.57 (m, 4H). HRMS (ESI): m/z [M + Na]+ calcd for C16H10NF17NaO2 594.0338; found 594.0333.Patterning
Electrical properties
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Samsung Research Funding & Incubation Center for Future Technology (Project No. SRFC-TA1703-05) and National Research Foundation of Korea (NRF) grants funded by the Korean government (MEST, NRF-2015R1D1A1A01060256). The authors thank Mr Warren Holcomb and Mr Chanin King for their kind EUV evaluation.Notes and references
- F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003–1022 RSC.
- H. Teng, Appl. Sci., 2012, 2, 496–512 CrossRef.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- S. Tan, J. Li, G. Gao, H. Li and Z. Zhang, J. Mater. Chem., 2012, 22, 18496–18504 RSC.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- A. Oshima, T. Seguchi and Y. Tabata, Radiat. Phys. Chem., 1999, 55, 61–71 CrossRef CAS.
- S.-H. Jung, K. Chuluunbandi, Y. Kim, J. Son, H.-T. Oh, J. H. Lee, J.-K. Lee and O. Beom-Hoan, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2672–2680 CrossRef CAS.
- H. H. Fong, J. K. Lee, Y. F. Lim, A. A. Zakhidov, W. W. Wong, A. B. Holmes, C. K. Ober and G. G. Malliaras, Adv. Mater., 2011, 23, 735–739 CrossRef CAS.
- J. H. Beck, R. A. Barton, M. P. Cox, K. Alexandrou, N. Petrone, G. Olivieri, S. Yang, J. Hone and I. Kymissis, Nano Lett., 2015, 15, 2555–2561 CrossRef CAS.
- J. Son, H.-T. Oh, O. J. Kwon, J.-M. Lim, H. Jung, B. J. Jung, D.-H. Hwang, C. Lee, J.-K. Lee, J. G. Yoon and S. Y. Yoon, J. Mater. Chem. C, 2017, 5, 926–930 RSC.
- D. P. Churchley, E. Barbu, R. J. Ewen, Z. Shen, Y. Kim, M. A. McHugh, Z. Y. Zhang, T. G. Nevell, G. D. Rees and J. Tsibouklis, J. Biomed. Mater. Res., Part A, 2008, 84, 994–1005 CrossRef.
- M.-S. Kim, D.-H. Lee, Y.-H. Cha, K.-B. Kim, S.-H. Jung, J.-K. Lee, B.-H. O, S.-G. Lee and S.-G. Park, Microelectron. Eng., 2014, 123, 33–37 CrossRef CAS.
- D. Yang, S. Chang and C.K. Ober, J. Mater. Chem., 2006, 16, 1693–1696 RSC.
- A. Bruno, Macromolecules, 2010, 43, 10163–10184 CrossRef.
- M. Zong, K. J. Thurecht and S. M. Howdle, Chem. Commun., 2008, 5942–5944 RSC.
- N. M. Hansen, K. Jankova and S. Hvilsted, Eur. Polym. J., 2007, 43, 255–293 CrossRef CAS.
- J. Ma, C. Cheng and K. L. Wooley, Macromolecules, 2009, 42, 1565–1573 CrossRef CAS.
- T. Isemura, R. Kakita and K. Kawahara, J. Chromatogr. A, 2004, 1026, 109–116 CrossRef CAS.
- K. Nishimori and M. Ouchi, Chem. Commun., 2020, 56, 3473–3483 RSC.
- K. Grundke, S. Zschoche, K. Pöschel, T. Gietzelt, S. Michel, P. Friedel, D. Jehnichen and A. Neumann, Macromolecules, 2001, 34, 6768–6775 CrossRef CAS.
- M. A. Walker, J. Org. Chem., 1995, 60, 5352–5355 CrossRef CAS.
- L. A. Galuska, W. W. McNutt, Z. Qian, S. Zhang, D. W. Weller, S. Dhakal, E. R. King, S. E. Morgan, J. D. Azoulay, J. Mei and X. Gu, Macromolecules, 2020, 53, 6032–6042 CrossRef CAS.
- G. n. Dlubek, J. r. Pionteck, K. Rätzke, J. Kruse and F. Faupel, Macromolecules, 2008, 41, 6125–6133 CrossRef CAS.
- J. Park, J. Ho, H. Yun, M. Park, J. H. Lee, M. Seo, E. E. Campbell, C. Lee, S. Pyo and S. W. Lee, Appl. Phys. A, 2013, 111, 1051–1056 CrossRef CAS.
- H. Hartl, C. East, Y. Xu, S. D. Yambem, K. E. Fairfull-Smith and J. MacLeod, Nanotechnology, 2019, 30, 335301 CrossRef CAS.
- M. Lazzari, M. Aglietto, V. Castelvetro and O. Chiantore, Chem. Mater., 2001, 13, 2843–2849 CrossRef CAS.
- A. Rathore, I. Pollentier, H. Singh, R. Fallica, D. De Simone and S. De Gendt, J. Mater. Chem. C, 2020, 8, 5958–5966 RSC.
- L. Li, X. Liu, S. Pal, S. Wang, C. K. Ober and E. P. Giannelis, Chem. Soc. Rev., 2017, 46, 4855–4866 RSC.
- R. M. M. Hasan and X. Luo, Nanomanuf. Metrol., 2018, 1, 67–81 CrossRef.
- C. Wagner and N. Harned, Nat. Photonics, 2010, 4, 24–26 CrossRef CAS.
- S. Fujii, T. Kozawa, K. Okamoto, J. J. Santillan and T. Itani, Jpn. J. Appl. Phys., 2015, 54, 116501 CrossRef.
- A. De Silva, J.-K. Lee, X. André, N. M. Felix, H. B. Cao, H. Deng and C. K. Ober, Chem. Mater., 2008, 20, 1606–1613 CrossRef CAS.
- M. S. Ober, D. R. Romer, J. Etienne, P. Thomas, V. Jain, J. F. Cameron and J. W. Thackeray, Macromolecules, 2019, 52, 886–895 CrossRef.
- C. Popescu, Y. Vesters, A. McClelland, D. De Simone, G. Dawson, J. Roth, W. Theis, G. Vandenberghe and A. P. Robinson, J. Photopolym. Sci. Technol., 2018, 31, 227–232 CrossRef CAS.
- C. Luo, C. Xu, L. Lv, H. Li, X. Huang and W. Liu, RSC Adv., 2020, 10, 8385–8395 RSC.
- M. Murphy, A. Narasimhan, S. Grzeskowiak, J. Sitterly, P. Schuler, J. Richards, G. Denbeaux and R. L. Brainard, J. Photopolym. Sci. Technol., 2017, 30, 121–131 CrossRef CAS.
- I. Bespalov, Y. Zhang, J. Haitjema, R. M. Tromp, S. J. van der Molen, A. M. Brouwer, J. Jobst and S. Castellanos, ACS Appl. Mater. Interfaces, 2020, 12, 9881–9889 CrossRef CAS.
- B. Cardineau, R. Del Re, M. Marnell, H. Al-Mashat, M. Vockenhuber, Y. Ekinci, C. Sarma, D. A. Freedman and R. L. Brainard, Microelectron. Eng., 2014, 127, 44–50 CrossRef CAS.
- H. Xu, K. Sakai, K. Kasahara, V. Kosma, K. Yang, H. C. Herbol, J. Odent, P. Clancy, E. P. Giannelis and C. K. Ober, Chem. Mater., 2018, 30, 4124–4133 CrossRef CAS.
- W. J. Bae, M. Trikeriotis, J. Sha, E. L. Schwartz, R. Rodriguez, P. Zimmerman, E. P. Giannelis and C. K. Ober, J. Mater. Chem., 2010, 20, 5186–5189 RSC.
- P. G. Reddy, N. Mamidi, P. Kumar, S. K. Sharma, S. Ghosh, K. E. Gonsalves and C. P. Pradeep, RSC Adv., 2016, 6, 67143–67149 RSC.
- J. J. Biafore, M. D. Smith, C. A. Mack, J. W. Thackeray, R. Gronheid, S. A. Robertson, T. Graves and D. Blankenship, Advances in Resist Materials and Processing Technology XXVI, 2009 Search PubMed.
- O. Yildirim, E. Buitrago, R. Hoefnagels, M. Meeuwissen, S. Wuister, G. Rispens, A. van Oosten, P. Derks, J. Finders and M. Vockenhuber, Extreme Ultraviolet (EUV) Lithography VIII, 2017 Search PubMed.
- T. Kozawa and S. Tagawa, Jpn. J. Appl. Phys., 2010, 49, 030001 CrossRef.
- A. N. Banerjee, S. Qian and S. W. Joo, J. Colloid Interface Sci., 2011, 362, 567–574 CrossRef CAS.
- M. Gruber, S.-H. Jung, S. Schott, D. Venkateshvaran, A. J. Kronemeijer, J. W. Andreasen, C. R. McNeill, W. W. Wong, M. Shahid and M. Heeney, Chem. Sci., 2015, 6, 6949–6960 RSC.
- S. Schott, E. Gann, L. Thomsen, S. H. Jung, J. K. Lee, C. R. McNeill and H. Sirringhaus, Adv. Mater., 2015, 27, 7356–7364 CrossRef CAS.
- G. Moad, Y. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- K. B. Woody, R. Nambiar, G. L. Brizius and D. M. Collard, Macromolecules, 2009, 42, 8102–8111 CrossRef CAS.
- B. J. Jung, K. Lee, J. Sun, A. G. Andreou and H. E. Katz, Adv. Funct. Mater., 2010, 20, 2930–2944 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08539a |
| ‡ Current address: Heeger Center for Advanced Materials (HCAM), Gwangju Institute of Science and Technology (GIST), Gwangju 61005, Republic of Korea |
| This journal is © The Royal Society of Chemistry 2021 |