DOI:
10.1039/D0RA08534K
(Paper)
RSC Adv., 2021,
11, 6035-6048
Investigation of interactions between binding residues and solubility of grafted humanized anti-VEGF IgG antibodies expressed as full-length format in the cytoplasm of a novel engineered E. coli SHuffle strain
Received
7th October 2020
, Accepted 25th January 2021
First published on 2nd February 2021
Abstract
Monoclonal antibodies (mAbs) are one of the fastest-growing areas of biopharmaceutical industry and have been widely used for a broad spectrum of diseases. Meanwhile, the immunogenicity of non-human derived antibodies can generate side effects by inducing the human immune response to produce human anti-mouse-immunoglobulin antibody (HAMA). In this work, we aim to reduce the immunogenicity of muMAb A.4.6.1 by substitute human sequences for murine sequences. Humanized antibodies are constructed by grafting, specificity determining residues (SDR), complementary determining regions (CDR), and chimeric region of muMAb A.4.6.1, onto variable domain of Trastuzumab (Herceptin). The interactions between grafted antibodies and their target, Vascular endothelial growth factor (VEGF), were theoretically investigated by molecular dynamics simulation in order to evaluate the antibodies–antigen binding behavior. The obtained protein–protein interactions and calculated binding free energy suggested that the SDR–VEGF complex presented a significantly greater binding affinity, number of contact and total number of H-bonds compared to CDR and chimeric mAbs, significantly. Moreover, the Camsol program predicted that the solubility of SDR mAb exhibits the greatest solubility. This result was supported by performing a western blot analysis of the grafted mAbs with soluble and insoluble fractions in order to evaluate their solubility, in which SDR was found to have a much lower amount of insoluble proteins. Consequently, the enhanced binding affinity and solubility of the designed SDR was achieved by the single S106D mutation using computational methods. With the aim of low immunogenicity, high solubility, and high affinity, this SDR humanized antibody was expected to have greater efficacy than murine or chimeric antibodies for future use in humans.
1. Introduction
Antibodies are glycosylated proteins (immunoglobulins [IgG]) produced as a part of the immune system response to foreign substances in vertebrates. Due to their high specificity and affinity, they play an important role in the treatment of many human diseases. However, murine monoclonal antibody can trigger the human immune system leading to adverse effects such as allergic reactions, nausea, fatigue, fever, diarrhea and fatal immune side effect by induces human immune responses to produce human anti-mouse-immunoglobulin antibody (HAMA).1,2 Therefore, development of monoclonal antibody (mAb) with less immunogenicity and a high antibody–antigen affinity is of prime concern. The level of immunogenicity of mAb is related to the content of murine sequences in its structure. Grafting technology remains the most popular and effective method for the production of therapeutic antibodies, although several techniques have since been developed which reduce antibody immunogenicity.3 A chimeric antibody is the initial modification, in which variable regions have murine sequences and the remaining sequences are human. This modified antibody could subsequently partially reduce immunogenicity compared to murine antibody.4 A humanized chimeric antibody can be generated by grafting amino acid residues responsible for antibody binding sites (ABS) or complementarity determining regions (CDRs), of murine antibody onto the frameworks of variable light and variable heavy chain domains of human antibodies.5 Numerous studies into CDRs, demonstrate that some residues in CDRs may evoke anti-idiotypic (anti-Id) responses in patients. Thus, CDR grafting is not a perfect method of overcoming the immunogenicity of the humanized antibody.6 The analysis of 3-D structures of the antibody combining sites suggested that only 20–30% of CDR residues are essential for an antibody–antigen interaction. These residues are located in the regions of high variability and are named as specificity determining residues (SDRs).
Natural antibodies are large multimeric proteins containing many disulfide bonds and require posttranslational modifications such as glycosylation. Currently, mAbs are largely produced in mammalian expression systems. This technique is attractive in terms of its high expression efficiency and its ability to produce large, complex proteins.
Antibody production requires a large number of cultured mammalian cells, resulting in low production rate, extremely high production costs, and technically challenging fermentation and purification steps.7,8 Alternative mAbs production systems in microorganisms such as Escherichia coli (E. coli) is more desirable due to its simplicity and reduced production time and costs.9 Currently, two antigen-binding fragments (Fab)–Lucentis® and Cimzia®–are commercially available and produced in the E. coli system, indicating that E. coli may be useful for industrial scale antibody production.10 Early attempts to produce full-length IgG in E. coli was in the form of inclusion bodies in the cytoplasmic space11 or in the soluble form in the periplasmic space.12,13 Drastic improvements have recently been made to engineer E. coli strains which are suitable for the expression of multi-disulfide bond proteins, called SHuffle strains.14 In 2015, Robinson et al. reported the first soluble expression of active, full-length IgGs in the cytoplasm of SHuffle E. coli cells, an achievement that was not possible in wild-type E. coli.15 Moreover, they showed that these cytoplasmic IgGs produced in E. coli, termed ‘cyclonals’ could be redirect to different target antigens by simply swapping the Fv domains while keeping the same scaffold. Genetic manipulation is also easier and more beneficial when using E. coli as a host, since many antibody engineering techniques such as grafting alternative epitope recognition domains, humanizing, and Fc engineering can be utilized to rapidly create custom mouse, rabbit, or humanized antibodies on demand.
Antibody efficacy is largely dependent upon their atomic interactions with an antigen. A longer binding time of antibody–antigen results in a higher antibody efficacy, resulting in the use of a lower drug dose. In the present study, to determine an effective humanized mAb specific for VEGF, three grafted antibodies (chimeric, CDR and SDR-grafting) were designed and constructed. All-atom molecular dynamics (MD) simulations were performed on the three complexes to investigate the structure and dynamics properties as well as the protein–protein interactions between the humanized antibodies and VEGF. The solubility of all grafted mAbs was then predicted using the Camsol program. To support the predicted solubility result and their expression in E. coli SHuffle strain, western blot analysis of three mAbs were performed. The most potent modeled mAb with a more favorable binding free energy and greatest solubility was then used as a template to further design improved the mab with binding interaction and solubility using computer modeling.
2. Materials and methods
2.1 Experimental methods
2.1.1 Bacterial strains and growth conditions. The E. coli DH5α strain was used for the cloning of all the grafted mAbs. For antibody expression, the SHuffle T7 Express (NEB) cells transformed with one of the pMAZ360-cIgG expression vectors which were grown in a Luria–Bertani (LB) medium supplemented with 100 μg mL−1 ampicillin and 25 μg mL−1 spectinomycin overnight at 30 °C. The following day, 1% v/v of the overnight cultured cells were subcultured into fresh LB supplemented with antibiotics and grown at 30 °C until the absorbance at 600 nm reached 0.7. Subsequently, protein expression was induced by the addition of 0.1 mM of isopropyl β-D-thiogalactopyranoside (IPTG) and allowed to grow for an additional 16 hours at 30 °C. Finally, the cells were harvested by centrifugation at 4 °C and 3500 rpm for 15 minutes, collected as pellets and kept at 4 °C prior to cell lysis.
2.1.2 Construction of recombinant plasmid. Chimeric anti-VEGF was created by replacing the Fv of anti-HER2 (trastuzumab) with the Fv of anti-VEGF antibody. To generate pMAZ360-anti-VEGF (mouse/human chimeric Ab), E. coli codon optimized VH and VL domains of anti-VEGF murine mAb (muMAb A.4.6.1)16 were chemically synthesized by GenScript (USA) and cloned into pMAZ360-Herceptin. Briefly, murine anti-VEGF VL domain was PCR amplified and relegated into NdeI-NotI digested pMAZ360-Herceptin, thus replacing the original anti-HER2 VL domain and creating pMAZ360-anti-VEGF VL/Herceptin plasmid. To replace the anti-HER2 VH domain, murine anti-VEGF VH domain was PCR amplified using primers that appended a 5′ RBS sequence with Hind III cut site flanking on both ends and relegated into HindIII treated pMAZ360-anti-VEGF VL/Herceptin plasmid to obtain pMAZ360-anti-VEGF (mouse/human chimeric Ab). Sequencing analysis confirmed the correct sequences and directions of both VH and VL.
2.1.3 Preparation of soluble and insoluble fractions. The cell pellets were resuspended in 3 mL of a Lysis buffer (1 mg lysozyme, 50 mM Tris–HCl, 1 mM EDTA, and 25 mM NaCl) and normalized to a final OD600 of 100. The suspended cell was aliquoted equally into six 1.5 mL microcentrifuge tubes. Sonification was performed using a vertical sonifier (Branson 150, USA) with a 1/8-inch probe and 40% amplitude. Supernatant was collected as a soluble fraction by centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and 4 °C for 10 minutes. To prepare insoluble fraction, the pellet was washed twice with 50 mM Tris–HCl and 1 mM EDTA, and then resuspended with 2% SDS in a PBS buffer. The suspension was then boiled at 100 °C for 10 minutes and centrifuged at 13000 rpm for 10 minutes. The supernatant was obtained as an insoluble fraction. The total protein of the soluble fraction was determined using a Bradford assay. Protein concentration determination was performed using 5 μL of 20-fold dilution of protein sample with 200 μL of Quick Start™ Bradford 1× dye reagent and detected at 595 nm. The soluble and insoluble fractions were further analyzed using the western blot technique.
000 rpm and 4 °C for 10 minutes. To prepare insoluble fraction, the pellet was washed twice with 50 mM Tris–HCl and 1 mM EDTA, and then resuspended with 2% SDS in a PBS buffer. The suspension was then boiled at 100 °C for 10 minutes and centrifuged at 13000 rpm for 10 minutes. The supernatant was obtained as an insoluble fraction. The total protein of the soluble fraction was determined using a Bradford assay. Protein concentration determination was performed using 5 μL of 20-fold dilution of protein sample with 200 μL of Quick Start™ Bradford 1× dye reagent and detected at 595 nm. The soluble and insoluble fractions were further analyzed using the western blot technique.
2.1.4 Protein A chromatography purification of the antibodies. The NAb™ Spin columns were used for IgG purification. The column was first equilibrated at room temperature. The bottom closure was snapped off and the column was centrifuged at 5000 g for 1 minute to remove the storage buffer. The immobilized protein A agarose beads were equilibrated twice with 400 μL of binding buffer (PBS) and centrifuged to discard the buffer. The bottom column was then capped and 500 μL of lysate soluble fraction was added to the column. The cell lysate (prepared using the same method as the soluble fraction) containing IgG was allowed to bind with protein A on agarose beads at room temperature with end-over-end mixing for 10 minutes. The column was then centrifuged at 5000 g for 1 minute and the beads were washed with 400 μL PBS three times to remove impurities. Binding was performed eight times to provide a total lysate volume of 4 mL. The antibodies were eluted with a 400 μL elution buffer (0.1 M glycine-HCl, pH 2.8) into 2 mL collection tubes containing 40 μL neutralization buffer (1 M Tris, pH 9.0). The elution was repeated three times to obtain four eluate fractions. The quantity of purified antibody in each eluate was analyzed using NanoDropTM 1000 Spectrophotometer (Thermo Fisher Scientific, US) by utilizing the relative absorbance at 280 nm with a 1% extinction coefficient of 13.7.
2.1.5 Western blot analysis. To analyze the grafted mAb expression, SDS-PAGE analysis was performed under non-reducing conditions. The samples were diluted 1:5 in a 6× Laemmli sample buffer without addition of 2-mercaptoethanol and heated at 100 °C for 10 minutes and then loaded on 4–20% Tris–HCl gradient gels. All samples were normalized (1 μg of total protein for purified protein, 20 μg of total protein for lysate and equal volume to lysate for insoluble fraction) as determined by the Bradford assay. The electrophoresis was performed at 120 volts for 1 hour. Following electrophoresis, resolved proteins were transferred to polyvinylidene fluoride membranes (Millipore). The membrane was washed three times with TBS buffer and then blocked with 5% milk (w/v) in TBS containing 0.1% Tween 20 (TBS-T) overnight at 4 °C. On the next day, the membrane was washed three times with TBS-T then Herceptin or grafted mAbs were probed with 1:10000-diluted anti-human IgG (H + L)-HRP conjugate (Abcam). After washing five more times with TBS-T, the membrane was incubated with Clarity MaxTM Western ECL substrate (Bio-Rad) and then visualized using a Bio-rad ChemiDoc XRS+.
2.2 Computational methods
2.2.1 System preparation and molecular dynamics simulation. Since the patents on Herceptin have already expired in the United States and Europe, the Herceptin sequence was selected as a human framework for humanized antibodies. To date, there is no available 3D structure of the Herceptin-VEGF complex. To construct the Herceptin-VEGF model, the sequence of Avastin was replaced with the sequence of Herceptin on the variable and constant region of light and heavy chains via sequence alignment using the BioEdit program17 and the crystal structure of Avastin was complexed with VEGF (PDB code: 1BJ1).18 The Herceptin sequence was selected from Michael Paul Robinson et al.,16 while the sequences of SDR, CDR, and the variable domains of muMAb A.4.6.1 were obtained in the previous study.19,20 To generate chimeric and humanized antibodies, the sequences of SDR, CDR, and variable domains of muMAb A.4.6.1 were grafted onto the VL and VH chains of the human framework. The protonation state of all the ionizable amino acids (Asp, Glu, Lys Arg and His) in the antibody-VEGF complex were assigned at pH 7.0 using PROPKA 3.0 program.21 The missing hydrogen atoms and a pair of disulfide bonds in the antibody-VEGF complex were added by the LEaP module in AMBER 16,22 and the whole complexes were then minimized with 1000 of steepest descents (SD) and followed by 2000 steps of conjugated gradients (CG) using the Sander module to reduce bad contact and steric hindrance. The antibody-VEGF complex was solvated by TIP3P water molecules23 within 12 Å around the system surface. Chloride ions were randomly introduced to neutralize the total positive charge of the antibody-VEGF complex. Water molecules and ions were then minimized with 500 steps of SD followed by 500 steps of CG, while a 500 kcal mol−1 Å2 force constant was applied to restrain the protein–protein complex. The whole system was fully minimized with 1000 steps of SD and CG. All the covalent bonds involving hydrogen atoms were constrained by the SHAKE algorithm (Amber, San Francisco, CA).24 Long-range electrostatic interactions were calculated according to the Particle Mesh Ewald (PME) approach25 with a cutoff distance of 12 Å for non-bonded interactions. After structure preparation, all three grafted mAbs-VEGF systems were performed by all-atom MD simulations under a periodic boundary condition using the PMEMD.cuda module. The system was heated to 310 K for 100 ps and then simulated at the same temperature for 200 ns in the NPT ensemble, using a time step of 2 fs. The MD trajectories from the last 10 ns of each system in the production phase were extracted for analysis in terms of binding free energy (ΔGbind) and per-residue decomposition free energy using the MM-GBSA and MM-PBSA methods.26 Moreover, the cpptraj module was used to analyze the root mean square deviation, number of contact residues, and the intermolecular hydrogen bonds between the protein–protein interfaces. The percentage of H-bond occupation was calculated using the criteria: (i) distance between the proton donor (HD) and the acceptor (HA) atoms being below 3.0 Å; and (ii) whether the angle of HD-H…HA was above 120°.
2.2.2 Designed antibody and solubility prediction. The grafted mAb with the highest binding efficiency with the VEGF target was selected for the newly designed antibody. To computationally design a good candidate for antibody therapy, single mutations were introduced to the mAb residue located at the protein–protein interface. The designed mAbs were separately docked to VEGF using ZDOCK version 3.0.2 on a ZDOCK server27 with 2000 runs by default. The docked mAb–VEGF complex with the highest ZDOCK score was selected to calculate the intrinsic solubility profile and CamSol score using Camsol version 2 according to the method described by Sormanni et al.28,29 The intrinsic solubility profile per amino acid and the overall solubility score of the whole protein, including the light and heavy chains of all the grafted mAbs, were predicted and compared. Note that the order of the light or heavy chains does not affect the resulting solubility score. Residues with scores below −1 have poor solubility, while those with scores above 1 are more soluble. Additionally, amino acid residue scores close to 0 indicate a negligible effect on protein solubility.
3. Results
3.1 Antibody grafting and sequence alignment
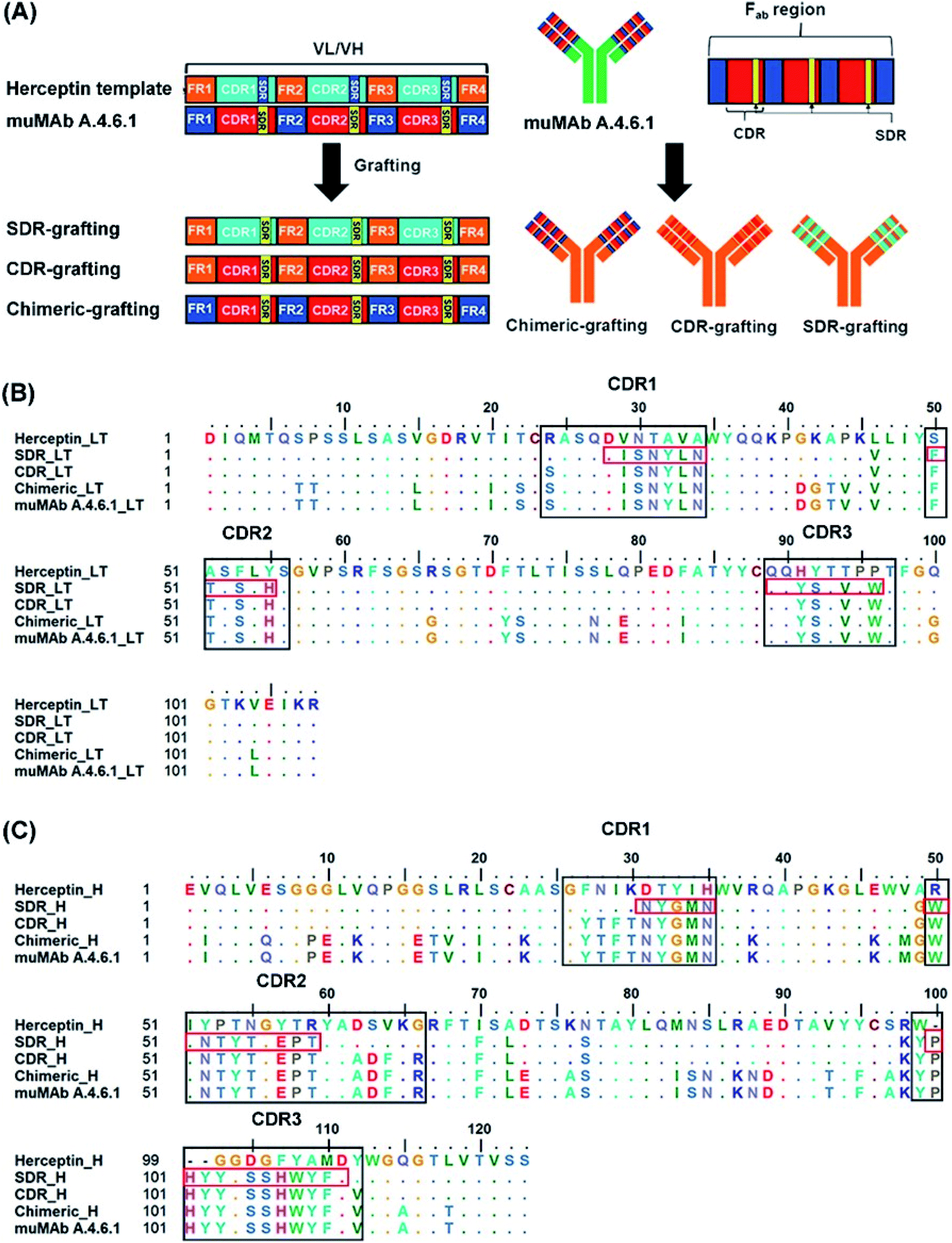
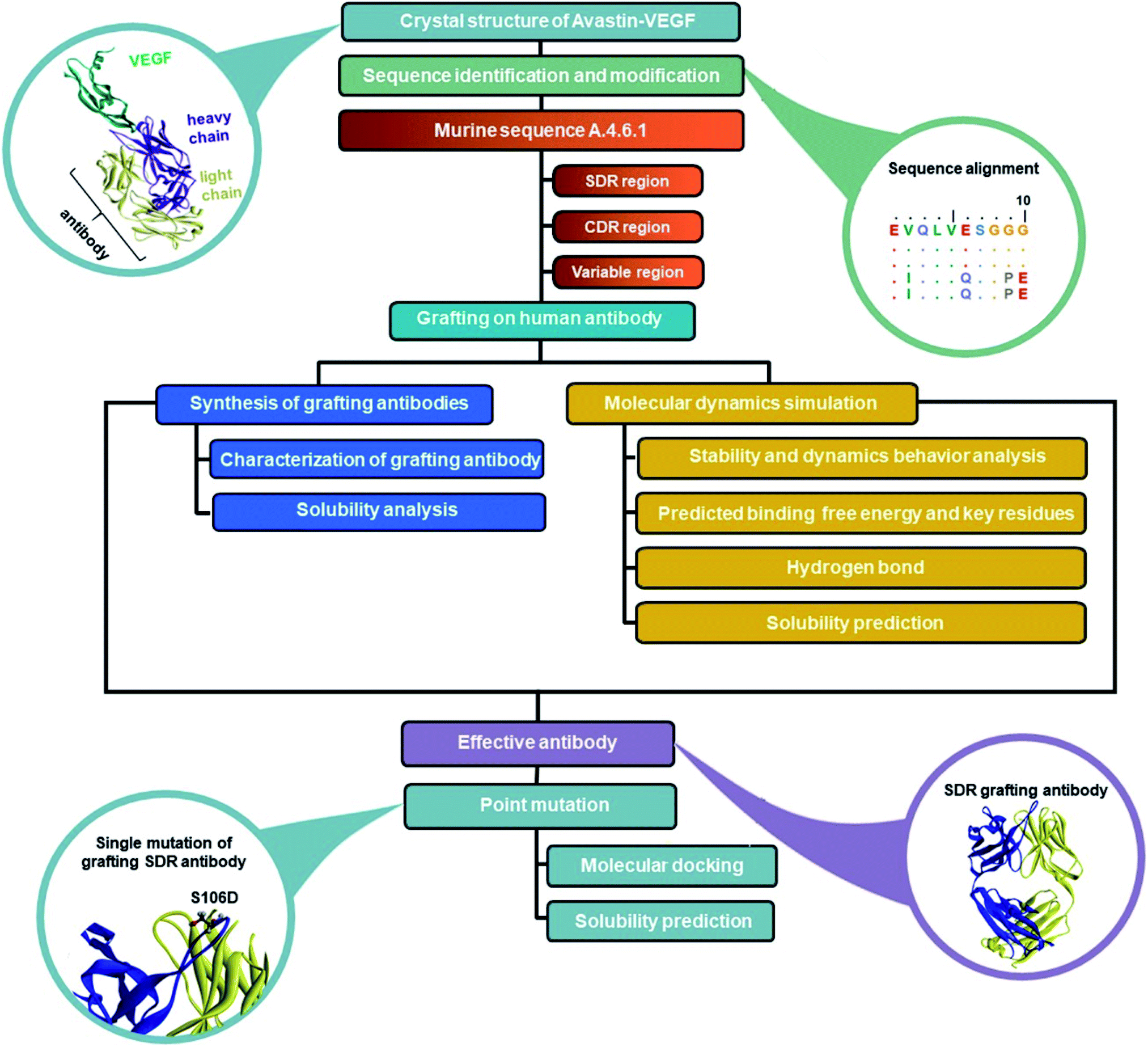
In order to reduce the immunogenicity of muMAb A.4.6.1, humanized mAbs were designed and produced by genetic engineering to remove murine amino acid sequences. Herceptin—a humanized IgG1 kappa mAbs—was selected as a human framework. Since the patents on Herceptin have already expired in the United States and Europe, several humanized antibodies are based on Herceptin's framework.30,31 To humanize muMAb A.4.6.1, we designed the sequence of muMAb A.4.6.1 in two ways. First, the Fv domains of Herceptin was simply replaced by the variable domains of muMAb A.4.6.1 to create a chimeric antibody. Nonetheless, this type of antibody still carries immunological risk since only the constant regions, representing approximately 70% of the antibody, are human sequences.32 Second, humanization of muMAb A.4.6.1 was performed by grafting murine CDR sequences onto the Herceptin framework. ‘Humanized’ antibodies typically contain 85–90% human sequence which further reduces the immunogenicity, but the process of humanizing is more technically demanding than the mouse-human fusions used to generate chimeric antibodies.32 The CDRs on VH and VL of muMAb A.4.6.1 were identified based on sequence hypervariability, in accordance with Kabat and Chothia's definition.33,34 The CDRs in the light chain were defined as the residues CDR_L1 (24–34), CDR_L2 (50–56), and CDR_L3 (89–97), while the CDRs in the heavy chain were composed of the residues CDR_H1 (26–35), CDR_H2 (50–66), and CDR_H3 (99–112). Since the humanized antibodies produced by CDR grafting contain some sequences that may not be important to antigen binding and can still induce immunogenicity, specificity determining residue (SDR) grafting was proposed. SDR grafting was expected to possess lower immunogenicity compared to CDR grafting due to the lower number of mouse residues. Padlan et al. proposed a method to identify SDR residues within the CDR regions of five antibody–antigen complexes.35 The present study used the same methods to identify the residues in the SDR regions of muMab A.4.6.1. The SDR residues in the light chain were defined as SDR_L1 (28–34), SDR_L2 (50–55), and SDR_L3 (89–96). In the heavy chain, the SDR_H1 (31–35), SDR_H2 (50–59), and SDR_H3 (100–111) were identified. Fig. 1A presents an illustration of the chimeric and humanized muMAb A.4.6.1 anti-VEGF design. The alignment of the VL and VH sequences of Herceptin, muMAb A. 4.6.1, and the three grafted mAbs are depicted in Fig. 1B and C, respectively. The three dimensional structures of the grafted mAbs were then generated using the 3D structure of Avastin complex with VEGF from the Protein Data Bank (PDB; 1BJ1) which was used as the grafting template. First, the Avastin sequence in the 3D structure was replaced with the Herceptin sequence on the variable and constant regions of the light and heavy chains. Second, to generate the chimeric and humanized antibodies, the sequences of SDR, CDR, or Fv domains of muMAb A.4.6.1 were grafted onto the VL and VH chain of the humanized Herceptin framework. The protein–protein complexes were then studied using all-atom molecular dynamics (MD) simulations in an aqueous solution in order to provide additional information about the inhibition of VEGF by the grafted mAbs and to investigate the detailed binding and interaction of these three complexes. The workflow of the in vitro and in silico study of chimeric and humanized mAbs is depicted in Fig. 2.
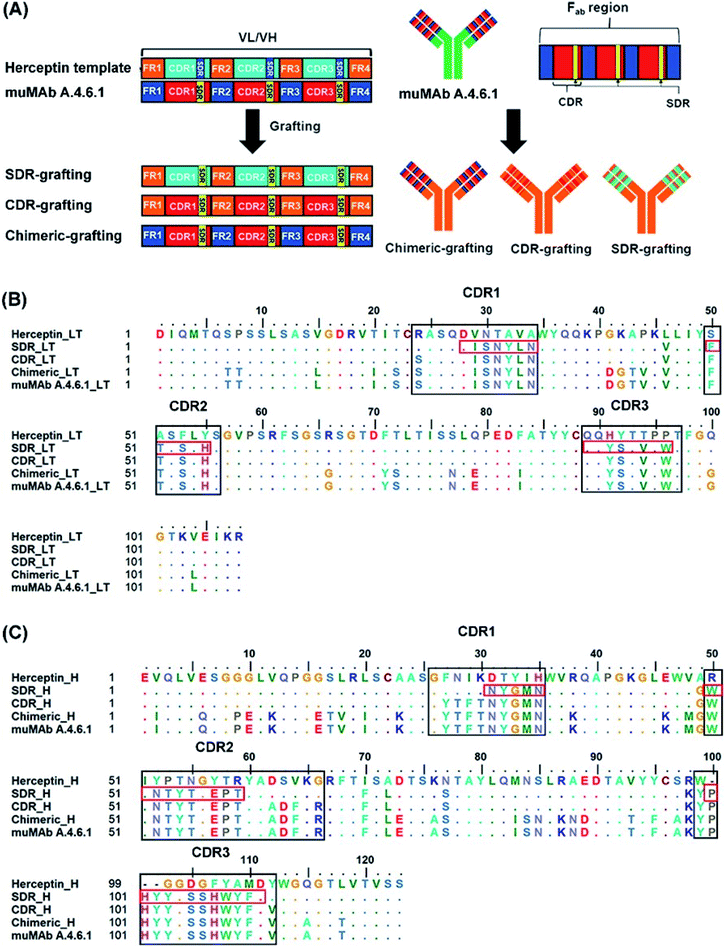 |
| | Fig. 1 Grafting of chimeric and humanized anti-VEGF mAbs. (A) Illustrative schematic of the SDR-, CDR-, and chimeric-grafting of the muMAb A.4.6.1 onto Herceptin's scaffold, the amino acid sequence of (B), variable light chain (VL), and (C) the variable heavy chain (VH) of muMAb A.4.6.1 and humanized antibodies (SDR-, CDR-, and chimeric-). The CDR and SDR regions are indicated by the black and red boxes, respectively, while the dots show identical amino acid residues. | |
 |
| | Fig. 2 Workflow of the in vitro and in silico study of chimeric and humanized mAbs. | |
3.2 Dynamic behaviors of three grafted mAbs-VEGF complexes
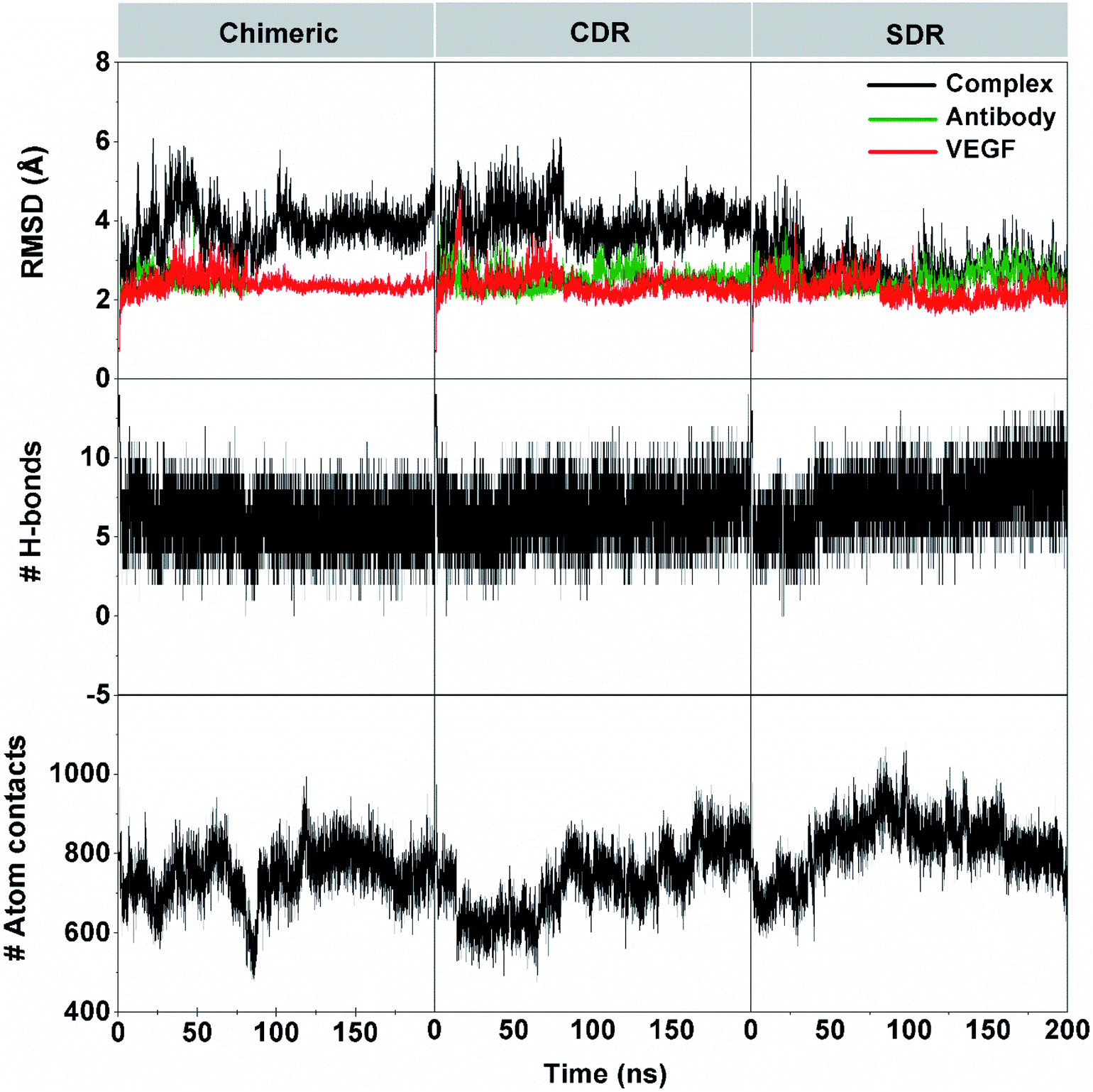
The 200 ns MD simulations were performed on each complex of the three grafted mAbs (chimeric-, CDR-, and SDR-) binding to VEGF. The conformation stability of the three simulated complexes in the solution was monitored using the root-mean square displacement (RMSD) calculation (Fig. 3). The RMSD plots of all atoms of complex (black), grafted mAb (green), and VEGF (red) were evaluated from the protein coordinates relative to those of the starting structure. The RMSD values of the complex of chimeric and CDR systems fluctuated by ∼3.1–4.9 Å along simulation time, while the SDR system continuously increased at the first 35 ns with RMSD values of ∼3.3–3.8 Å before reducing to ∼2.7–3.2 Å until 200 ns. Considering the RMSDs of either the grafted mAb or VEGF at ∼2.8 Å together, all the complexes reached equilibrium after ∼60 ns. The number of intermolecular hydrogen bonds (H-bonds) with VEGF and the number of atom contacts within the 5 Å sphere of each grafted mAb plotted over the simulation period well supports the convergence to equilibrium. Of the considered grafting antibodies, the SDR complex formed the strongest H-bond interactions with VEGF (9–10 H-bonds), while only 7–8 H-bonds were detected in the other systems (discussed further in section 3.4). Additionally, higher contact atoms in the SDR complex (∼820 atoms over the last 100 ns) suggested that the SDR moved closer to better interact with VEGF compared to chimeric and CDR (∼760 and ∼780 atoms). Although all the grafted mAbs had the same CDR-H3 (binding region) amino acid sequences, the CDR-H3 conformation is controlled by both its sequence as well as its environment.36 To gather detailed information of the binding free energy and the interaction of the grafted mAbs towards VEGF, the last 10 ns MD trajectories of each model were extracted for further analysis.
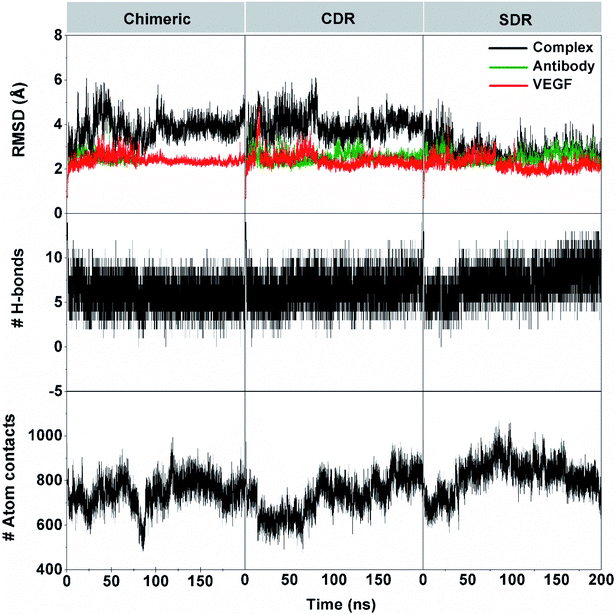 |
| | Fig. 3 System stability was investigated by RMSD, the number of intermolecular hydrogen bonds, and the number of atoms contact on time series of chimeric-, CDR-, and SDR–VEGF complexes. The RMSD for all atoms of the grafted mAb–VEGF complex (black), grafted mAb (green), and VEGF (red). | |
3.3 Binding affinity of the mAb–VEGF complex
The MM-GBSA and MM-PBSA methods are commonly used to calculate the binding free energy of protein–protein complexes37–41 and were applied to evaluate the binding affinity of three grafted mAbs against VEGF. The binding free energy (ΔGbind (MMGB/PBSA)) and the energy components of the 100 snapshots over the last 10 ns simulation of the three grafted mAbs are summarized in Table 1. It is evident that electrostatic interaction (ΔEeleMM) was the primary contribution to the binding between the three grafted mAbs and VEGF, which is over twice as strong as the van der Waals interaction (ΔEvdWMM). When including entropy term and the solvation effect, the ΔGbind (MMGB/PBSA) values suggest that the binding affinity of the grafted mAbs to-wards VEGF are in the following order: SDR mAb (−54.20 kcal mol−1) > CDR mAb (−35.32 kcal mol−1) > chimeric mAb (−31.22 kcal mol−1). The MM-GBSA calculation also provides a similar binding affinity trend: SDR mAb (−49.99 kcal mol−1) > CDR mAb (−28.63 kcal mol−1) > chimeric mAb (−28.14 kcal mol−1). Note that both methods predicted that all three grafted mAbs could give somewhat a stronger binding affinity towards VEGF compared to muMAb A.4.6.1 and Avastin (ΔGbind(experiment) of −12.44 and −12.28 kcal mol−1 converted from the KD values42).
Table 1 MM-PB(GB)SA binding free energies and their energy components (kcal mol−1) for three grafted mAbs in complex with VEGF, compared to the experimental binding free energy of muMAb A.4.6.1 and Avastina
| Energy component |
Chimeric |
CDR |
SDR |
muMAb A.4.6.1 |
Avastin |
| The experimental binding free energy was converted from the reported KD using the equation ΔGbind(experiment) = RTlnKD, where R is the ideal gas constant (1.987 cal mol−1 K−1), T is temperature in K, KD = Koff/Kon. |
| Gas term |
| ΔGvdWMM |
−106.21 ± 0.53 |
−99.78 ± 1.99 |
−119.32 ± 1.20 |
— |
— |
| ΔGeleMM |
−149.92 ± 1.54 |
−285.31 ± 2.21 |
−208.63 ± 2.09 |
— |
— |
| ΔEMM |
−256.13 ± 1.87 |
−385.10 ± 3.66 |
−327.96 ± 2.80 |
— |
— |
| TΔS |
−40.93 ± 3.67 |
−39.56 ± 2.79 |
−38.59 ± 2.61 |
— |
— |
| Solvation term |
| PBSA |
| ΔGpolarsolv (PBSA) |
196.98 ± 1.42 |
322.65 ± 2.61 |
249.01 ± 2.07 |
— |
— |
| ΔGnonpolarsolv (PBSA) |
−13.00 ± 0.06 |
−12.44 ± 0.20 |
−13.83 ± 0.13 |
— |
— |
| ΔGsolv (PBSA) |
183.98 ± 1.41 |
310.21 ± 2.46 |
235.17 ± 1.98 |
— |
— |
|
| GBSA |
| ΔGpolarsolv (GBSA) |
199.86 ± 1.43 |
329.32 ± 2.53 |
253.28 ± 2.01 |
— |
— |
| ΔGnonpolarsolv (GBSA) |
−12.80 ± 0.06 |
−12.40 ± 0.22 |
−13.90 ± 1.46 |
— |
— |
| ΔGsolv (GBSA) |
187.07 ± 1.48 |
316.91 ± 2.37 |
239.38 ± 1.90 |
— |
— |
|
| Binding free energy |
| ΔGbind (MM-PBSA) |
−31.22 ± 1.64 |
−35.32 ± 1.67 |
−54.20 ± 1.25 |
— |
— |
| ΔGbind (MM-GBSA) |
−28.14 ± 1.95 |
−28.63 ± 1.59 |
−49.99 ± 1.20 |
— |
— |
| ΔGbind (experiment) |
— |
— |
— |
−12.44 42 |
−12.28 42 |
3.4 Key residues of grafted mAbs and VEGF
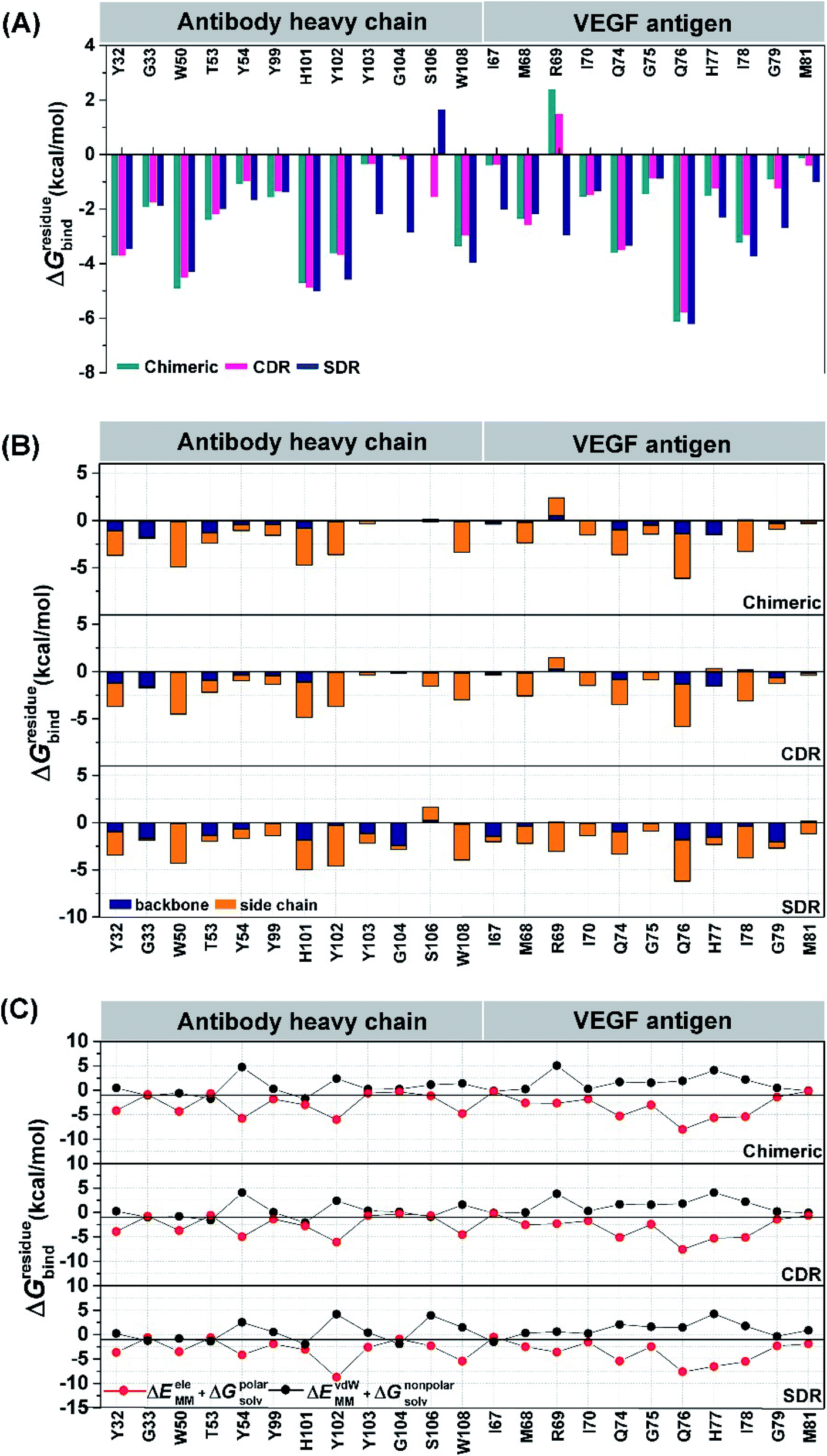
To elucidate the residue contribution in the protein–protein binding, the per-residue decomposition free energy (ΔGresiduebind) was calculated by the MM-PBSA method using the same set of snapshots used above. Since the binding interactions of the three systems are largely derived from the heavy chain of antibodies, their energy contributions are plotted to the left of Fig. 4, while those of the contacted VEGF residues are illustrated to the right of Fig. 4. At the protein–protein interface, there are 12 mAb residues, namely Y32, G33, W50, T53, Y54, Y99, H101, Y102, Y103, G104, S106, and W108, in addition to 11 VEGF residues, namely I67, M68, R69, I70, Q74, G75, Q76, H77, I78, G79, and M81 (ΔGresiduebind ≤−1 kcal mol−1 from at least one system). In the Fig. 4A, the residues S106 (1.64 kcal mol−1) destabilized protein–protein binding in SDR while the residue R69 was found to be destabilizing residue in chimeric (2.40 kcal mol−1) and CDR (1.48 kcal mol−1). These residues tend to stabilize protein–protein binding mostly through their side chains, as evidenced by the lowered free energy value (orange bars in Fig. 4B) relative to those of backbones (blue bars), except for the mAb residues G33, T53 and G104 and the VEGF residues I67, H77 and G79. Moreover, the results reveal that the major energy contribution for protein–protein binding from each residue comes from the nonpolar term (ΔEvdWMM + ΔGnonpolarsolv, see the red line in Fig. 4C). Some grafted mAb binding residues obtained from this work, such as H101 were also found in the Avastin-VEGF system, while the vdW force had an important role in the complexation of Avastin and Lucentis binding with this antigen.41
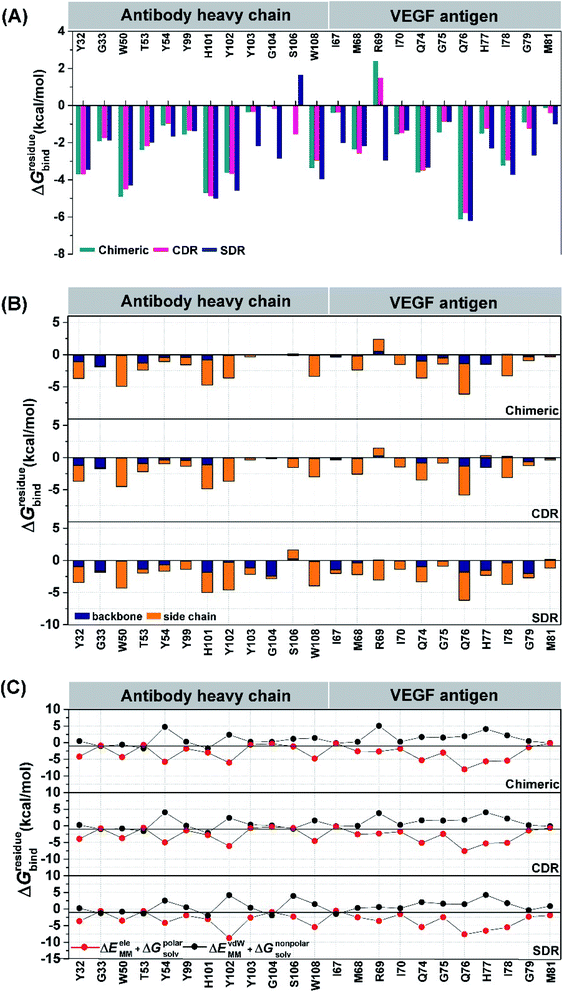 |
| | Fig. 4 Comparison of the crucial residues from the three studied mAbs binding to VEGF as a result of the MM-PBSA per-residue decomposition free energy calculation. The energy contribution from (A) residue and its backbone and side chain in (B) (C) Nonpolar (ΔEvdWMM + ΔGnonpolarsolv) and (ΔEeleMM + ΔGpolarnonsolv) components of each residue are also given. | |
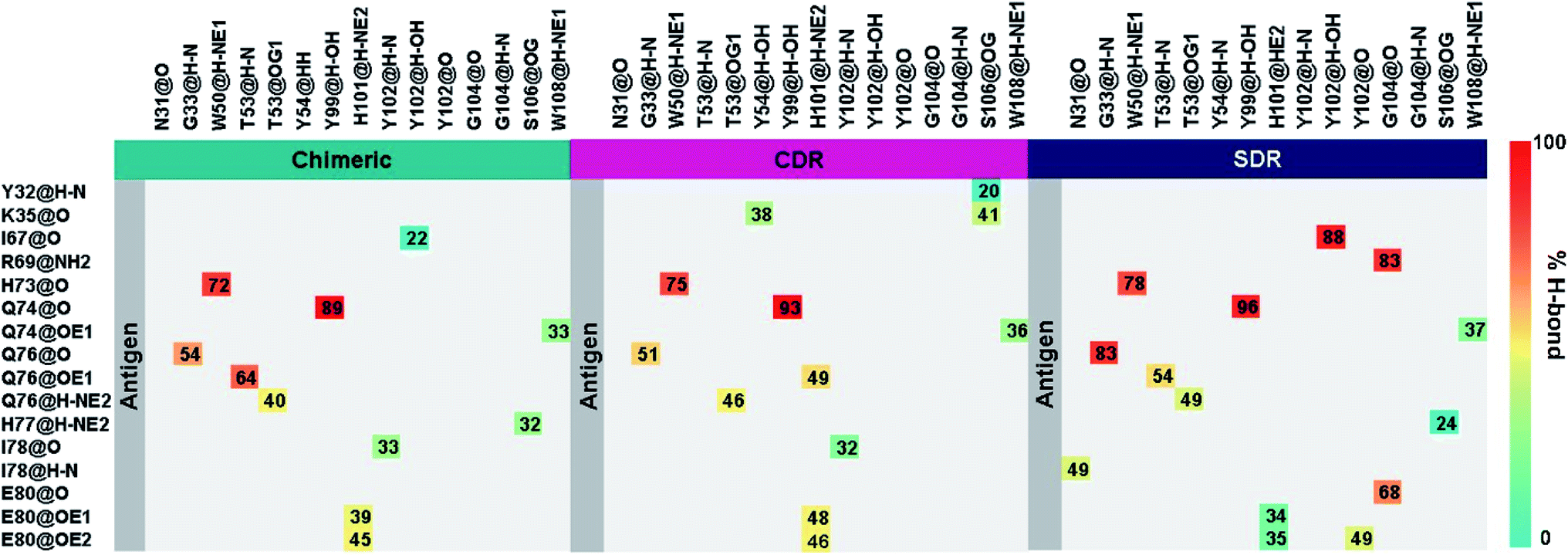
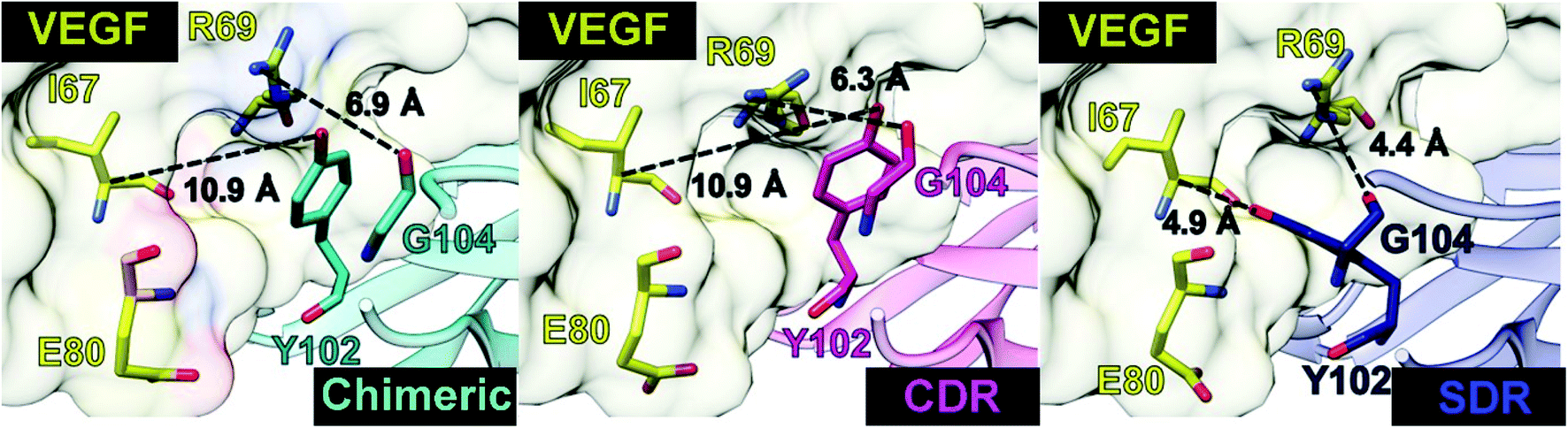
Intermolecular hydrogen bonding is also a significant factor in the formation of an antibody–antigen complex. All systems share a similar hydrogen bond pattern across the protein–protein interface, with 1 to 4 strong hydrogen bonds (≥ 80% occupation, red gradient in Fig. 5). The G33, W50, Y99, Y102, and G104 of SDR mAb strongly interacts with VEGF at the residues I67, R69, H73, Q74, and Q76. Meanwhile, the strong interactions with the last two residues dramatically reduced in chimeric and CDR systems. The Y102 nitrogen backbone of chimeric only formed weak hydrogen bonds (22 and 33% occupation) with I67 and I78 residues of VEGF, while Y102 and G104 of CDR mAbs totally lost their hydrogen bond interactions with I67 and R69 residues of VEGF (also see the averaged distance between the mAb Y102 phenolic oxygen and the VEGF I67 Cα atom, and the averaged distance between the mAb G104 carbonyl oxygen and the VEGF R69 NH group in Fig. 6). This information corresponds with the ΔGresiduebind data (Fig. 4A), in which of the three studied mAbs, most of the important residues in the heavy chain of SDR presented a relatively lower energy contribution. In particular, Y102 and G104 (∼1 and ∼3 kcal mol−1 energy stabilization higher than the rest of the systems) and their hydrogen bond pairs (I67 and R69 residues of VEGF, respectively) provided a greater energy stabilization compared to those of chimeric and CDR.
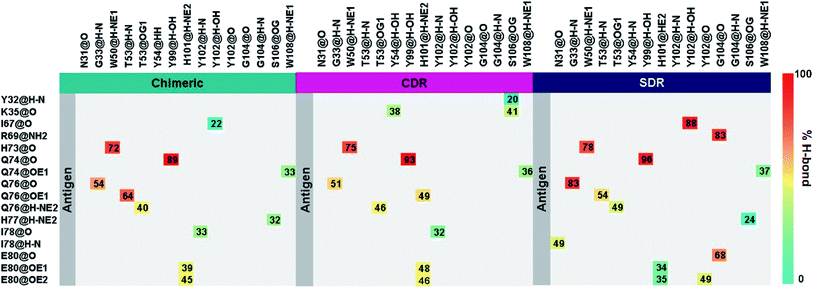 |
| | Fig. 5 Percentage of intermolecular hydrogen bond occupation of the three grafted mAbs-VEGF complexes, as determined using the following criteria: (i) Distance between the hydrogen donor (HD) and acceptor (HA) ≤ 3.0 Å; and (ii) the angle of HD-H…HA ≥120°. | |
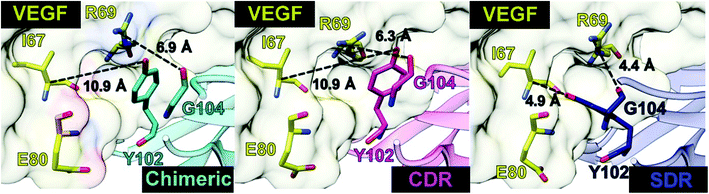 |
| | Fig. 6 Distances related to the two hydrogen bond pairs—Y102 (mAbs)—I67(VEGF) and G104 (mAbs)—R69(VEGF)—established on the protein–protein interface. | |
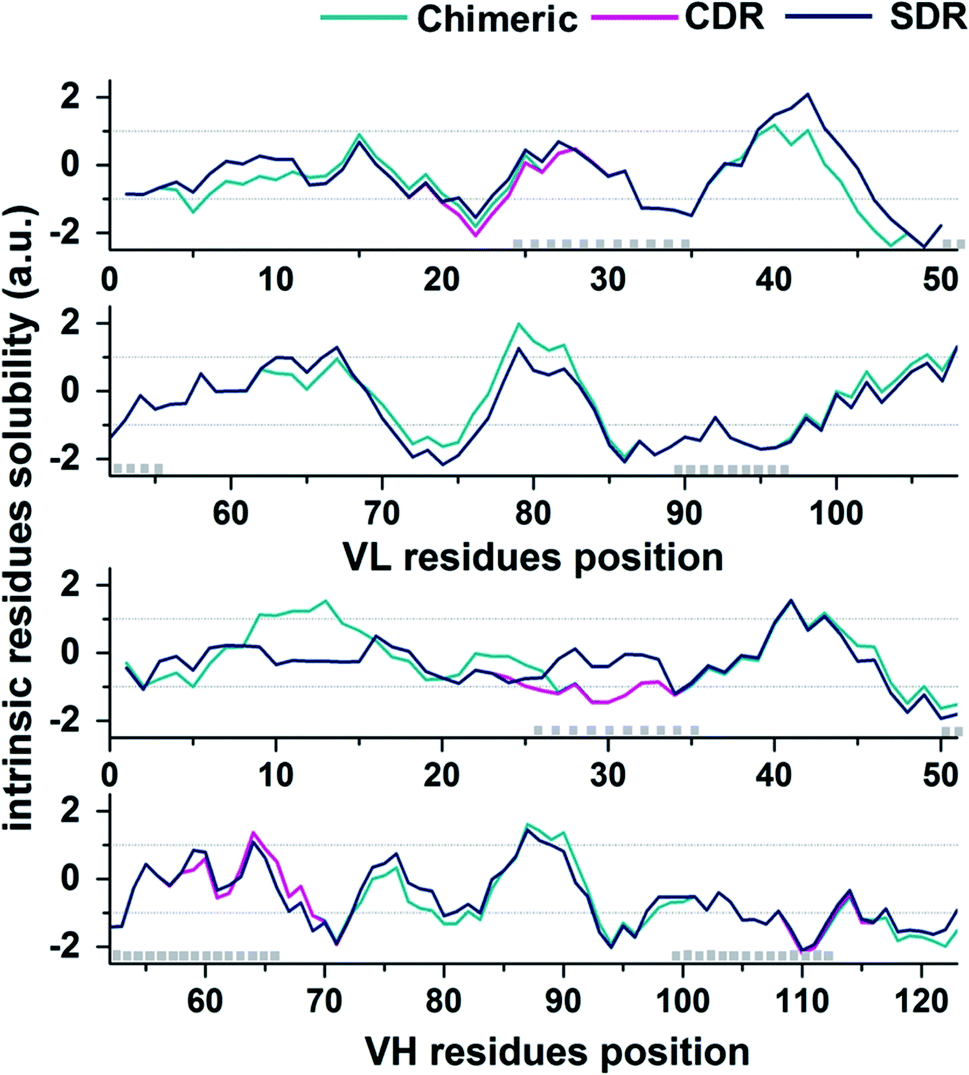
In the present study, the prediction of the solubility for the three grafted mAbs against VEGF was performed by the Camsol method29 using protein primary sequence information as the input. The accuracy of the Camsol method accuracy is highest compared to SOLpro46 and PROSO II.47 This method was used to successfully predict the solubility of three IgG mAbs (WFL, STT, and mAb1).48 Moreover, the algorithm has also been used to suggest structure mutations of β2-microglobulin to enhance solubility without impacting its structure or stability.49 Since the constant domains of all three grafted mAb are based on the same scaffold, meaning that they contain the same amino acid sequences, only VL and VH domains sequences were used to predict the solubility in each system. The intrinsic solubility profiles for the VL and VH domain of chimeric (green), CDR (pink), and SDR (blue) are shown in Fig. 7. Scores higher than +1 indicate higher protein solubility, values close to 0 are predicted to exert a negligible impact, while values less than −1 are considered to be proteins with poor solubility. The intrinsic solubility profiles of the three grafted antibodies on VL, VH, and combined VL and VH were calculated to generate the CamSol intrinsic scores (a.u.) (Table 2). As expected, the profile for chimeric antibody is quite different from the humanized antibodies since a large number of amino acid residues are different. Since humanized antibodies are more clinically desirable, we focused on comparing the CDR and SDR intrinsic solubility profiles. It can be observed that the CamSol intrinsic solubility profiles of the VL domains of CDR and SDR are very similar, except for the region closed to the CDR-L1, where S24 in the CDR graft was changed to R24 in SDR graft, causing the score VL score differences in CDR and SDR. Interestingly, the VH domains showed large differences, especially in the residue solubility profiles (−1.05 a.u.).
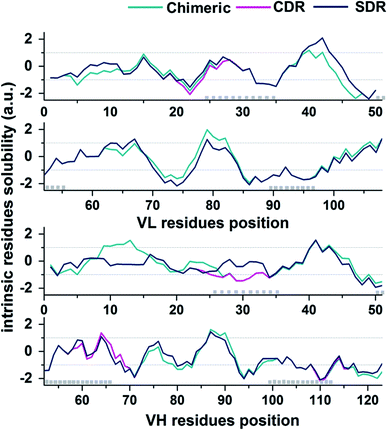 |
| | Fig. 7 CamSol solubility profiles for the VL (top plot) and VH (lower plot) domains of chimeric (green), CDK (pink), and SDR (blue). CDR positions are represented by grey boxes (Kabat annotation). | |
Table 2 Solubility scores for the sequences of the VH domain, VL domain, and combined VH-VL complex of the studied mAbs
| Grafted mAbs |
CamSol intrinsic score (a.u.) |
| VL |
VH |
VH-VL |
| Chimeric |
−0.057 |
−0.113 |
−0.825 |
| CDR |
−0.152 |
−0.097 |
−0.878 |
| SDR |
−0.040 |
−0.115 |
−0.586 |
3.5 Solubility of chimeric and humanized antibodies
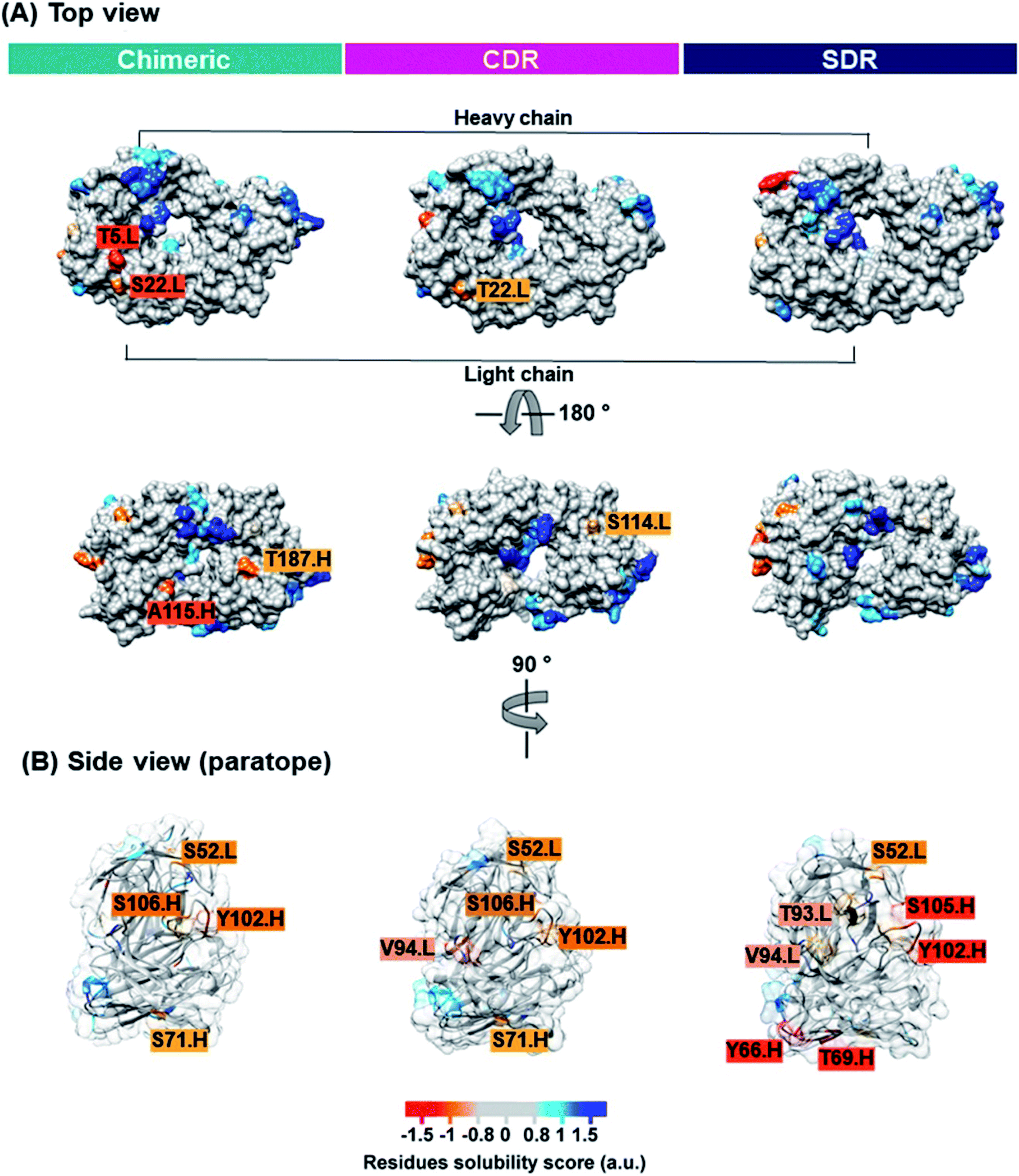
Solubility is a key property that is typically determined during mAb development. Inadequate mAb solubility can result in aggregation under typical storage conditions. Furthermore, self-aggregation of mAbs can result in the loss of function and can also be associated with patient immune responses.43 Many intrinsic properties of proteins can affect protein solubility, such as size, hydrophobicity, electrostatics, and charge distribution.44 Therefore, the computational prediction of solubility of the developed antibodies should be investigated prior to beginning a cost- and-time experiment. Several methods for protein solubility prediction have been reviewed.45 Compared to SDR (0.10 a.u.). The side chains of lysine (a positively charged amino acid), and asparagine (a polar amino acid found in the CDR-H1 of SDR) may have preferential interactions with water, therefore resulting in increased solubility. The obtained result concurs well with previous studies of amino acid contributions to protein solubility.50 Overall, SDR (−0.586 a.u.) exhibited the highest solubility, followed by chimeric (−0.825 a.u.) and CDR (−0.878 a.u.), as represented by the VL-VH combined CamSol intrinsic score. To predict the tendency to aggregate the residues of individual conformations, we ran the Camsol structurally corrected algorithm on the final snapshot structure from the MD simulation of the variable, as well as the constant domain of the three complexes (Fig. 8). The analysis suggests that CDR and SDR decreased the number of aggregation-prone residues compared to chimeric. In particular, the residues from the light chain (T5 and S114) and from the heavy chain (A115 and T187) increased solubility in SDR (Fig. 8A). This result is in accordance with the Camsol intrinsic solubility score. The aggregation-prone residues of all the systems are mostly located in CDR3 of the paratope region (Fig. 8B). However, these aggregation-prone residues in the paratope region require further improvement.
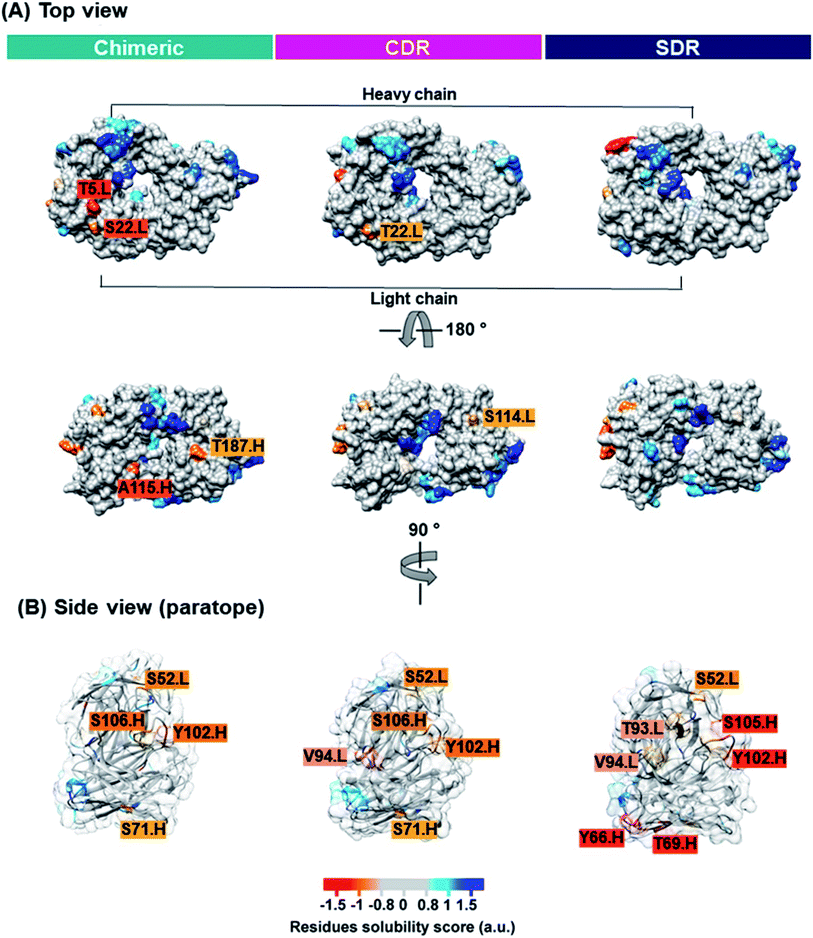 |
| | Fig. 8 The CamSol structurally corrected profile is color-coded on the surface of the three grafted mAbs inside view (A) and paratope region (B) to identify potential self-assembly hotspots. Residue solubility scores with values over 1 (blue) indicate highly soluble proteins, values close to 0 (grey) are predicted to exert a negligible impact, while values below −1 (red) are considered to have a negative effect on solubility. | |
3.6 Cytoplasmic expression analysis of the three grafted mAbs in the E. coli SHuffle strain
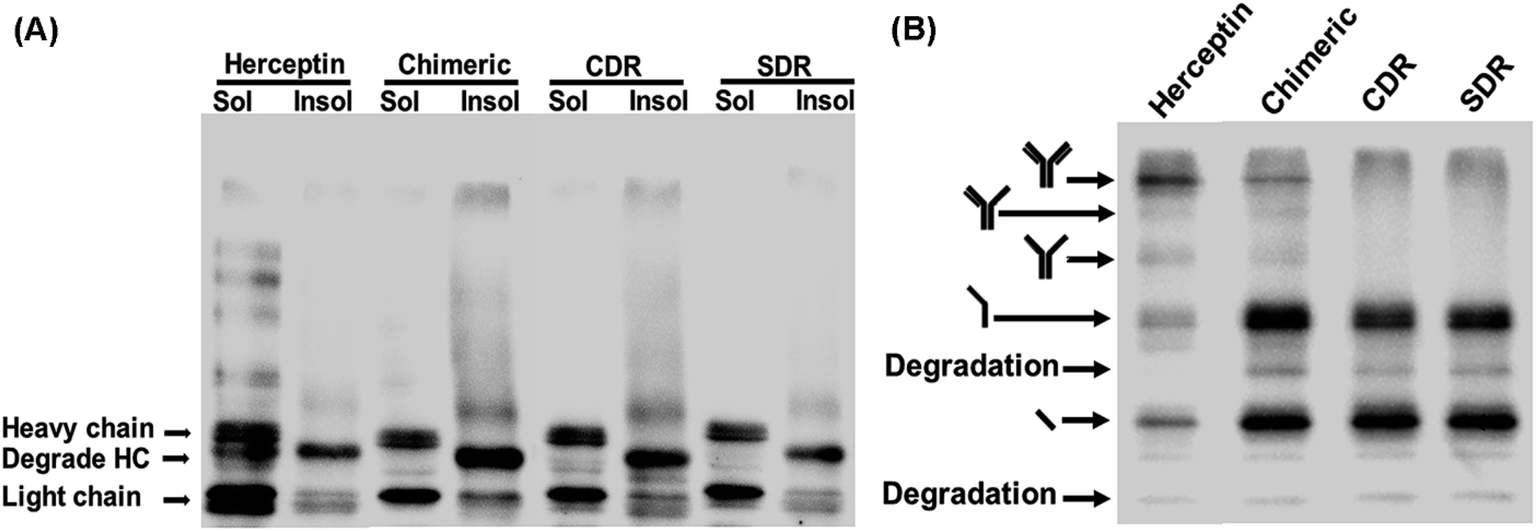
To investigate the expression of the designed grafted mAbs in the cytoplasm of the engineered SHuffle strain, gene synthesis corresponding to the amino acid sequences designed in Section 2.1 were ordered from GenScript (USA) with codon optimization for E. coli expression and cloned into pMAZ360-cIgG-Herceptin, which was used as template for all the grafted mAbs 16. This plasmid was designed for cytoplasmic IgG expression as cyclonal, so the full-length and properly assembled IgGs were produced from a bicistronic operon comprised of light (VL-CL) and heavy (VH-CH1-CH2-CH3) chain genes without signal peptide used for periplasmic export, and were under an IPTG inducible promoter. Western blot analysis was performed with soluble and insoluble fractions to evaluate the solubility of the grafted mAbs. Food and Drug Administration approved humanized IgG, Trastuzumab or Herceptin, was used as a template and was also investigated. It is found that Chimeric and CDR showed a higher amount of insoluble proteins which was observed as a smear on the western blot, while SDR had a much lower amount of insoluble proteins (Fig. 9A), which concurs with the prediction that SDR is the most soluble of all the studied mAbs. Unfortunately, the ladder pattern showing various assembled constructs were not observed in the grafted mAbs, while the ladder pattern was clearly observed for Herceptin. These cyclonals were further investigated by purifying the antibodies using protein A chromatography, while the eluates where subjected to western blot analysis. After purification small amounts of full-length IgG from Chimeric antibody were observed, while CDR and SDR where not observed (Fig. 9B). The inability to spontaneously form full-length IgGs of the grafted mAbs may be due to incompatibility between the template (Herceptin) and the grafted sequences. These results suggest that grafting by only replacing the variable domain, CDR, or SDR may not result in a compatible structure when produced experimentally inside the cell, thus emphasizing the usefulness and practicality of computer modeling to improve antibody humanization.
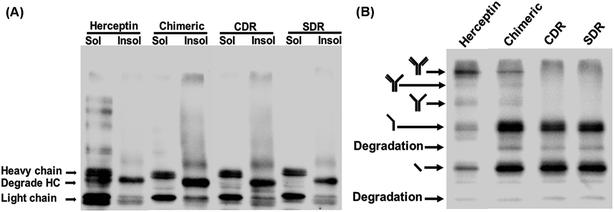 |
| | Fig. 9 Western blot analysis of SDR-, CDR-, and chimeric-grafted anti-VEGF antibodies compared to Herceptin. Representative non-reducing western blot of IgGs expressed at 30 °C showing (A) soluble and insoluble fractions and (B) purified samples probed with rabbit anti-human IgG H&L HRP. | |
3.7 Binding affinity and solubility improvement
From the above computational investigation into the binding affinity of the three grafted mAbs, the SDR mAb exhibited the strongest binding to VEGF. Nonetheless, the residue S106 on the protein–protein interface of SDR mAb showed a destabilization in complex with VEGF (1.64 kcal mol−1, Fig. 4A). To improve the SDR binding efficiency, this residue was rationally mutated to the negatively charged residues as S106D and S106E in order to increase electrostatic attraction with the surface residue H77 of VEGF. The docking score in Table 3 suggests that the S106D substitution in SDR mAb improved the binding affinity by up to 78.6% higher when compared to WT, while the change of this residue to glutamate means that it has a comparative binding affinity with that of WT. This could be due to the suitable size of the side chain of aspartate which interacts with the imidazole ring of VEGF H77. Additionally, the S106D mutation in SDR decreased the tendency to aggregate as seen by CamSol intrinsic score increasing from −0.649 a.u. (WT) to −0.555 a.u (S106D).
Table 3 Predicted binding affinity of single and double mutations of SDR antibody binding to VEGF antigen compared to the wild type (WT) using ZDOCK server,27 and their solubility in terms of the CamSol intrinsic score determined by the Camsol program51
| Variant name |
ZDOCK score |
CamSol intrinsic score (a.u.) |
| WT |
1431.74 |
−0.649 |
|
| Single mutation |
| S106D |
2556.38 |
−0.555 |
| S106E |
1752.13 |
−0.547 |
4. Conclusions
The chimeric and two humanized antibodies (CDR and SDR) were designed to remove murine amino acid sequences. The variable regions, CDR and SDR and muMAb A.4.6.1 was grafted onto the amino acid sequence of Herceptin. Then, the mechanism of these grafted antibodies in complex with VEGF were studies at the molecular level by all-atom MD simulation in an aqueous solution. From the MM-PB (GB)SA binding free energy results, the SDR–VEGF complex showed a significantly greater binding affinity than chimeric and CDR. The key residues responsible for protein–protein binding were the 12 mAb residues, namely Y32, G33, W50, T53, Y54, Y99, H101, Y102, Y103, G104, S106, and W108, and the 11 VEGF residues, namely I67, M68, R69, I70, Q74, G75, Q76, H77, I78, G79, and M81. Interestingly, Y102 and G104 residues were found to strongly interact with VEGF at the residues I67, R69 whereas these interaction dramatically reduced in chimeric and completely lost their hydrogen bond interactions in CDR systems. Solubility is an important property in the process of developing antibody therapeutic. All grafted antibodies were predicted and confirmed by computational and experimental method. SDR mAb exhibited the highest solubility. However, the residue S106 on the protein–protein interface of SDR mAb showed a destabilization in complex with VEGF and appeared to have aggregation-prone residues in the paratope region. From this point, the SDR was improved by S106D mutation to have high solubility and maintain its binding affinity. The S106D mAb improved solubility by up to 16% and binding affinity by up to 78.6% higher than WT.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Thailand Research Fund (TRF) [grant number RSA6280008], the Research Strengthening Project of the Faculty of Engineering, King Mongkut's University of Technology Thonburi, and the Thailand Science Research and Innovation (TSRI), Basic Research Fund: Fiscal year 2021 under project number 64A306000003.
References
- E. Seccamani, M. Tattanelli, M. Mariani, E. Spranzi, G. Scassellati and A. Siccardi, Int. J. Radiat. Appl. Instrum., Part B, 1989, 16, 167–170 CrossRef CAS.
- D. Colcher, D. E. Milenic, P. Ferroni, J. A. Carrasquillo, J. C. Reynolds, M. Roselli, S. M. Larson and J. Schlom, J. Nucl. Med., 1990, 31, 1133–1142 CAS.
- D. G. Williams, D. J. Matthews and T. Jones, Antibody Engineering, Springer, 2010, pp. 319–339 Search PubMed.
- B. W. Newsome and M. S. Ernstoff, Br. J. Clin. Pharmacol., 2008, 66, 6–19 CrossRef CAS.
- D. Colcher, D. E. Milenic, P. Ferroni, J. A. Carrasquillo, J. C. Reynolds, M. Roselli, S. M. Larson and J. Schlom, J. Nucl. Med., 1990, 31, 1133–1142 CAS.
- S. V. Kashmiri, R. De Pascalis, N. R. Gonzales and J. Schlom, Methods, 2005, 36, 25–34 CrossRef CAS.
- P. Chames, M. Van Regenmortel, E. Weiss and D. Baty, Br. J. Pharmacol., 2009, 157, 220–233 CrossRef CAS.
- A. R. Aricescu, W. Lu and E. Y. Jones, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 1243–1250 CrossRef.
- R. Hakim and I. Benhar, mAbs, 2009, 1, 281–287 CrossRef.
- C.-J. Huang, H. Lin and X. Yang, J. Ind. Microbiol. Biotechnol., 2012, 39, 383–399 CrossRef CAS.
- R. Hakim and I. Benhar, mAbs, 2009, 1, 281–287 CrossRef.
- C. E. Z. Chan, A. P. C. Lim, A. H. Y. Chan, P. A. MacAry and B. J. Hanson, PLoS One, 2010, 5(5), e10261 CrossRef.
- T. Makino, G. Skretas, T.-H. Kang and G. Georgiou, Metab. Eng., 2011, 13, 241–251 CrossRef CAS.
- J. Lobstein, C. A. Emrich, C. Jeans, M. Faulkner, P. Riggs and M. Berkmen, Microb. Cell Fact., 2012, 11, 753 CrossRef.
- M.-P. Robinson, N. Ke, J. Lobstein, C. Peterson, A. Szkodny, T. J. Mansell, C. Tuckey, P. D. Riggs, P. A. Colussi and C. J. Noren, Nat. Commun., 2015, 6, 1–9 CAS.
- M.-P. Robinson, N. Ke, J. Lobstein, C. Peterson, A. Szkodny, T. J. Mansell, C. Tuckey, P. D. Riggs, P. A. Colussi, C. J. Noren, C. H. Taron, M. P. DeLisa and M. Berkmen, Nat. Commun., 2015, 6, 8072 CrossRef CAS.
- T. A. Hall, Nucleic Acids Symp. Ser., 1999, 41, 95–98 CAS.
- Y. A. Muller, Y. Chen, H. W. Christinger, B. Li, B. C. Cunningham, H. B. Lowman and A. M. de Vos, Structure, 1998, 6, 1153–1167 CrossRef CAS.
- L. G. Presta, H. Chen, S. J. O'connor, V. Chisholm, Y. G. Meng, L. Krummen, M. Winkler and N. Ferrara, Cancer Res., 1997, 57, 4593–4599 CAS.
- E. A. Padlan, C. Abergel and J. P. Tipper, FASEB J., 1995, 9, 133–139 CrossRef CAS.
- M. H. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS.
- D. Case, R. Betz, D. S. Cerutti, T. Cheatham, T. Darden, R. Duke, T. J. Giese, H. Gohlke, A. Götz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T.-S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko and P. Kollman, Amber 16, University of California, San Francisco, 2016 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- D. M. York, T. A. Darden and L. G. Pedersen, J. Chem. Phys., 1993, 99, 8345–8348 CrossRef CAS.
- J. Phanich, T. Rungrotmongkol, N. Kungwan and S. Hannongbua, J. Comput.-Aided Mol. Des., 2016, 30, 917–926 CrossRef CAS.
- F. Fanelli and S. Ferrari, J. Struct. Biol., 2006, 153, 278–283 Search PubMed.
- P. Sormanni, L. Amery, S. Ekizoglou, M. Vendruscolo and B. Popovic, Sci. Rep., 2017, 7, 8200 CrossRef.
- P. Sormanni, F. A. Aprile and M. Vendruscolo, J. Mol. Biol., 2015, 427, 478–490 CrossRef CAS.
- W.-L. Ling, W.-H. Lua, J.-J. Poh, J. Y. Yeo, D. P. Lane and S. K.-E. Gan, Front. Immunol., 2018, 9, 469 CrossRef.
- Y.-F. Zhang and M. Ho, Sci. Rep., 2017, 6, 33878 CrossRef.
- M. Duvall, N. Bradley and R. N. Fiorini, mAbs, 2011, 3(2), 203–208 CrossRef.
- E. A. Kabat, T. Te Wu, H. M. Perry, C. Foeller and K. S. Gottesman, Sequences of proteins of immunological interest, DIANE publishing, 1992 Search PubMed.
- C. Chothia and A. M. Lesk, J. Mol. Biol., 1987, 196, 901–917 CrossRef CAS.
- E. A. Padlan, C. Abergel and J. Tipper, FASEB J., 1995, 9, 133–139 CrossRef CAS.
- B. D. Weitzner, R. L. Dunbrack Jr and J. J. Gray, Structure, 2015, 23, 302–311 CrossRef CAS.
- B. Jawad, L. Poudel, R. Podgornik, N. F. Steinmetz and W.-Y. Ching, Phys. Chem. Chem. Phys., 2019, 21, 3877–3893 RSC.
- G. Weng, E. Wang, F. Chen, H. Sun, Z. Wang and T. Hou, Phys. Chem. Chem. Phys., 2019, 21, 10135–10145 RSC.
- L. Wang, Q.-C. Bao, X.-L. Xu, F. Jiang, K. Gu, Z.-Y. Jiang, X.-J. Zhang, X.-K. Guo, Q.-D. You and H.-P. Sun, RSC Adv., 2015, 5, 96138–96145 RSC.
- Y. Sun and Q. Liu, RSC Adv., 2014, 5, 2318–2327 RSC.
- C. B. M. Platania, L. Di Paola, G. M. Leggio, G. L. Romano, F. Drago, S. Salomone and C. Bucolo, Front. Pharmacol., 2015, 6, 248 CrossRef.
- W.-C. Liang, X. Wu, F. V. Peale, C. V. Lee, Y. G. Meng, J. Gutierrez, L. Fu, A. K. Malik, H.-P. Gerber and N. Ferrara, J. Biol. Chem., 2006, 281, 951–961 CrossRef CAS.
- K. D. Ratanji, J. P. Derrick, R. J. Dearman and I. Kimber, J. Immunotoxicol., 2014, 11, 99–109 CrossRef CAS.
- S. S. Strickler, A. V. Gribenko, A. V. Gribenko, T. R. Keiffer, J. Tomlinson, T. Reihle, V. V. Loladze and G. I. Makhatadze, Biochemistry, 2006, 45, 2761–2766 CrossRef CAS.
- S. Navarro and S. Ventura, Expert Opin. Drug Discovery, 2019, 14, 1077–1088 CrossRef CAS.
- C. N. Magnan, A. Randall and P. Baldi, Bioinformatics, 2009, 25, 2200–2207 CrossRef CAS.
- P. Smialowski, G. Doose, P. Torkler, S. Kaufmann and D. Frishman, FEBS J., 2012, 279, 2192–2200 CrossRef CAS.
- L. F. Willis, A. Kumar, J. Dobson, N. J. Bond, D. Lowe, R. Turner, S. E. Radford, N. Kapur and D. J. Brockwell, Biotechnol. Bioeng., 2018, 115, 1216–1225 CrossRef CAS.
- C. Camilloni, B. M. Sala, P. Sormanni, R. Porcari, A. Corazza, M. De Rosa, S. Zanini, A. Barbiroli, G. Esposito and M. Bolognesi, Sci. Rep., 2016, 6, 25559 CrossRef CAS.
- L. K. Mosavi and Z. y. Peng, Protein Eng., Des. Sel., 2003, 16, 739–745 CrossRef CAS.
- P. Sormanni, L. Amery, S. Ekizoglou, M. Vendruscolo and B. Popovic, Sci. Rep., 2017, 7, 1–9 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Dujduan Waraho-zhmayev*a,
Korakod Haonoob,
Sarun Torpaiboonb,
Tarin Teacharsripaiboonb,
Thanyada Rungrotmongkol
a,
Dujduan Waraho-zhmayev*a,
Korakod Haonoob,
Sarun Torpaiboonb,
Tarin Teacharsripaiboonb,
Thanyada Rungrotmongkol