
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrospun collagen core/poly-L-lactic acid shell nanofibers for prolonged release of hydrophilic drug†
Wan-Ying Huanga,
Toshiya Hibinoa,
Shin-ichiro Suyeab and
Satoshi Fujita *ab
*ab
aDepartment of Frontier Fiber Technology and Science, Graduate School of Engineering, University of Fukui, 3-9-1 Bunkyo, Fukui, 910-8507, Japan. E-mail: fujitas@u-fukui.ac.jp
bLife Science Innovation Center, University of Fukui, Fukui, 910-8507, Japan
First published on 2nd February 2021
Abstract
The development of sustained control drug release for delivering hydrophilic drugs has been challenging due to a burst release. Nanofibers are used as materials that enable efficient drug delivery systems. In this study, we designed drug-encapsulated core–shell nanofibers comprising a hydrophilic core of collagen (Col) incorporated with berberine chloride (BC), an anti-inflammatory and anti-cancer agent used as a model drug, and a hydrophobic shell of poly-L-lactic acid (PLLA). Long-term drug release profiles under both the physiological and hydrolysis-accelerated conditions were measured and analyzed using a Korsmeyer–Peppas kinetics model. We found that the Col/PLLA core–shell fiber achieved a controllable long-term release of the hydrophilic drug incorporated inside the core by the slow degradation of the PLLA shell to prevent the burst release while PLLA monolithic fibers showed early release due to the dissolution of drug and the following rapid hydrolysis of fibers. As shown by the results of Col/PLLA core–shell fiber under a hydrolysis-accelerated condition to promote the release of drugs test, it would provide sustained release over 16 days under physiological conditions. Here, the development of the nanomaterial for the long-term drug release of hydrophilic drugs was achieved, leading to its potential medical application including cancer treatment.
1. Introduction
Drug delivery systems (DDS) were advocated to describe new formulations by numerous researchers.1–4 DDS provides a great channel for the combination of an effective active pharmaceutical ingredient and a tailored controlled-release rate.5,6 It provides advantages over conventional dosage forms, such as an optimum drug concentration released at the desired site, reduced fluctuation of the drug concentration in vivo to improve the efficiency of treatment with a minimal amount of drug, and the prevention of drug exposure to other parts of the organism.7–11 In DDS, ideal drug carriers used to deliver a drug should be spatially and temporally regulated.12 Nanofibers are one of the most promising tools to serve as a DDS carrier. Electrospun nanofibers have an edge over current traditional methods of drug encapsulation because they provide a high surface area to volume ratio, which facilitates loading of high amount of drug on these fibers.13–17 Nanofibrous mats allow a favorable control over porosity and exhibit architectural similarity to fibrous structures in the extracellular matrix,18 which can assist in the production of flexible sheets for drug loading and modulated release.19–21 Nanofibers can be exploited for the delivery of hydrophobic molecules loaded in their matrices; however, numerous available hydrophilic drugs, including natural-derived small molecule compounds, saccharides, peptides, and proteins, are difficult to accumulate and retain within the nanofiber because this biomaterial is usually produced with hydrophobic polymers.22–26 Recently, electrospinning is developing various types of nanofibers, including coaxial,27 side-by-side,28,29 tri-axial,30,31 multifluidic,32,33 and nanostructured fibers,34,35 but the encapsulation of hydrophilic drugs are still difficult because of their immiscibility with the matrix polymers.In this study, we developed biodegradable collagen (Col)/poly-L-lactic acid (PLLA) core–shell nanofiber. We expected that the hydrophobic shell would protect the hydrophilic drug against in vivo degradation to achieve long-term release of the hydrophilic drugs and avoid protracted tailing of the drug release. Collagen, a fibrous structure in the extracellular matrix, is one of the most abundant proteins in the living body. It is highly expected to be used as a medical material in vivo after surgery due to the high biocompatibility.36,37 Berberine chloride (BC), a hydrophilic drug, was incorporated in a hydrophilic core of collagen, which was covered with a hydrophobic shell of poly-L-lactic acid (PLLA), to obtain a drug-loaded core–shell fiber. BC is an active ingredient extracted from the natural plants, and reportedly shows anti-microbial, anti-inflammatory, anti-protozoal,38–40 as well as anti-cancer properties, inducing apoptosis by inhibiting the G0/G1 cell cycle in MDA-MB-231, breast cancer cells.41 The PLLA shell is expected to inhibit the rapid release of BC and finally get degraded and metabolized. To reveal the availability of core–shell fibers for long-term release, we compared the drug release mechanism of PLLA monolithic nanofibers and Col/PLLA core–shell nanofibers. The long-term release of hydrophilic drugs is still an unmet requirement. This system could be applied to the treatment of cancer, e.g., as a strategy for sustained release of the chemotherapeutic drug post-operation.
2. Materials and methods
2.1. Materials
Poly-L-lactic acid (PLLA; MW = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Cosmo Bio (Tokyo, Japan); berberine chloride (BC) was purchased from Sigma-Aldrich (MO, USA); 1,1,1,3,3,3-hexafluoro-2-propanol (HFP), chloroform (CHCl3) and dimethyl sulfoxide (DMSO) was purchased from FUJIFILM Wako Pure Chemical (Osaka, Japan); 2,2,2-trifluoroethanol (TFE) was purchased from Tokyo Chemical Industry (Tokyo, Japan); cell count reagent SF was purchased from NacalaiTesque (Kyoto, Japan); collagen (bovine hide) was purchased from Kaneka (Osaka, Japan); MDA-MB-231 was purchased from ATCC (VA, USA). All the other chemicals and reagents were of analytical grade and were used without further purification.
000) was purchased from Cosmo Bio (Tokyo, Japan); berberine chloride (BC) was purchased from Sigma-Aldrich (MO, USA); 1,1,1,3,3,3-hexafluoro-2-propanol (HFP), chloroform (CHCl3) and dimethyl sulfoxide (DMSO) was purchased from FUJIFILM Wako Pure Chemical (Osaka, Japan); 2,2,2-trifluoroethanol (TFE) was purchased from Tokyo Chemical Industry (Tokyo, Japan); cell count reagent SF was purchased from NacalaiTesque (Kyoto, Japan); collagen (bovine hide) was purchased from Kaneka (Osaka, Japan); MDA-MB-231 was purchased from ATCC (VA, USA). All the other chemicals and reagents were of analytical grade and were used without further purification.
2.2. Fabrication of Col/PLLA core–shell nanofiber
The drug-loaded Col/PLLA core–shell nanofiber was fabricated using the coaxial electrospinning setup (MECC, Fukuoka, Japan),42,43 which consists of an ultra-coaxial nozzle (core diameter of 0.2 mm; shell diameter of 0.8 mm) and a rotating collector, as shown in Fig. 1. Electrospinning conditions were as follows. Core solution: 10 w/v% collagen added with 2 w/v% BC dissolved in TFE; shell solution: 10 w/v% PLLA dissolved in HFP; flow rates of core and shell solution: 0.3 and 2.4 mL h−1, respectively; electric field: 2.5 kV cm−1; rotation speed of the collector: 1500 rpm; spinning time: 30 min to obtain a robust fiber sheet. As a comparison, a drug-loaded PLLA monolithic nanofiber was fabricated from 10 w/v% PLLA added with 2 w/v% BC dissolved in HFP using a single-nozzle electrospinning setup.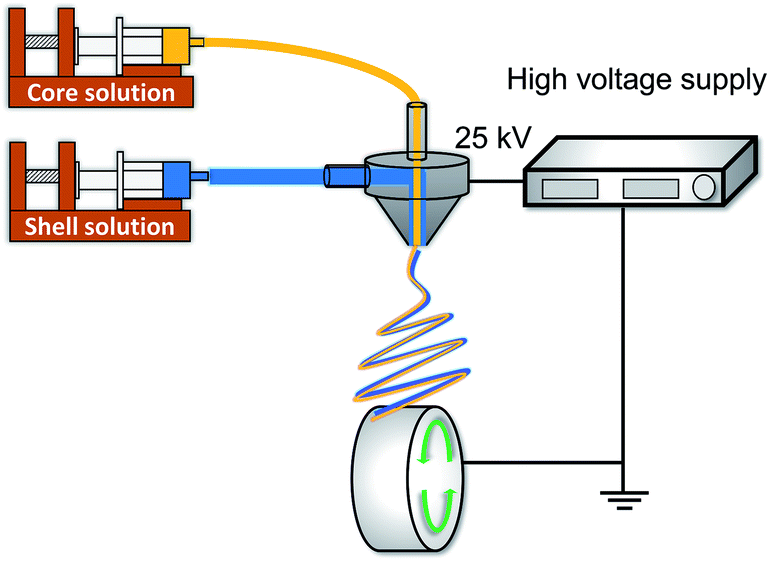 | ||
| Fig. 1 Schematic of the coaxial electrospinning setup. | ||
2.3. Scanning electron microscopy (SEM)
For observation, the fabricated nanofibers were rolled up along the nanofiber axis, immersed in liquid nitrogen, cut into small pieces, washed with water to dissolve the collagen in the core layer, and then dried at 37 °C overnight. The samples were sputtered with Pt/Pd using an ion sputter (MSP-1S, Vacuum Device Inc., Japan) for 120 s. The observation was carried out by SEM (S-2600H, HITACHI, Japan) at an accelerating voltage of 8.0 kV. The nanofiber diameter was measured from the SEM images using Fiji (Fiji.sc; Ver 2.0.0).2.4. Characterization of nanofibers
Wide-angle X-ray diffraction (WAXD) was executed with Ultima IV (Rigaku, Tokyo, Japan) using CuKα radiation (0.1542 nm) at 2θ scanning angle ranging from 10° to 35° with a scanning speed of 1° min−1. In differential scanning calorimetry (DSC), the samples sealed in Al pans were measured using DSC-60 Plus (SHIMADZU, Kyoto, Japan) at a rate of 10 °C min−1 from 0 °C to 200 °C. Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy was performed on the Nicolet 6700 system (Thermo Scientific, MA, USA) in the range of 500 to 4000 cm−1 with a resolution of 4 cm−1 and an average of 64 scans using the KBr disk method.2.5. In vitro weight loss and drug release test
In the weight loss test, the nanofibers (3 mg) were treated with oxygen plasma for 30 s then immersed in 3 mL of phosphate-buffered saline (PBS, pH 7.0) or pH 11.0 glycine–NaOH buffer solution and stored at 37 °C until the measurement. Upon centrifugation of these samples, PBS or pH 11.0 glycine–NaOH buffer solution was removed from the tube and was washed thrice with 3 mL DI water. The nanofibers were frozen in liquid nitrogen and dried in a lyophilizer (LABCONCO, America) for two days. The weight loss A% was calculated from the initial weight (wi) and the weight after decomposition (wd):| A% = (wi − wd)/wi × 100 | (1) |
For the evaluation of drug release, 5.0 mg drug-loaded nanofiber mat was hydrophilized by oxygen plasma treatment (Dinner, Germany; 40 kHz, 100 W, 30 s, 0.3 MPa). The specimen was put into glass vials with 5.0 mL PBS for the release test at the physiological condition of pH 7.0 or at the hydrolysis-accelerated condition of pH 11.0 using glycine–NaOH buffer solution kept at 37 °C in an incubator, following the collection of the drug released at 1.5, 3, 6, 12, 24, 72, and 192 h. For each sampling time, 100 μL of the sample solution was added to each well of a 96-well plate to determine the amount of drug released. The BC content in the collected samples was determined using a microplate reader (Thermo Scientific, Japan) by measuring the absorbance at 340 nm. After the last sampling, the remaining nanofiber was dissolved in HFP and the amount of drug inside was measured by spectrometry at 420 nm.
2.6. Statistical analysis
All the experiments were executed at least three times, and all the values were expressed as mean and standard deviation.3. Results
3.1. Fabrication of core–shell nanofibers
PLLA monolithic and Col/PLLA core–shell nanofibers were fabricated by electrospinning using a coaxial nozzle. All the fabrication conditions and schematics are as seen in Table 1 and Fig. 1. The Col/PLLA core–shell nanofibers fabricated under the optimized condition44 were observed by SEM. The top view showed a smooth surface without a bead structure (Fig. S1A and B†). The cross-section was prepared by cutting in liquid nitrogen and washing with water to remove the collagen layer that was encapsulated into the PLLA layer to form the core–shell structure (Fig. 2A). The shell and core measured 678 ± 104 nm and 147 ± 40 nm, respectively (Fig. S1C†). The diameter of the shell was sufficiently larger than that of the core to wrap it fully. However, we also observed incomplete Col/PLLA core–shell nanofibers, including a crescent-shaped nanofiber structure and a PLLA monolithic nanofiber that did not contain the collagen core (Fig. 2B). The collagen monolithic nanofibers were washed out with water during SEM sample preparation. The crescent-shaped nanofibers were observed with grooves on its surface after collagen removal due to washing (Fig. 2C). These incomplete-structured nanofibers would cause a burst in the early stage of drug release. In the optimized electrospinning conditions, the percentages of the Col/PLLA core–shell nanofibers, the crescent nanofibers and the monolithic nanofibers with no core were 44%, 36%, and 20%, respectively. However, diameter of the drug-loaded PLLA monolithic nanofibers fabricated with single-nozzle electrospinning was 386 ± 151 nm (Fig. S1D†).| Condition | PLLA monolithic | Col/PLLA core–shell | |
|---|---|---|---|
| Shell | Core | ||
| Solution | 10% PLLA, 2% BC in HFP | 10% PLLA in HFP | 10% collagen, 2% BC in TFE |
| Electric field (kV cm−1) | 2.5 | 2.5 | 2.5 |
| Flow rate (mL h−1) | 2.4 | 2.4 | 0.3 |
| Time (min) | 30 | 30 | 30 |
| Linear velocity (m s−1) | 1.6 | 1.6 | 1.6 |
 | ||
| Fig. 2 SEM images of core–shell nanofibers structure. (A) Cross-section view. (B) Grooves appeared after washing with water. (C) Representative images of core–shell nanofibers morphology, including complete core–shell structure (left), incomplete crescent structure (middle) and monolithic fiber which has no core (right). | ||
3.2. Drug distribution on nanofibers
To analyze the incorporation of the drug in the nanofiber, ATR-FTIR was carried out. The ATR-FTIR spectra of drug-loaded PLLA monolithic nanofiber and Col/PLLA core–shell nanofiber, PLLA, BC, and collagen are shown in Fig. 3. Sharp and steep peaks were observed for PLLA at 1755 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) and 1450 cm−1 (C–H bending) in both the nanofibers. The peaks derived from collagen were not observed in the Col/PLLA core–shell nanofibers. The peak at 1506 cm−1 for the CC stretching of BC was observed in the PLLA monolithic fiber, but not in the Col/PLLA core–shell nanofiber. These observations suggest that a high amount of drug was distributed near the surface in the drug-loaded PLLA monolithic fibers, while the collagen core containing the drug was encapsulated and shielded with a shell of PLLA in the Col/PLLA core–shell nanofibers.
O stretching) and 1450 cm−1 (C–H bending) in both the nanofibers. The peaks derived from collagen were not observed in the Col/PLLA core–shell nanofibers. The peak at 1506 cm−1 for the CC stretching of BC was observed in the PLLA monolithic fiber, but not in the Col/PLLA core–shell nanofiber. These observations suggest that a high amount of drug was distributed near the surface in the drug-loaded PLLA monolithic fibers, while the collagen core containing the drug was encapsulated and shielded with a shell of PLLA in the Col/PLLA core–shell nanofibers.
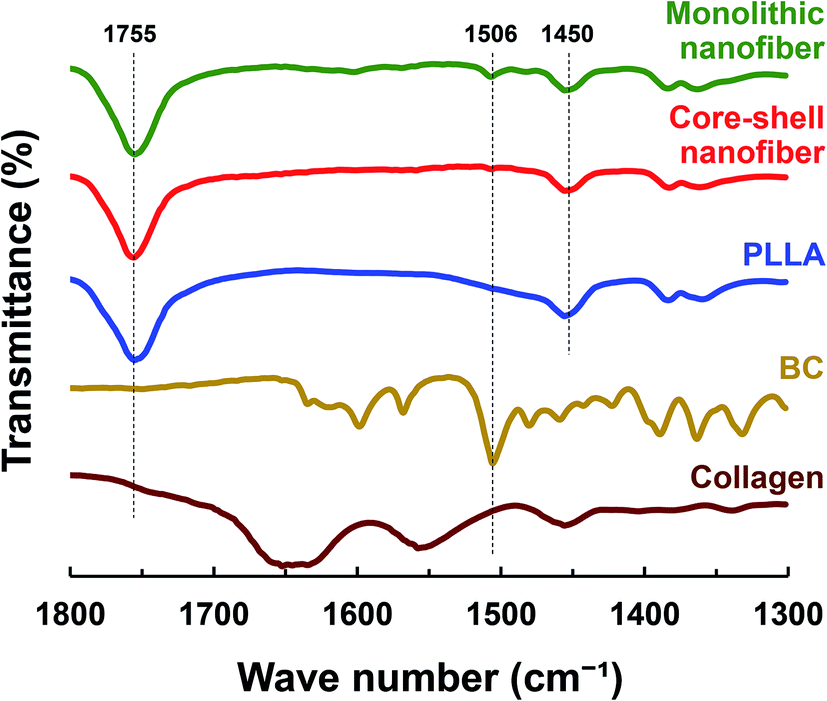 | ||
| Fig. 3 ATR-FTIR spectra of drug-loaded PLLA monolithic nanofiber, drug-loaded Col/PLLA core–shell nanofiber, PLLA, BC, and collagen. | ||
3.3. Crystallinity of BC incorporated in nanofibers
To analyze whether BC is in a crystal or amorphous state, DSC and WAXD were carried out. As shown in the DSC curves (Fig. 4A), the PLLA monolithic nanofiber showed two endothermic peaks, representing the Tm of PLLA at 178 °C (ref. 45) and the Tm of crystalized BC at 195 °C.46 However, the Col/PLLA core–shell nanofiber showed a single endothermic peak for the Tm of PLLA at 178 °C.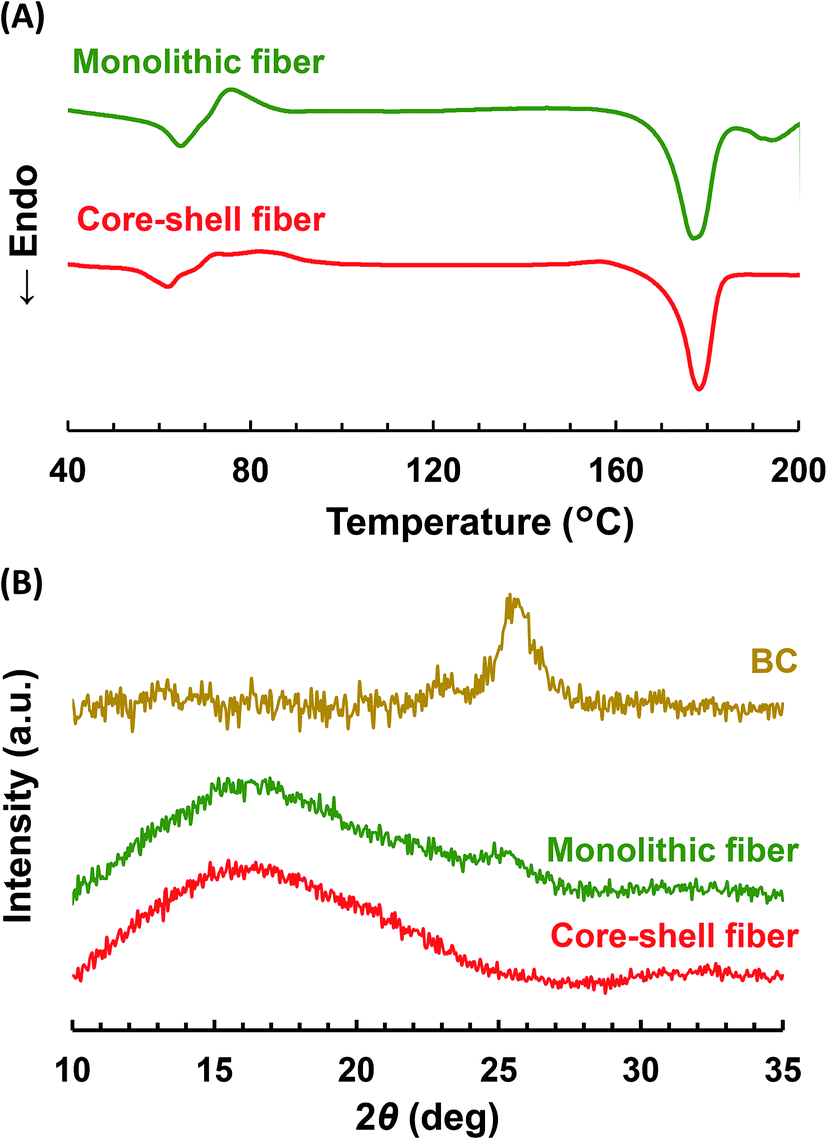 | ||
| Fig. 4 (A) DSC thermograms and (B) WAXD profiles of drug-loaded PLLA monolithic nanofibers and Col/PLLA core–shell nanofibers. | ||
The WAXD profiles (Fig. 4B) showed that the broad peaks attributed to the amorphous structure of PLLA were found around at 16° in both the fibers. This result would be due to high beam sensitivity and a small total end-point dose.47,48 In the PLLA monolithic nanofibers, the peak observed at 25° was derived from the crystalline BC.49 However, it was not visible in the Col/PLLA core–shell nanofiber. This result suggests that BC was in a crystalline structure in the PLLA monolithic nanofibers; however, it was distributed in the Col/PLLA core–shell nanofiber without forming crystalline structures. These different crystalline states are critical to note because they may affect the degradation and drug release behaviors.
3.4. Degradability of nanofibers
The ways of drug release incorporated in the nanofiber include the dissolution of the drug deposited on the nanofiber surface, diffusion of the drug through the polymer matrix, and elution from the collapsed or degraded polymer matrix. To investigate the drug release profile from the fabricated nanofibers, we first examined its degradation in the physiological condition. The change in the weight loss of the whole scaffold and the molecular weight of PLLA were measured. In the physiological PBS solution (pH 7.0), there was no significant change between the weight of PLLA monolithic and Col/PLLA core–shell nanofibers, and no swelling of the nanofibers. The shape of the scaffold was retained (Fig. 5A). The molecular weight of the PLLA measured by gel permeation chromatography (GPC) showed no change in the Col/PLLA core–shell nanofiber but gradually decreased in the monolithic nanofiber after a 30 day immersion in PBS by non-enzymatic hydrolysis (Fig. S2A†).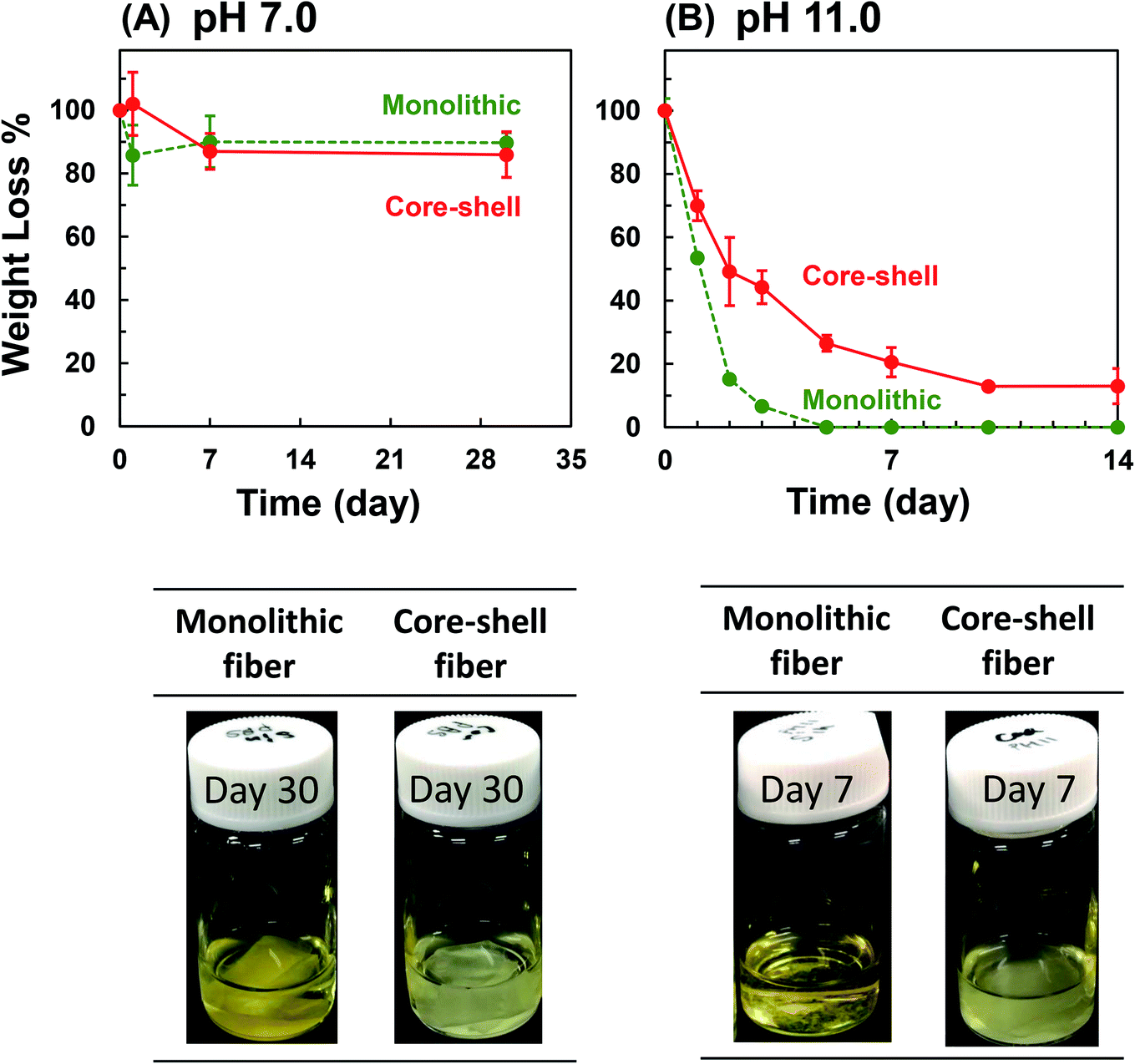 | ||
| Fig. 5 (A) Degradation of the fiber sheet under physiological (PBS, pH 7.0) and (B) hydrolysis-accelerated conditions (glycine–NaOH buffer, pH 11.0). (Upper panels) Time course of the weight loss of the sheet. (Lower panels) The morphological changes of the sheet. Cadmium yellow canary representing degradation; light yellow representing dissolution (n = 3). | ||
To investigate the long-term effect of non-enzymatic hydrolysis, we measured under a hydrolysis-accelerated condition using an alkaline buffer (pH 11.0), which can promote the release of drugs from the nanofibers to shorten the observation time for mimicking the long-term release in vivo under physiological conditions. Both the nanofibers showed remarkable weight loss under pH 11.0 (Fig. 5B). The PLLA monolithic nanofibers completely collapsed within 5 days, but the Col/PLLA core–shell nanofibers kept gradually degrading over 14 days. Fig. S3† corresponds to the SEM images of the nanofiber after immersing in the hydrolysis-accelerated conditions after 1, 2, and 5 days. The fiber structure of core–shell nanofiber was maintained, while the monolithic fiber morphology was disintegrated. By GPC analysis, PLLA chain of the monolithic nanofibers was digested into smaller chains after 3 days (Fig. S2B†) and the result was consistent with the changes in the observed morphology. However, the molecular weight of PLLA in Col/PLLA core–shell nanofibers remained unaffected following a 14 day immersion in an alkaline solution (Fig. S2B†). This suggests that the early drug release from the PLLA monolithic fiber would cause the penetration of water into the fibers, leading to the erosion after the physical break down of the PLLA fibers, while sustained release from the Col/PLLA core–shell fibers would be attributed to the diffusion through the PLLA shell.
3.5. Drug release test
The amount of drug released by dissolution and diffusion from the nanofiber under the physiological condition was tested. As shown in Fig. 6A, 10% and 26% of the drug were immediately released from the PLLA monolithic and Col/PLLA core–shell nanofibers, respectively, which represented the dissolution of the drugs deposited on the nanofiber surface. The reason the amount of drug released from the Col/PLLA core–shell was higher than that of the PLLA monolithic nanofiber was the release of collagen monolithic nanofiber, which was formed as a by-product during electrospinning of the Col/PLLA core–shell nanofiber. After the initial burst release, the accumulated amount of drug released was almost constant.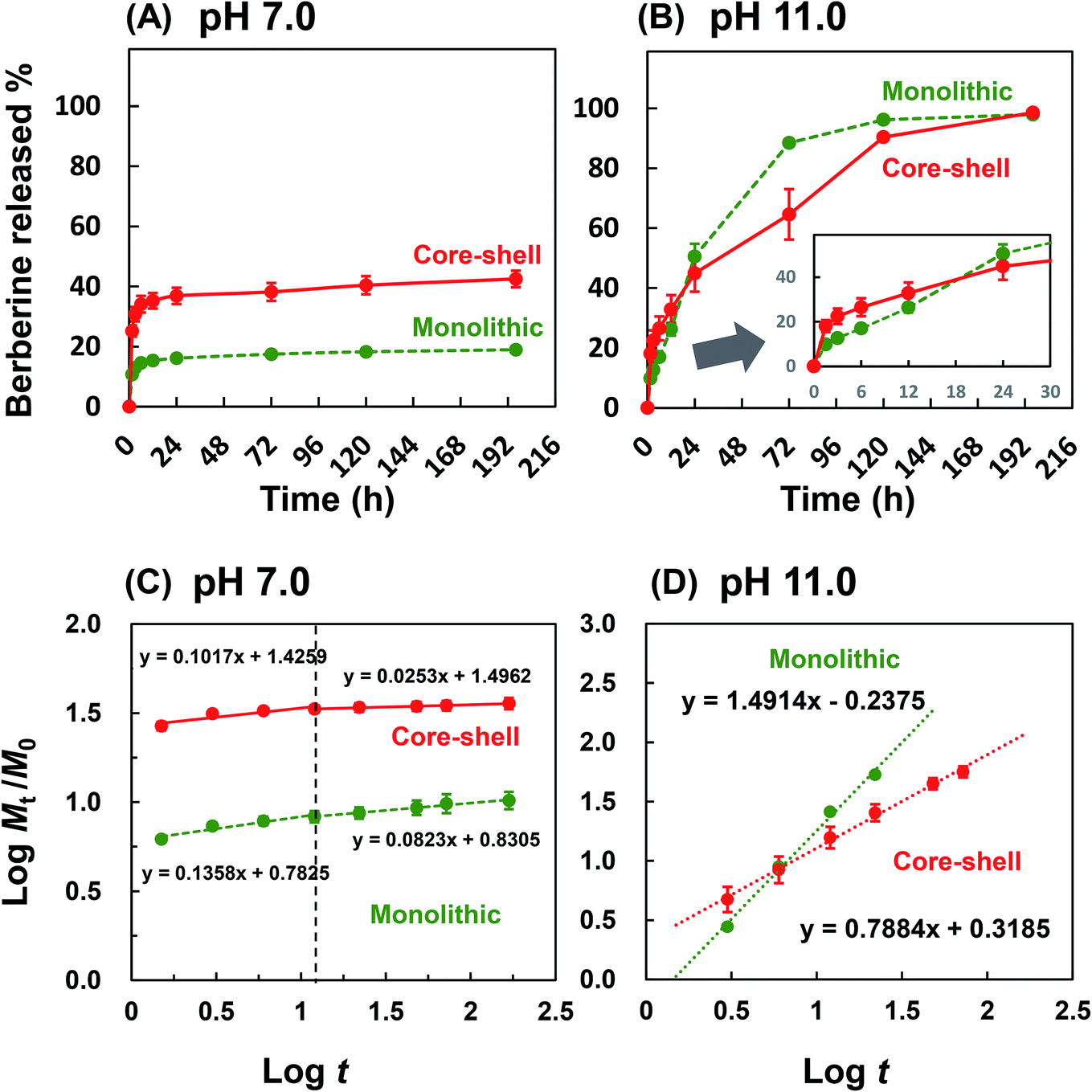 | ||
| Fig. 6 Drug release profile from monolithic nanofiber and core–shell nanofiber under (A) physiological (PBS, pH 7.0) and (B) hydrolysis-accelerated conditions (glycine–NaOH buffer, pH 11.0). Inset represents the magnified plot in the early stage (n = 3). (C and D). The Korsmeyer–Peppas model fitting of drug release profiles. | ||
To estimate the amount of drug released due to the degradation of the nanofiber after long-term immersion, the drug release test was carried out under accelerated hydrolysis in alkaline conditions. As shown in Fig. 6B, the release profiles of PLLA monolithic fiber and Col/PLLA fiber under alkaline conditions were completely different. The PLLA monolithic nanofibers exhibited a higher percentage release (51%) than the Col/PLLA core–shell nanofiber (45%) after 24 h. The amount of drug released from the PLLA monolithic nanofiber (88%) greatly exceeded by 23% that from the Col/PLLA core–shell (65%) nanofiber at 72 h, demonstrating that the Col/PLLA core–shell nanofiber release rate was controlled. These results clearly demonstrated that the burst release from PLLA monolithic nanofibers was from the surface of nanofibers and the degradation was enhanced as the drug was released, and long-term release from Col/PLLA core–shell nanofibers was achieved by the hydrophobic shell.
Besides, the bioactivity of BC interacting with the collagen in core–shell fibers was investigated. To examine whether the released BC from the core–shell fiber was not inactivated because of the interaction with collagen, MTT assay using a cancer cell line was performed. The dose-dependency of BC released from the core–shell nanofibers and the BC powder over the drug concentration range of 5 to 150 μg mL−1 on the viability of MDA-MB-231 cells are shown in Fig. S4.† As a result, LC50 (semi-lethal dose) of the BC powder and the one released from core–shell nanofibers was 95.4 and 94.3 μg mL−1, respectively, where there was no significant difference. This result suggests that BC released from the collagen core maintained its pharmacological bioactivity even if it was encapsulated in collagen.
3.6. Drug release mechanism
Note that additional analysis of the drug release mechanism was calculated using the Korsmeyer–Peppas equation,
 | (2) |
In the physiological condition, it was observed that the Col/PLLA core–shell nanofiber caused a higher burst release than the PLLA monolithic nanofibers within 1.5 h (Fig. 6C), possibly because of the dissolution of the drug from the collagen uncovered with the PLLA shell of incomplete core–shell and collagen monolithic nanofiber as an electrospinning by-product. To compare the release mechanism of the two nanofibers by ignoring the effect of the burst release from the by-product nanofibers, the drug release profile after 1.5 h was analyzed by fitting with eqn (2). By this calculation, the release profile from the core–shell fiber was correctly analyzed. As a result, the drug release profile in the first 12 h was different from the later time points. The n value of the early 12 h time point of the PLLA monolithic and Col/PLLA core–shell nanofibers was 0.136 and 0.102 and the n value after 12 h was 0.082 and 0.025, thus suggesting that the Col/PLLA core–shell nanofibers have suppressed release more efficiently than the PLLA monolithic nanofibers. In the hydrolysis-accelerated condition, the release profiles of both the nanofibers were highly correlated with the Korsmeyer–Peppas model, but the n values were different (Fig. 6D). In the PLLA monolithic nanofibers, n was 1.491, which shows the drug was extraordinarily released due to the degradation of matrix PLLA.
4. Discussion
Two main reasons can be suggested to explain why the Col/PLLA core–shell nanofibers decrease the drug release rate. First, the drug release rates were closely correlated with the BC crystalline state. Collagen and BC solutions were mixed to fabricate the core–shell nanofiber by electrospinning. During fabrication, there would be an interaction between the BC and collagen and therefore BC did not form a crystalline state in the Col/PLLA core–shell nanofibers, as it formed in the PLLA monolithic nanofiber. Second, the localization and crystallization of BC affected the release behavior. In PLLA monolithic nanofibers, the quick dissolution of BC deposited on the surface contributed significantly to the fast release, and the degradation was enhanced as BC was released. The Korsmeyer–Peppas analysis showed that uniform blending of BC in the hydrophilic polymer of the Col/PLLA core–shell nanofiber led to the drug release behavior induced by not only diffusion but also low degradation. According to the results of WAXD and DSC, in the PLLA monolithic nanofibers, the drug existed in a crystalline state. Therefore, the rapid release in the PLLA monolithic nanofiber would have been caused by the dissolution of crystalline BC and the following penetration of water into the vacant pore of the dissolute drug to enhance the matrix hydrolysis. However, in the Col/PLLA core–shell fibers, the hydrophobic PLLA was BC clad in the core–shell structure. Thus, we concluded that in the Col/PLLA core–shell fibers the drug release was primarily by diffusion (Fig. 7). However, the value of the Col/PLLA core–shell nanofibers was n = 0.788, which is higher than 0.5 suggesting that the drug release behavior could have been induced by not only diffusion but also low degradation.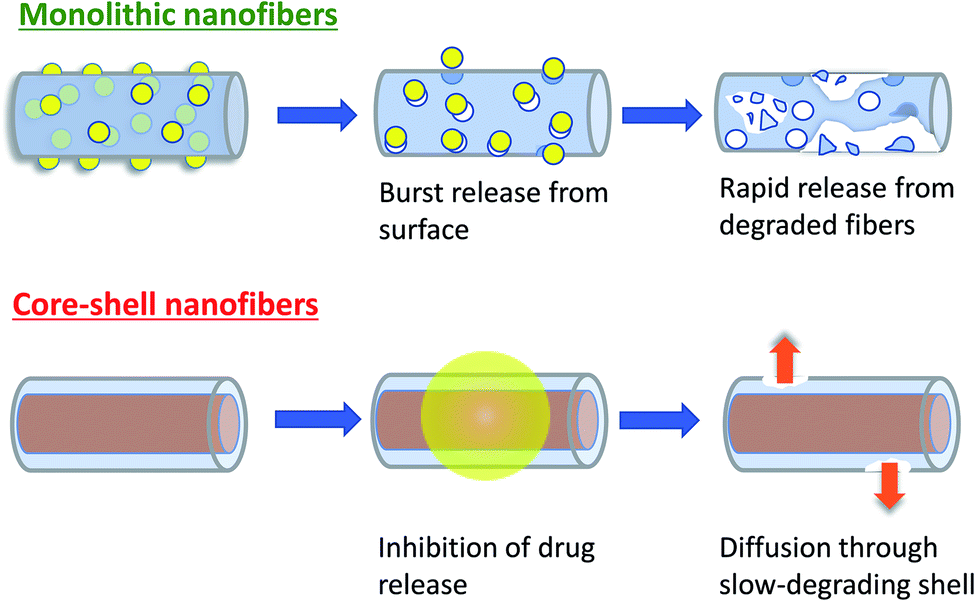 | ||
| Fig. 7 Schematic illustration of the proposed drug release mechanism from PLLA monolithic and Col/PLLA core–shell nanofiber. | ||
Regarding the estimated release duration using the core–shell fibers, other reports described that the rate of PLLA degradation at pH 10.0 is around two times faster than that at pH 7.4,52 corresponding to our results obtained from the testing at pH 11.0 and 7.0. The amount of drug released at the physiological condition was quite low, which shows that long-term release over 192 h was achieved. The Col/PLLA core–shell nanofibers showed complete release after 192 h in the hydrolysis-accelerated condition, implying that it would provide sustained release over 384 h (16 days) under physiological conditions. Thus, the Col/PLLA core–shell nanofibers not only achieved the desired long-term release but also precluded prolonged tailing toward the end of the degradation.
BC, a plant alkaloid, possesses a wide range of pharmacological effects, including anti-inflammatory and anti-cancer effects by inhibiting the growth, invasion, and metastatic progression. BC is currently well known to exert these effects in various cancer cell lines, such as hepatoma,53 lung cancer,54 esophageal cancer,55 bladder cancer56 and breast cancer,57 and to display low-toxicity to the normal cells.58 Thus, BC-loaded Col/PLLA core–shell nanofibers with high permeability and high surface area employed for the treatment of cancer to reduce less adverse effects would be a significant strategy for anti-cancer drug delivery in post-operation.
5. Conclusions
The main problem of long-term release of hydrophilic drugs using nanofibers fabricated with conventional electrospinning methods persists due to the burst release of the incorporated drug in the early stage. To address it, we successfully fabricated the anti-cancer Col/PLLA core–shell nanofiber where the collagen of the core layer plays an important role to retain the drug, and the hydrophobic shell of PLLA would sustain a long-term release over 16 days of the incorporated hydrophilic drug while degrading. By analyzing the drug release profile, the PLLA shell was maintained from the fiber digestion and suppressed release. Under conditions of accelerated degradation, the Col/PLLA core–shell and PLLA monolithic nanofibers showed a significant difference in the degradation rates. PLLA monolithic nanofiber illustrated a large burst release effect up to 51% during 24 h to 72 h. The slower degradation of Col/PLLA core–shell nanofiber compared to the PLLA monolithic fiber would be due to higher crystallinity. We also showed that the drug distribution in drug-loaded nanofibers influenced the drug release rate associated with diffusion or degradation in both the nanofibers. These results illustrate that the manipulation of drug–polymer miscibility, localization of drug deposit, and polymer degradability are key factors in modulating the drug release profile. As shown herein, the blending of drugs with a hydrophilic polymer as a core layer and hydrophobic polymer as a shell layer can enable an effectively controlled drug release profile and facilitate an ideal strategy for sustained drug release.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by Foundation of Maedakosen Group.References
- M. R. Ki, J. K. Kim, S. H. Kim, T. K. M. Nguyen, K. H. Kim and S. P. Pack, J. Ind. Eng. Chem., 2020, 81, 367–374 CrossRef CAS.
- S. Khaledian, M. Abdoli, M. Shahlaei, L. Behbood, D. Kahrizi, E. Arkan and S. Moradi, Colloids Surf., A, 2020, 585, 124077 CrossRef CAS.
- S. Yang, J. Fan, S. Lin, Y. Wang and C. Liu, Colloids Surf., A, 2019, 585, 124133 CrossRef.
- Y. Xiao, T. Gong, Y. Jiang, C. Bao and S. Zhou, J. Mater. Chem. B, 2019, 7, 4347–4360 RSC.
- S. Chakraborty, I.-C. Liao, A. Adler and K. W. Leong, Adv. Drug Delivery Rev., 2009, 61, 1043–1054 CrossRef CAS.
- R. Sridhar, R. Lakshminarayanan, K. Madhaiyan, V. A. Barathi, K. H. C. Limh and S. Ramakrishna, Chem. Soc. Rev., 2015, 44, 790–814 RSC.
- R. Wang, L. Zhou, W. Wang, X. Li and F. Zhang, Nat. Commun., 2017, 8, 1–12 CrossRef CAS.
- Y. Zhai, X. Bai, J. Zhu, X. Sun, G. Pan, B. Dong, L. Xu, W. Xu, S. Zhang and H. Song, J. Mater. Chem. B, 2018, 6, 3579–3585 RSC.
- S. Edsbäcker, B. Bengtsson, P. Larsson, P. Lundin, Å. Nilsson, J. Ulmius and P. Wollmer, Aliment. Pharmacol. Ther., 2003, 17, 525–536 CrossRef.
- D. G. Yu, J. J. Li, G. R. Williams and M. Zhao, J. Controlled Release, 2018, 292, 91–110 CrossRef CAS.
- D. Carson, Y. Jiang and K. A. Woodrow, Pharm. Res., 2016, 33, 125–136 CrossRef CAS.
- L. Palanikumar, M. T. Jeena, K. Kim, J. Yong Oh, C. Kim, M.-H. Park and J.-H. Ryu, Sci. Rep., 2017, 7, 46540 CrossRef CAS.
- S.-F. Chou, D. Carson and K. A. Woodrow, J. Controlled Release, 2015, 220, 584–591 CrossRef CAS.
- Y. Lu, J. Huang, G. Yu, R. Cardenas, S. Wei, E. K. Wujcik and Z. Guo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 654–677 CAS.
- A. K. Blakney, E. A. Krogstad, Y. H. Jiang and K. A. Woodrow, Int. J. Nanomed., 2014, 9, 2967–2978 CrossRef.
- A. K. Moghe and B. S. Gupta, Polym. Rev., 2008, 48, 353–377 CrossRef CAS.
- G. C. Rutledge and S. V. Fridrikh, Adv. Drug Delivery Rev., 2007, 59, 1384–1391 CrossRef CAS.
- W. Friess, Eur. J. Pharm. Biopharm., 1998, 45, 113–136 CrossRef CAS.
- C. Cleeton, A. Keirouz, X. Chen and N. Radacsi, ACS Biomater. Sci. Eng., 2019, 5, 4183–4205 CrossRef CAS.
- N. Bhardwaj and S. C. Kundu, Biotechnol. Adv., 2010, 28, 325–347 CrossRef CAS.
- N. G. Rim, C. S. Shin and H. Shin, Biomed. Mater., 2013, 8, 014102 CrossRef.
- S. Arpicco, L. Battaglia, P. Brusa, R. Cavalli, D. Chirio, F. Dosio, M. Gallarate, P. Milla, E. Peira, F. Rocco, S. Sapino, B. Stella, E. Ugazio and M. Ceruti, J. Drug Delivery Sci. Technol., 2016, 32, 298–312 CrossRef CAS.
- Y. Fu and W. J. Kao, Expert Opin. Drug Delivery, 2010, 7, 429–444 CrossRef CAS.
- J. K. W. Lam, Y. Xu, A. Worsley and I. C. K. Wong, Adv. Drug Delivery Rev., 2014, 73, 50–62 CrossRef CAS.
- S. T. Yohe, Y. L. Colson and M. W. Grinstaff, J. Am. Chem. Soc., 2012, 134, 2016–2019 CrossRef CAS.
- A. D. Woolfson, R. K. Malcolm, R. J. Morrow, C. F. Toner and S. D. McCullagh, Int. J. Pharm., 2006, 325, 82–89 CrossRef CAS.
- M. Danilo, P. A. M. Chagas, I. S. Leite, N. M. Inada, S. R. De Annunzio, C. R. Fontana, S. P. Campana-filho and D. S. Correa, Int. J. Biol. Macromol., 2020, 142, 521–534 CrossRef.
- J. Yang, K. Wang, D. G. Yu, Y. Yang, S. W. A. Bligh and G. R. Williams, Mater. Sci. Eng., C, 2020, 111, 110805 CrossRef CAS.
- K. Wang, X. K. Liu, X. H. Chen, D. G. Yu, Y. Y. Yang and P. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 2859–2867 CrossRef CAS.
- C. K. Huang, K. Zhang, Q. Gong, D. G. Yu, J. Wang, X. Tan and H. Quan, Int. J. Biol. Macromol., 2020, 152, 68–76 CrossRef CAS.
- Y. Yang, S. Chang, Y. Bai, Y. Du and D. G. Yu, Carbohydr. Polym., 2020, 243, 116477 CrossRef CAS.
- M. Wang, D.-G. Yu, X. Li and G. R. Williams, Mater. Highlights, 2020, 1, 1–13 Search PubMed.
- S. Chang, M. Wang, F. Zhang, Y. Liu, X. Liu, D. G. Yu and H. Shen, Mater. Des., 2020, 192, 108782 CrossRef CAS.
- J. Hou, J. Yang, X. Zheng, M. Wang, Y. Liu and D. G. Yu, Int. J. Pharm., 2020, 583, 119397 CrossRef CAS.
- K. Wang, P. Wang, M. Wang, D.-G. Yu, F. Wan and S. W. A. Bligh, Mater. Sci. Eng., C, 2020, 113, 110988 CrossRef CAS.
- Z. Bazrafshan and G. K. Stylios, Int. J. Biol. Macromol., 2019, 129, 693–705 CrossRef CAS.
- X. Gong, J. Kulwatno and K. L. Mills, Acta Biomater., 2020, 108, 128–141 CrossRef CAS.
- Y. Gao, X. Jin, Y. Sun, F. F. Xu and M. Zhang, Adv. Powder Technol., 2018, 29, 682–691 CrossRef CAS.
- S. Wang, Y. Hou, S. Zhang, J. Li, Q. Chen, M. Yu and W. Li, J. Mater. Chem. B, 2018, 6, 4972–4984 RSC.
- E. Mirhadi, M. Rezaee and B. Malaekeh-Nikouei, Biomed. Pharmacother., 2018, 104, 465–473 CrossRef CAS.
- D. Liu, X. Meng, D. Wu, Z. Qiu and H. Luo, Front. Pharmacol., 2019, 10, 1–12 CrossRef.
- S. Fujita, Y. Wakuda, M. Matsumura and S. Suye, J. Mater. Chem. B, 2019, 7, 6556–6563 RSC.
- Y. Wakuda, S. Nishimoto, S. Suye and S. Fujita, Sci. Rep., 2018, 8, 6248 CrossRef.
- A. Kato, S. Suye and S. Fujita, Kobunshi Ronbunshu, 2016, 73, 366–369 CrossRef CAS.
- E. Y. Kang, E. Lih, I. H. Kim, Y. K. Joung and D. K. Han, Biomater. Res., 2016, 20, 1–9 CrossRef.
- S. K. Battu, M. A. Repka, S. Maddineni, A. G. Chittiboyina, M. A. Avery and S. Majumdar, AAPS PharmSciTech, 2010, 11, 1466–1475 CrossRef CAS.
- L. Su, J. Zou, S. Dong, N. Hao and H. Xu, RSC Adv., 2017, 7, 55364–55370 RSC.
- X. Ma, J. Liu, C. Ni, D. C. Martin, D. B. Chase and J. F. Rabolt, ACS Macro Lett., 2012, 1, 428–431 CrossRef CAS.
- J. Jia, K. Zhang, X. Zhou, D. Zhou and F. Ge, Polymers, 2018, 10, 1–16 Search PubMed.
- P. Costa and J. M. Sousa Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- J.-W. Lee and J. A. Gardella, Appl. Surf. Sci., 2004, 231–232, 442–446 CrossRef CAS.
- Q. Hou, X. Tang, H. Liu, J. Tang, Y. Yang, X. Jing, Q. Xiao, W. Wang, X. Gou and Z. Wang, Cancer Sci., 2011, 102, 1287–1292 CrossRef CAS.
- T. P. Hamsa and G. Kuttan, Phytother. Res., 2012, 26, 568–578 CrossRef CAS.
- Q. Liu, H. Jiang, Z. Liu, Y. Wang, M. Zhao, C. Hao, S. Feng, H. Guo, B. Xu, Q. Yang, Y. Gong and C. Shao, PLoS One, 2011, 6, e23427 CrossRef CAS.
- K. Yan, C. Zhang, J. Feng, L. Hou, L. Yan, Z. Zhou, Z. Liu, C. Liu, Y. Fan, B. Zheng and Z. Xu, Eur. J. Pharmacol., 2011, 661, 1–7 CrossRef CAS.
- J. B. Kim, J.-H. Yu, E. Ko, K.-W. Lee, A. K. Song, S. Y. Park, I. Shin, W. Han and D. Y. Noh, Phytomedicine, 2010, 17, 436–440 CrossRef CAS.
- A. Agnarelli, M. Natali, M. Garcia-Gil, R. Pesi, M. G. Tozzi, C. Ippolito, N. Bernardini, R. Vignali, R. Batistoni, A. M. Bianucci and S. Marracci, Sci. Rep., 2018, 8, 10599 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08353d |
| This journal is © The Royal Society of Chemistry 2021 |