
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient synthetic route to L-γ-methyleneglutamine and its amide derivatives, and their selective anticancer activity†
Md Imran Hossaina,
Ajit G. Thomasb,
Fakhri Mahdia,
Amna T. Adama,
Nicholas S. Akins a,
Morgan M. Woodarda,
Jason J. Parisa,
Barbara S. Slusherb and
Hoang V. Le*a
a,
Morgan M. Woodarda,
Jason J. Parisa,
Barbara S. Slusherb and
Hoang V. Le*a
aDepartment of BioMolecular Sciences, Research Institute of Pharmaceutical Sciences, School of Pharmacy, University of Mississippi, Mississippi 38677, USA. E-mail: hle@olemiss.edu
bJohns Hopkins Drug Discovery, Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
First published on 10th February 2021
Abstract
In cancer cells, glutaminolysis is the primary source of biosynthetic precursors, fueling the TCA cycle with glutamine-derived α-ketoglutarate. The enhanced production of α-ketoglutarate is critical to cancer cells as it provides carbons for the TCA cycle to produce glutathione, fatty acids, and nucleotides, and contributes nitrogens to produce hexosamines, nucleotides, and many nonessential amino acids. Efforts to inhibit glutamine metabolism in cancer using amino acid analogs have been extensive. L-γ-Methyleneglutamine was shown to be of considerable biochemical importance, playing a major role in nitrogen transport in Arachis and Amorpha plants. Herein we report for the first time an efficient synthetic route to L-γ-methyleneglutamine and its amide derivatives. Many of these L-γ-methyleneglutamic acid amides were shown to be as efficacious as tamoxifen or olaparib at arresting cell growth among MCF-7 (ER+/PR+/HER2−), and SK-BR-3 (ER−/PR−/HER2+) breast cancer cells at 24 or 72 h of treatment. Several of these compounds exerted similar efficacy to olaparib at arresting cell growth among triple-negative MDA-MB-231 breast cancer cells by 72 h of treatment. None of the compounds inhibited cell growth in benign MCF-10A breast cells. Overall, N-phenyl amides and N-benzyl amides, such as 3, 5, 9, and 10, arrested the growth of all three (MCF-7, SK-BR-3, and MDA-MB-231) cell lines for 72 h and were devoid of cytotoxicity on MCF-10A control cells; N-benzyl amides with an electron withdrawing group at the para position, such as 5 and 6, inhibited the growth of triple-negative MDA-MB-231 cells commensurate to olaparib. These compounds hold promise as novel therapeutics for the treatment of multiple breast cancer subtypes.
1. Introduction
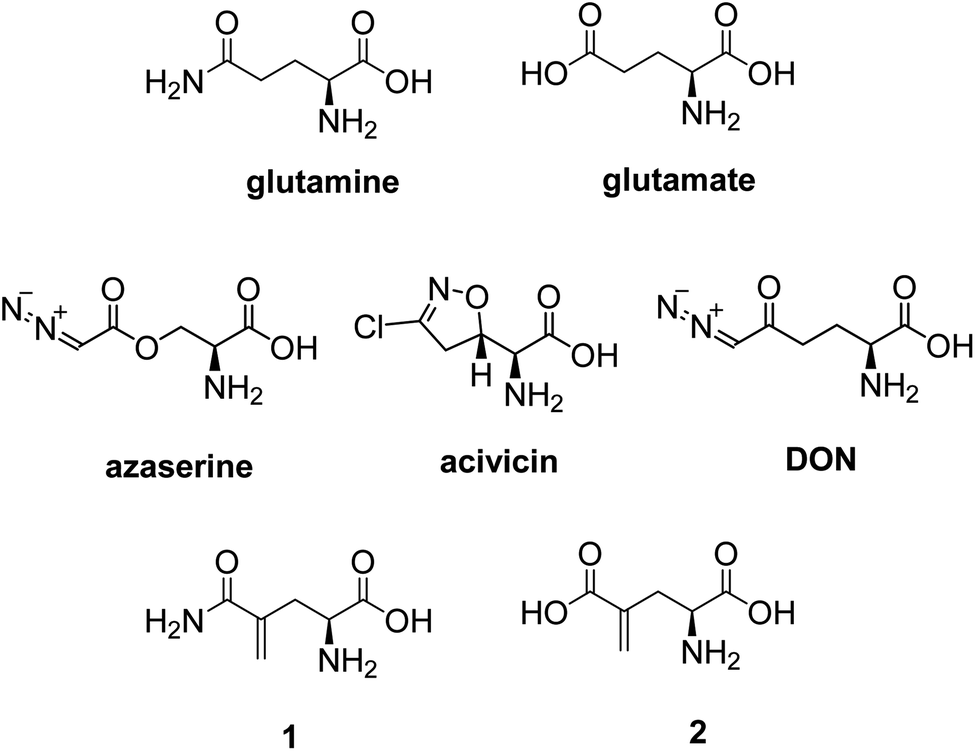
While glucose is the primary nutrient for the maintenance and promotion of cell function, glutamine and glutamate are considered to be equally important.1 Glutamine participates in numerous functional activities in cells, including being a substrate for protein synthesis, ureogenesis in the liver, and for hepatic and renal gluconeogenesis.1 Glutamine has been shown to be a precursor for neurotransmitter synthesis, nucleotide and nucleic acid synthesis, and glutathione production.2 In addition, glutamine is an oxidative fuel for the immune system, a major source of nitrogen for purine and pyrimidine biosynthesis, and a nitrogen transporter between organs.1 Many of these functions are done via the formation of glutamate from glutamine. In cancer cells, glutaminolysis is the primary source of biosynthetic precursors, fueling the TCA cycle with glutamine-derived α-ketoglutarate.3,4 The enhanced production of α-ketoglutarate is critical to cancer cells as it provides carbons for the citric acid cycle to produce glutathione, fatty acids, and nucleotides, and contributes nitrogens to produce hexosamines, nucleotides, and many nonessential amino acids.5Efforts to inhibit glutamine metabolism in cancer using amino acid analogs have been extensive.6 There are a number of naturally occurring glutamine analogues, such as azaserine,6 acivicin,6 and 6-diazo-5-oxo-L-norleucine (DON)7 (Fig. 1), that are inhibitors of glutaminase,8,9 NAD synthase,10 CTP synthetase,11 FGAR aminotransferase,12 and many other glutamine-dependent enzymes.5 These compounds have been shown to suppress the growth of a variety of tumors and demonstrate their activity in some clinical trials.6,7 However, they have also demonstrated variable degrees of gastrointestinal toxicity, myelosuppression, and neurotoxicity, due to their non-selectivity. In recent years, considerable interest has focused on the discovery of new agents that selectively target glutamine-consuming processes, as well as the development of methods directed at specific nodes of glutamine metabolism.
 | ||
| Fig. 1 Structures of important naturally occurring glutamine derivatives. | ||
L-γ-Methyleneglutamine (1) and L-γ-methyleneglutamic acid (2) (Fig. 1) were first isolated from groundnut seedling (Arachis hypogaea) in 1952.13,15 These compounds were also later found in several other quite unrelated species, including tulip bulbs (Tulipa gerneriana),16 hops,17 and Amorpha fruticosa (a species of flowering plant in the legume family, Fabaceae).18 Both 1 and 2 were shown to be of considerable biochemical importance, playing a major role in nitrogen transport in Arachis and Amorpha plants.18,19 Compound 2 was later shown to exhibit strong central nervous system (CNS) inhibitory activity20 as well as to be 10 times more potent than L-glutamate as a depolarizing agent on the newborn rat spinal cord.21 The syntheses of racemic mixture of 1 and racemic mixture of 2 were reported in 1955 (Scheme 1),13,14 starting from diethyl 1-formamidobut-3-yne-1,1-dicarboxylate (11) that had been prepared in advance from another publication.22 The synthesis of the biological relevant isomer 2 was reported several times in the literature by a reaction of homochiral aziridine-2-carboxylates with stabilized Wittig reagents,23 a 15-step synthesis starting from N-Boc-L-aspartate acid γ-benzyl ester,21 a reduction of the enaminone product of the reaction between tert-butyl-N-Boc-pyroglutamate and the Bredereck reagent,24 or a synthetic route via a reaction between the lithium lactam enolate of ethyl N-Boc-pyroglutamate and the Eschenmoser's salt.25 In addition, the synthesis of C3-deuterium-labeled L-γ-methyleneglutamic acid 2 from L-pyroglutamic acid was also reported.26 However, no synthesis of L-γ-methyleneglutamine 1, which is the biological relevant isomer, nor further research on its biological activity has been reported. Herein we report an efficient synthetic route to L-γ-methyleneglutamine (1) and its amide derivatives (3–10, Fig. 2). These compounds were evaluated for their anticancer activity on three different breast cancer cell lines: MCF-7 (ER+/PR+/HER2−), SK-BR-3 (ER−/PR−/HER2+), and triple negative MDA-MB-231. The compounds were also evaluated for their activity on a non-cancerous breast cell line, MCF-10A, as a control.
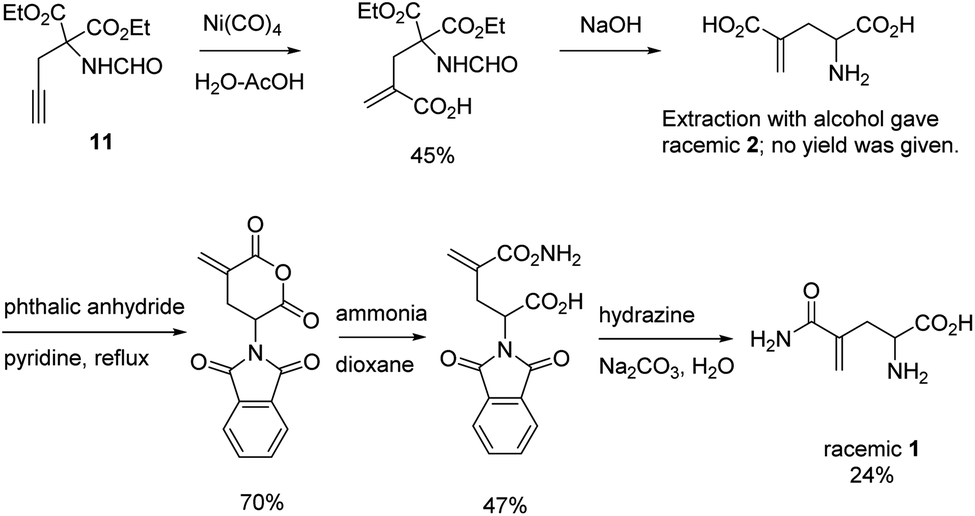 | ||
| Scheme 1 Synthesis of racemic mixture of 1 and racemic mixture of 2.13,14 | ||
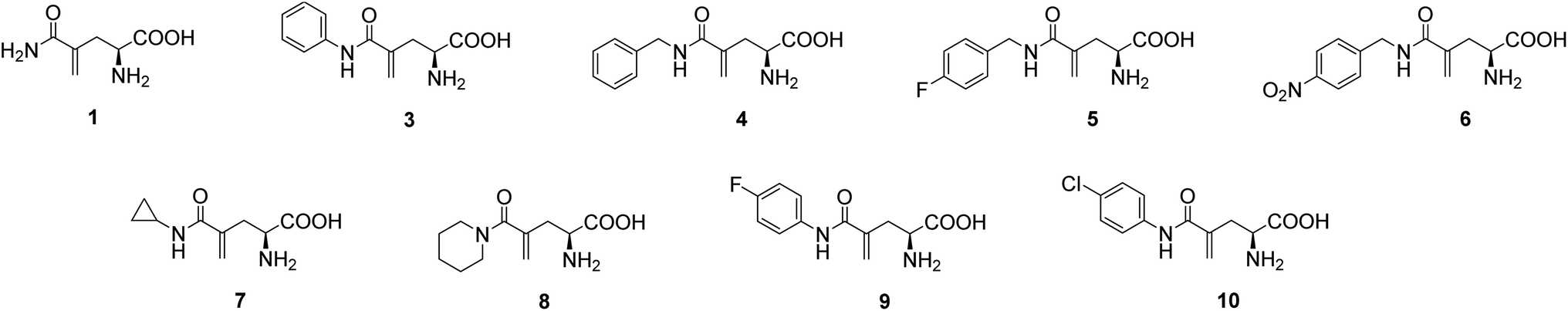 | ||
| Fig. 2 Structures of L-γ-methyleneglutamine (1) and its amide derivatives (3–10). | ||
2. Results and discussion
2.1. Synthesis of L-γ-methyleneglutamine and its amide derivatives
In our initial attempt to synthesize L-γ-methyleneglutamine (1), we decided to base part of the synthetic route on the previously reported synthesis of the racemic mixture of 1,13,14 going through the formation of the corresponding phthalimido-anhydride 15 (Fig. 3). We started the synthetic route with the commercially available L-pyroglutamic acid (12). The carboxylic acid and amide groups of 12 were first protected by ethyl ester and Boc group, respectively. Then, the methylene group was introduced at C4 via a α-methylenation of the sterically hindered carbonyl group27 to give 13. Hydrolysis of 13 with LiOMe opened the lactam ring; however, an esterification also occurred at the newly formed acid group and gave us 14. Hydrolysis of 14 with 9 M HBr resulted in 2 in a mixture with other side products that were very difficult to purify 2 from. We then used the crude mixture of 2 to react with phthalic anhydride to try to synthesize the phthalimido-anhydride 15. Despite our effort with many different reaction conditions, the formation of 15 was not detected. Therefore, our initial synthetic plan to synthesize 1, which is shown in Fig. 3, was abandoned.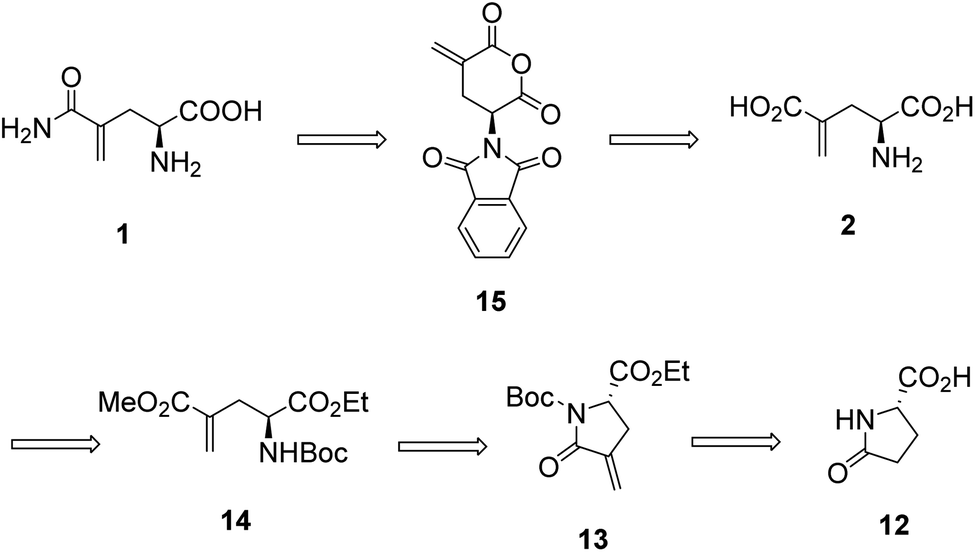 | ||
| Fig. 3 Our original synthetic plan for L-γ-methyleneglutamine (1). | ||
We then decided to utilize an installment of the amide group directly from the corresponding carboxylic acid using an amide coupling protocol in our synthetic route to 1. We also decided to utilize tert-butyl group as the protecting group for the carboxylic acid, so that we can use an acid to remove it later, instead of a base. We were successful in synthesizing 1 from the commercially available L-pyroglutamic acid (12) (Scheme 2). The carboxylic acid and amide groups of 12 were first protected by tert-butyl ester and Boc group, respectively, giving 17. Then, the methylene group was introduced at C4 via a α-methylenation of the sterically hindered carbonyl group27 to give 18 in 66% yield. The cyclic amide ring was selectively opened with LiOH28 to afford the common intermediate 19 in 70% yield. Amide was then installed to 19 from ammonium chloride using a published amide coupling protocol,29 resulting in 20 in 85% yield. Treatment of 20 with TFA removed both the tert-butyl and Boc protecting groups, affording the desired L-γ-methyleneglutamine 1 in 80% yield.
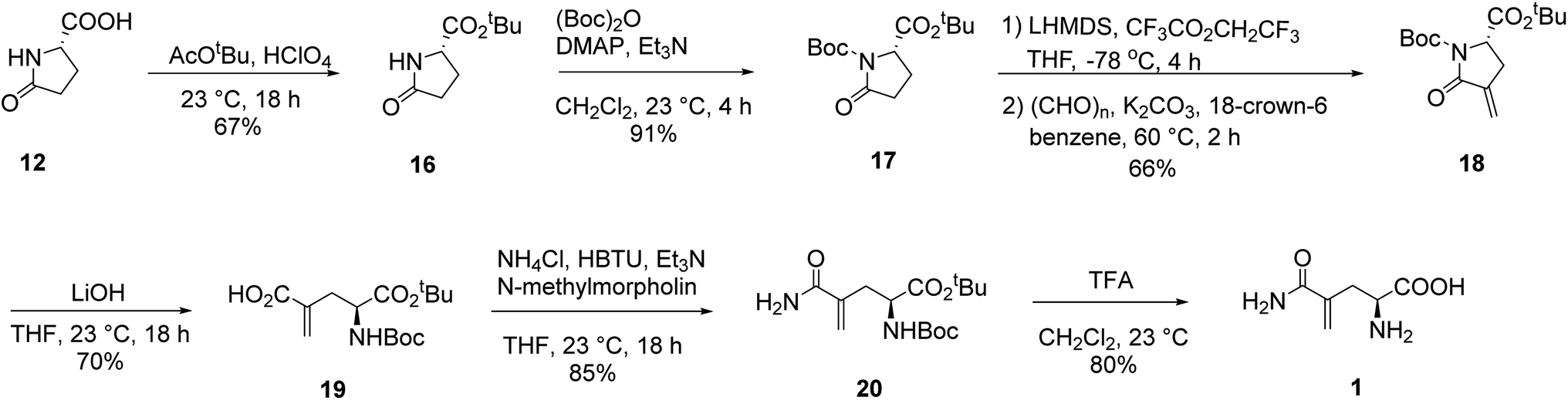 | ||
| Scheme 2 Synthesis of L-γ-methyleneglutamine 1. | ||
In a similar fashion, a library of L-γ-methyleneglutamine amide derivatives (3–10, Fig. 2), varying from aromatic amides (3, 9, 10) and non-aromatic secondary amides (4, 5, 6, 7) to tertiary amide (8), was quickly generated using the common intermediate 19 and the corresponding amines. The amide coupling protocol was observed to accommodate a wide range of functional groups. Protected amides 22–29 were synthesized via the published amide coupling protocol29 using 19 and the corresponding amines (21) (Scheme 3). The removal of the tert-butyl and Boc protecting groups using TFA was less accommodating. While the desired deprotected amides 3–8 were obtained in good yields from treating the corresponding 22–27 with TFA (Scheme 3), the desired deprotected 4-substituted phenyl amides 9–10 were not formed. Upon treating with TFA, the corresponding 28–29 quickly underwent cyclization, producing 30 instead (Scheme 4). This cyclization turned out to be an example that follows the 5-exo-trig of Baldwin's rules for ring closure reactions.30,31 When a milder acid, H3PO4, was used, some 9–10 were detected by LC-MS, but 30 was still the major product. When a neutral deprotection condition, ZnBr2 in DCM, was used, the desired deprotected amides 9–10 were obtained in 11–13% yields after purification via both silica gel flash column and HPLC.
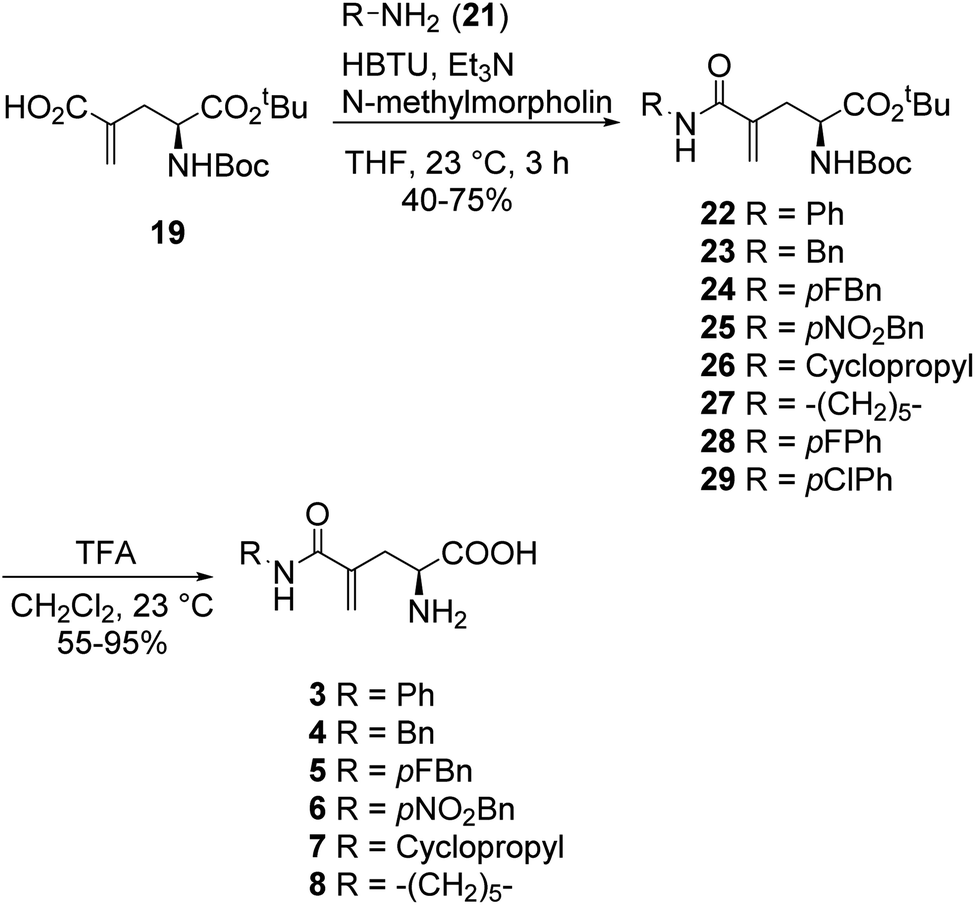 | ||
| Scheme 3 Synthesis of L-γ-methyleneglutamine amide derivatives 3–8. | ||
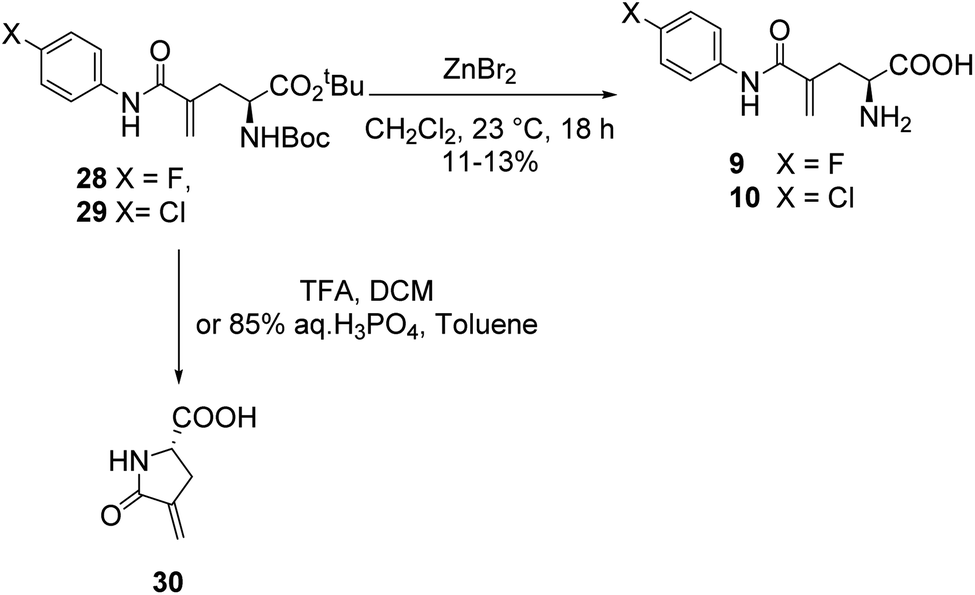 | ||
| Scheme 4 Deprotection of compounds 28 and 29 via acidic and neutral conditions. The formation of 30 turned out to be an example that follows the 5-exo-trig of Baldwin's rules for ring closure reactions. | ||
We suspected that the cyclization of 28 and 29 upon deprotection in acidic conditions was due to the electron withdrawing effect of the substituted phenyl groups. We decided to make the corresponding protected amide with 3,5-dichloroaniline (31, Scheme 5) to test its cyclization in a neutral deprotection condition. However, only the cyclized product 30 was obtained. The desired amide 31 was detected in trace amount and observed to be very unstable. It is very likely that 31 was formed from the coupling reaction, but then quickly underwent cyclization, even in the coupling reaction condition. We then tried the reaction again with several other coupling reaction conditions using different amide coupling reagents, including HATU, EDC, HOBt. Similar results were observed with 30 being the major product and 31 being detected only in trace amounts.
 | ||
| Scheme 5 Result of the coupling reaction between 19 and 3,5-dichloroaniline. | ||
2.2. Evaluation of L-γ-methyleneglutamine and its amide derivatives for their anticancer activity
In order to assess their capacity for inhibition of tumor growth, compounds 1 and 3–10 were screened for 24 h and 72 h growth of MCF-7 (ER+/PR+/HER2−), SK-BR-3 (ER−/PR−/HER2+), and MDA-MB-231 (triple negative) breast cancer cell lines. Inhibition of cell growth was assessed as the proportional change from vehicle-treated cells (negative controls) (Fig. 4 and Table 1). Compound potency was also assessed in comparison to tamoxifen and olaparib (positive controls). Tamoxifen was chosen given its capacity to suppress the proliferation of not only ER+ cancers, but also ER-negative cells via the inhibition of glutamine uptake.32 Olaparib was chosen because it is one of a few FDA-approved chemotherapeutics for triple-negative breast cancer.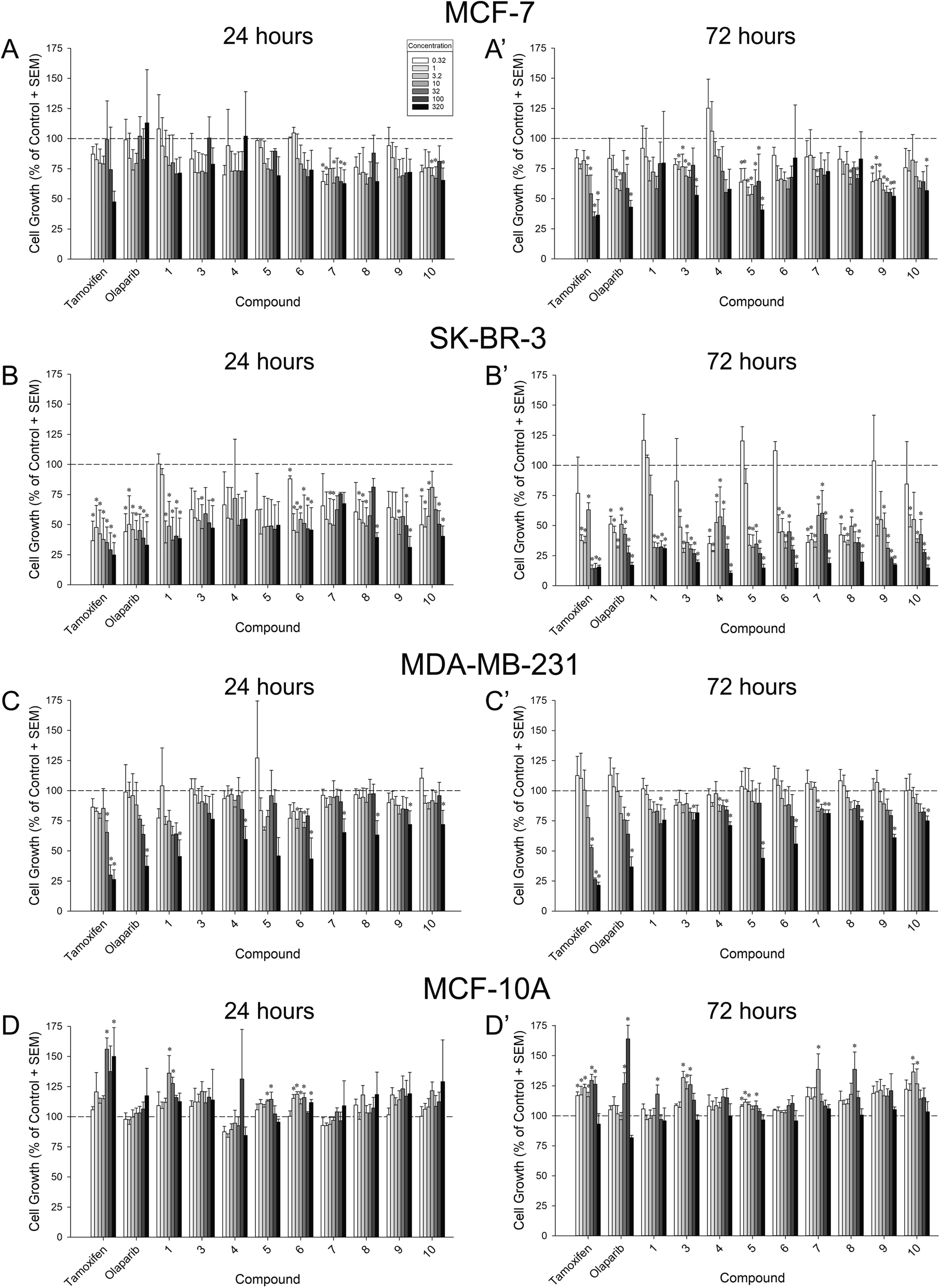 | ||
| Fig. 4 Proportional change from vehicle-treated MCF-7 (A–A′), SK-BR-3 (B–B′), MDA-MB-231 (C–C′), or MCF-10A (D–D′) cells after 24 h (left) or 72 h (right) exposure to a concentration-response regimen (0.32–320 μM) of compounds 1, 3–10, or positive controls (tamoxifen or olaparib). Dotted line indicates vehicle-treated control values. * significantly different from vehicle-treated control, p ≤ 0.05. | ||
| Growth inhibition [log(IC50 [μM]) ± SEM] | ||||||||
|---|---|---|---|---|---|---|---|---|
| MCF-7 | SK-BR-3 | MDA-MB-231 | MCF-10A | |||||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |
| a Significantly different from tamoxifen.b Significantly different from olaparib, p ≤ 0.05. | ||||||||
| Tamoxifen | 2.46 ± 0.24 | 1.62 ± 0.14 | −0.06 ± 0.25 | 0.24 ± 0.19 | 1.73 ± 0.12 | 1.62 ± 0.13 | >2.50 | >2.50 |
| Olaparib | >2.50 | 2.03 ± 0.22 | 0.21 ± 0.27 | 0.02 ± 0.19 | 2.21 ± 0.15 | 2.19 ± 0.14 | >2.50 | >2.50 |
| 1 | 2.66 ± 0.30 | 2.79 ± 0.39a | 0.90 ± 0.23a,b | 1.05 ± 0.16a,b | 2.15 ± 0.19 | 2.77 ± 0.17a,b | >2.50 | >2.50 |
| 3 | 3.03 ± 0.42 | 2.40 ± 0.18a | 1.57 ± 0.29a | 0.22 ± 0.19 | 2.89 ± 0.22a,b | 2.91 ± 0.18a,b | >2.50 | >2.50 |
| 4 | 3.41 ± 0.94 | 2.29 ± 0.22a | 1.87 ± 0.36a,b | −0.25 ± 0.28 | 2.68 ± 0.14a,b | 2.81 ± 0.12a,b | >2.50 | >2.50 |
| 5 | 2.77 ± 0.21 | 1.93 ± 0.24 | 0.59 ± 0.32 | 0.65 ± 0.16b | 2.54 ± 0.29a | 2.53 ± 0.18a,b | >2.50 | >2.50 |
| 6 | 2.66 ± 0.21 | 2.73 ± 0.36a | 1.11 ± 0.27 | 0.50 ± 0.18b | 2.34 ± 0.17a | 2.57 ± 0.18a,b | >2.50 | >2.50 |
| 7 | 2.36 ± 0.24 | 2.62 ± 0.24a,b | 2.50 ± 0.35a,b | −0.24 ± 0.28 | 2.81 ± 0.15a,b | 2.97 ± 0.15a,b | >2.50 | >2.50 |
| 8 | 2.64 ± 0.28 | 2.78 ± 0.28a,b | 2.13 ± 0.30a,b | −0.21 ± 0.22 | 2.84 ± 0.15a,b | 2.91 ± 0.14a,b | >2.50 | >2.50 |
| 9 | 2.62 ± 0.25 | 1.88 ± 0.21 | 1.06 ± 0.27 | 0.68 ± 0.22b | 2.81 ± 0.16a,b | 2.62 ± 0.12a,b | >2.50 | >2.50 |
| 10 | 2.60 ± 0.25 | 2.32 ± 0.22 | 1.90 ± 0.27a,b | 0.60 ± 0.21 | 2.94 ± 0.16a,b | 2.85 ± 0.15a,b | >2.50 | >2.50 |
When assessed in MCF-7 cells, six compounds exerted a significant inhibition of cell growth lasting at least 72 h, apart from tamoxifen and olaparib: 3, 5, 7, 8, 9, and 10 (Fig. 4A–A′). Compared to vehicle-treated cells, compounds 5 and 9 significantly inhibited MCF-7 growth at any concentration assessed at 72 h (0.32 μM–320 μM; p = 0.0002–0.05; Fig. 4A′). Similarly, compound 3 significantly inhibited MCF-7 growth by 72 h when administered at 1 μM or any greater concentration (with the exception of 100 μM; p = 0.0008–0.05; Fig. 4A′). Compounds 7, 8, and 10 exerted concentration-dependent effects to inhibit MCF-7 growth at 72 h (7 inhibiting only at 10 μM, 8 inhibiting only at 10 and 100 μM, and 10 inhibiting only at 320 μM; p = 0.02–0.05; Fig. 4A′). Only compounds 7 and 10 exerted early inhibition of MCF-7 growth as measured at 24 h (p = 0.03–0.05; Fig. 4A). When compared to positive controls, compounds 5, 9, and 10 exerted potency that did not differ from tamoxifen or olaparib at 72 h (Table 1). Conversely, compounds 1, 3, 4, and 6 were significantly less potent than tamoxifen (p = 0.0008–0.02), and compounds 7 and 8 were less potent than tamoxifen or olaparib at 72 h (p = 0.01–0.04; Table 1). No significant differences in potency were observed at 24 h (Table 1).
All compounds exerted an apparent broad capacity to inhibit growth in SK-BR-3 cells (Fig. 4B–B′), but this was driven by cytotoxicity for most compounds (including the positive control, tamoxifen as described in Section 2.3. below). By 72 h, compounds 4, 7, and 8 significantly inhibited SK-BR-3 growth at any concentration assessed (0.32 μM–320 μM; p < 0.0001–0.02; Fig. 4B′). Similarly, compound 3, 6, 9, and 10 significantly inhibited SK-BR-3 growth at all but the lowest concentration assessed (1 μM–320 μM; p < 0.0001–0.04; Fig. 4B′). Compound 5 significantly inhibited growth at 3.2 μM or greater concentrations (p < 0.0001), and 1 significantly inhibited growth at 10 μM or greater concentrations (p = 0.0002; Fig. 4B′). All compounds showed the capacity for early inhibition of growth at 24 h (p = 0.01–0.05) with the exceptions of 4 and 5 (Fig. 4B). In comparison to positive controls, compounds 5, 6, and 9 exerted potency that did not differ from tamoxifen or olaparib at 24 h (Table 1), all other compounds were significantly less potent than tamoxifen and/or olaparib at this time-point (p = 0.002–0.04; Table 1). However, by 72 h, all compounds were equipotent to tamoxifen (except 1 which was less potent; p < 0.0001), and compounds 3, 4, 7, 8, and 10 were equipotent to olaparib (all others were significantly less potent; p < 0.0001–0.04; Table 1).
As expected, the triple-negative MDA-MB-231 cell line provoked the greatest variance in compound effectiveness (Fig. 4C–C′) and potency (Table 1). After 24 h, only the highest concentration (320 μM) of compounds 1, 4, 7, 8, 9, and 10 significantly inhibited MDA-MB-231 growth (p = 0.009–0.04; Fig. 4C). Several concentrations of 6 significantly inhibited growth at this time-point (3.2, 32, and 320 μM; p = 0.0003–0.02; Fig. 4C). Neither 3 nor 5 significantly influenced MDA-MB-231 growth after only 24 h (Fig. 4C). However, by 72 h, compounds 4 and 7 significantly inhibited MDA-MB-231 growth at 10 μM, 100 μM, or 320 μM concentrations (p = 0.009–0.04), and compounds 5, 6, 8, 9, and 10 caused significant inhibition at the highest concentration (320 μM; p = 0.003–0.04; Fig. 4C′). Compounds 1 and 3 inhibited MDA-MB-231 growth only at the 100 μM concentration (p = 0.02–0.04; Fig. 4C′). Compared to olaparib, only 1 was equipotent when assessed at 24 h, all other compounds exerted significantly reduced potency at 24 h or 72 h (p < 0.001–0.005; Table 1).
As a control, all compounds were assessed against benign MCF-10A breast cells (Fig. 4D–D′). None of the compounds assessed significantly inhibited the growth of MCF-10A cells (Fig. 4D–D′; Table 1). However, significant increases were observed at 24 h with compounds 1, 5, and 6 (p = 0.0005–0.04; Fig. 4D) as well as at 72 h with compounds 1, 3, 5, 7, 8, and 10 (p = 0.001–0.05; Fig. 4D′).
2.3. Evaluation of L-γ-methyleneglutamine and its amide derivatives for their cytotoxic activity
To assess whether cytotoxicity contributed to reductions in cell growth, positive controls and compounds were screened for viability at 24 h and 72 h using the necrosis indicator, propidium iodide (Fig. 5; Table 2).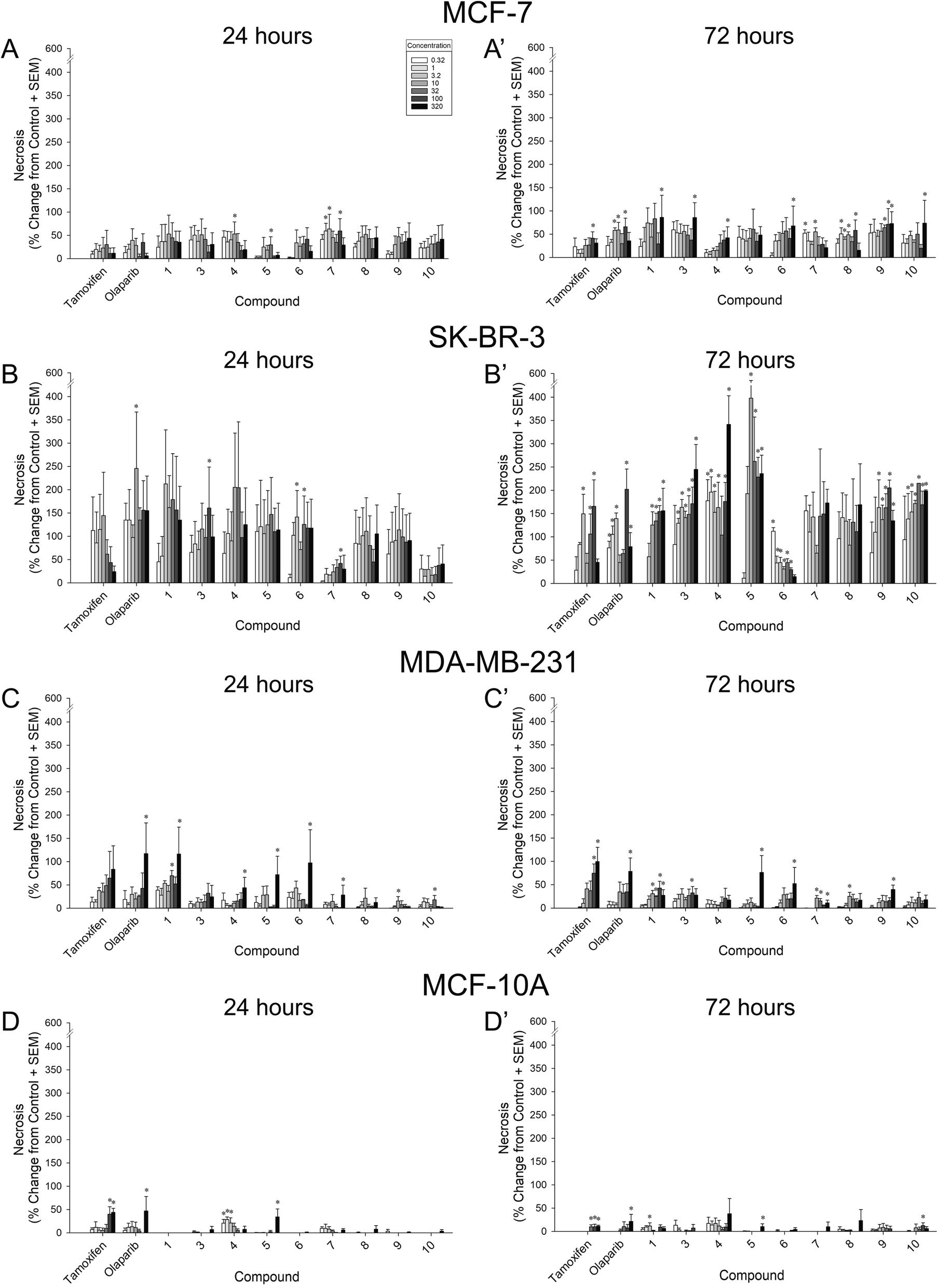 | ||
| Fig. 5 Proportional increase from vehicle-treated MCF-7 (A–A′), SK-BR-3 (B–B′), MDA-MB-231 (C–C′), or MCF-10A (D–D′) cells after 24 h (left) or 72 h (right) exposure to a concentration-response regimen (0.32–320 μM) of compounds 1, 3–10, or positive controls (tamoxifen or olaparib). * significantly different from vehicle-treated control, p ≤ 0.05. | ||
| Cytotoxicity [log(EC50 [μM]) ± SEM] | ||||||||
|---|---|---|---|---|---|---|---|---|
| MCF-7 | SK-BR-3 | MDA-MB-231 | MCF-10A | |||||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |
| a Significantly different from tamoxifen.b Significantly different from olaparib, p ≤ 0.05. | ||||||||
| Tamoxifen | >2.50 | 2.46 ± 0.20 | >2.50 | −0.48 ± 0.52 | 1.42 ± 0.32 | 1.49 ± 0.17 | 2.43 ± 0.12 | >2.50 |
| Olaparib | >2.50 | 1.42 ± 0.29 | >2.50 | −1.22 ± 1.05 | 1.79 ± 0.32 | 1.99 ± 0.19 | >2.50 | >2.50 |
| 1 | 2.18 ± 0.39 | 0.47 ± 0.37a | −0.73 ± 1.28a,b | 0.31 ± 0.36a,b | 0.95 ± 0.29b | >2.50a,b | >2.50 | >2.50 |
| 3 | >2.50 | 0.31 ± 0.33a | −0.71 ± 0.75a,b | >2.50 | >2.50a,b | >2.50a,b | >2.50 | >2.50 |
| 4 | >2.50 | 2.34 ± 0.18b | −1.01 ± 1.73a,b | >2.50 | >2.50a,b | >2.50a,b | >2.50 | >2.50 |
| 5 | >2.50 | 1.74 ± 0.33 | >2.50 | −0.41 ± 1.47 | 2.42 ± 0.25a,b | 2.42 ± 0.19a,b | >2.50 | >2.50 |
| 6 | >2.50 | 1.31 ± 0.29a | −0.47 ± 0.57a,b | −0.36 ± 0.88 | 2.18 ± 0.34a | 2.43 ± 0.19a,b | >2.50 | >2.50 |
| 7 | 1.81 ± 0.35 | >2.50b | 2.44 ± 0.23 | >2.50 | >2.50a,b | >2.50a,b | >2.50 | >2.50 |
| 8 | 2.29 ± 0.32 | 2.30 ± 0.31 | −1.14 ± 1.38a,b | >2.50 | >2.50a,b | >2.50a,b | >2.50 | >2.50 |
| 9 | 2.20 ± 0.25a,b | 0.36 ± 0.28a,b | −0.74 ± 0.87a,b | −1.22 ± 1.58 | >2.50a,b | >2.50a,b | >2.50 | >2.50 |
| 10 | 2.31 ± 0.28 | 1.97 ± 0.30 | 2.42 ± 0.32 | >2.50 | >2.50a,b | >2.50a,b | >2.50 | >2.50 |
Compared to vehicle-treated MCF-7 cells, compounds 4 (10 μM; p = 0.05), 5 (32 μM; p = 0.05), and 7 (1, 3.2, and 100 μM; p = 0.03–0.04) exerted some early concentration-dependent toxicity when assessed at 24 h (Fig. 5A). After 72 h, compounds 7 (0.32 and 10 μM; p = 0.006–0.009), 8 (1–10 and 100 μM; p = 0.004–0.04), 9 (32–100 μM; p = 0.02–0.04), and olaparib (3.2–10 and 100 μM; p = 0.007–0.02) significantly increased toxicity at multiple concentrations (Fig. 5A′). Only the highest concentration (320 μM) of compounds 1, 3, 4, 6, and 10 caused a significant increase in cytotoxicity to MCF-7 cells (p = 0.01–0.05; Fig. 5A′). Of note, the 100 μM concentration of tamoxifen was significantly more toxic to MCF-7 cells than vehicle (p = 0.03; Fig. 5A′). No significant increase in cytotoxicity was observed for compound 5 at 72 h. When compared to the cytotoxic potency of tamoxifen and/or olaparib, 9 was significantly more potent at 24 h and 72 h (p = 0.03–0.05), whereas 1, 3, and 6 were significantly more potent only at 72 h (p = 0.01–0.04; Table 2). Compounds 4 and 7 were less potent than olaparib at promoting 72 h MCF-7 cytotoxity (p = 0.0003–0.009; Table 2).
The greatest observations of cytotoxicity occurred in SK-BR-3 cells and were found in response to most compounds (including the control compound, tamoxifen). In SK-BR-3 cells, some early concentration-dependent increases in cytotoxicity were observed for compounds 3 (100 μM; p = 0.04), 6 (3.2 and 32 μM; p = 0.03–0.05), 7 (100 μM; p = 0.05), and olaparib (10 μM; p = 0.01) compared to 24 h vehicle treatment (Fig. 5B). Moreover, several compounds demonstrated significantly more cytotoxic potency after 24 h than did tamoxifen or olaparib, including 1, 3, 4, 6, 8, and 9 (p < 0.0001–0.001; Table 2). After 72 h, all compounds with the exceptions of 7 and 8 exerted concentration-dependent increases in cytotoxicity, including 1 (10–320 μM; p = 0.0002–0.002), 3 (1–320 μM; p = 0.0006–0.04), 4 (0.32–10, 100–320 μM; p = 0.002–0.05), 5 (10–320 μM; p = 0.002–0.04), 6 (0.32–100 μM; p < 0.0001–0.02), 9 (3.2–320 μM; p = 0.005–0.05), 10 (1–320 μM; p = 0.003–0.04), tamoxifen (3.2, 32–100 μM; p = 0.002–0.03), and olaparib (3.2–3.2, 100–320 μM; p < 0.0001–0.02; Fig. 5B′). However, only 1 exerted more cytotoxic potency than tamoxifen or olaparib (p = 0.001–0.02; Table 2).
When assessed in MDA-MB-231 cells after 24 h, all compounds with the exceptions of 3, 8, and tamoxifen exerted some concentration-dependent toxicity including 1 (32 and 320 μM; p = 0.002–0.05), 4 (320 μM; p = 0.02), 5 (320 μM; p = 0.01), 6 (320 μM; p = 0.02), 7 (320 μM; p = 0.05), 9 (10 μM; p = 0.03), 10 (32 μM; p = 0.04), and olaparib (320 μM; p = 0.009; Fig. 5C). Compound 1 exerted significantly more potent cytotoxic effects than did olaparib (p = 0.05), whereas all other compounds were less potent than tamoxifen and/or olaparib (p < 0.0001–0.04; Table 2). By 72 h, the highest concentrations of compounds 3 (100 μM; p = 0.04), 5 (320 μM; p = 0.001), 6 (320 μM; p = 0.02), 9 (320 μM; p = 0.003), tamoxifen (100–320 μM; p = 0.0004–0.004), and olaparib (320 μM; p = 0.003) were more cytotoxic than vehicle (Fig. 5C′). However, compounds 1 (10–320 μM; p = 0.001–0.04), 7 (10–32 and 320 μM; p = 0.0004–0.04), and 8 (10 μM; p = 0.03) demonstrated greater toxicity at lower concentrations (Fig. 5C′). Compounds 4 and 10 did not increase toxicity (Fig. 5C′). When compared to positive controls for potency, all compounds exerted less potent cytotoxicity than did tamoxifen or olaparib at this time-point (p < 0.0001–0.03; Table 2).
Our criteria for identifying leads required that efficacious concentrations occur without cytotoxicity. Despite the cytotoxic effects observed on cancer cell lines, compounds exerted far less toxicity when assessed on benign MCF-10A cells (Fig. 5D–D′). Only compounds 4 (0.32–3.2 μM; p = 0.006–0.03) and 5 (320 μM; p = 0.001) exerted any cytotoxicity after 24 h of treatment, as did tamoxifen (100–320 μM; p = 0.002–0.005) and olaparib (320 μM; p = 0.02; Fig. 5D). After 72 h, compounds 1 (3.2 μM; p = 0.03), 5 (320 μM; p = 0.005), 10 (100 μM; p = 0.05), tamoxifen (32–320 μM; p = 0.007–0.02), and olaparib (320 μM; p = 0.04) exerted significant toxicity compared to vehicle (Fig. 5D′). Concentration-response shifts could not be calculated given that all EC50s exceeded 2.5 logarithmic units (Table 2).
2.4. Evaluation of L-γ-methyleneglutamine and its amide derivatives for their possible inhibitory activity on glutaminase
As mentioned earlier, azaserine, acivicin, and DON failed to advance in clinical trials due to their non-selectivity on glutamine-dependent enzymes. We were wondering whether the suppression activity of these compounds on the growth of MCF-7, SK-BR-3, and MDA-MB-231 breast cancer cell lines followed a similar mechanism to that of azaserine, acivicin, and DON. These compounds were tested for their potential inhibitory activity on glutaminase, the enzyme that catalyzes the conversion of glutamine in the cytosol to glutamate in the mitochondria. However, these compounds did not show any inhibitory activity on kidney-type glutaminase (GLS1), with or without pre-incubation (2 h) with the enzyme. This result suggested that these compounds may follow a different mechanism to that of azaserine, acivicin, and DON in suppressing the growth of MCF-7, SK-BR-3, and MDA-MB-231 breast cancer cells, which is good news as azaserine, acivicin, and DON failed to advance in clinical trials. One possible biological target of these L-γ-methyleneglutamic acid amides is glutamine transporters, rather than glutamine-dependent enzymes. Recent studies have highlighted the importance of glutamine transporters in physiology and cancer cell growth.33,34 Given that these compounds are unsaturated amides, which are significantly less reactive than typical Michael acceptors like unsaturated ketones, and that these compounds did not inhibit the activity of GLS1 with pre-incubation, and that these compounds did not significantly inhibit the growth and exerted far less toxicity when assessed on benign MCF-10A cells, it is very likely that these compounds would act as reversible inhibitors of glutamine transporters, rather than irreversible inhibitors. Future experiments will aim to further characterize lead compounds to determine their mechanism(s) of action.3. Conclusion
We have reported for the first time an efficient synthetic route to the biologically-relevant L-γ-methyleneglutamine (1) and its amide derivatives (3–10). These compounds were evaluated for their anticancer activity on three different breast cancer cell lines: MCF-7 (ER+/PR+/HER2−), SK-BR-3 (ER−/PR−/HER2+), and triple negative MDA-MB-231. The compounds were also evaluated for their activity on a benign cell line, MCF-10A, as a control. For MCF-7 cancer cells, L-γ-methyleneglutamic acid amides with secondary amine, like 8, and branched alkyl amine, like 7, are less potent in inhibiting the growth than amides with primary amine, like 4, 5, and 6, and aromatic amine, like 3, 9, and 10. Within each subset, the amines with a stronger electron withdrawing group exhibit better potency. For example, 5 and 6 with a fluoro and a nitro groups, respectively, at the para position of the aromatic ring are more potent than 4. Similarly, 9 is more potent than 10, which is more potent than 3. Amides with aromatic amine are more potent than primary amine; for example 9 is more potent than 5. Notably, compounds 5 and 9 carry a fluorine atom at the para position of the aromatic ring, and they are the most potent compounds of the series. This may be due to the hydrophobic nature of fluorine that enhances the binding and its potential formation of special interactions with the biological target. These structure–activity relationships (SARs) are generally also true for the SK-BR-3 and MDA-MB-231 cancer cells. Overall, N-phenyl amides and N-benzyl amides, such as 3, 5, 9, and 10, exerted concentration-dependent inhibition of growth by 72 h across all three cancer cell lines. Additionally, N-benzyl amides with an electron withdrawing group at the para position, such as 5 and 6, were the only compounds to inhibit the growth of triple-negative MDA-MB-231 cells commensurate to olaparib. Unlike azaserine, acivicin, and DON, these compounds did not inhibit GLS1, suggesting that they may follow a different mechanism to that of azaserine, acivicin, and DON in exerting commensurate cytotoxic effects to cancer cell lines, which is good news as azaserine, acivicin, and DON failed to advance in clinical trials. One possible biological target of these L-γ-methyleneglutamic acid amides is glutamine transporters, rather than glutamine-dependent enzymes. In support, these amides were largely devoid of cytotoxicity in benign MCF-10A cells and the modest toxicity observed at the highest concentrations of 5 and 10 did not reach that produced by tamoxifen or olaparib. Notably, some compounds demonstrated an apparent reverse dose-dependency (exerting greater effects at lower dosing, such as 4 and 7; Fig. 5A–A′). Given that the pharmacodynamic profiles are not yet known, it cannot be ruled out whether greater dosing produces an upregulation of biological targets, thereby reducing the efficacy of some compounds at higher concentrations.These compounds hold promise for the development of novel anticancer therapeutics given their capacity for cancer-selective necrosis across breast cancer subtypes. Future experiments will aim to further characterize lead compounds to determine their mechanism(s) of action, including their capacity to arrest the cell cycle or promote apoptosis, as well as their pharmacokinetic profiles.
4. Experimental section
4.1. General chemical synthesis procedures
All chemicals were obtained from Sigma-Aldrich or Fisher Scientific and used as received unless specified. All syntheses were conducted with anhydrous conditions under an atmosphere of argon, using flame-dried glassware and employing standard techniques for handling air-sensitive materials unless otherwise noted. All solvents were distilled and stored under an argon or nitrogen atmosphere before use. 1H NMR and 13C NMR spectra were recorded on a Bruker-400 and or a Bruker-500 spectrometer using CDCl3, MeOD, or D2O as the solvent. Chemical shifts (δ) were recorded in parts per million and referenced to CDCl3 (7.26 ppm for 1H NMR and 77.16 ppm for 13C NMR), MeOD (3.31 ppm for 1H NMR and 49.00 ppm for 13C NMR), or D2O (4.79 ppm for 1H NMR). 19F NMR spectra were recorded on a Bruker-400 spectrometer. Coupling constants (J) are in Hz. The following abbreviations were used to designate the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintuplet, m = multiplet, br = broad. Melting points were measured using an OptiMelt automated melting point system. LC-MS were measured using an ACQUITY-Waters micromass (ESCi) system. High-resolution mass spectra (HRMS) were measured using a Synapt Q-TOF ESI-MS.4.2. Synthetic procedures
4.3. General procedure for amide coupling
To a solution of the acid compound (1 equiv.) in anhydrous THF in argon condition was added HBTU (1.5 equiv.), Et3N (1.5 equiv.), 4-methylmorpholin (1.5 equiv.), and amine (1.5 equiv.) at rt. The reaction mixture was stirred for 3 hours at rt. The solid was filtered off, and the filtrate was concentrated. The crude mixture was purified via silica column (ethyl acetate and hexane) to afford the amides.4.4. General method for deprotection of amide compounds
tert-Butyl and Boc protected amides were dissolved in a DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TFA = 4:1 mixture at rt and stirred until all the starting material was fully consumed. After the reaction was complete, the solvents were evaporated. The crude mixture was purified via silica gel column chromatography (DCM and MeOH) or HPLC (H2O and ACN).
:TFA = 4:1 mixture at rt and stirred until all the starting material was fully consumed. After the reaction was complete, the solvents were evaporated. The crude mixture was purified via silica gel column chromatography (DCM and MeOH) or HPLC (H2O and ACN).
4.5. Evaluation of growth inhibition and viability of cancer cell lines
Compounds 1 and 3–10 were purified by HPLC (7.8 × 30 mm, 7 μm, C18, gradient 98% water in acetonitrile to 80% water in acetonitrile, flow rate 2 mL min−1, retention time 13 min) until their purities were higher than 95% before being evaluated in cell assays; purities were measured using a Waters 2695 analytical HPLC system. All cell lines were all obtained from the American Type Culture Collection (ATCC; Manassas, VA) for the purpose of this study. All cells used were passaged less than 10 times. Cells were seeded onto 96-well plates at a density of 2 × 104 cells per well for assessment of growth and cell death. MCF-7, SK-BR-3, and MDA-MB-231 cells were maintained in DMEM/F12 media (#11320-033, Life Technologies, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; #SH30071.03, Thermo Scientific Hyclone, Logan, UT), and 0.5% antibiotic/antimycotic mixture (#15240-062, Life Technologies). MCF-10A cells were maintained in sterile, unfiltered MEBM growth media supplemented with all components of a MEGM kit (#CC-3150, Lonza Group Ltd, Switzerland), with exception of #GA-1000 (gentamycin–amphotericin-B mixture). In addition, 0.5% penicillin–streptomycin mixture (#15-140-163, Thermo Fisher Scientific, Waltham, MA) and Cholera toxin (100 ng mL−1; #C8052, Sigma) were added to the media. All compounds were dissolved in 50% DMSO with the exception of tamoxifen which was dissolved in 90% EtOH. All compounds were then diluted to concentration in media (DMSO final concentration < 0.8%; EtOH final concentration < 0.6%). Control cells were incubated with the same concentrations of DMSO or EtOH and used as a negative control for statistical comparison. Cells were incubated with all compounds in a concentration-response regimen (0.32, 1, 3.2, 10, 32, 100, 320 μM) for 24 h or 72 h in a 37 °C humidified incubator (5% CO2). Media was not changed over the course of the experiment. On the day of assessment, a working solution of propidium iodide (ex/em: 536/617 nm) and Hoechst 33342 (ex/em: 350/461 nm) was prepared by diluting stocks in Hank's Balanced Salt Solution (HBSS; 1:50 dilution for propidium iodide and 1/10000 for Hoescht). Media in the 96-well plates were removed and 100 μL of HBSS containing fluorophores was added to each well. Cells were incubated for 15 minutes at 37 °C (5% CO2) and fluorescent emissions were read on a CLARIOstar plate reader (BMG Labtech, Cary, NC). Relative fluorescent units (RFU) for treatment wells were calculated as a proportion of non-treated control wells in order to assess growth (negative controls indicate by dashed line at 100% in Fig. 4). Viability of the cells was assessed by calculating the proportion of necrotic cells as a function of the total cell RFU per well (data depicted as % increase from negative control wells in Fig. 5). All experiments were independently replicated 3 times and each treatment was run in technical duplicate for each experiment.
4.6. Statistical analyses
To delineate differences in cell growth and necrosis in comparison to negative controls (vehicle-treated cells), separate two-way analyses of variance (ANOVA) were conducted with treatment exposure time (24 or 72 h) and compound concentration as the between-subjects factors (see Fig. 4 and 5). For each compound, simple main effects and planned post hoc contrasts were conducted to reveal dosing that significantly differed from controls. All post hoc comparisons were corrected for family-wise error and considered significant when p ≤ 0.05. To assess comparative changes in potency from positive controls (tamoxifen and olaparib), median inhibitory and effective concentrations (logIC50, logEC50) were determined via non-linear regression (sigmoidal curvilinear modeling with variable slope; Prism 7, GraphPad Software, La Jolla, CA) using a least-squares fit for each treatment group (bottom values constrained to 0; see Tables 1 and 2). For each cell type (MCF-7, SK-BR-3, MDA-MB-231, or MCF-10A) and treatment time (24 or 72 h), logIC50/logEC50 values were compared to those obtained for tamoxifen and olaparib via extra sum-of-squares F-test. Median shifts were considered significant when p ≤ 0.05.
4.7. Evaluation for possible inhibitory activity on kidney-type glutaminase
Compounds 1 and 3–10 were purified by HPLC (7.8 × 30 mm, 7 μm, C18, gradient 98% water in acetonitrile to 80% water in acetonitrile, flow rate 2 mL min−1, retention time 13 min) until their purities were higher than 95% before being evaluated in GLS1 assays; purities were measured using a Waters 2695 analytical HPLC system. A published procedure was followed.35 hKGAd1 construct was prepared from the hKGA cDNA by deleting the sequence encoded in exon 1 and cloning into pET15b. Purified human GLS1 (hKGA124–669; 250 nM) was used as the source of enzyme, and radiolabeled glutamine (L-[3H]-glutamine) was used as the substrate. The assay was conducted in the presence and absence of the compound, at rt (with and without 2 h incubation), in phosphate buffer (pH 8.2). All compounds were tested on an 7-point concentration response curve, with 10-fold dilution between concentrations and starting at the highest concentrations determined by the solubility of the compounds. In all cases, the highest concentration tested was equal to or greater than 100 μM. At the end of the reaction period, the assay was terminated upon the addition of imidazole buffer (pH 7). 96-well spin columns packed with strong anion ion-exchange resin was used to separate the substrate and the reaction product. Unreacted [3H]-glutamine was removed by washing with imidazole buffer. [3H]-Glutamate, the reaction product, was eluted with diluted HCl and analyzed for radioactivity.Author contributions
H. V. L. designed the molecules and supervised the overall coordination of the research. A. T. A., M. I. H., and M. M. W. worked on the original synthetic plan for 1 (Fig. 3). M. I. H. synthesized compounds 1–10 and studied the formation of 30 (Schemes 2–5). N. S. A. measured melting points of the compounds and took 19F NMR spectra. F. M. and J. J. P. carried out the evaluation of compounds 1, 3–10 for their antigrowth/necrotic activity. A. G. T. and B. S. S. carried out the evaluation of compounds 1, 3–10 for their possible inhibitory activity on glutaminase. M. I. H., J. J. P., and H. V. L. wrote the manuscript. All authors approved the final version.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Work was supported by the American Association of Colleges of Pharmacy (2018 New Investigator Award to H. V. L.), National Institute on Drug Abuse (R00 DA039791 to J. J. P.), National Institute of General Medical Sciences (P30GM122733 pilot project awards to H. V. L. and J. J. P.), National Cancer Institute (R01 CA193895 to B. S. S.), and funds from the Department of BioMolecular Sciences at the University of Mississippi, School of Pharmacy. The content is solely the responsibility of the authors and does not necessarily represent the official views of these funders.References
- P. Newsholme, J. Procopio, M. M. R. Lima, T. C. Pithon-Curi and R. Curi, Glutamine and Glutamate – Their Central Role in Cell Metabolism and Function, Cell Biochem. Funct., 2003, 21(1), 1–9 CrossRef CAS.
- P. Newsholme, M. M. R. Lima, J. Procopio, T. C. Pithon-Curi, S. Q. Doi, R. B. Bazotte and R. Curi, Glutamine and Glutamate as Vital Metabolites, Braz. J. Med. Biol. Res., 2003, 36(2), 153–163 CrossRef CAS.
- N. S. Akins, T. C. Nielson and H. V. Le, Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer, Curr. Top. Med. Chem., 2018, 18(6), 494–504 CrossRef CAS.
- J. W. Erickson and R. A. Cerione, Glutaminase: A Hot Spot For Regulation Of Cancer Cell Metabolism?, Oncotarget, 2010, 1(8), 734–740 CrossRef.
- Y. Xiang, Z. E. Stine, J. Xia, Y. Lu, R. S. O'Connor, B. J. Altman, A. L. Hsieh, A. M. Gouw, A. G. Thomas, P. Gao, L. Sun, L. Song, B. Yan, B. S. Slusher, J. Zhuo, L. L. Ooi, C. G. L. Lee, A. Mancuso, A. S. McCallion, A. Le, M. C. Milone, S. Rayport, D. W. Felsher and C. V. Dang, Targeted Inhibition of Tumor-Specific Glutaminase Diminishes Cell-Autonomous Tumorigenesis, J. Clin. Invest., 2015, 125(6), 2293–2306 CrossRef.
- C. T. Hensley, A. T. Wasti and R. J. DeBerardinis, Glutamine and Cancer: Cell Biology, Physiology, and Clinical Opportunities, J. Clin. Invest., 2013, 123(9), 3678–3684 CrossRef CAS.
- R. A. Shapiro, V. M. Clark and N. P. Curthoys, Inactivation of Rat Renal Phosphate-Dependent Glutaminase with 6-Diazo-5-Oxo-L-Norleucine. Evidence for Interaction at the Glutamine Binding Site, J. Biol. Chem., 1979, 254(8), 2835–2838 CrossRef CAS.
- K. Thangavelu, C. Q. Pan, T. Karlberg, G. Balaji, M. Uttamchandani, V. Suresh, H. Schüler, B. C. Low and J. Sivaraman, Structural Basis for the Allosteric Inhibitory Mechanism of Human Kidney-Type Glutaminase (KGA) and Its Regulation by Raf-Mek-Erk Signaling in Cancer Cell Metabolism, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(20), 7705–7710 CrossRef CAS.
- K. Thangavelu, Q. Y. Chong, B. C. Low and J. Sivaraman, Structural Basis for the Active Site Inhibition Mechanism of Human Kidney-Type Glutaminase (KGA), Sci. Rep., 2014, 4, 3827 CrossRef CAS.
- R. K. Barclay and M. A. Phillipps, Effects of 6-Diazo-5-Oxol-Norleucine and Other Tumor Inhibitors on the Biosynthesis of Nicotinamide Adenine Dinucleotide in Mice, Cancer Res., 1966, 26(2), 282–286 CAS.
- A. Hofer, D. Steverding, A. Chabes, R. Brun and L. Thelander, Trypanosoma brucei CTP Synthetase: A Target for the Treatment of African Sleeping Sickness, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(11), 6412–6416 CrossRef CAS.
- A. I. Grayzel, J. E. Seegmiller and E. Love, Suppression of Uric Acid Synthesis in the Gouty Human by the Use of 6-Diazo-5-Oxo-L-Norleucine, J. Clin. Invest., 1960, 39(3), 447–454 CrossRef CAS.
- P. Wailes, M. C. Whitting and L. Fowden, Synthesis of γ-Methyleneglutamic Acid and γ-Methyleneglutamine, Nature, 1954, 174(4420), 130–131 CrossRef CAS.
- P. C. Wailes and M. C. Whiting, Researches on Acetylenic Compounds. Part LI. The Syntheses of γ-Methyleneglutamic Acid and γ-Methyleneglutamine, J. Chem. Soc., 1955, 3636–3641 RSC.
- J. Done and L. Fowden, A New Amino Acid Amide in the Groundnut Plant (Arachis Hypogaea); Evidence of the Occurrence of Gamma-Methyleneglutamine and Gamma-Methyleneglutamic Acid, Biochem. J., 1952, 51(4), 451–458 CrossRef CAS.
- R. M. Zacharius, J. K. Pollard and F. C. Steward, γ-Methyleneglutamine and γ-Methyleneglutamic Acid in the Tulip (Tulipa gesneriana), J. Am. Chem. Soc., 1954, 76(7), 1961–1962 CrossRef CAS.
- G. Harris and A. R. Tatchell, Amino Acids and Peptides of Hops and Wort III. The Amino Acids of Fresh Hops, J. Inst. Brew., 1953, 59(5), 371–377 CrossRef CAS.
- B. Tschiersch, Über γ-Methylenglutamin Und γ-Methylenglutaminsäure in Keimlingen von Amorpha fruticosa L, Phytochemistry, 1962, 1(2), 103–105 CrossRef CAS.
- L. Fowden, The Nitrogen Metabolism of Groundnut Plants: The Role of γ-Methyleneglutamine and γ-Methyleneglutamic Acid, Ann. Bot., 1954, 18(4), 417–440 CrossRef.
- G. K. Powell and E. E. Dekker, A Modified, High Yield Procedure for the Synthesis of Unlabeled and 14C-Labeled 4-Methylene-DL-Glutamic Acid, Prep. Biochem. Biotechnol., 1981, 11(3), 339–350 CrossRef CAS.
- O. Ouerfelli, M. Ishida, H. Shinozaki, K. Nakanishi and Y. Ohfune, Efficient Synthesis of 4-Methylene-L-Glutamic Acid and Its Analogues, Synlett, 1993, 1993(6), 409–410 CrossRef.
- H. Gershon, J. S. Meek and K. Dittmer, Propargylglycine: An Acetylenic Amino Acid Antagonist, J. Am. Chem. Soc., 1949, 71(10), 3573–3574 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington and N. G. Robinson, Nucleophilic Ring Opening of Aziridine-2-Carboxylates with Wittig Reagents; an Enantioefficient Synthesis of Unsaturated Amino Acids, J. Chem. Soc., Chem. Commun., 1987,(3), 153–155 RSC.
- C. M. Moody and D. W. Young, Synthesis of Naturally-Occurring 4-Alkylideneglutamic Acids, Tetrahedron Lett., 1993, 34(29), 4667–4670 CrossRef CAS.
- J. Eacquerra, C. Pedregal, I. Micó and C. Nájera, Efficient Synthesis of 4-Methylene-L-Glutamic Acid and Its Cyclopropyl Analogue, Tetrahedron: Asymmetry, 1994, 5(5), 921–926 CrossRef.
- P. Dieterich and D. W. Young, Synthesis of (2S,3S)-[3-2H1]-4-Methyleneglutamic Acid and (2S,3R)-[2,3-2H2]-4-Methyleneglutamic Acid, Org. Biomol. Chem., 2006, 4(8), 1492–1496 RSC.
- M. V. Riofski, J. P. John, M. M. Zheng, J. Kirshner and D. A. Colby, Exploiting the Facile Release of Trifluoroacetate for the α-Methylenation of the Sterically Hindered Carbonyl Groups on (+)-Sclareolide and (−)-Eburnamonine, J. Org. Chem., 2011, 76(10), 3676–3683 CrossRef CAS.
- X. Durand, P. Hudhomme, J. A. Khan and D. W. Young, Two Independent Syntheses of (2S,4S)- and (2S,4R)-[5,5-2H2]-5,5′-Dihydroxyleucine, Tetrahedron Lett., 1995, 36(8), 1351–1354 CrossRef CAS.
- M. I. Hossain, S. Hanashima, T. Nomura, S. Lethu, H. Tsuchikawa, M. Murata, H. Kusaka, S. Kita and K. Maenaka, Synthesis and Th1-Immunostimulatory Activity of α-Galactosylceramide Analogues Bearing a Halogen-Containing or Selenium-Containing Acyl Chain, Bioorg. Med. Chem., 2016, 24(16), 3687–3695 CrossRef CAS.
- J. E. Baldwin, Rules for Ring Closure, J. Chem. Soc., Chem. Commun., 1976,(18), 734–736 RSC.
- J. E. Baldwin, R. C. Thomas, L. I. Kruse and L. Silberman, Rules for Ring Closure: Ring Formation by Conjugate Addition of Oxygen Nucleophiles, J. Org. Chem., 1977, 42(24), 3846–3852 CrossRef CAS.
- V. K. Todorova, Y. Kaufmann, S. Luo and V. S. Klimberg, Tamoxifen and Raloxifene Suppress the Proliferation of Estrogen Receptor-Negative Cells through Inhibition of Glutamine Uptake, Cancer Chemother. Pharmacol., 2011, 67(2), 285–291 CrossRef CAS.
- Y. D. Bhutia and V. Ganapathy, Glutamine Transporters in Mammalian Cells and Their Functions in Physiology and Cancer, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863(10), 2531–2539 CrossRef CAS.
- M. Scalise, L. Pochini, M. Galluccio, L. Console and C. Indiveri, Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth, Front. Oncol., 2017, 7, 306 CrossRef.
- K. Shukla, D. V. Ferraris, A. G. Thomas, M. Stathis, B. Duvall, G. Delahanty, J. Alt, R. Rais, C. Rojas, P. Gao, Y. Xiang, C. V. Dang, B. S. Slusher and T. Tsukamoto, Design, Synthesis, and Pharmacological Evaluation of Bis-2-(5-Phenylacetamido-1,2,4-Thiadiazol-2-Yl)Ethyl Sulfide 3 (BPTES) Analogs as Glutaminase Inhibitors, J. Med. Chem., 2012, 55(23), 10551–10563 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: More experimental procedures, NMR spectroscopy, and cell assay data. See DOI: 10.1039/d0ra08249j |
| This journal is © The Royal Society of Chemistry 2021 |