
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-small Fe3O4 nanodots encapsulated in layered carbon nanosheets with fast kinetics for lithium/potassium-ion battery anodes†
Qianqian Peng‡
a,
Chuan Guo‡a,
Shuo Qia,
Weiwei Suna,
Li-Ping Lv a,
Fei-Hu Dua,
Baofeng Wangb,
Shuangqiang Chen*a and
Yong Wang*a
a,
Fei-Hu Dua,
Baofeng Wangb,
Shuangqiang Chen*a and
Yong Wang*a
aDepartment of Chemical Engineering, School of Environmental and Chemical Engineering, Shanghai University, 99 Shangda Road, Shanghai, 200444, P. R. China. E-mail: chensq@shu.edu.cn; yongwang@shu.edu.cn; Tel: +86-21-66136598
bShanghai Key Laboratory of Materials Protection and Advanced Materials in Electric Power, Shanghai University of Electric Power, Shanghai 200090, China
First published on 4th January 2021
Abstract
Iron oxides are regarded as promising anodes for both lithium-ion batteries (LIBs) and potassium-ion batteries (KIBs) due to their high theoretical capacity, abundant reserves, and low cost, but they are also facing great challenges due to the sluggish reaction kinetics, low electronic conductivity, huge volume change, and unstable electrode interphases. Moreover, iron oxides are normally prepared at high temperature, forming large particles because of Ostwald ripening, and exhibiting low electronic/ionic conductivity and unfavorable mechanical stability. To address those issues, herein, we have synthesized ultra-small Fe3O4 nanodots encapsulated in layered carbon nanosheets (Fe3O4@LCS), using the coordination interaction between catechol and Fe3+, demonstrating fast reaction kinetics, high capacity, and typical capacitive-controlled electrochemical behaviors. Such Fe3O4@LCS nanocomposites were derived from coordination compounds with layered structures via van der Waals's force. Fe3O4@LCS-500 (annealed at 500 °C) nanocomposites have displayed attractive features of ultra-small particle size (∼5 nm), high surface area, mesoporous and layered feature. When used as anodes, Fe3O4@LCS-500 nanocomposites delivered exceptional electrochemical performances of high reversible capacity, excellent cycle stability and rate performance for both LIBs and KIBs. Such exceptional performances are highly associated with features of Fe3O4@LCS-500 nanocomposites in shortening Li/K ion diffusion length, fast reaction kinetics, high electronic/ionic conductivity, and robust electrode interphase stability.
Introduction
In the last three decades, rechargeable lithium-ion batteries (LIBs) have been widely used in portable electronic devices, stationary energy storage stations, and electric vehicles due to their high energy density, high coulombic efficiency, and long cycle life.1–5 However, the gap between limited element reserves and vast demands for Li resources is getting larger because of the large-scale application of LIBs.6–9 Therefore, the development of an alternative energy storage system is quite urgent.10 Potassium ion batteries (KIBs) have attracted wide interest due to even distribution, low cost, and similar electrochemical behaviors.11 At first glance, KIBs have no advantage over LIBs, as the cation radius of K (0.138 nm) is larger than that of Li (0.076 nm), and has higher redox potential (−2.93 V) compared to Li (−3.04 V).12–14 However, the abundance of K (2.09 wt%) on Earth is thousands of times higher than that of Li (0.0017 wt%), and more even distribution.15,16 In the last few years, extensive efforts have been devoted to demonstrate the application possibilities for both LIBs and KIBs, achieving many important progresses.11,17–19 However, great challenges are remained to develop suitable and cheap anode materials for both LIBs and KIBs with features of high capacity, good rate capability, and long cycle life.Transition metal oxides (TMOs, M = Mn, Fe, Ni, Co, Cu, etc.) have attracted many attentions due to their high capacities (around 1000 mA h g−1), natural abundance, and environmental friendliness.12,20–23 Among them, Fe3O4 is regarded as one of the most promising anode candidates for both LIBs and KIBs due to the high theoretical capacity (926 mA h g−1),4 natural abundance, low cost, and non-toxic feature.24,25 However, there are still many challenges remained for Fe3O4 materials in LIBs and KIBs on the following aspects: (1) large volume changes during conversion reactions with Li/K cations, resulting in severe electrode pulverization and contact loss from current collectors; (2) poor ionic and electronic conductivity, leading to the low ionic diffusion coefficient and high electrical resistance; (3) unstable solid electrolyte interface (SEI), giving low coulombic efficiencies; (4) large particles size, relating to long durations of chemical diffusion.26,27 To overcome these drawbacks, several effective strategies are reported to enhance the electrochemical performances, including nanometerization,19,28 carbon coating,29,30 heteroatom-doping,31–34 and surface-engineering.35–37 Nanometerization is normally applied for minimizing particle size and shortening diffusion pathways, but Ostwald ripening inevitably happens during hydrothermal and annealing treatments, leading to particle aggregations.38 Carbon coating is one kind of universal route to immensely enhance the electronic conductivity, providing fast charge transferring and robust structural stability.39 Heteroatom-doping is adopted on enhancing ionic conductivity for better rate capability.32,40 Surface-engineering works on the SEI layer with better mechanical stability, delivering long cycle life.3,41 Metal coordination compounds with high surface area and porous feature are considered as good precursors to synthesize electrode materials.3,42 Various TMOs materials with mesoporous and graphitic carbon skeleton, derived from metal coordination compound's precursors, are synthesized for anodes of LIBs/KIBs.43,44
To enhance the rate capability and cycling performance of Fe3O4-based material, layered iron coordination compound with the catechol–iron coordination were synthesized to further prepare ultra-small Fe3O4 nanodots (∼5 nm in average particle size) encapsulated in thin layered carbon sheets (∼4 nm in thickness, labeled as Fe3O4@LCS). Such Fe3O4@LCS nanocomposites have displayed high surface area, thin-layered, and mesoporous structure. Different Fe3O4@LCS nanocomposites, obtained at different temperatures, were applied as anodes in LIBs/KIBs, demonstrating high reversible capacity, good cycle stability, and rate performance. These exceptional electrochemical behaviors are highly ascribed to the elaborately designed material features on shortening Li/K ion's diffusion length, fast reaction kinetics, high electronic conductivity, and robust structure stability.29,45
Experimental
Chemicals
Catechol (C6H4(OH)2, >98%), iron(III) nitrate hexahydrate (Fe(NO3)3·9H2O, >98.5%), N,N-dimethylformamide (C3H7NO, DMF, >99.5%), ethanol (ET, >95%) were purchased from Sinopharm Chemical Reagent Co. Ltd. All the chemicals were used without further purification. Deionized water (DI) was obtained from a Milli-Q System (Millipore).Preparation of metal coordination precursor
In a typical synthesis procedure, catechol (0.11 g) was added into mixed solvent (9 mL) of DI/ET/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, v/v/v) with continuous stirrings for 15 min as solution A. Iron(III) nitrate hexahydrate (0.808 g) was dissolved in mixed solvents with the same composition as solution B. Solution B was added drop by drop in solution A with continuous stirrings for another 30 min, and then treated at 150 °C for 1 h using a single-mode microwave (NOVA-2S, 1000 W) radiation reactor with digital-controlled temperature and pressure apparatus. The products were collected by centrifugation, washed with deionized water and ethanol for 3 times, and then dried at 60 °C overnight.
:1:1, v/v/v) with continuous stirrings for 15 min as solution A. Iron(III) nitrate hexahydrate (0.808 g) was dissolved in mixed solvents with the same composition as solution B. Solution B was added drop by drop in solution A with continuous stirrings for another 30 min, and then treated at 150 °C for 1 h using a single-mode microwave (NOVA-2S, 1000 W) radiation reactor with digital-controlled temperature and pressure apparatus. The products were collected by centrifugation, washed with deionized water and ethanol for 3 times, and then dried at 60 °C overnight.
Preparation of Fe3O4/LCS nanomaterials
The precursors were placed in a tube furnace and heated to 400, 500, 600 °C for 2 h in N2 with a ramp rate of 2 °C min−1 respectively. After cooling, ultra-small Fe3O4 nanodots were successfully encapsulated in porous carbon nanosheets and marked as Fe3O4@LCS-400/500/600. As a comparison, Fe3O4 bulk was prepared via similar procedures without the addition of catechol.Materials characterizations
The structural information and morphology were characterized by an X-ray diffractometer (XRD, Rigaku D/max-2550V) and field emission scanning electron microscope (FE-SEM, JSM-6700F) as well as transmission electron microscope (TEM, JEM-2010F). Raman spectra were collected on a laser Raman spectrometer (Renishaw). Elemental analysis was measured by an Elemental Analyzer (Vario MICRO cube). Surface area and porous properties were measured on a Micromeritics ASAP 2460 analyzer. X-ray photoelectron spectrometer (XPS, PHI ESCA-5000C) was utilized to evaluate the surface compositions of composites before and after cycles.Electrochemical measurement
CR2032 coin cells were assembled in an Ar-filled glove box where the concentrations of H2O and O2 are kept below 0.1 ppm. Active materials (Fe3O4@LCS), conductive agent (acetylene black), and binder (polyvinylidene difluoride, PVDF) were uniformly dispersed in N-methyl pyrrolidone (NMP) (8:1:1, w/w/w) and coated onto copper foil, then dried at 60 °C overnight. The mass loading of working electrodes was about 1 mg cm−2. All capacities were calculated based on the whole weight of electrodes. For LIBs, lithium foil was used as the counter electrode. 1 M LiPF6 was dissolved in the mixed solvent (1:1, v/v) of ethylene carbonate (EC) and diethyl carbonate (DEC) as the electrolyte, and the microporous polypropylene (Celgard 2400) film was applied as the separator. However, potassium ion batteries were composed of pure potassium foil (counter electrode), 0.8 M KPF6 in a mixed solvent of EC/DEC (1:1, v/v) as the electrolyte, and glass microfiber filter membrane (Whatman, Grade GF/A) as the separator.
The galvanostatic charge/discharge and the galvanostatic intermittent titration technique (GITT) measurements were evaluated by LAND-CT2001A at different current densities (from 100 mA g−1 to 2000 mA g−1) with a voltage range of 5 mV to 3 V. Cyclic voltammetry (CV) was measured on a CHI 660D electrochemical working station. Electrochemical impedance spectroscopy (EIS) was performed on an Autolab PGDTAT101 electrochemical workstation with a frequency range of 0.1–1 × 106 Hz.
Results and discussion
Structure and morphology investigation
A series of Fe3O4@LCS nanocomposites were successfully prepared via a two-step process, as shown schematically in Fig. 1a. The Fe-precursor was firstly prepared by chemical coordination assisted by the microwave radiation at 150 °C, which can be detected by XRD and FTIR as shown in Fig. S2.† Iron cations performed as coordinator center binding with catechol as a ligand, forming a layered and stacked structure via van der Waals' force.25 With the homogenously distributed iron coordinator, ultra-small Fe3O4 nanodots were eventually isolated and encapsulated in porous carbon nanosheets, which were derived from carbonized catechol during the annealing process in N2 at 400 °C, 500 °C and 600 °C, avoiding the particle growth of Ostwald ripening. Moreover, there are many voids in carbon sheets containing Fe3O4 nanodots formed by adjacent atom losses, providing pre-reserved spaces for volume expansions.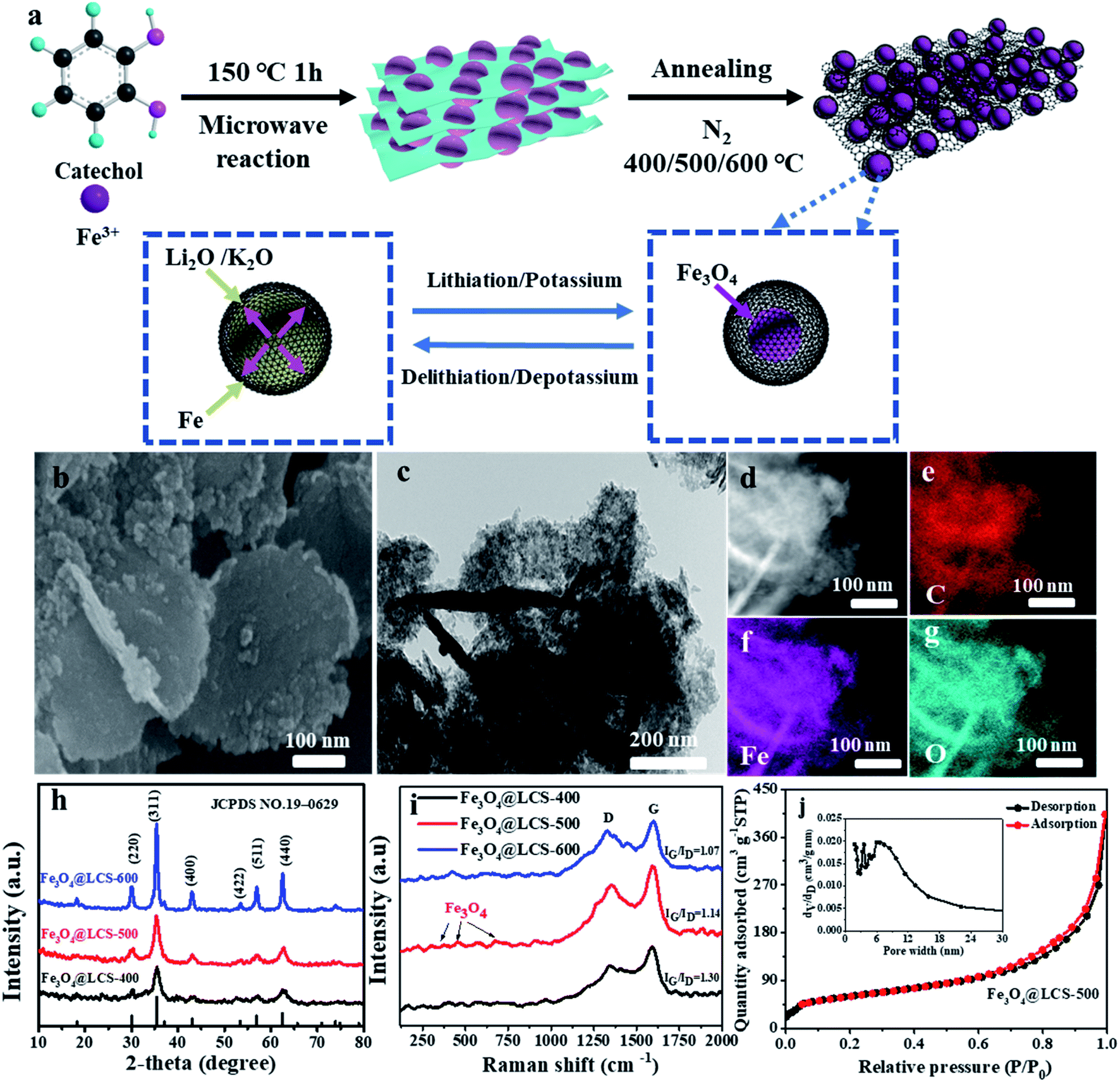 | ||
| Fig. 1 (a) The synthesis schematic of the derived Fe3O4@LCS (400/500/600) nanocomposites; (b) SEM images of Fe3O4@LCS-500; (c) TEM images of Fe3O4@LCS-500; (d–g) corresponding elemental maps of C, Fe, and O; (h) XRD patterns and (i) Raman spectra of Fe3O4@LCS (400/500/600) nanocomposites; (j) nitrogen absorption isotherm and the corresponding average pore size distribution of Fe3O4@LCS-500. | ||
Fe3O4@LCS-500 nanocomposites display a typical layered morphology with mutually perpendicular structures in Fig. 1b and c. No apparent particle aggregation is observed in the TEM image, demonstrating the ideal particle distributions of Fe3O4 nanodots. More morphology information is shown in the ESI (Fig. S1†) for a whole glance in different conditions. The elemental mapping images in Fig. 1d–g further indicate the homogenous distributions of C, Fe, and O in the Fe3O4@LCS-500 sample, illustrating the elaborately inherited architecture from layered Fe-coordination compound. The atomic percentages of all elements in Fe3O4@LCS (400/500/600) nanocomposites are shown in Table S1† for elemental comparison.
The phase information and purity of the as-prepared samples were shown in XRD patterns in Fig. 1h. The diffraction peaks of Fe3O4@LCS-400/500/600 samples are almost in the same positions. Specifically, all characteristic diffraction peaks at 30.1°, 35.6°, 43.3°, 53.4°, 57.3°, 62.6° are well assigned to the (220), (311), (400), (422), (511) and (220) planes of Fe3O4 (JCPDS: 19-0629), which implies the full phase transformations of the precursor into Fe3O4-based materials. Meanwhile, the XRD patterns of Fe-coordination compound and Fe3O4 bulk are shown in Fig. S2a and b.† The Raman spectra of all composites are shown in Fig. 1i. Typical peaks at 383, 460, and 684 cm−1 are assigned to Eg, T2g and A1g of Fe3O4.25,41 And peak positions of D band and G band are at ∼1351 cm−1 and 1601 cm−1, in which D band stands for the disordered structure or defects in carbon and G band represents for the graphitic structure. Besides the intensity ratios of ID/IG of Fe3O4@LCS (400/500/600) samples are 1.30, 1.14, and 1.07, respectively. As a higher ID/IG value means a higher defect level, a lower value will result in a better electronic conductivity and higher mechanical stability. Therefore, a higher annealing temperature will give a lower ID/IG ratio, relating to a better electronic conductivity and good mechanical stability as well as less defects.46 Furthermore, nitrogen absorption isotherms with pore size distributions of Fe3O4@LCS (400/500/600) nanocomposites were investigated by the BET (Brunauer, Emmett and Teller) method. As shown in Fig. 1j, the curve is coincided to the typical type IV H4 isothermal adsorption curve with an apparent hysteresis loop, displaying a high surface area of 213.84 m2 g−1 for Fe3O4@LCS-500 nanocomposites. Simultaneously, the relative average pore diameter was around 11.65 nm, leaving sufficient voids for volume changes during cycles. In contrast, the surface areas of Fe3O4@LCS (400/600) in Fig. S3a and b† were 158.22 m2 g−1 and 250.50 m2 g−1, respectively. There is an apparent trend between specific surface area and annealing temperature, namely a higher surface area at higher annealing temperature.
The fine structure of Fe3O4@LCS-500 nanocomposites was further analyzed by high-resolution TEM (HRTEM) in Fig. 2a, displaying good crystallization of Fe3O4 nanodots. The evenly distributed Fe3O4 nanodots with an average particle size of around 5 nm, verified by the statistical plot in Fig. 2b, are homogenously encapsulated by porous carbon matrix. The interplanar d-spacings (0.256 nm, and 0.296 nm) are corresponding to the (311) and (220) lattice planes of Fe3O4 in Fig. 2c and d. Other phase planes of (400), (511), and (440) are shown in the SAED pattern in Fig. 2e, exhibiting a polycrystalline nature. The average thickness of the Fe3O4@LCS-500 nanocomposites is around 4 nm based on atomic force microscopy (AFM) images in Fig. 2f–i, displaying an encapsulation structure because of the existence of single carbon shell with Fe3O4 nanodots. XPS technique was used to analyze the surficial valence state of Fe3O4@LCS-500 nanocomposites in Fig. 2j, illustrating the presence of Fe, C and O elements. The Fe 2p spectrum can be divided into five major peaks in Fig. 2k. The peaks of binding energy at 711.91 and 710.2 eV are to 2p3/2 of Fe3+ and Fe2+. And peaks at ∼724.9 eV and 722.2 eV are referring to 2p1/2 of Fe3+ and Fe2+, respectively. Meanwhile, the peak at 719.02 eV is regarded as a satellite peak for the above four peaks, demonstrating the formation of Fe3O4.24,25,41,46 The C 1s spectrum is shown in Fig. 2l, divided into three sub-peaks at about 284.1, 285.6, and 286.9 eV. Those peaks are assigned to the covalent bonds of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C, and C–O, respectively,3,47 indicating the existence of defects, in-plane structure, and surficial oxygen absorption. The O 1s spectrum in Fig. 2m shows peaks at about 529.6 and 531.6 eV, which are assigned to the oxygen states in Fe3O4 and C–O, and the peak at ∼533.7 eV is attributed to the surface adsorbed oxygen.
C, C–C, and C–O, respectively,3,47 indicating the existence of defects, in-plane structure, and surficial oxygen absorption. The O 1s spectrum in Fig. 2m shows peaks at about 529.6 and 531.6 eV, which are assigned to the oxygen states in Fe3O4 and C–O, and the peak at ∼533.7 eV is attributed to the surface adsorbed oxygen.
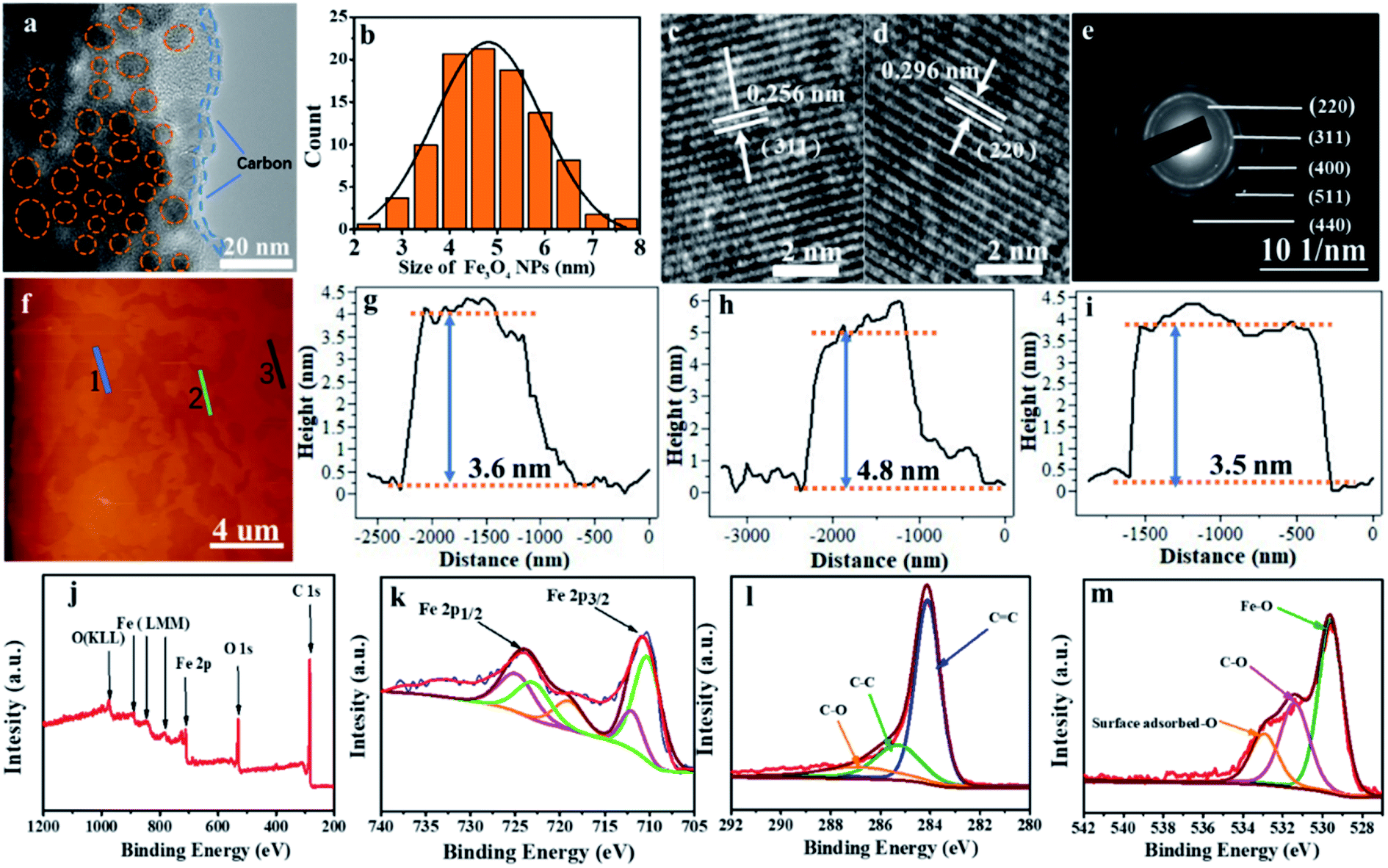 | ||
| Fig. 2 HRTEM images (a), showing an apparent amorphous carbon layer and tiny Fe3O4 nanodots. The size distribution (b), and the crystal fringes (c and d), selected area electron diffraction (SAED) pattern (e); and the AFM (f) and the thickness profiles (g–i); and XPS spectra (j): Fe 2p (k); C 1s (l) and O 1s (m) of the Fe3O4@LCS-500 composites. | ||
The electrochemical performances of Fe3O4@LCS composites were investigated as anodes of LIBs. The CV curves in the first four cycles are presented in Fig. 3a. The small peaks above 1.1 V in the first reduction (cathodic) scan are associated with the formation of Li2Fe3O4, which are disappeared in the following cycles because of the thermodynamics reason and the good stability of SEI layer. A special in situ XRD cell was assembled by directly coating the slurry on an ultra-thin Be foil to illustrate the generation of Li2Fe3O4 phase. The diffraction peaks of Fe3O4 could be observed obviously at the initial state, and then gradually disappeared in Fig. S10.† Diffraction peaks at 36.75° and 42.7°, corresponding to the (222) and (400) planes of Li2Fe3O4, appeared when the voltage dropped to ∼1.1 V, and then disappeared in following discharge process, confirming Li2Fe3O4 was truly generated in the reduction process. Those following current may come from the lithium storage behavior of porous carbon layers. The anodic processes occurred at about 1.8 V due to the reformation of Fe3O4 and the delithiation of carbon layers. Apparently, in the following cathodic sweeps, the reduction peak is slightly shifted to ∼0.74 V, which is mainly ascribed to the formation of the SEI film and slightly increased inner resistance. Obviously, the almost overlapped curves in the subsequent scanning cycles imply highly reversible capability. The reversible reactions are given as follows:
| Fe3O4 + 2Li+ + 2e− → Li2Fe3O4 | (1) |
| Li2Fe3O4 + 6Li+ + 6e− → 3Fe + 4Li2O | (2) |
| 3Fe + 4Li2O ↔ Fe3O4 + 8Li+ + 8e− | (3) |
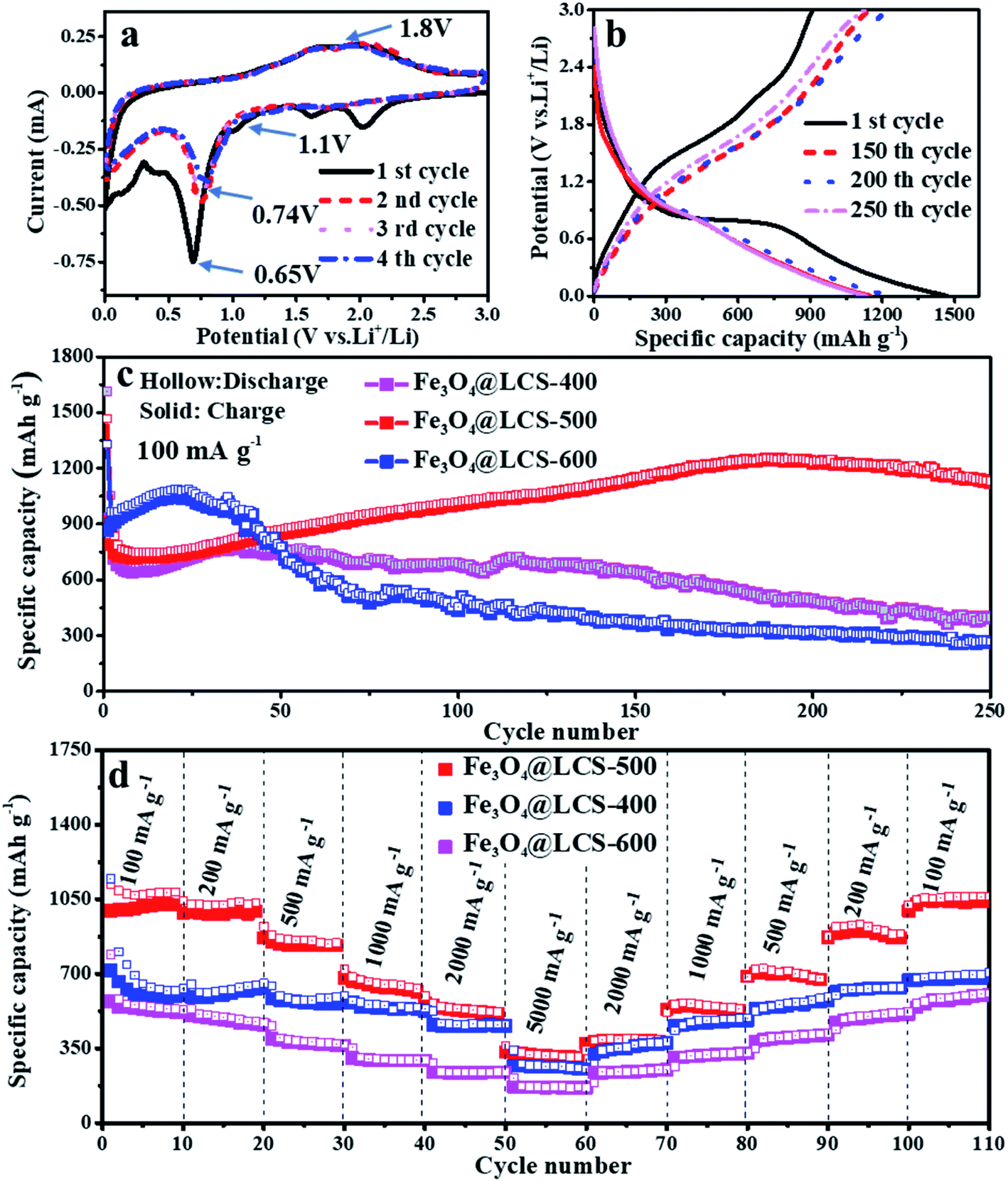 | ||
| Fig. 3 Lithium storage performances of Fe3O4@LCS-(400/500/600) electrodes: (a) CV curves in the first four cycles at a scan rate of 0.1 mV s−1; (b) charge/discharge profiles at 100 mA g−1; (c) cycle performances of Fe3O4@LCS-(400/500/600) at the current density of 100 mA g−1 for 250 cycles; (d) the rate performance of Fe3O4@LCS-(400/500/600) at various current densities. | ||
Fe3O4@LCS-500 nanocomposites delivered a high reversible capacity of 908 mA h g−1 with a coulombic efficiency of 65.76% at 100 mA g−1 in Fig. 3b. The irreversible capacity loss can be ascribed to the formation of the SEI layer and the decomposition of the electrolyte.4 Fig. 3c shows long-term cycling performances of Fe3O4@LCS (400/500/600) electrodes at 100 mA g−1 for 250 cycles. The Fe3O4@LCS-400/600 electrodes delivered a reversible capacity of 929 mA h g−1 and 868 mA h g−1, respectively. Those electrodes have shown a similar capacity trend of a slight decrease in the first several cycles and gradual increase afterward, which is normally seen in many reports about the activation process of transition metal oxides during cycles.42,48 However, the Fe3O4 bulk electrode only delivered a reversible capacity of 729.2 mA h g−1 and no such phenomenon as shown in Fig. S8.† Fe3O4@LCS-500 electrode maintained a reversible capacity of 1126.2 mA h g−1 after 250 cycles. It exhibited anomalously high capacities beyond their theoretical value, which was probably related to the strong surface capacitance around metal nanoparticles and reversible storage of spin-polarized electrons in the already-reduced iron nanodots.4 By contrast, other electrodes, including Fe3O4@LCS-400, Fe3O4@LCS-600, and bulk Fe3O4, only displayed reversible capacities of 395.5, 265.9 mA h g−1, and 257.5 mA h g−1 after 250 cycles, respectively. That is mainly associated to features of Fe3O4@LCS-500 nanocomposites on (i) the higher specific surface area and fewer defects in carbon layers, offering more active sites for reversible cation's accommodations, (ii) its lower inner resistance, facilitating to charge transfer and fast ionic diffusion coefficient. Fe3O4@LCS-500 electrode exhibited better rate performance than the others in Fig. 3d, delivering rechargeable capacities of 996, 988, 848, 666, 438 and 210 mA h g−1 at stepwise current densities of 100, 200, 500, 1000, 2000 and 5000 mA g−1, respectively. When the current density was returned to 100 mA g−1, the reversible capacity was recovered to around 1000 mA h g−1, implying a fast lithium diffusion coefficient and kinetics. On the contrast, the capacities of Fe3O4@LCS-(400/600) and bulk Fe3O4 electrodes (Fig. S8b†) were stable at low current densities, but decreased at high rates, displaying poor rate performances. Moreover, electrochemical performances of Fe3O4@LCS nanocomposites as anodes for KIBs were also measured and presented in Fig. S4, S5 and S9,† demonstrating the application possibility in KIBs.
To measure the specific values of the lithium diffusion coefficient and quantify the capacitive characterization, we utilized the CV method cycled at different scan rates to measure lithium cation's behaviors and diffusion coefficients. Fig. 4a depicts CV curves at different scan rates (from 0.1 to 0.5 mV s−1). During scans, the intensities of redox peaks are increased with scan rates, which might be assigned to the different storage behaviors (the pseudo-capacitive contribution or cation-diffusion contribution). That can be estimated by the following equation:39,49,50
| i = k1v + k2v1/2 = avb | (4) |
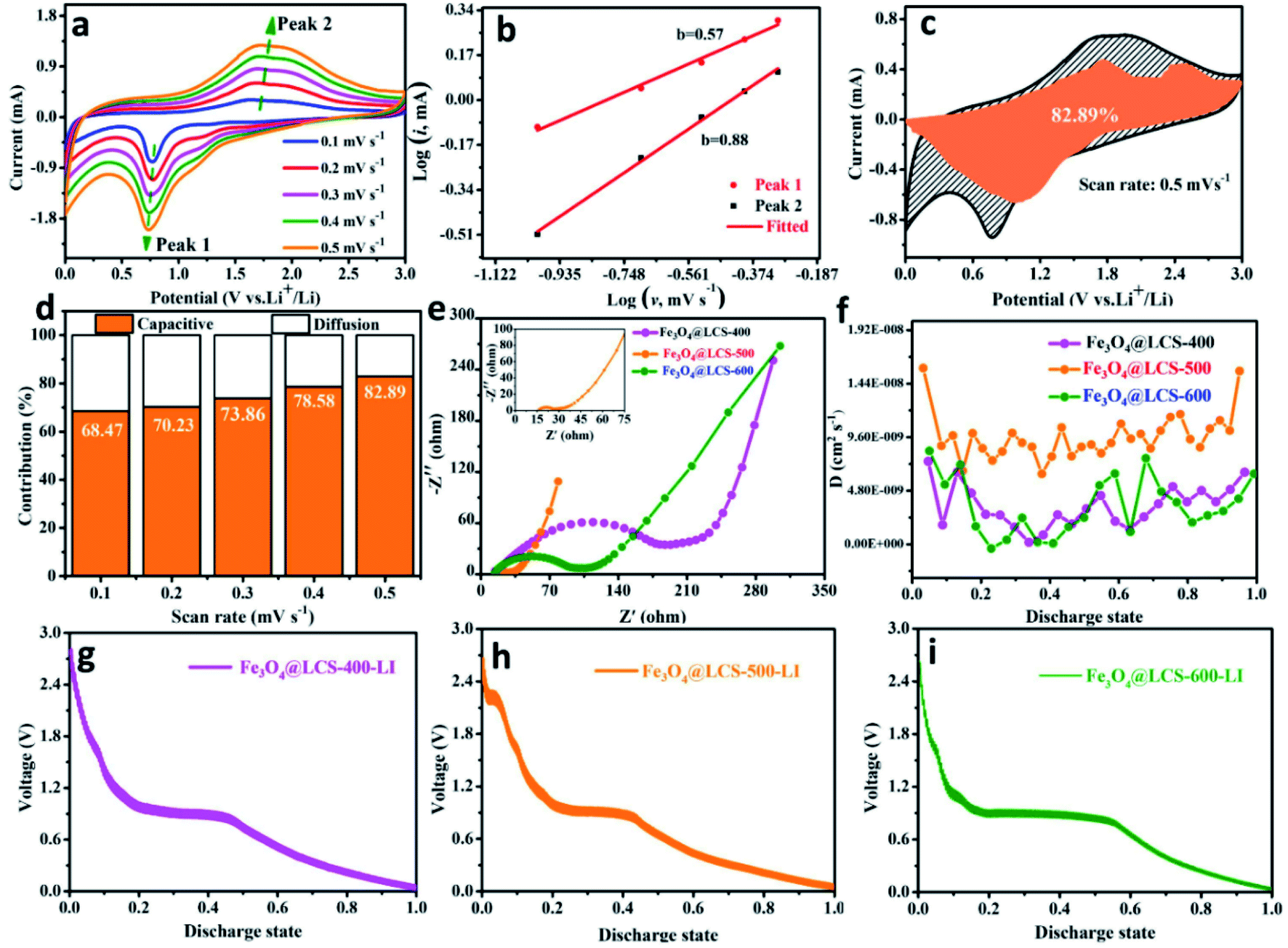 | ||
| Fig. 4 Capacity contribution and lithium diffusion coefficients: (a) CV curves of the Fe3O4@LCS-500 electrode at different scan rates from 0.1 to 0.5 mV s−1; (b) b value with linear relationship between log(i) and log(v); (c) capacitive contributions of Fe3O4@LCS-500 electrode at the scan rate of 0.5 mV s−1; (d) contribution ratios of capacitive capacities at various scan rates; (e) the Nyquist plots of the Fe3O4@LCS-400/500/600 electrodes; (f) the calculated Li chemical diffusion coefficients; (g–i) GITT curves of Fe3O4@LCS-400/500/600 electrodes. | ||
When the value of the slope (b) approaches 0.5, the storage behavior is dominated by diffusion-type reactions; when it is close to 1.0, it is inclined to a capacitive controlled process.25,51
Fig. 4b exhibits the relationship between log(i) and log(v), where peak 1 (cathodic peak) and peak 2 (anodic peak) of the slope b were calculated to be 0.57 and 0.88, respectively. That means it was dominated by capacitive-controlled behavior. Fig. 4c presents the portion of the capacitive contribution (82.92%) against the whole scan area at 0.5 mV s−1. Contribution ratios of capacitive-controlled behaviors in Fig. 4d were calculated to be 68.47%, 70.23%, 73.86%, 78.56% and 82.92% at different scan rates. It is noted that the capacitive contribution is increased with the increase of scan rates. Furthermore, the electrochemical impedance spectra of Fe3O4@LCS-(400/500/600) electrodes are shown in Fig. 4e, S7 and Table S2,† in which the depressed semi-circle in the high and middle-frequency region is related to charge-transfer, and the inclined line in the low-frequency region is associated to ionic diffusion. Apparently, the semi-circle's radius of Fe3O4@LCS-500 is much smaller than other electrodes, implying a lowest electrochemical resistance and best electronic conductivity. The thermodynamic or kinetic behaviors were further analyzed by the galvanostatic intermittent titration technique (GITT) in Fig. 4f–i. The diffusion coefficient of Li+ ion is acquired by the equation as follows:35,52
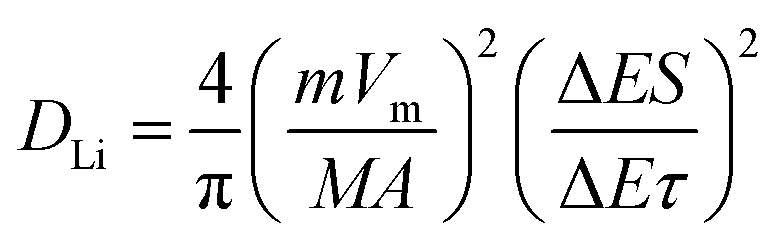 | (5) |
In which m and M mass and molecular weight of the active substance; Vm: active substance's molar volume; τ: constant titration time; A: electrode surface area; ΔES: steady-state voltage variation under current disturbance; ΔEτ: voltage variation under constant current titration.53 The diffusion coefficient values of lithium (DLi) of Fe3O4@LCS-(400/500/600) electrodes are shown in Fig. 4f. During the whole discharging process, we can obviously observe that the diffusion coefficients of the Fe3O4@LCS-500 was the highest (∼1.04 × 10−8 cm2 S−1), which is much higher than others (Fe3O4@LCS-400: 3.49 × 10−9 cm2 s−1; Fe3O4@LCS-600: 6.29 × 10−9 cm2 s−1). The information of capacity contribution and diffusion coefficients of KIBs was shown in Fig. S5,† giving similar electrochemical behaviours.
The ex situ XPS patterns of Fe3O4@LCS-500 sample, acquired at different reaction states, were examined to verify the redox reaction mechanism in LIBs. The main peak of carbon in Fig. 5a can be divided into three peaks at ∼284.1, 285.6, and 286.9 eV, corresponding to CC, C–C, and C–O, respectively. After the full lithiation, a new peak of 289.62 eV appears in Fig. 5b, which is assigned to the C–Li bond, demonstrating porous carbon nanosheets have contributed some capacity. When it is fully delithiated, the peak intensity is obviously decreased in Fig. 5c, indicating the partially reversible behavior of porous carbon nanosheets.11,47 The XPS spectrum of iron can be divided into five major peaks in Fig. 5d. The configuration of Fe 2p3/2 is composed by peaks at ∼711.91 and 710.2 eV, relating to a mixed valence state of +3 and +2. The peak at about 724.9 eV is referring to Fe 2p1/2 (a valence state of +3). Meanwhile, the peaks at 722.2 eV is referring to Fe 2p1/2 (a valence state of +2). After the full lithiation process, a new peak at 709.02 eV appears in Fig. 5e, which is ascribed to the formation of Fe0. In the fully delithiated process, the peak intensity is obviously weakened in Fig. 5f, indicating the reversible conversion reactions between Fe3O4 and Li.54 The spectra of carbon and iron in different valence states of Fe3O4@LCS-500 electrodes for KIBs were measured by ex situ XPS as shown in Fig. S6.†
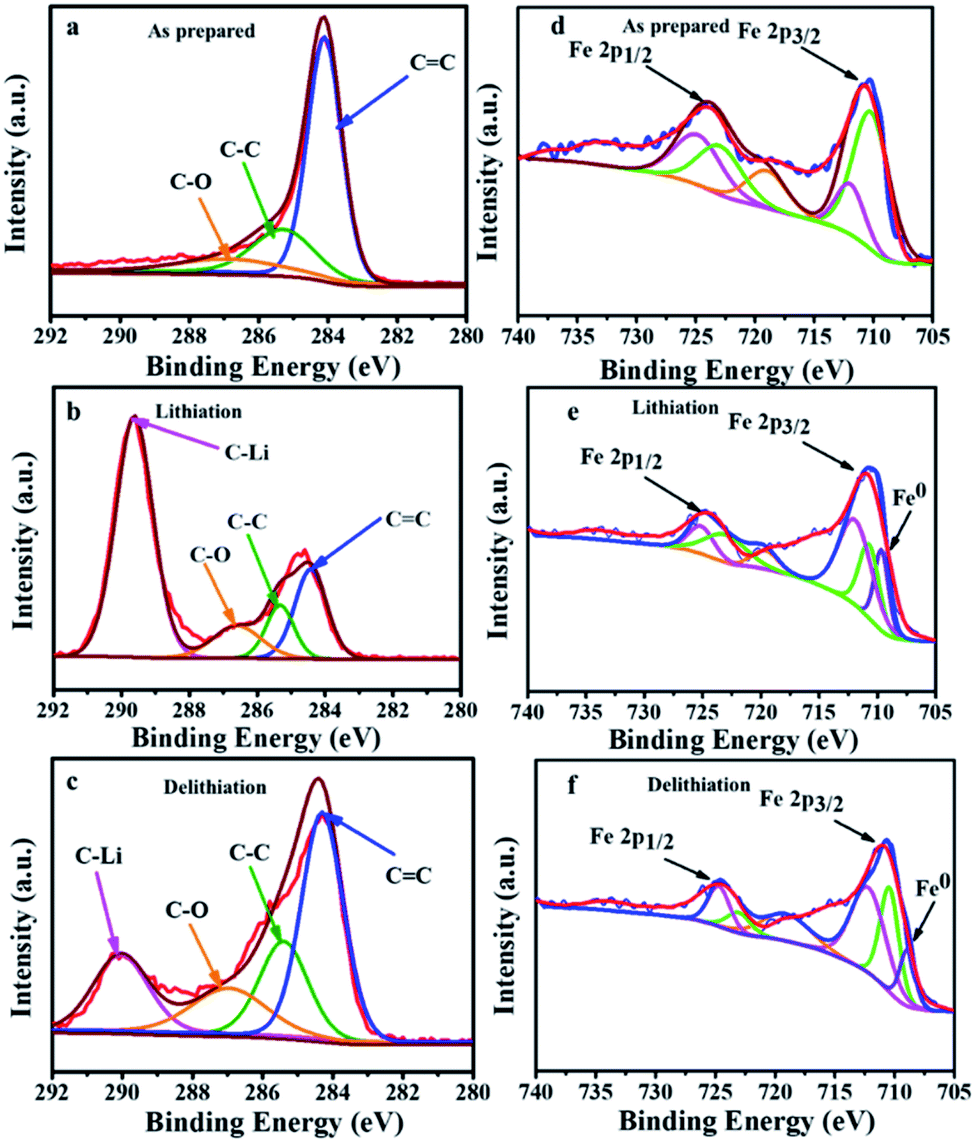 | ||
| Fig. 5 Ex situ XPS spectra of carbon and iron in the different states of Fe3O4@LCS-500 electrodes in LIBs. (a) Carbon spectrum; (b) carbon spectrum in the fully lithiated state; (c) carbon spectrum in the fully delithiated state; (d) iron spectrum; (e) iron spectrum in the fully lithiated state; (f) iron spectrum in the fully delithiated state is close to 1.0, it is inclined to a capacitive controlled process.25,51 | ||
The superior electrochemical performances of Fe3O4@LCS electrodes for LIBs/KIBs are attributed to their unique features of the thin carbon layer, pre-reserved space, and ultra-small particle size as well as flexible capability. Carbon coated Fe3O4 (Fe3O4–C) bulk in Fig. 6a, perplexed by Ostwald ripening, are normally experiencing the growth of SEI film, and cracks on carbon shells after cycles. Even though electron and ion can easily transfer among particles.
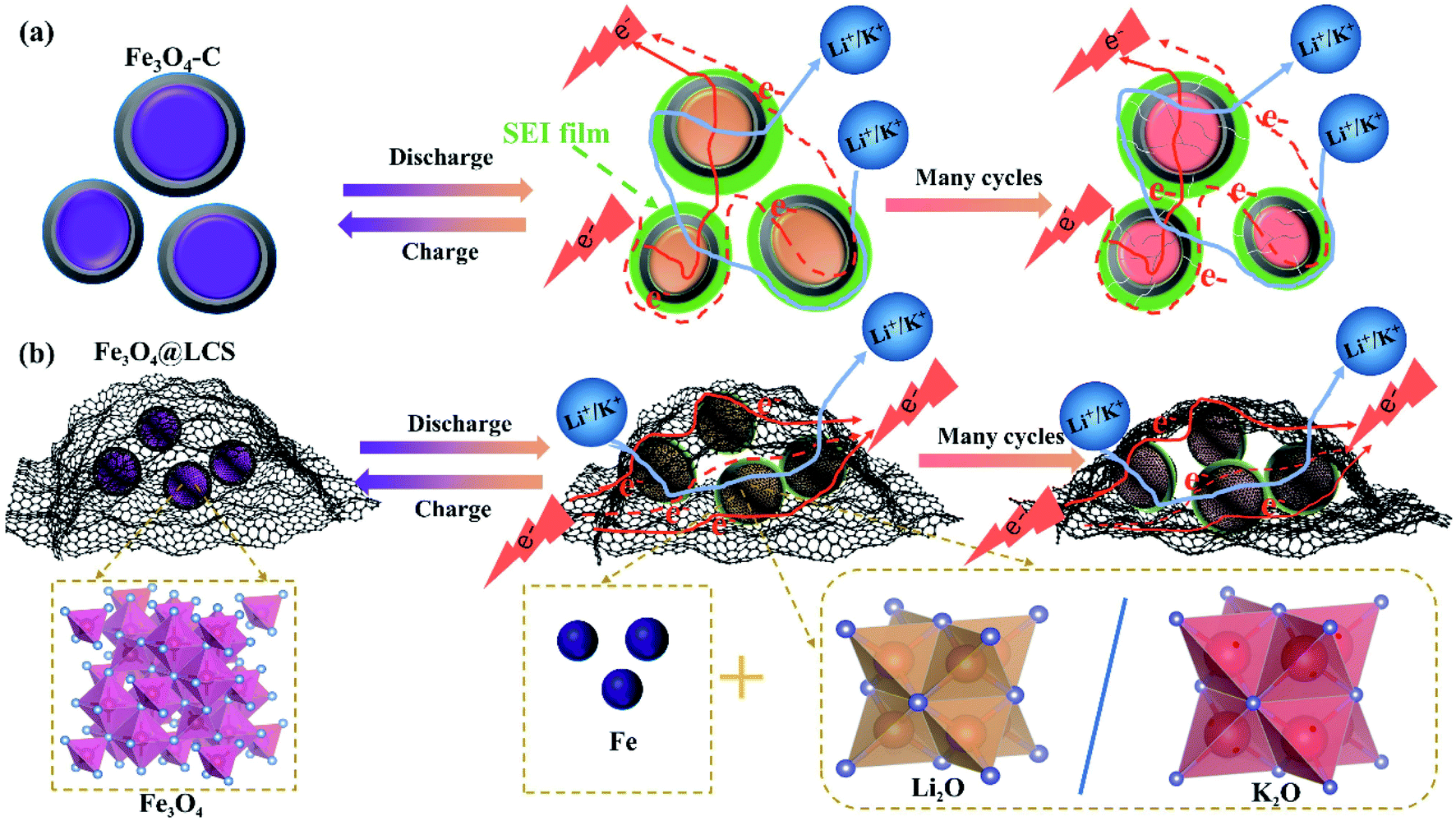 | ||
| Fig. 6 Schematic illustration of the morphology evolutions of Fe3O4–C (a) and Fe3O4@LCS (b) materials. | ||
Without the pre-reserved space, the rigid carbon shell is tended to crack after many cycles due to large volume changes and side reactions. Unlike Fe3O4–C bulk, Fe3O4@LCS materials are composed of ultra-small Fe3O4 nanodots and layered carbon nanosheets with the flexible feature and pre-reserved spaces in Fig. 6b. Layered carbon sheets are functioned as an elastic buffer with pre-reserved spaces to prevent pulverization or aggregation of encapsulated ultra-small Fe3O4 nanodots during cycles, maintaining stable and unobstructed channels for the fast electronic and ionic conductivity.54–56 The thin and layered carbon nanosheets not only enable full utilization of active ultra-small Fe3O4 nanodots and abundant accessibility of electrolyte, but also accommodate the mechanical stress from the volume variations, as shown in Fig. S11a–i† in both LIBs and KIBs.57,58 Therefore, the unique features of the ultra-small particle size, less defects and stable layered structures in Fe3O4@LCS composites are jointly contributed to the high capacity, good rate capability, and long cycle life as well as high diffusion coefficients.
Conclusion
In summary, Fe3O4@LCS nanocomposites were derived from metal coordination compound with stacked layered structure, avoiding the Ostwald ripening phenomenon. This kind of materials has unique features of ultra-small particle size, high surface area, pre-reserved spaces, flexibility. When applied as anodes for LIBs/KIBs, Fe3O4@LCS-500 nanocomposites with less defects and short ionic diffusion length, and high electronic conductivity have delivered exceptional electrochemical performances, which are largely ascribed to the unique properties of high diffusion coefficients, fast reaction kinetics, pre-reserved volume variation spaces, flexible carbon sheets and capacitive-controlled electrochemical behaviors. Such material design strategy and characterization methods can be extended to other anodes for rechargeable Li/Na/K ion batteries.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (21975154, 52073170), Shanghai Municipal Education Commission (Innovation Program (2019-01-07-00-09-E00021)) and Innovative Research Team of High-level Local Universities in Shanghai. Research is also supported by The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and Shanghai Key Laboratory of Materials Protection and Advanced Materials in Electric Power. The authors thank Laboratory for Microstructures, Instrumental Analysis, and Research Center of Shanghai University for offering access to material characterizations.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- M. Chen, J. Liu, D. Chao, J. Wang, J. Yin, J. Lin, H. J. Fan and Z. X. Shen, Nano Energy, 2014, 9, 364–372 CrossRef CAS.
- M. Wang, M. Lin, J. Li, L. Huang, Z. Zhuang, C. Lin, L. Zhou and L. Mai, Chem. Commun., 2017, 53, 8372–8375 RSC.
- Q. Li, H. Li, Q. Xia, Z. Hu, Y. Zhu, S. Yan, C. Ge, Q. Zhang, X. Wang, X. Shang, S. Fan, Y. Long, L. Gu, G. X. Miao, G. Yu and J. S. Moodera, Nat. Mater., 2021, 20, 76–83 CrossRef CAS.
- T. Jin, H. Li, Y. Li, L. Jiao and J. Chen, Nano Energy, 2018, 50, 462–467 CrossRef CAS.
- H. Dai, K. Xi, X. Liu, C. Lai and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17515–17521 CrossRef CAS.
- Y. Yu, L. Gu, C. Wang, A. Dhanabalan, P. A. van Aken and J. Maier, Angew. Chem., Int. Ed., 2009, 48, 6485–6489 CrossRef CAS.
- X. Wang, X. Xu, C. Niu, J. Meng, M. Huang, X. Liu, Z. Liu and L. Mai, Nano Lett., 2017, 17, 544–550 CrossRef CAS.
- B. Mao, D. Guo, J. Qin, T. Meng, X. Wang and M. Cao, Angew. Chem., Int. Ed., 2018, 57, 446–450 CrossRef CAS.
- P. Lian, Y. Dong, Z.-S. Wu, S. Zheng, X. Wang, S. Wang, C. Sun, J. Qin, X. Shi and X. Bao, Nano Energy, 2017, 40, 1–8 CrossRef CAS.
- H. Zhang, W. Sun, X. Chen and Y. Wang, ACS Nano, 2019, 13, 14252–14261 CrossRef CAS.
- K. Cao, H. Liu, W. Li, Q. Han, Z. Zhang, K. Huang, Q. Jing and L. Jiao, Small, 2019, 15, e1901775 CrossRef.
- H. Z. Lin, M. L. Li, X. Yang, D. X. Yu, Y. Zeng, C. Z. Wang, G. Chen and F. Du, Adv. Energy Mater., 2019, 9, e1900323 CrossRef.
- W. Zhang, Z. Wu, J. Zhang, G. Liu, N.-H. Yang, R.-S. Liu, W. K. Pang, W. Li and Z. Guo, Nano Energy, 2018, 53, 967–974 CrossRef CAS.
- S. Chong, Y. Wu, C. Liu, Y. Chen, S. Guo, Y. Liu and G. Cao, Nano Energy, 2018, 54, 106–115 CrossRef CAS.
- Q. Tan, P. Li, K. Han, Z. Liu, Y. Li, W. Zhao, D. He, F. An, M. Qin and X. Qu, J. Mater. Chem. A, 2019, 7, 744–754 RSC.
- R. Qiu, R. Fei, T. Zhang, X. Liu, J. Jin, H. Fan, R. Wang, B. He, Y. Gong and H. Wang, Electrochim. Acta, 2020, 356, 136832 CrossRef CAS.
- R. Kanakaraj and C. Sudakar, J. Power Sources, 2020, 458, 228064 CrossRef CAS.
- M. Mao, C. Cui, M. Wu, M. Zhang, T. Gao, X. Fan, J. Chen, T. Wang, J. Ma and C. Wang, Nano Energy, 2018, 45, 346–352 CrossRef CAS.
- Z. Fan, J. Liang, W. Yu, S. Ding, S. Cheng, G. Yang, Y. Wang, Y. Xi, K. Xi and R. V. Kumar, Nano Energy, 2015, 16, 152–162 CrossRef CAS.
- J. Liang, K. Xi, G. Tan, S. Chen, T. Zhao, P. R. Coxon, H.-K. Kim, S. Ding, Y. Yang, R. V. Kumar and J. Lu, Nano Energy, 2016, 27, 457–465 CrossRef CAS.
- G. Gao, S. Lu, B. Dong, Y. Xiang, K. Xi and S. Ding, J. Mater. Chem. A, 2016, 4, 6264–6270 RSC.
- Y. Zou and Y. Wang, Nanoscale, 2011, 3, 2615–2620 RSC.
- G. Zhong, K. Qu, C. Ren, Y. Su, B. Fu, M. Zi, L. Dai, Q. Xiao, J. Xu, X. Zhong, F. An, M. Ye, S. Ke, S. Xie, J. Wang, P. Gao and J. Li, Nano Energy, 2020, 74, 104876 CrossRef CAS.
- L. Wan, D. Yan, X. Xu, J. Li, T. Lu, Y. Gao, Y. Yao and L. Pan, J. Mater. Chem. A, 2018, 6, 24940–24948 RSC.
- X. Song, X. Li, Z. Chen and Z. Wang, Mater. Lett., 2020, 275, 128109 CrossRef CAS.
- H. Li, J. Wang, Y. Zhang, Y. Wang, A. Mentbayeva and Z. Bakenov, J. Power Sources, 2019, 437, 226901 CrossRef CAS.
- J. Liang, C. Yuan, H. Li, K. Fan, Z. Wei, H. Sun and J. Ma, Nano-Micro Lett., 2018, 10, 21 CrossRef.
- S. Chen, F. Wu, L. Shen, Y. Huang, S. K. Sinha, V. Srot, P. A. van Aken, J. Maier and Y. Yu, ACS Nano, 2018, 12, 7018–7027 CrossRef CAS.
- S. Chen, L. Shen, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1605650 CrossRef.
- J. Wang, X. Yan, Z. Zhang, H. Ying, R. Guo, W. Yang and W.-Q. Han, Adv. Funct. Mater., 2019, 29, 1904819 CrossRef.
- W. Yang, N. Luo, C. Zheng, S. Huang and M. Wei, Small, 2019, 15, e1903904 CrossRef.
- J. Bai, B. Xi, H. Mao, Y. Lin, X. Ma, J. Feng and S. Xiong, Adv. Mater., 2018, 30, e1802310 CrossRef.
- X. Cheng, D. Li, F. Liu, R. Xu and Y. Yu, Small Methods, 2018, 3, 1800170 CrossRef.
- W. Sun, C. Cai, X. Tang, L.-P. Lv and Y. Wang, Chem. Eng. J., 2018, 351, 169–176 CrossRef CAS.
- H. Dai, K. Xi, X. Liu, C. Lai and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17515–17521 CrossRef CAS.
- X. Wu, Z. Han, X. Zheng, S. Yao, X. Yang and T. Zhai, Nano Energy, 2017, 31, 410–417 CrossRef CAS.
- F. L. Li, P. Wang, X. Huang, D. J. Young, H. F. Wang, P. Braunstein and J. P. Lang, Angew. Chem., Int. Ed., 2019, 58, 7051–7056 CrossRef CAS.
- B. Liu, Q. Zhang, L. Li, Z. Jin, C. Wang, L. Zhang and Z. M. Su, ACS Nano, 2019, 13, 13513–13523 CrossRef CAS.
- X. Chang, Y. Ma, M. Yang, T. Xing, L. Tang, T. Chen, Q. Guo, X. Zhu, J. Liu and H. Xia, Energy Storage Materials, 2019, 23, 358–366 CrossRef.
- Z. S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS.
- W. Sun, S. Chen and Y. Wang, Dalton Trans., 2019, 48, 2019–2027 RSC.
- W. Guo, W. Sun and Y. Wang, ACS Nano, 2015, 9, 11462–11471 CrossRef CAS.
- X. Zhou, L. Chen, W. Zhang, J. Wang, Z. Liu, S. Zeng, R. Xu, Y. Wu, S. Ye, Y. Feng, X. Cheng, Z. Peng, X. Li and Y. Yu, Nano Lett., 2019, 19, 4965–4973 CrossRef CAS.
- Z. Liu, A. Daali, G.-L. Xu, M. Zhuang, X. Zuo, C.-J. Sun, Y. Liu, Y. Cai, M. D. Hossain, H. Liu, K. Amine and Z. Luo, Nano Lett., 2020, 20, 3844–3851 CrossRef CAS.
- X. Chen, L.-P. Lv, W. Sun, Y. Hu, X. Tao and Y. Wang, J. Mater. Chem. A, 2018, 6, 13705–13716 RSC.
- W. Sun, X. Tang, Q. Yang, Y. Xu, F. Wu, S. Guo, Y. Zhang, M. Wu and Y. Wang, Adv. Mater., 2019, 31, e1903176 CrossRef.
- S. Ru, H. Xiao, G. Ma, J. Tan, X. Wang and Z. Ai, Mater. Lett., 2020, 276, 128244 CrossRef CAS.
- G. Fang, Z. Wu, J. Zhou, C. Zhu, X. Cao, T. Lin, Y. Chen, C. Wang, A. Pan and S. Liang, Adv. Energy Mater., 2018, 8, 1703155 CrossRef.
- X. Yin, X. Chen, W. Sun, L.-P. Lv and Y. Wang, Energy Storage Materials, 2020, 25, 846–857 CrossRef.
- M. Ye, J.-Y. Hwang and Y.-K. Sun, ACS Nano, 2019, 13, 9306–9314 CrossRef CAS.
- Q. Gan, H. He, Y. Zhu, Z. Wang, N. Qin, S. Gu, Z. Li, W. Luo and Z. Lu, ACS Nano, 2019, 13, 9247–9258 CrossRef CAS.
- X. Tang, M. Liang, Y. Zhang, W. Sun and Y. Wang, Dalton Trans., 2019, 48, 4413–4419 RSC.
- Z. Zheng, H. H. Wu, H. Liu, Q. Zhang, X. He, S. Yu, V. Petrova, J. Feng, R. Kostecki, P. Liu, D. L. Peng, M. Liu and M. S. Wang, ACS Nano, 2020, 14, 9545–9561 CrossRef CAS.
- F. Wu, V. Srot, S. Chen, S. Lorger, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2019, 31, 1905146 CrossRef CAS.
- J. Chen, X. Fan, X. Ji, T. Gao, S. Hou, X. Zhou, L. Wang, F. Wang, C. Yang, L. Chen and C. Wang, Energy Environ. Sci., 2018, 11, 1218–1225 RSC.
- P. Xiong, P. Bai, S. Tu, M. Cheng, J. Zhang, J. Sun and Y. Xu, Small, 2018, 14, 1802140 CrossRef.
- C. Wu, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1604015 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08503k |
| ‡ The authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2021 |