
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConjugate between hydrolyzed collagen from defatted seabass skin and epigallocatechin gallate (EGCG): characteristics, antioxidant activity and in vitro cellular bioactivity
Lalita Chotphruethiponga,
Wanida Sukketsirib,
Maurizio Battino c and
Soottawat Benjakul*a
c and
Soottawat Benjakul*a
aInternational Center of Excellence in Seafood Science and Innovation, Faculty of Agro-Industry, Prince of Songkla University, Hat Yai, Songkhla, 90110, Thailand. E-mail: soottawat.b@psu.ac.th; Fax: +66-7455-8866; Tel: +66-7428-6334
bDepartment of Pharmacology, Faculty of Science, Prince of Songkla University, Hat Yai, Songkhla 90110, Thailand
cDipartimento di Scienze Cliniche Specialistiche ed Odontostomatologiche (DISCO)-Sez. Biochimica, Facolt`a di Medicina, Universit`a Politecnica delle Marche, Ancona, Italy
First published on 8th January 2021
Abstract
Conjugation between peptides and polyphenols, especially epigallocatechin gallate (EGCG) using covalent grafting, is a promising method that can modify peptides or augment their antioxidant activities. Moreover, the resulting conjugates can be intensively served as functional ingredient or supplement. Thus, the objectives of the present study were to investigate the grafting between hydrolyzed collagen (HC) from defatted seabass skin and EGCG and to study characteristics as well as bioactivities of the obtained HC–EGCG conjugate. Levels of EGCG used (1–5%, w/w) affected surface hydrophobicity (SH) and antioxidant activities of the conjugates. Overall, the addition of EGCG at 3% to HC (HC–3% EGCG) increased SH, ABTS radical scavenging and metal chelating activities (p < 0.05). FTIR spectra of HC–3% EGCG revealed the interaction between HC and EGCG via H-bonding and covalent interaction. Sephadex G-25 fraction of conjugate with molecular weight (MW) of 2771 Da rendered the highest redox ability. When HC–3% EGCG was applied in fibroblast (MRC-5) and keratinocyte (HaCaT) cells, all levels tested (125–1000 μg mL−1) had no toxicity on both cells. Higher proliferation of both cells were attained with increasing levels of HC–3% EGCG, particularly at 500 and 1000 μg mL−1 (p < 0.05). Moreover, both levels used had cytoprotective ability against reactive oxygen species (ROS) as evidenced by lowered ROS and cell death detected as compared to those found in cells induced with H2O2 or AAPH alone (p < 0.05) for both cells. HC–3% EGCG could serve as an effective antioxidant for application in foods or as supplement for skin nourishment.
1. Introduction
Reactive oxygen species (ROS) are generally produced as a consequence of cellular metabolism.1 Overproduction of ROS causes oxidative stress in cells, leading to numerous diseases such as cancer and neurological diseases and induces the aging process.2,3 Additionally, ROS play a crucial role in foods, especially food rich in lipids, which are susceptible to oxidation.4 These species are associated with the development of offensive odors/flavors and texture in food products. Additionally, the toxic compounds can be formed.5 Several natural antioxidants including plant or animal protein hydrolysates are proven to defend oxidative stress and retard the deterioration of food products by providing their proton/electron to ROS, thus lowering the adverse effects of ROS.6–8 Hydrolyzed collagen (HC) from seabass skin containing peptides was an alternative antioxidant that could scavenge ROS.9,10 Furthermore, it was able to promote proliferation of fibroblast cells.11,12 Although HC had several benefits, its activity, especially antioxidant activity, was still low. Therefore, the development of the new peptides with strong antioxidant activity is required.Covalent grafting between proteins and polyphenols is a promising approach for modifying or increasing the functional properties of proteins and their antioxidant activities, in which a number of hydroxyl groups and hydrophobic domains were augmented.13,14 Epigallocatechin gallate (EGCG) is an excellent antioxidant and is able to inhibit lipid oxidation.15 Previously, Quan and Benjakul15 reported that conjugation between duck albumen hydrolysate (DAH) and EGCG enhanced antioxidant activities and emulsifying property of proteins. Also, DAH–EGCG conjugate could retard oxidation of fish oil emulsion effectively.15 Chanphai et al.14 documented that EGCG had higher binding affinity than other derivatives in grafting with β-lactoglobulin. Furthermore, the incorporation of EGCG in protein-based film could increase water vapor barrier property and tensile strength of the resulting film.16 Those information suggested that proteins added with EGCG can be intensively used as additive in diverse food products. Since no information on the grafting of HC from defatted seabass skin with EGCG is available, this study aimed to examine the characteristics and antioxidant activities of HC–EGCG conjugate and its impact on proliferation of keratinocyte and fibroblast cells as well as cytoprotective ability against ROS generation.
2. Material and methods
2′,7′-Dichlorofluorescein, 8-anilo-1-naphthalenesulfonic acid (ANS), lipase from porcine pancreas type II, 2,4,6-tripyridyltriazin (TPTZ) and Sephadex G-25 resin as well as other chemicals were procured from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Papaya papain was acquired from Siam Victory Chemicals Co, Ltd (Bangkok, Thailand). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco BRL Life Technologies (Grand Island, NY, USA). The MRC-5 fibroblast cell line (human fetal lung) and HaCaT keratinocyte cell were procured from the Cell Line Service, Heidelberg, Germany. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was purchased from Invitrogen, Carlsbad, CA.2.1 Enzyme assays
Lipase activity was determined using p-NPP as a substrate at pH 8.0 and 50 °C.12 One unit (U) of activity was defined as the amount of lipase producing 1 μmol p-NP per min under the assay condition. Papain activity was examined using casein as a substrate at pH 7.0 and 40 °C for 15 min.12 One unit of activity was defined as the amount of papain that released 0.01 μmol of tyrosine equivalent per min (μmol Tyr equivalent per min).2.2 Pretreatment and defatting of Asian sea bass skin
Frozen Asian sea bass skins were procured from King-fisher Holdings Co., Ltd, Songkhla, Thailand. Pretreatment of the skins was performed following the procedure reported previously.17 Thawed skins were subjected to non-collagenous protein removal using 0.1 M NaOH, followed by washing. Subsequently, PEF-assisted process was applied to the prepared skins, in which electric field strengths used was 24 kV cm−1 for 72 ms with 135 kJ kg−1 specific energy input. Pulse repetition times and pulse width were 20 ms and 0.1 ms, respectively. PEF-treated skins were soaked in 0.05 M citric acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, w/v) for 2 h. Thereafter, the swollen skins were washed until neutral pH was attained and used for further defatting using vacuum impregnation (VI) process in combination with PPL at 42.36 unit per g skin dry matter (VI-PPL process). Resulting defatted skins were used as a raw material for preparation of hydrolyzed collagen.
:10, w/v) for 2 h. Thereafter, the swollen skins were washed until neutral pH was attained and used for further defatting using vacuum impregnation (VI) process in combination with PPL at 42.36 unit per g skin dry matter (VI-PPL process). Resulting defatted skins were used as a raw material for preparation of hydrolyzed collagen.
2.3 Preparation of hydrolyzed collagen (HC) from defatted Asian sea bass skin
HC production from defatted skins obtained from VI-PPL process was performed using papain at 0.3 U g−1 dry matter. Hydrolysis was done at 40 °C for 90 min. After hydrolysis, enzyme was inactivated at 90 °C for 15 min.17 The resulting HC was mixed with activated charcoal (0.05%, w/v), filtered and lyophilized.172.4 Preparation of HC–EGCG conjugates
Free radical grafting method was used.15 Firstly, the lyophilized HC was solubilized in distilled water (DW) to obtain the concentration of 1% (w/v). Subsequently, the solution was oxidized by adding 0.15 g of ascorbic acid and 0.5 mL of 5 M H2O2, followed by stirring for 2 h at 25 ± 2 °C. The resulting mixture was named as ‘oxidized HC, OHC’. Thereafter, EGCG at different levels (0, 1, 2, 3, 4 and 5%, based on HC weight) was added into OHC solution and stirred at 4 °C for 24 h. Unconjugated EGCG was removed by dialysis (3500 MWCO) at 4 °C for 24 h against 10 volumes of DW with 4 changes. After dialysis, the percentage of conjugation of all treatments was calculated using the following equation:% conjugation of HC–EGCG conjugates using EGCG at 1%, 2%, 3%, 4% and 5% were 96.74%, 96.85%, 97.26%, 96.52% and 96.02%, respectively. All the HC–EGCG conjugates were lyophilized and kept at −40 °C until analysis.
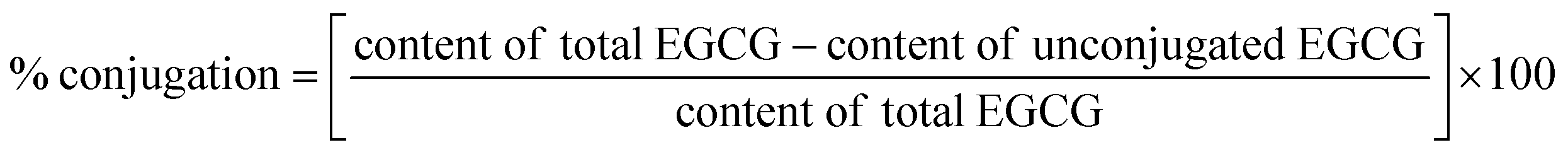
2.5 Analyses
2.6 Characterization of the selected HC–EGCG conjugate
The conjugate rendering the highest antioxidant activities and surface hydrophobicity was selected and subjected to FTIR analysis and size exclusion chromatography in comparison with OHC and HC.2.7 Effects of HC–EGCG conjugate on cell proliferation and the ability in inhibiting ROS generation
2.8 Statistical analysis
Completely randomized design (CRD) was used for all studies, which were run in triplicate. Analysis of variance (ANOVA) was conducted for all the data. Means were compared by the Duncan's multiple range test. Statistical Package for Social Science (SPSS 11.0 for windows, SPSS Inc., Chicago, IL, USA) was used.3. Results and discussion
3.1 Characteristics of HC–EGCG conjugate prepared using EGCG at various levels
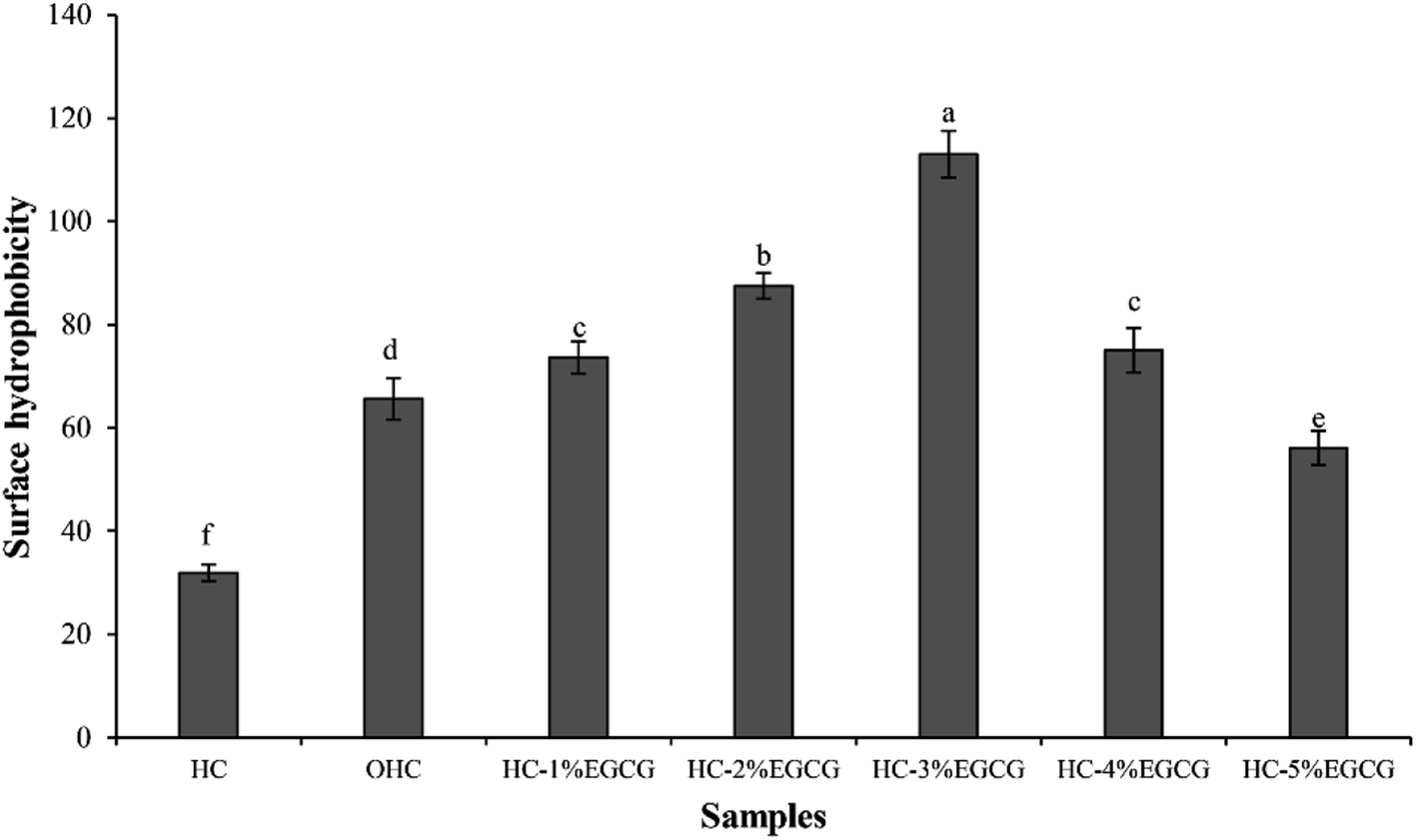 | ||
| Fig. 1 Surface hydrophobicity of hydrolyzed collagen (HC) from defatted seabass skin, oxidized HC (OHC) and HC–EGCG conjugates prepared using EGCG at different levels. Bars represent the standard deviation (n = 3). Different lowercase letters on bars indicate significant differences between samples (p < 0.05). | ||
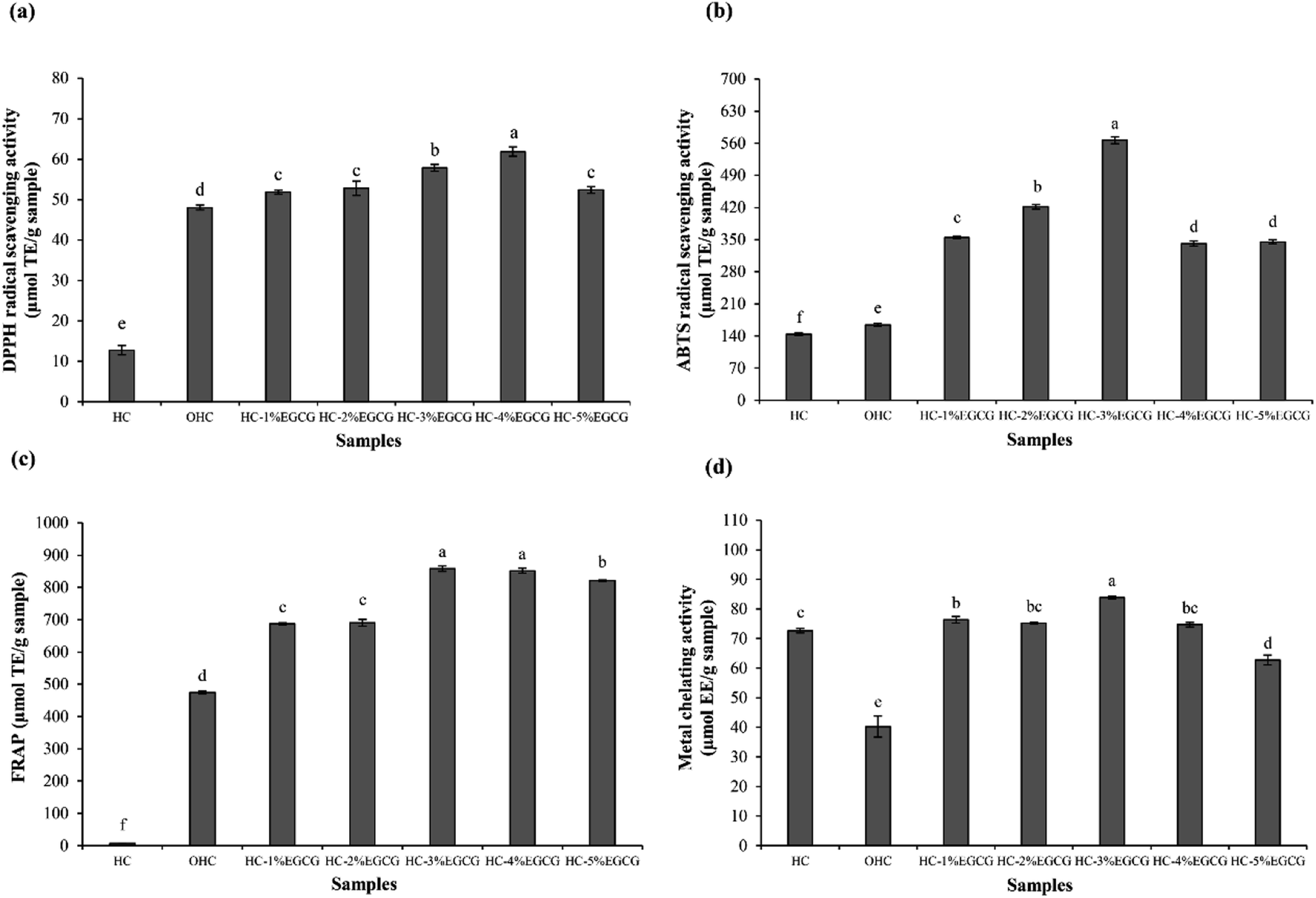 | ||
| Fig. 2 DPPH radical scavenging activity (a), ABTS radical scavenging activity (b), FRAP (c) and metal chelating activity (d) of HC, OHC and HC–EGCG conjugates prepared using EGCG at different levels. Bars represent the standard deviation (n = 3). Different lowercase letters on bars indicate significant differences between samples (p < 0.05). | ||
The highest FRAP (Fig. 2c) was detected in HC–3% EGCG, compared to others (p < 0.05). Nevertheless, similar FRAP between HC–3% EGCG and HC–4% EGCG was found (p > 0.05). In addition, high level of EGCG (5%) drastically decreased FRAP of the obtained conjugate. HC showed lower activity than OHC and HC–EGCG at all levels of EGCG used for conjugate preparation (p < 0.05). After being oxidized with H2O2 and ascorbic acid, the activity in reducing TPTZ–Fe(III) to TPTZ–Fe(II) was rapidly increased. This was plausibly related to the exposure of some amino acids in peptides with reducing power, as related with the increased SH (Fig. 1). Tkaczewska et al.27 documented that peptides with Ala and Tyr could promote FRAP and these amino acids were found in HC from defatted seabass skin.12 Apart from Ala and Try, higher activity of OHC might be owing to the remaining ascorbic acid in solution (reduced form), which possessed strong reducing power and led to enhanced FRAP. However, FRAP of OHC was lower than that of the conjugates prepared with all EGCG levels used (p < 0.05), indicating that the protein–polyphenol conjugates could be used to increase AAs of HC.
For metal chelating activities (Fig. 2d), the result was not similar to AAs tested by other assays. HC had higher activity than OHC, suggesting that the oxidation of HC resulted in the lowered chelating activity. Some polar amino acids with chelating activity such as Glu, Asp and Ser,12,28 might lose activity after oxidation process. As a consequence, the ability in inhibition of ferrozine–Fe2+ was decreased. However, the augmented activity was observed when EGCG was incorporated, especially at 3% EGCG, which showed the highest activity. High OH groups in EGCG of conjugate might form ligand with Fe2+. As a whole, Fe2+ was not able to bind to Ferrozine. Thus, the results revealed that HC–EGCG conjugate could act as potential metal chelators. With the highest ABTS-RSA and metal chelating activity, along with high DPPH-RSA and FRAP of HC–3% EGCG, compared to other samples, this conjugate was selected for molecular characterization.
3.2 Characteristics of the selected HC–EGCG conjugate
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching/H-bonding). Moreover, the peaks of amide A (∼3373 cm−1), amide II (1546 cm−1) and amide III (1254 cm−1) were also detected in HC, related to N–H stretching coupled with H-bonding, N–H bending combined with C–N stretching and C–N stretching coupled with N–H deformation, respectively. In general, those peaks were found in seabass gelatin-derived peptides.29 When HC was oxidized, the peaks at wavenumbers of 1790 cm−1 and 1745 cm−1 (CO stretching) appeared in OHC sample (Fig. 3a), while these wavenumbers were absent in HC sample. This result demonstrated that the peptides in HC underwent oxidation, thus generating carbonyl groups (CO group). This reaction was mainly induced by hydroxyl radical. The addition of H2O2 to blood plasma caused protein oxidation, which was evidenced by the increased absorbance of CO group (1740 cm−1).30 Additionally, higher intensities of all amide peaks were found in OHC sample, compared to those of HC sample. Modifications of protein or peptide with hydroxyl radicals (OH˙) generated from redox reaction caused increasing intensities of the amide peaks.31,32 Also, the shift to a lower wavenumber of amide II (1546 cm−1 to 1541 cm−1) and amide III (1254 cm−1 to 1209 cm−1) observed (Fig. 3a) in OHC sample might be related to interaction between OH˙ radical and peptides of HC via covalent bond, especially at N–H groups of peptides. Via conjugation of HC with EGCG, no shift of amide I and amide II peaks in HC–EGCG conjugate was observed as compared to OHC sample. Thus, there was no interaction between OHC and EGCG via CO or N–H in the peptide backbone. Nevertheless, lower wavenumber of amide III peak was found in HC–EGCG, compared to OHC. This suggested the interaction between EGCG and peptide to some degree. Moreover, lower intensity of all amide peaks were found in HC–EGCG conjugate sample as compared to other samples more likely due to the dilution effect by EGCG incorporated. Also, peak of O–H stretching vibration shifted to a higher wavenumber as EGCG was conjugated (3373 cm−1 to 3400 cm−1) (Fig. 3b). This might be related to increasing OH group, mainly from EGCG into HC sample via conjugation. In general, the wavenumber of O–H group of EGCG was found at broad spectrum ranging from 3200 to 3570 cm−1.33 The vibration of O–H group of amide A peak to higher wavenumber in HC–EGCG sample was possibly due to the coupling between EGCG and N–H group of peptides via hydrogen linkage, resulting in broadening and shift to higher wavenumber. Also, new peak appeared at wavenumber of 1740 cm−1 for HC–EGCG conjugate as compared to HC sample (Fig. 3a). This peak also had lower wavenumber (1740 cm−1) than that of OHC sample (1745 cm−1), suggesting the interaction between EGCG and carbonyl group of oxidized peptides in OHC. EGCG might act as nucleophile via OH group, which could form H-bonds with CO of oxidized peptides (electrophile).
O stretching/H-bonding). Moreover, the peaks of amide A (∼3373 cm−1), amide II (1546 cm−1) and amide III (1254 cm−1) were also detected in HC, related to N–H stretching coupled with H-bonding, N–H bending combined with C–N stretching and C–N stretching coupled with N–H deformation, respectively. In general, those peaks were found in seabass gelatin-derived peptides.29 When HC was oxidized, the peaks at wavenumbers of 1790 cm−1 and 1745 cm−1 (CO stretching) appeared in OHC sample (Fig. 3a), while these wavenumbers were absent in HC sample. This result demonstrated that the peptides in HC underwent oxidation, thus generating carbonyl groups (CO group). This reaction was mainly induced by hydroxyl radical. The addition of H2O2 to blood plasma caused protein oxidation, which was evidenced by the increased absorbance of CO group (1740 cm−1).30 Additionally, higher intensities of all amide peaks were found in OHC sample, compared to those of HC sample. Modifications of protein or peptide with hydroxyl radicals (OH˙) generated from redox reaction caused increasing intensities of the amide peaks.31,32 Also, the shift to a lower wavenumber of amide II (1546 cm−1 to 1541 cm−1) and amide III (1254 cm−1 to 1209 cm−1) observed (Fig. 3a) in OHC sample might be related to interaction between OH˙ radical and peptides of HC via covalent bond, especially at N–H groups of peptides. Via conjugation of HC with EGCG, no shift of amide I and amide II peaks in HC–EGCG conjugate was observed as compared to OHC sample. Thus, there was no interaction between OHC and EGCG via CO or N–H in the peptide backbone. Nevertheless, lower wavenumber of amide III peak was found in HC–EGCG, compared to OHC. This suggested the interaction between EGCG and peptide to some degree. Moreover, lower intensity of all amide peaks were found in HC–EGCG conjugate sample as compared to other samples more likely due to the dilution effect by EGCG incorporated. Also, peak of O–H stretching vibration shifted to a higher wavenumber as EGCG was conjugated (3373 cm−1 to 3400 cm−1) (Fig. 3b). This might be related to increasing OH group, mainly from EGCG into HC sample via conjugation. In general, the wavenumber of O–H group of EGCG was found at broad spectrum ranging from 3200 to 3570 cm−1.33 The vibration of O–H group of amide A peak to higher wavenumber in HC–EGCG sample was possibly due to the coupling between EGCG and N–H group of peptides via hydrogen linkage, resulting in broadening and shift to higher wavenumber. Also, new peak appeared at wavenumber of 1740 cm−1 for HC–EGCG conjugate as compared to HC sample (Fig. 3a). This peak also had lower wavenumber (1740 cm−1) than that of OHC sample (1745 cm−1), suggesting the interaction between EGCG and carbonyl group of oxidized peptides in OHC. EGCG might act as nucleophile via OH group, which could form H-bonds with CO of oxidized peptides (electrophile).
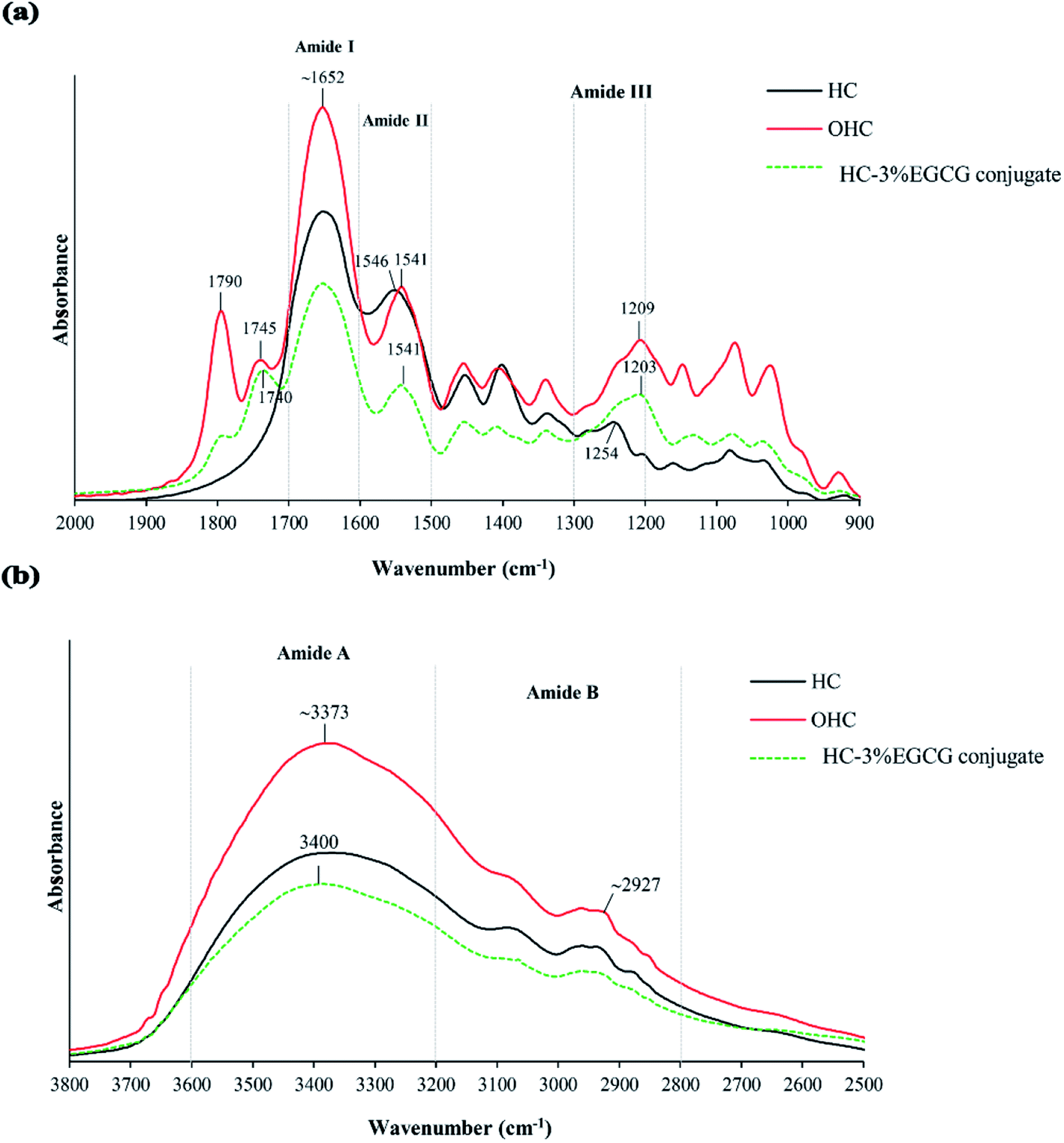 | ||
| Fig. 3 FTIR spectra in wavenumber region of 2000–900 cm−1 (a) and 2500–3800 cm−1 (b) of HC, OHC and HC–3% EGCG conjugate. | ||
524 Da. After grafting, peptide with MW of 2771 Da became predominant, while the peaks of peptide with high MW were absent in HC–3% EGCG conjugate. Additionally, there was a peak having MW of 585 Da in OHC. For EGCG, the peak with MW of 452 Da was noticeable. Peak with MW of 452 Da disappeared in HC–3% EGCG conjugate. The result confirmed that EGCG was totally bound with both low and high MW peptides of OHC, resulting in the peak of new peptides generated with higher MW.
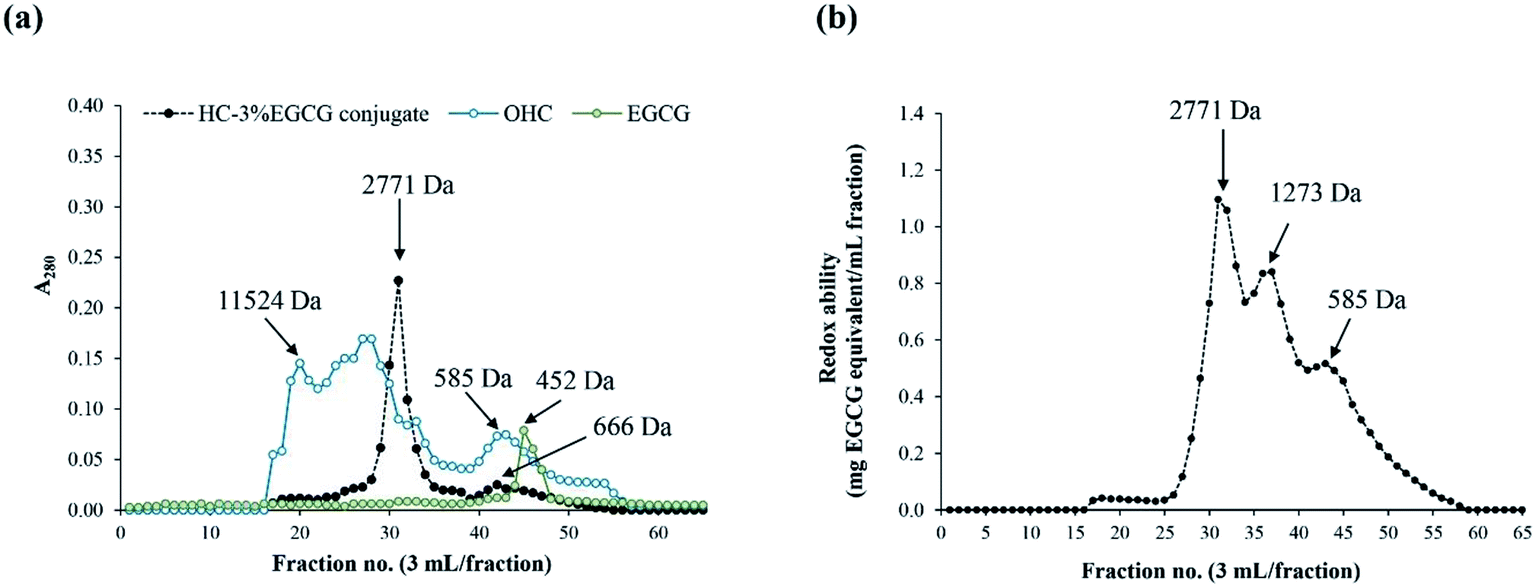 | ||
| Fig. 4 Elution profiles of OHC, HC–3% EGCG conjugate and EGCG using SephadexTM G-25 gel filtration chromatography (a) and redox ability of all fractions of HC–3% EGCG (b). | ||
Redox ability of HC–EGCG conjugate was determined in all fractions using the Folin–Ciocalteu assay (Fig. 4b). The fraction with MW of 2771 Da of HC–EGCG conjugate exhibited the highest redox ability, followed by fractions with MW of 1273 and 585 Da. Peak of EGCG (452 Da) was not detected in HC–3% EGCG conjugate. This confirmed that no free EGCG was present in the resulting conjugate. Similar result was documented in duck egg albumin (DEA) conjugated with EGCG,15 which indicated that increased MW of DEA was achieved after being conjugation with EGCG. Moreover, Li et al.35 also reported that increased MW of β-lactoglobulin was noted when catechin was conjugated. Thus, the modification of HC with EGCG using covalent grafting method increased MW of HC, and had the influence on physiochemical and antioxidant properties of HC–EGCG conjugate.
3.3 Effect of HC–3% EGCG conjugate on cell proliferation and ROS inhibition of MRC-5 and HaCaT cells
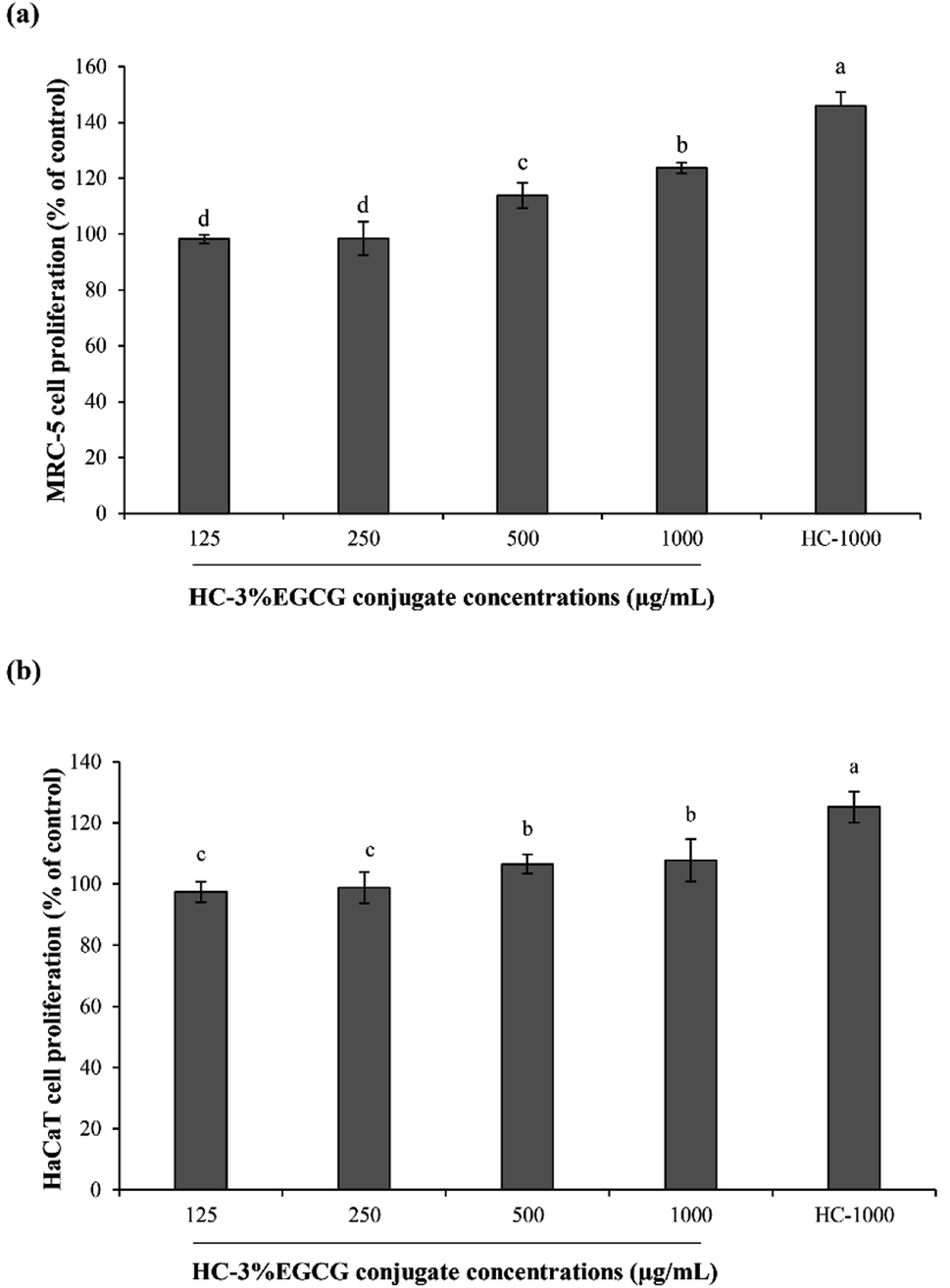 | ||
| Fig. 5 Effect of HC–3% EGCG conjugate at different levels on proliferation of MRC-5 (a) and HaCaT (b) cells. HC-1000: HC at 1000 μg mL−1. Bars represent the standard deviation (n = 3). Different lowercase letters on bars indicate significant differences between samples (p < 0.05). | ||
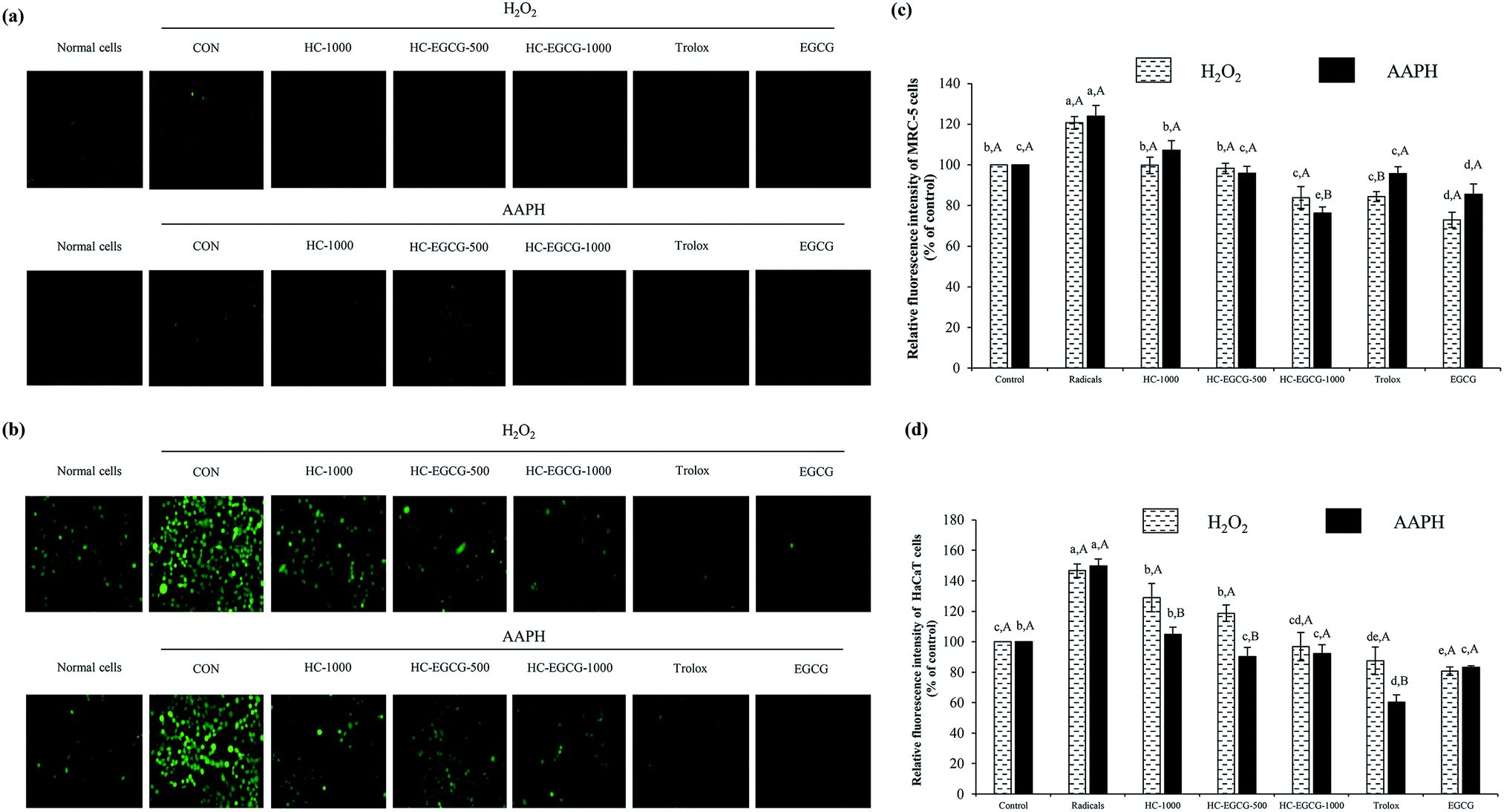 | ||
| Fig. 6 Morphology of MRC-5 cells (a) HaCaT cells (b) and the relative fluorescence intensity after treatment with oxidative stressors (c and d). Bars represent the standard deviation (n = 3). Different lowercase letters on bars within the same stressor tested indicate significant differences (p < 0.05). Different uppercase letters on bars within the same compounds tested indicate significant differences (p < 0.05). CON: cells treated with 0.1 mM H2O2 or 0.1 mM AAPH alone, HC-1000: cells treated with HC at 1000 μg mL−1, HC–3% EGCG-500 and HC–3% EGCG-1000: cells treated with HC–3% EGCG conjugate at 500 and 1000 μg mL−1, respectively. Trolox: cells treated with trolox at 50 μM. EGCG: cells treated with EGCG at 50 μM. | ||
When HaCaT cells were exposed to H2O2 (Fig. 6b and d), the lowest ROS generation was found in cells treated with EGCG as compared to other samples (p < 0.05). Also, no difference in protective ability between HC–EGCG-1000 and trolox was noticeable (p > 0.05). For AAPH-induced HaCaT cells (Fig. 6b and d), the cytoprotective ability of conjugate at both concentrations was comparable to EGCG (p > 0.05). This suggested that conjugates had a cellular radical scavenging activity. However, the activity of both samples was less than trolox as evidenced by higher ROS generation. When comparing between cells treated with H2O2 and AAPH, lower ROS generation was observed in the latter for all samples tested (p > 0.05), except cells treated with HC–3% EGCG-1000 and EGCG (p > 0.05). ROS is generated during metabolic processes of all cells, especially respiration process.1 Among ROS, hydroxyl radical (OH˙) is strongly ROS, which causes oxidative damage of cells. It is produced by the reaction between metal ions (Fe2+ and Cu+) in intracellular proteins and H2O2 via Fenton reaction.38 Furthermore, OH˙ is also generated from the reaction of superoxide anion (˙O2−) with H2O2 in Haber–Weiss reaction,38 while peroxyl radical (ROO˙) is produced via lipid peroxidation, particularly polyunsaturated fatty acids (PUFA) of cell membranes.39 This radical can induce the production of lipid hydroperoxides, leading to the formation of aldehydes.39 In general, cellular homeostasis is maintained by enzymatic pathways and antioxidants.40 When excessive ROS occurs, it can stimulate oxidative stress in normal cells and results in the aggravation of pathologic processes.41 Thus, the treatment with the potential antioxidants such as polyphenols and tocopherols can prevent skin damage associated with ROS, especially skin-aging.41 According to the aforementioned results, it confirmed that the use of HC–3% EGCG conjugate, especially at high concentration could alleviate ROS-induced skin cell damage by reducing intracellular reactive oxygen species levels more effectively than HC alone.
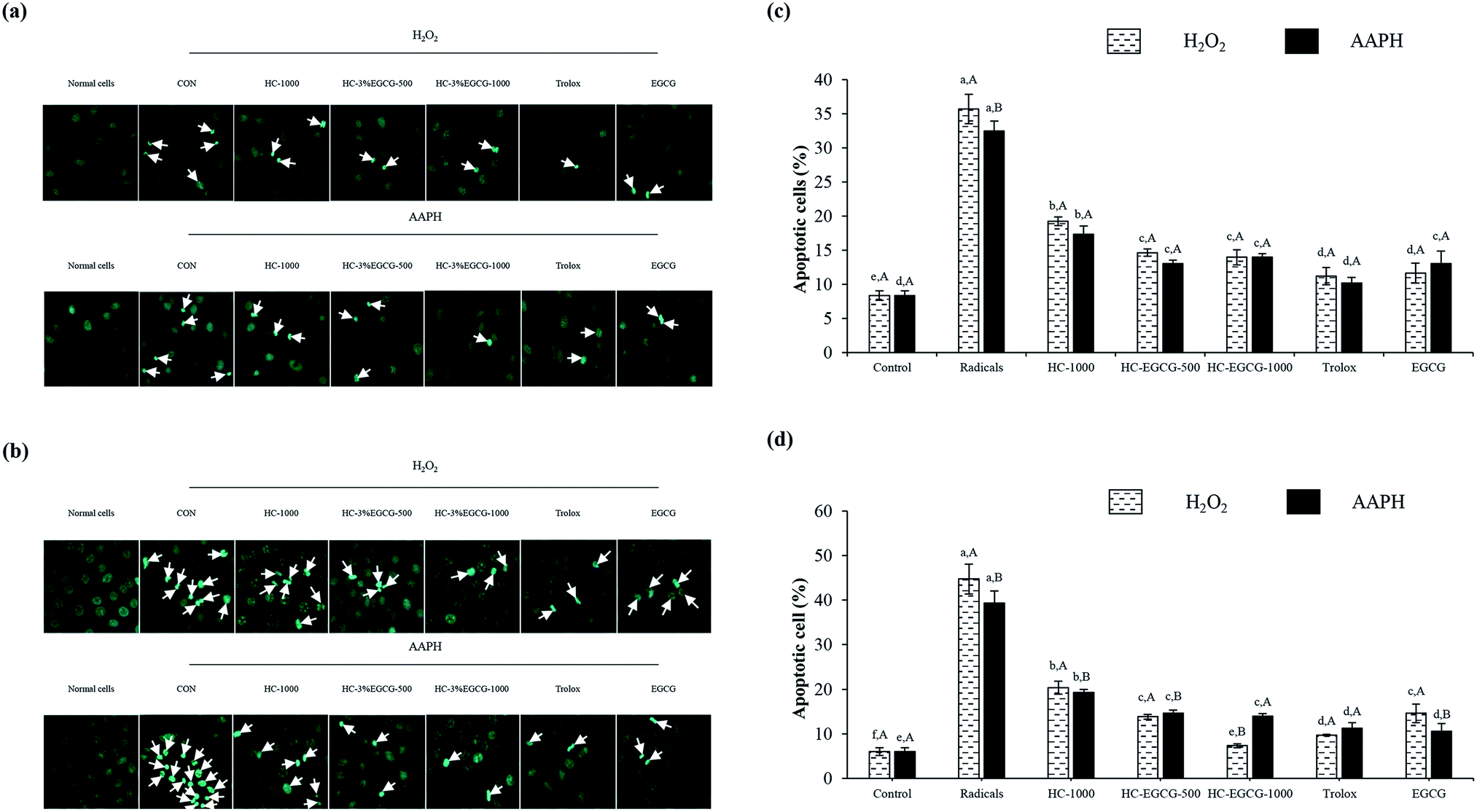 | ||
| Fig. 7 Nuclear morphology of MRC-5 cells (a) HaCaT cells (b) and amount of apoptotic cells after treatment with oxidative stressors (c and d). Bars represent the standard deviation (n = 3). Different lowercase letters on bars within the same stressor tested indicate significant differences (p < 0.05). Different uppercase letters on bars within the same compounds tested indicate significant differences (p < 0.05). CON: cells treated with 0.1 mM H2O2 or 0.1 mM AAPH alone, HC-1000: cells treated with HC at 1000 μg mL−1, HC–3% EGCG-500 and HC–3% EGCG-1000: cells treated with HC–3% EGCG conjugate at 500 and 1000 μg mL−1, respectively. Trolox: cells treated with trolox at 50 μM. EGCG: cells treated with EGCG at 50 μM. | ||
4. Conclusions
Conjugation of hydrolyzed collagen (HC) from defatted seabass skin with EGCG could increase surface hydrophobicity (SH) and antioxidant activities (AAs) of the resulting conjugates. Overall, SH and AAs were increased as HC was grafted with 3% EGCG. HC–3% EGCG conjugate with MW of 2771 Da provided the highest reducing ability. Moreover, the resulting conjugate could promote the proliferation of MRC-5 and HaCaT cells. More importantly, it could inhibit the ROS production and death of both cells, especially when HC–3% EGCG conjugate at high level was used. Therefore, the HC–EGCG conjugate could be considered as alternative potential antioxidant for protecting skin cell damage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Thailand Research Fund under the Royal Golden Jubilee PhD Program to Lalita Chotphruethipong (PHD/0183/2560) and Prince of Songkla University, Thailand (Grant No. AGR6302013N).References
- S. J. Forrester, D. S. Kikuchi, M. S. Hernandes, Q. Xu and K. K. Griendling, Circ. Res., 2018, 122, 877–902 CrossRef CAS.
- B. Uttara, A. V. Singh, P. Zamboni and R. Mahajan, Curr. Neuropharmacol., 2009, 7, 65–74 CAS.
- C.-Q. Luo, L. Xing, P.-F. Cui, J.-B. Qiao, Y.-J. He, B.-A. Chen, L. Jin and H.-L. Jiang, Int. J. Nanomedicine., 2017, 12, 855 CrossRef CAS.
- V. Lobo, A. Patil, A. Phatak and N. Chandra, Pharmacogn. Rev., 2010, 4, 118–126 CrossRef CAS.
- A. Drewnowski and E. Almiron-Roig, 11 Human perceptions and preferences for fat-rich foods, 2009 Search PubMed.
- M. Logarušić, I. Slivac, K. Radošević, M. Bagović, I. R. Redovniković and V. G. Srček, Mol. Biol. Rep., 2019, 46, 6079–6085 CrossRef.
- S. Y. Lee and S. J. Hur, Compr. Rev. Food Sci. Food Saf., 2019, 18, 923–935 CrossRef CAS.
- M. Nikoo, S. Benjakul, M. Yasemi, H. A. Gavlighi and X. Xu, LWT, 2019, 108, 120–128 CrossRef CAS.
- T. Sae-leaw, Y. C. O'Callaghan, S. Benjakul and N. M. O'Brien, J. Food Sci. Technol., 2016b, 53, 197–208 Search PubMed.
- T. Senphan and S. Benjakul, J. Funct. Foods., 2014, 6, 147–156 CrossRef CAS.
- S. Benjakul, S. Karnjanapratum and W. Visessanguan, Int. J. Food Sci. Technol., 2018, 53, 1871–1879 CAS.
- L. Chotphruethipong, R. E. Aluko and S. Benjakul, J. Food Biochem., 2019a, 43, 1–13 Search PubMed.
- U. G. Spizzirri, F. Iemma, F. Puoci, G. Cirillo, M. Curcio, O. I. Parisi and N. Picci, Biomacromolecules, 2009, 10, 1923–1930 CrossRef CAS.
- P. Chanphai, P. Bourassa, C. Kanakis, P. Tarantilis, M. Polissiou and H. Tajmir-Riahi, Food Hydrocoll., 2018, 77, 322–328 CrossRef CAS.
- T. H. Quan and S. Benjakul, Colloids Surf. A Physicochem. Eng. Asp., 2019, 579, 123711 CAS.
- K. Nilsuwan, S. Benjakul, T. Prodpran and K. de la Caba, Food Hydrocoll., 2019, 89, 783–791 CrossRef CAS.
- L. Chotphruethipong, R. E. Aluko and S. Benjakul, Process Biochem., 2019b, 86, 58–64 Search PubMed.
- A. Thanonkaew, S. Benjakul, W. Visessanguan and E. A. Decker, Food Chem., 2006, 95, 591–599 CrossRef CAS.
- L. Chotphruethipong, S. Benjakul and K. Kijroongrojana, J. Food Biochem., 2017, 41, 1–10 CrossRef.
- S. Singkhorn, M. H. Tantisira, S. Tanasawet, P. Hutamekalin, T. Wongtawatchai and W. Sukketsiri, Phytother. Res., 2018, 32, 1397–1403 CAS.
- L.-C. Chen, Y.-C. Chen, C.-Y. Su, C.-S. Hong, H.-O. Ho and M.-T. Sheu, Int. J. Nanomedicine., 2016, 11, 1557 CAS.
- G. Reina, A. Gismondi, R. Carcione, V. Nanni, C. Peruzzi, M. Angjellari, N. D. Q. Chau, A. Canini, M. L. Terranova and E. Tamburri, Appl. Surf. Sci., 2019, 470, 744–754 CrossRef CAS.
- J. Feng, H. Cai, H. Wang, C. Li and S. Liu, Food Chem., 2018, 241, 60–69 CrossRef CAS.
- F. Liu, C. Sun, W. Yang, F. Yuan and Y. Gao, RSC Adv., 2015, 5, 15641–15651 RSC.
- M. Fingerman, Biomaterials from aquatic and terrestrial organisms, CRC Press, 2006 Search PubMed.
- S. Legeay, M. Rodier, L. Fillon, S. Faure and N. Clere, Nutrients, 2015, 7, 5443–5468 CrossRef CAS.
- J. Tkaczewska, M. Bukowski and P. Mak, Molecules, 2019, 24, 97 CrossRef.
- Q. Liu, B. Kong, Y. L. Xiong and X. Xia, Food Chem., 2010, 118, 403–410 CrossRef CAS.
- S. Sinthusamran, S. Benjakul and H. Kishimura, Food Chem., 2014, 152, 276–284 CrossRef CAS.
- J. Bujok, M. Gąsior-Głogowska, M. Marszałek, N. Trochanowska-Pauk, F. Zigo, A. Pavľak, M. Komorowska and T. Walski, Biomed Res. Int., 2019, 2019, 1–9 CrossRef.
- W. Theerawitayaart, T. Prodpran and S. Benjakul, J. Chem., 2019, 2019, 1–11 CrossRef.
- X. Guo, H. Qiu, X. Deng, X. Mao, X. Guo, C. Xu and J. Zhang, Molecules, 2019, 24, 3205 CrossRef CAS.
- K. S. Avadhani, J. Manikkath, M. Tiwari, M. Chandrasekhar, A. Godavarthi, S. M. Vidya, R. C. Hariharapura, G. Kalthur, N. Udupa and S. Mutalik, Drug Deliv, 2017, 24, 61–74 CrossRef CAS.
- Z. Jia, M. Zheng, F. Tao, W. Chen, G. Huang and J. Jiang, LWT--Food Sci. Technol., 2016, 66, 305–310 CrossRef CAS.
- J. Yi, Y. Zhang, R. Liang, F. Zhong and J. Ma, J. Agric. Food Chem., 2015, 63, 297–303 CrossRef CAS.
- F. Subhan, H. Y. Kang, Y. Lim, M. Ikram, S.-Y. Baek, S. Jin, Y. H. Jeong, J. Y. Kwak and S. Yoon, Oxidative med. cell. longev., 2017, 2017, 1–17 CrossRef.
- S. Yarnpakdee, S. Benjakul, H. G. Kristinsson and H. E. Bakken, J. Food Sci. Technol., 2015, 52, 6194–6205 CrossRef CAS.
- A. Phaniendra, D. B. Jestadi and L. Periyasamy, Indian J. Clin. Biochem., 2015, 30, 11–26 CrossRef CAS.
- S. B. Nimse and D. Pal, RSC Adv., 2015, 5, 27986–28006 RSC.
- M. Rinnerthaler, J. Bischof, M. K. Streubel, A. Trost and K. Richter, Biomolecules, 2015, 5, 545–589 CrossRef CAS.
- H. Xu, Y.-W. Zheng, Q. Liu, L.-P. Liu, F.-L. Luo, H.-C. Zhou, H. Isoda, N. Ohkohchi and Y.-M. Li, Reactive oxygen species ROS living cells. London, UK: IntechOpen. 2018, pp. 69–87 Search PubMed.
- M. Redza-Dutordoir and D. A. Averill-Bates, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2977–2992 CrossRef CAS.
- M. Savitskaya and G. Onishchenko, Biochemistry, 2015, 80, 1393–1405 CAS.
| This journal is © The Royal Society of Chemistry 2021 |