
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbout the mechanism of ultrasonically induced protein capsule formation†
Ulrike Doeringa,
Dmitry Grigoriev *b,
Kosti Tapioa,
Sophia Rosencrantzb,
Ruben R. Rosencrantzb,
Ilko Balda and
Alexander Bökerab
*b,
Kosti Tapioa,
Sophia Rosencrantzb,
Ruben R. Rosencrantzb,
Ilko Balda and
Alexander Bökerab
aInstitute of Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany
bFraunhofer Institute for Applied Polymer Research IAP, Geiselbergstr. 69, 14476 Potsdam, Germany. E-mail: dmitry.grigoriev@iap.fraunhofer.de
First published on 30th April 2021
Abstract
In this paper, we propose a consistent mechanism of protein microcapsule formation upon ultrasound treatment. Aqueous suspensions of bovine serum albumin (BSA) microcapsules filled with toluene are prepared by use of high-intensity ultrasound following a reported method. Stabilization of the oil-in-water emulsion by the adsorption of the protein molecules at the interface of the emulsion droplets is accompanied by the creation of the cross-linked capsule shell due to formation of intermolecular disulfide bonds caused by highly reactive species like superoxide radicals generated sonochemically. The evidence for this mechanism, which until now remained elusive and was not proven properly, is presented based on experimental data from SDS-PAGE, Raman spectroscopy and dynamic light scattering.
In the past three decades, protein microcapsules have been attracting increasing interest due to their potential applications as drug carriers in the fields of biomedicine and pharmaceuticals.1–5 Suslick and coworkers have developed a method6 to prepare aqueous suspensions of protein-based microcapsules filled with water-insoluble liquids using high-intensity ultrasound including bovine serum albumin (BSA), human serum albumin (HSA), hemoglobin (HB) and myoglobin (MB).6–8 In the early 2000's, Gedanken and coworkers again demonstrated the effectiveness of the sonochemical method for the synthesis of protein microcapsules, which does not utilize any toxic ingredients and is fast and economical.9,10 It is widely accepted that the capsule formation is a result of two very fast subsequently occurring phenomena: emulsification and shell crosslinking.6,11–13 The ultrasonic treatment of a two-phase liquid system leads to the formation of oil-in-water (O/W) emulsion stabilized by protein molecules adsorbed at the interface of emulsion droplets. Additionally, the propagation of the ultrasound wave through a liquid medium produces acoustic cavitations, i.e. the formation, growth and collapse of bubbles. The collapse is a very fast adiabatic process and yields highly reactive radicals11,14,15 like superoxide. Suslick et al. proposed that the superoxide radicals cause the reaction between thiol groups in cysteines, which are present in BSA, HSA and HB, and that the protein capsule shells are held together by cross-linking through interprotein disulfide linkages arising from this oxidation of cysteine. Additionally, they compared MB, which does not possess cysteine, and HB. A substantial decrease in protein capsule yield was revealed for Mb relative to HB.6–8 However, the BSA structure possesses only one thiol group enabling solely the formation of dimers but not a cross-linked network. Moreover, Gedanken et al. demonstrated in addition to the findings of Suslick et al. the possibility of ultrasonically driven formation of microcapsules made of streptavidin, which does not contain any cysteine.16 The authors hypothesize that hydrophobic interactions or thermal denaturation of the protein after the initial ultrasonic emulsification support the microcapsule formation. Further development of these ideas continued more recently in the works of Cavaco-Paulo and collaborators,17,18 where the cysteine-free mechanism of ultrasonically driven protein capsule formation was clearly demonstrated. At the same time, these publications do not contain any evidences refuting unambiguously the mechanism suggesting the formation of intermolecular disulfide bridges upon high-intensity ultrasound treatment, proposed earlier by Suslick et al.6–8,11 Moreover, there are until now no scientific studies, which either present an unequivocal substantiation or demonstrate a clear rebuttal of this mechanism. In particular chemical processes behind it remain elusive.
In this work, we present several experimental proofs pointing towards the formation of intermolecular S–S bonds in the capsule shell and corresponding structural changes of the protein during this process.
We followed Suslick's method and successfully synthesized BSA microcapsules loaded with toluene by using high-intensity ultrasound (see ESI†), verified by Cryo-SEM. Microcapsules exhibit a spherical morphology and have protein shells with the average thickness of several tens of nanometers (Fig. 1A). Fig. 1B shows the toluene-loaded capsules with fluorescently labelled shells after one-week storage at standard laboratory conditions. The capsule cores made of volatile toluene were evaporated and only the thin protein-based capsule shells and shell fragments remained. Clearly different results were obtained for capsules prepared by use of a vortex mixer (IKASONIC U50 control, IKA Werke Germany) (Fig. 1C). After a week, only residual protein debris with wide variability of shapes were observed without any evidences of empty capsule shells or their fragments.
 | ||
| Fig. 1 Cryo-SEM image of protein microcapsules loaded with toluene. Enlarged image of the cross section of a microcapsule presented in the right corner of the image (A); fluorescence microscopy images of collapsed RITC labelled BSA microcapsules after one week aging, prepared by sonication (B) and by means of vortex mixer (C). | ||
This essentially lower stability of capsules generated by vortex suggests that even a vigorous shaking of toluene with an aqueous protein solution still does not possesses an energy enough to induce permanent covalent crosslinking of the capsule protein shells.
To examine the protein cross-linking in the microcapsule shell via disulfide bonds and to shed more light on the mechanism of their formation, we performed SDS-PAGE (for more details see ESI†).
Fig. 2 shows the SDS-PAGE assay of empty BSA microcapsules and pristine BSA as control before and after the treatment with dithiothreitol (DTT). This reducing agent was used for the reductive cleavage of disulfide bonds within the protein as well as in the cross-linked proteinaceous shells. The empty BSA microcapsules in lanes 2 and 4 show numerous diffuse bands in the SDS-PAGE assay, especially at higher molecular weights in the range from 140 kDa to >310 kDa. This means that the microcapsule shells contain some proteinaceous components in this molecular weight range.
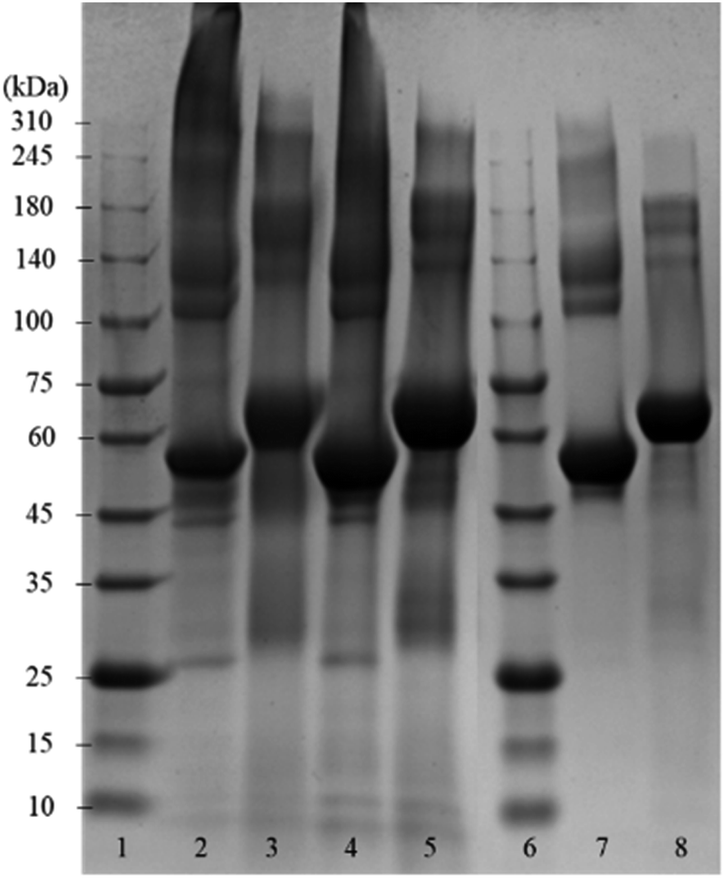 | ||
| Fig. 2 Coomassie-stained SDS-PAGE gel of empty BSA microcapsules, which were loaded with toluene, and pristine BSA. Lanes 1 and 6: reference marker; lane 2: empty capsules (dialyzed); lane 3: sample 2 treated with DTT; lane 4: empty capsules (washed with dropping funnel); lane 5: sample 4 treated with DTT; lane 7: pristine BSA; lane 8: pristine BSA treated with DTT. | ||
However, distinct bands at approximately 55 kDa can also be observed, still indicating the presence of a significant amount of monomeric BSA in the shells, as follows from the comparison with the pristine BSA in lane 7. Here, in addition to the prominent main band at 55 kDa, only a slight smearing between 140 kDa and 240 kDa can be seen but no smear or diffuse bands above 240 kDa are visible. The observed appearance of the lane 7 for the pristine BSA is in a good agreement with the data in the literature, both in well-known as well as in recently published works.19–27 Notably also, the view of the pristine BSA's lane practically did not depend on the protein concentration in a wide range (from 3 μg to 50 μg) of the concentrations studied, well coinciding with the formerly reported results.20,24,27 Especially in the parts of SDS-PAGE assays corresponding to higher Mw, neither essential changes nor an occurrence of additional bands indicating the presence of high-molecular components were detected.
Moreover, this property of BSA to maintain a well-resolved lane pattern over a broad concentration range has enabled its use as a standard protein in the SDS-PAGE assays, which utilize a BSA dilution series.28
Under the assumption that DTT can cleave both inter- and intramolecular S–S bonds, the comparison of lanes 3, 5 and 8 enables the conclusion that the shells of protein capsules, although to an essential extent cross-linked via intermolecular disulfide bonds between BSA may also contain molecules bound together by other forces such as hydrogen bonds or hydrophobic interactions.
Dynamic light scattering (DLS) is known as a simple and quick experimental technique to identify and measure the size of various, especially globular proteins27,29 in a non-invasive manner. Since the intensity weighted (Z-average) peak will change its mean size and its width even at small changes in population sizes in the sample, DLS is proven to be an extremely sensitive method enabling clear comparisons between populations existing in samples and identification of samples where essential dimerization, oligomerization or aggregation30 are happening. Therefore, to verify the SDS PAGE findings described above we carried out the DLS measurements for samples of pristine monomeric BSA, empty BSA capsules and BSA chemically cross-linked with glutaraldehyde (GA). The corresponding results are presented in the ESI for our manuscript (Fig. S1).† As one can see, the DLS spectrum of the pristine BSA displays only one peak around 6 nm, which correlates well with the hydrodynamic diameter of monomeric BSA.27,31–33 In contrast to this spectrum, the DLS-spectrum for ultrasonically cross-linked BSA demonstrates two very distinct peaks; one – in the same size range as for the pristine BSA and the second one – at around 130 nm. The first of these two peaks corresponds to the abovementioned BSA monomers. The second one can be attributed to the cross-linked BSA oligomers occurred due to ultrasound treatment. The DLS spectrum for the chemically cross-linked BSA only displays a single peak at approximately 80 nm, which reflects the presence of BSA in this sample practically solely in the oligomeric form resulted from chemical cross-linking. This set of results proves undoubtedly the formation of the BSA oligomers caused by sonication. In addition, a well-pronounced peak in the lower nanometer range for ultrasonically treated BSA indicates that monomeric BSA is still present in significant amounts. Taking into account the relative sizes of peaks for both monomeric and oligomeric fractions of BSA in this sample, one can estimate30 the amount of latter as high as several mol%. On the other hand, the low-molecular-weight oligomers whose presence in sonicated samples can be inferred from SDS-PAGE data, are probably either too small or form too minor fractions to cause additional peaks in the DLS spectrum. Moreover, the application of the empirical equation between the size and molecular weight of globular proteins34 yields an approximate molecular weight of oligomeric BSA of many millions of Da. Obviously, such large oligomers cannot be detected by analytical techniques like electrospray ionization mass-spectrometry (ESIMS) or Matrix Assisted Laser Desorption Ionization-Time-of-Flight mass spectrometry (MALDI-TOF).35–37 The low sensitivity of MALDI-TOF could probably be connected36 the fact that MALDI generates ions with essentially lower charge38 than ions obtained by electrospray ionization ESI,35 which could lead to very high values of m/z even in the case of tri- or tetramers of cross-linked BSA. For this reason, investigations of proteins/biopolymers of high molecular weight by means of MALDI-TOF are rare, especially at Mw ≥ 100 kDa. In this case, even the usage of very high acceleration voltage does not allow37 to achieve a detection efficiency higher than 25%.
To understand whether the capsule formation affects disulfide bonds in BSA and, if so, what transformations occur in protein molecules during this process, Raman spectra measurements were performed for the pristine BSA, toluene and dried BSA microcapsules (see Fig. 3A). Raman spectroscopy is sufficiently sensitive to detect and resolve the protein conformational changes on the basis of spectral changes in the low frequency range,39 in contrast to FTIR spectroscopy, which can be used to monitor changes in the three-dimensional structure of proteins as it was successfully done by Silva et al.17 The FTIR results and simulations presented in this paper substantiate that the capsule formation and their following stabilization can be caused by the spatial rearrangement of protein molecules at the oil–water interface upon ultrasound treatment and by following hydrophobic interactions of corresponding moieties in the protein molecules with changed conformation. With that, the presence of cysteine groups is not necessary for the formation of a stable capsule shell.17,18 On the other hand, the mechanism involving the ultrasonically induced covalent cross-linking of BSA molecules in the capsule shell via intermolecular S–S bridges formation is nowhere controverted or denied in the paper of Silva et al. and in other publications of the Cavaco-Paulo's group.17,18 FTIR used therein as a main analytical tool is simply not sensitive enough for undoubtful confirmation or refutation of this phenomenon.39 Thus, Cavaco-Paulo and co-authors did not exclude the mechanism of covalent cross-linking between proteins in capsules via disulfide bonding but admitted such a possibility along with other physically or physicochemically driven mechanisms of capsule stabilization. These authors have also at no point claimed that this mechanism is a major reason for proteinaceous capsule formation; on the contrary, they suggested the plurality of various mechanisms leading to the formation of protein capsules:18 “the possibility to form protein capsules from proteins like silk fibroin and cysteine-free peptides indicates the presence of further driving forces in addition to covalent cross-linking, namely hydrophobic, electrostatic interactions and enhanced mass transport, induced by ultrasound in liquid media”. The major bands that appear in the Raman spectrum of BSA are the amide I (∼1655 cm−1), CH2 and CH3 bending (∼1449 cm−1), amide III (∼1200–1300 cm−1) and S–S stretching vibration (∼510 cm−1). The amide I band around 1650 cm−1 along with the broad amide III band around 1300 cm−1 show that the secondary structure of BSA is mainly in α-helix form.40,41 The peak at 510 cm−1 refers to the disulfide bonds of the cystine in the pristine BSA. In general, the S–S stretching vibration can appear in the 450–550 cm−1 range depending on the surrounding molecular structure, which makes this part of Raman spectra useful for the characterization of structural changes in proteins. Nonetheless, the interpretation of the disulfide peak position is not simple since several competing models have been proposed explaining the correlation between the peak wavenumber position and conformation of the molecule containing the corresponding bond. One model proposes that the wavenumber position of the S–S stretching vibration is dependent on the torsional angles in the Cα–Cβ–S–S–Cβ′–Cα′ conformation.42,43 In this instance, the peak position of the S–S stretching vibration should appear at 510 cm−1 for the lowest energy gauche–gauche–gauche (g–g–g) conformation, at 525 cm−1 for the higher energy gauche–gauche–trans (g–g–t) conformation and at 540 cm−1 for the highest energy trans–gauche–trans (t–g–t) conformation. Accordingly, the disulfide bonds in BSA have a g–g–g conformation. However, this theory does not explain the shift of S–S stretching vibration to wavenumbers below 510 cm−1 as can be seen in the Raman spectrum of the dried BSA–toluene capsules at 487 cm−1 (Fig. 3A and B, red spectra). This band can also not be attributed to traces of toluene as follows from its Raman spectrum (Fig. 3A, green spectrum). In another approach, the Raman shift of the S–S stretching vibration is dependent on the dihedral angle between the planes containing the Cβ–S and the S–Cβ′ bonds of the Cα–Cβ–S–S–Cβ′–Cα′ group, respectively.44–46 Disulfide bonds with an S–S stretching vibration with a value near 510 cm−1 should have a Cβ–S–S–Cβ′ dihedral angle of ±90°.47 Although the mostly observed S–S stretching vibrations exist between 500 and 540 cm−1, the strained disulfide groups with dihedral angles less than ±65° are also possible.
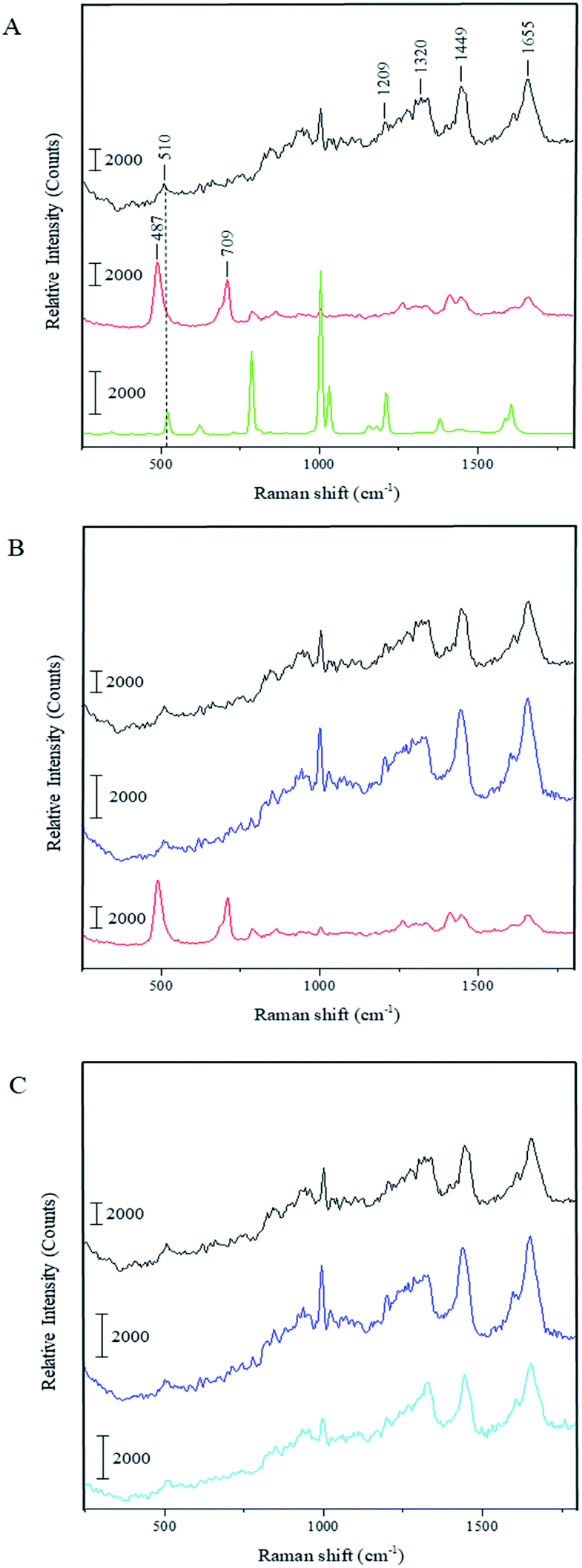 | ||
| Fig. 3 Raman spectra for: (A) pristine BSA (black), dried BSA–toluene-capsules (red), toluene (green), (B) capsules treated with DTT (blue) and (C) pristine BSA treated with DTT (light blue). Data acquisition conditions: excitation wavelength = 488 nm; laser power density = 3.7 × 104 W cm−2. | ||
Biswas and coworkers observed a peak at 486 cm−1 while investigating the surfactant protein B (dSP-B1–25) with Raman spectroscopy.47 This protein is a homodimer with three intramolecular disulfide bonds and one intermolecular disulfide bridge. On the basis of several interpretations proposed by these authors for the spectrum of protein B, we suggest the following explanation for our system: the peak position at 487 cm−1 indicates that there is a highly strained but stable disulfide bond conformation with a dihedral angle in the range of ±10° due to the crosslinking of the neighboring protein molecules in the capsule shell. We assume that the same strain could also lead to the increase of the intensity of this peak as can be observed in the spectrum of dried capsules in Fig. 3. Relative amounts of intramolecular disulfide bonds of BSA and intermolecular disulfide bonds occurred due to the ultrasound treatment can be compared as follows. Although the intensity of the same peaks in the different spectra cannot be compared quantitatively, it is possible to make an inherent comparison of different peaks in the same spectrum (inherent normalization of a spectrum)48 and then recognize these results when consider different spectra. In accordance with this, the relative intensity of the disulfide band at 510 cm−1 in the spectrum of the pristine BSA is quite low comparing with the intensity of the amide I peak at 1655 cm−1 (black spectrum in the Fig. 3A). On the other hand, the relative intensity of the band for the intermolecular disulfide bridges in the capsule shell (487 cm−1) is higher than the intensity of the amide I peak at 1655 cm−1 in the sample containing only BSA capsules (red spectra in the Fig. 3A and B). This qualitative comparison indicates that the relative intensity of the band for the residual intramolecular S–S bridges at 510 cm−1 in the spectrum for BSA capsules should be lower than the band corresponding to intermolecular S–S bridges in the capsule shell at 487 cm−1, i.e. the contribution of intermolecular S–S bridges to the resulting Raman spectrum in the low-frequency range is prevalent and the results suggest that a significant amount of intramolecular bridges is converted into intermolecular ones. Moreover, the right shoulder of the stronger peak at 487 cm−1 can cover the weak residual signal at 510 cm−1 almost completely. Furthermore, the Raman spectrum of the capsules shows an additional peak at 709 cm−1, which refers to the C–S stretching vibration of the N–C–C–S–S moieties with N and S–S in the trans-conformation.47 This is another hint for the intermolecular cross-linking in the shells causing the conformational changes in the BSA molecules after the capsule formation. Additionally, the capsules spectrum shows a band around 1450 cm−1, which is assigned to the CH2 and CH3 bending vibration, and one around 1410 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of dissociated carboxyl groups.49 On the contrary, this band appears only as a shoulder in the spectra of the pristine BSA and DTT treated capsules. The observed difference could be explained by the reduction of the band intensity at 1450 cm−1 due to sterical constraints in the local surrounding of CH2 moieties caused by the intermolecular crosslinking at the capsule formation. Thus, the CO stretching vibration around 1410 cm−1 becomes visible as an individual peak in the capsules' spectrum.
O stretching vibration of dissociated carboxyl groups.49 On the contrary, this band appears only as a shoulder in the spectra of the pristine BSA and DTT treated capsules. The observed difference could be explained by the reduction of the band intensity at 1450 cm−1 due to sterical constraints in the local surrounding of CH2 moieties caused by the intermolecular crosslinking at the capsule formation. Thus, the CO stretching vibration around 1410 cm−1 becomes visible as an individual peak in the capsules' spectrum.
Fig. 3B shows the Raman spectrum of the BSA capsules after the treatment with DTT and subsequent dialysis in comparison to the spectra of the capsules before the cleavage of the disulfide bonds and of the pristine BSA. The two additional peaks at 487 cm−1 and 709 cm−1 in the capsule spectrum are not present after the reaction with DTT. Furthermore, the peaks of the dissolved capsules correlate with the peaks of pristine BSA. This demonstrates that after the cleavage of the S–S bonds initial structure of BSA was regained. We also investigated the effect of DTT to pristine BSA (see Fig. 3C). The Raman spectra of BSA before and after the treatment with DTT are nearly identical, which allows the assumption that after removal of the DTT by dialysis, the thiol groups of the BSA molecules could be easily rearranged back to intramolecular S–S bonds, e.g. by an oxidative action of oxygen from the air.
The detailed analysis of Raman spectra for pristine BSA, empty BSA capsules and the samples cleaved with DTT brings additional evidences for the formation of intermolecular S–S bonds in the BSA composing the capsule shells and therefore confirms the findings of the SDS PAGE discussed above. However, the network formation requires the cross-linking of several groups whereas BSA in its native form possesses only one thiol group, which can be oxidized by superoxide radicals. Thus, BSA dimers and not a cross-linked network could theoretically be formed. This inconsistency can be resolved by taking into account the results of Zhang and coworkers, who investigated the effect of ultrasound on the thiol groups and disulfide bonds of wheat gluten.50 According to their findings, the ultrasound treatment can dynamically change the amount of thiol groups and disulfide bonds in the protein. After a few minutes of ultrasound treatment, the content of thiol groups was rapidly increased due to the sonochemical reduction of disulfide bonds and reached the value up to three times the initial amount. On the contrary, the content of disulfide bonds was quickly decreased up to 20% and then fluctuated within the same time interval. Assuming the similar effects in the case of BSA we can suggest that the cross-linking mechanism leading to the formation of a stable capsule shell is based on the rearrangement of disulfide bonds upon ultrasonication. Since the native BSA molecule contains 17 intramolecular disulfide bonds, it is reasonable to assume that ultrasonically induced reductive cleavage of approx. 20% of them should yield 3 to 4 thiol groups in addition to the single thiol group in the pristine BSA. These groups can be then easily oxidized by the superoxide radicals leading to the formation of new intermolecular disulfide bonds and a cross-linked network. The rearrangement of intramolecular disulfide bonds into intermolecular ones can explain how BSA with only one oxidizable native thiol group can be cross-linked and form the shell of the microcapsules.
In this work, the experimental evidences of ultrasonically induced cross-linking of the BSA in the shells of protein-based microcapsules are demonstrated. Comparison of the results of SDS-PAGE and Raman spectroscopy for the BSA in the capsule shells, for the products of their reductive cleavage and for the pristine BSA showed that the BSA molecules in the shells are to an essential extent cross-linked via disulfide bridges between neighboring molecules. Additionally, the presence of further effects like hydrogen bonds or hydrophobic interactions may also stabilize the protein capsules. Therefore, the concept proposed many years ago by Suslick and co-workers is confirmed analytically for the first time. Moreover, in contrast to the Suslick's insufficiently substantiated hypothesis, a consistent mechanism for the formation of intermolecular disulfide bonds in capsule shells is proposed based on experimental data on the redistribution of thiol and disulfide groups in BSA under the action of high-energy ultrasound.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Brigitte Tiersch and Sibylle Rüstig for the SEM images of the protein capsules. Additionally, we give thanks to Dr Ulrich Glebe for performing the MALDI-TOF measurements.References
- M. S. Latha, K. Rathinam, P. V. Mohanan and A. Jayakrishnan, J. Controlled Release, 1995, 34, 1–7 CrossRef CAS.
- B. A. Heelan and O. I. Corrigan, J. Microencapsulation, 1998, 15, 93–105 CrossRef CAS PubMed.
- S. J. Lee and M. Rosenberg, J. Controlled Release, 1999, 61, 123–136 CrossRef CAS PubMed.
- L. Duan, Q. He, X. Yan, Y. Cui, K. Wang and J. Li, Biochem. Biophys. Res. Commun., 2007, 354, 357–362 CrossRef CAS PubMed.
- Q. He, Y. Cui and J. Li, Chem. Soc. Rev., 2009, 38, 2292–2303 RSC.
- K. S. Suslick and M. W. Grinstaff, J. Am. Chem. Soc., 1990, 112, 7807–7809 CrossRef CAS.
- K. S. Suslick, M. W. Grinstaff, K. J. Kolbeck and M. Wong, Ultrason. Sonochem., 1994, 1, S65–S68 CrossRef CAS.
- M. W. Grinstaff and K. S. Suslick, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 7708–7710 CrossRef CAS PubMed.
- S. Avivi, I. Felner, I. Novik and A. Gedanken, Biochim. Biophys. Acta, 2001, 1527, 123–129 CrossRef CAS.
- S. Avivi, Y. Nitzan, R. Dror and A. Gedanken, J. Am. Chem. Soc., 2003, 125, 15712–15713 CrossRef CAS PubMed.
- K. S. Suslick and G. J. Price, Annu. Rev. Mater. Sci., 1999, 29, 295–326 CrossRef CAS.
- A. Gedanken, Chem.–Eur. J., 2008, 14, 3840–3853 CrossRef CAS PubMed.
- X. Cui, B. Wang, S. Zhong, Z. Li, Y. Han, H. Wang and H. Moehwald, Colloid Polym. Sci., 2013, 291, 2271–2278 CrossRef CAS.
- J. Rae, M. Ashokkumar, O. Eulaerts, C. von Sonntag, J. Reisse and F. Grieser, Ultrason. Sonochem., 2005, 12, 325–329 CrossRef CAS PubMed.
- C. Little, M. El-Sharif and M. J. Hepher, Ultrason. Sonochem., 2007, 14, 375–379 CrossRef CAS PubMed.
- S. Avivi and A. Gedanken, Biochem. J., 2002, 366, 705–707 CrossRef CAS PubMed.
- R. Silva, H. Ferreira, N. G. Azoia, U. Shimanovich, G. Freddi, A. Gedanken and A. Cavaco-Paulo, Mol. Pharmaceutics, 2012, 9, 3079–3088 CrossRef CAS PubMed.
- U. Shimanovich, G. J. L. Bernardes, T. P. J. Knowles and A. Cavaco-Paulo, Chem. Soc. Rev., 2014, 43, 1361–1371 RSC.
- T. Peters Jr and R. C. Feldhoff, Biochemistry, 1975, 14, 3384–3391 CrossRef CAS PubMed.
- R. C. Feldhoff and T. Peters, Biochemistry, 1975, 14, 4508–4514 CrossRef CAS PubMed.
- P. Restani, A. Fiocchi, B. Beretta, T. Velonà, M. Giovannini and C. L. Galli, Lett. Pept. Sci., 1997, 4, 269–273 CAS.
- P. Restani, T. Velonà, A. Plebani, A. G. Ugazio, C. Poiesi, A. Muraro and C. L. Galli, Clin. Exp. Allergy, 1995, 25, 651–658 CrossRef CAS PubMed.
- B. T. Kurien and R. H. Scofield, Methods Mol. Biol., 2012, 869, 471–479 CrossRef CAS PubMed.
- S. M. Alonso Villela, H. Kraïem, B. Bouhaouala-Zahar, C. Bideaux, C. A. Aceves Lara and L. Fillaudeau, MicrobiologyOpen, 2020, 9, 1–8 CrossRef PubMed.
- B. Liu, Y. Pang, R. Bouhenni, E. Duah, S. Paruchuri and L. McDonald, Chem. Commun., 2015, 51, 11060–11063 RSC.
- L.-J. Lai, F.-L. Huang, P.-H. Cheng and R. Y. Chiou, Int. J. Clin. Nutr. Diet., 2017, 3, 1–5 Search PubMed.
- C. Burgard, PhD thesis, University of Saarland, 2009.
- Bio-Rad, https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040.pdf, accessed March 2021.
- Malvern Panalytical, https://www.malvernpanalytical.com/en/academia/research-themes/biological-medical-science/index.html, accessed March 2021.
- Malvern Panalytical, https://www.malvernpanalytical.com/en/learn/knowledge-center/application-notes/AN120917SEC-LS-DLSQuantificationProteinAggregates, accessed March 2021.
- H. P. Erickson, Biol. Proced. Online, 2009, 11, 32–51 CrossRef CAS PubMed.
- Malvern Panalytical, https://www.malvernpanalytical.com/en/learn/knowledge-center/application-notes/AN101104ZetasizerAPSEliminatesCrossContamination, accessed March 2021.
- Malvern Panalytical, https://www.malvernpanalytical.com/en/learn/knowledge-center/application-notes/AN110707ProteinCharacterisationZetasizerMicroV, accessed March 2021.
- U. Nobbmann, in Mesoscale Phenomena in Fluid Systems, ed. F. Case and P. Alexandridis, American Chemical Society, Washington, 2003, ch. 4, pp. 44–59 Search PubMed.
- K. Hirayama, S. Akashi, M. Furuya and K. Fukuhara, Biochem. Biophys. Res. Commun., 1990, 173, 639–646 CrossRef CAS PubMed.
- L. Signor and E. Boeri Erba, J. Visualized Exp., 2013, 79, 1–7 Search PubMed.
- R. Liu, Q. Li and L. M. Smith, J. Am. Soc. Mass Spectrom., 2014, 25, 1374–1383 CrossRef CAS PubMed.
- J. Jacksén, PhD thesis, Royal Institute of Technology, 2011.
- N. N. Brandt, A. Y. Chikishev, A. A. Mankova and I. K. Sakodynskaya, J. Biomed. Opt., 2015, 20, 1–6 Search PubMed.
- C.-H. Wang, C.-C. Huang, L.-L. Lin and W. Chen, J. Raman Spectrosc., 2016, 47, 940–947 CrossRef CAS.
- M. Liang, V. Y. T. Chen, H.-L. Chen and W. Chen, Talanta, 2006, 69, 1269–1277 CrossRef CAS PubMed.
- S. Hirorau, G. Akikatsu and M. Tatsuo, Chem. Lett., 1972, 1, 83–86 CrossRef.
- S. Hiromu, G. Akikatsu and M. Tatsuo, Bull. Chem. Soc. Jpn., 1973, 46, 3407–3411 CrossRef.
- H. E. Van Wart and H. A. Scheraga, J. Phys. Chem., 1976, 80, 1823–1832 CrossRef CAS.
- H. E. Van Wart and H. A. Scheraga, J. Phys. Chem., 1976, 80, 1812–1823 CrossRef CAS.
- H. E. Van Wart, F. Cardinaux and H. A. Scheraga, J. Phys. Chem., 1976, 80, 625–630 CrossRef CAS.
- N. Biswas, A. J. Waring, F. J. Walther and R. A. Dluhy, Biochim. Biophys. Acta, 2007, 1768, 1070–1082 CrossRef CAS PubMed.
- J. R. Maher, M. Takahata, H. A. Awad and A. J. Berger, J. Biomed. Opt., 2011, 16, 1–6 CrossRef PubMed.
- N. Howell and E. Li-Chan, Int. J. Food Sci. Technol., 1996, 31, 439–451 CrossRef CAS.
- Y. Zhang, Y. Li, S. Li, H. Zhang and H. Ma, Molecules, 2018, 23, 1–13 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and analytic protocols. See DOI: 10.1039/d0ra08100k |
| This journal is © The Royal Society of Chemistry 2021 |