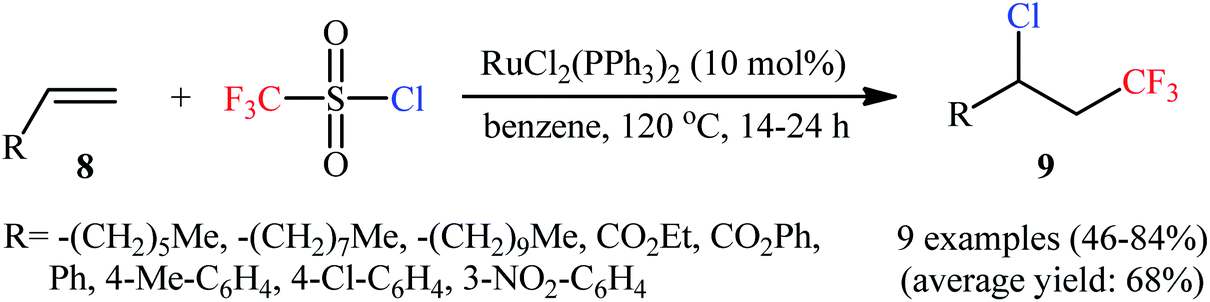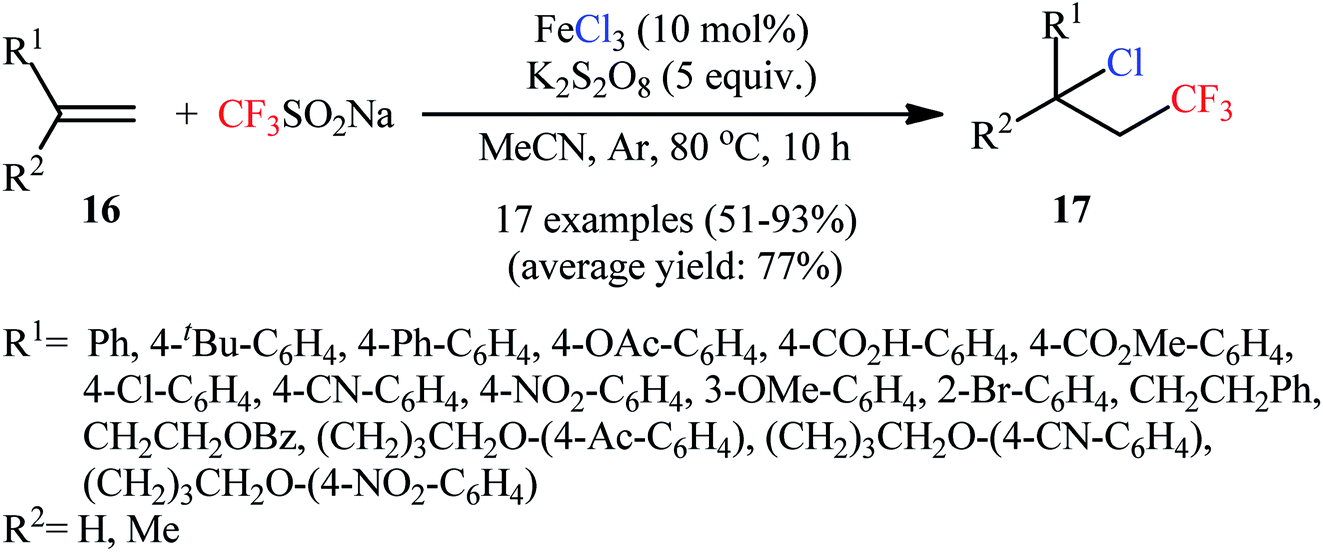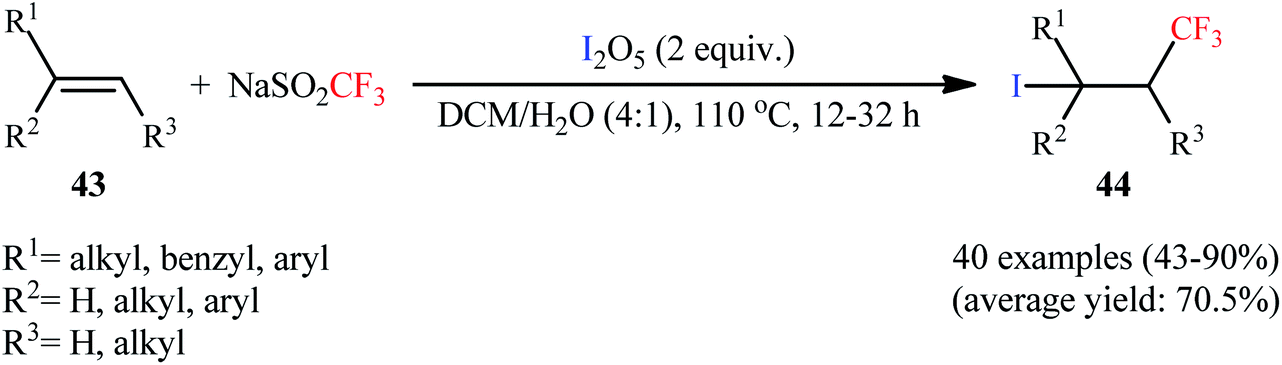DOI:
10.1039/D0RA06872A
(Review Article)
RSC Adv., 2021,
11, 14941-14955
Vicinal halo-trifluoromethylation of alkenes
Received
9th August 2020
, Accepted 23rd March 2021
First published on 21st April 2021
Abstract
Both trifluoromethyl and halide groups are widely found in medicinally and pharmaceutically important compounds and, moreover, organohalides are commonly used as versatile intermediates in synthetic organic chemistry. Due to their prevalence and easy accessibility, alkene halo-trifluoromethylation provides a convenient way to install these valuable functionalities in complex targets. In this review, we summarize recent advances and achievements in this fast-growing research field. For clarity, the reactions were classified according to the type of halogen atom.
 Rzgar Tawfeeq Kareem | Rzgar Tawfeeq Kareem was born in Sulaimaneyah, Iraq, in 1988. He received his B.S. degree in pure chemistry from Garmian University, Iraq, and his M.S. degree in analytical chemistry from the University of Siirt, Turkey. now, he is completing his PhD degree at the University of Bu Ali Sina. |
 Bayan Azizi | Bayan Azizi was born in Iraq, in 1986. She has received her bachelor degree in Applied Chemistry at Razi University, Iran in 2008. Also, she has received her master degree in Physical Chemistry at Kurdistan University in 2012. After graduation, she worked at the Quality Control Laboratory in Jahad Daneshgahi (Oslub Sanat Company), Iran and Payam Noor University, Kermanshah in 2013. She was lecturer at the Technical College of Applied Science, Sulaimani Polytechnic University, Kurdistan Region of Iraq between 2014 and 2018. Currently she is lecturer at the Department of Medical Laboratory Science, University of Human Development, Kurdistan Region of Iraq from 2018. Her research interests include nano-technology, quantum chemistry, physical chemistry, and clinical chemistry. |
 Manzarbanou Asnaashriisfahani | Manzarbanou Asnaashari Isfahani was born in Esfahan, Iran, in 1950. She received her B.S. degree in pure chemistry from the University of Esfahan, Esfahan Iran and her M.S. degree in organic chemistry from North Tehran Branch, Islamic Azad University, Tehran, Iran, in 1994 under the supervision of Prof. A. Roostaeyan. She completed her PhD degree in 2002 under the supervision of Prof. E. Yavary in Tehran Azad University of Science and Research, Tehran, Iran. Now, she is working at North Tehran Branch, Islamic Azad University as an Associate Professor in organic chemistry. Her research interests include theoretical organic chemistry, new methodologies in organic synthesis and organic heterocyclic synthesis. |
 Abdolghaffar Ebadi | Dr Abdol Ghaffar Ebadi finished his doctoral degree in Environmental Biotechnology (Algology) from Tajik Academy of Sciences. Now he is a researcher in TAS in Tajikistan and faculty member at the Islamic Azad University of Jouybar in Mazandaran. Dr Ebadi has published more than 400 scientific papers in qualified international journals and attended more than 50 international conferences. He has cooperation with many research project teams around world such as in China, Malaysia, and Thailand. His interests are environmental biotechnology, biochemistry, and gene pathways in the phytoremediation processes. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in pure chemistry from the University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran university, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as a Professor in organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
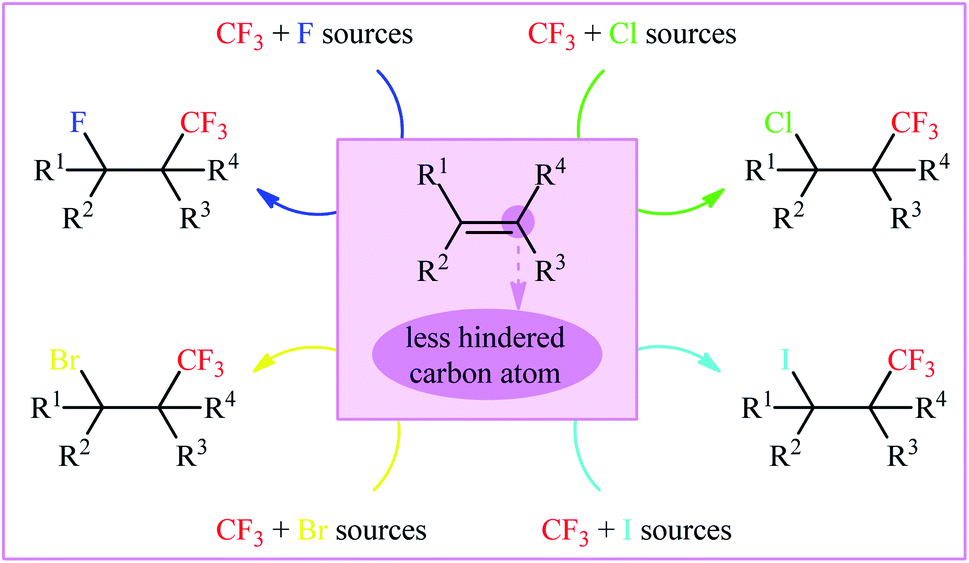
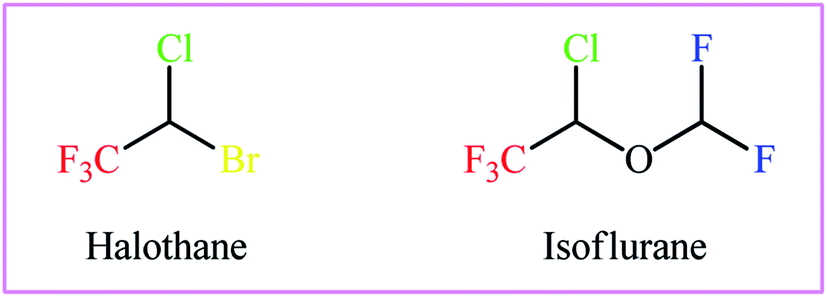
As a result of the unique physical, chemical, and physiological properties of the fluorine atom, fluoroorganic compounds have attracted considerable attention in diverse fields, ranging from pharmaceuticals and agrochemicals to refrigerants and advanced materials.1–3 Interestingly, over one-fourth of FDA-approved drugs and circa half of contemporary agrochemicals4 contain one or more fluorine atoms in their chemical structures. Among various fluorine containing substituents, the trifluoromethyl group (CF3) is highly valuable for applications in drug discovery, as the incorporation of this substituent often significantly improves the lipophilicity, bioavailability and protein-binding affinity of a molecule and suppresses metabolic detoxification processes to increase the in vivo lifetime of a drug.5 Therefore, the development of efficient and versatile strategies for introducing the CF3 moiety into various organic molecules has become one of the hottest topics in advanced organic synthesis in recent years.6 Among the many methods developed, particularly noteworthy is the vicinal halotrifluoromethylation of alkenes due to its high pot- and step-economy for the introduction of two versatile functional groups (i.e., trifluoromethyl and a halogen) into ubiquitous feedstock materials (Fig. 1). Noteworthy, two trifluoromethylated alkyl halides, halothane and isoflurane, are on the WHO's list of essential medicines (Scheme 1).7 Needless to say that halogen atoms in the titled compounds could serve as highly valuable synthons for the preparation of various types of compounds via the well-known nucleophilic substitution and cross-coupling reactions.
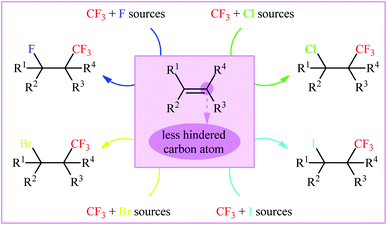 |
| | Fig. 1 Vicinal halotrifluoromethylation of alkenes. | |
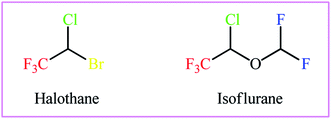 |
| | Scheme 1 The structures of halothane and isoflurane. | |
The recent growth on the trifluoromethylation reactions has led to a surprisingly large number of excellent reviews on this domain.6,8 However, the literature review reveals that no comprehensive review article is available covering vicinal halotrifluoromethylation of alkenes. In connection with our recent works on organofluorine chemistry9–13 and modern organic synthesis,14 herein, we will summarize recent discoveries and developments in the arena of halotrifluoromethylation of alkenes, although we occasionally mention earlier work when relevant. For clarity, the reactions were organized according to the type of halogen atoms.
2. Fluorotrifluoromethylation
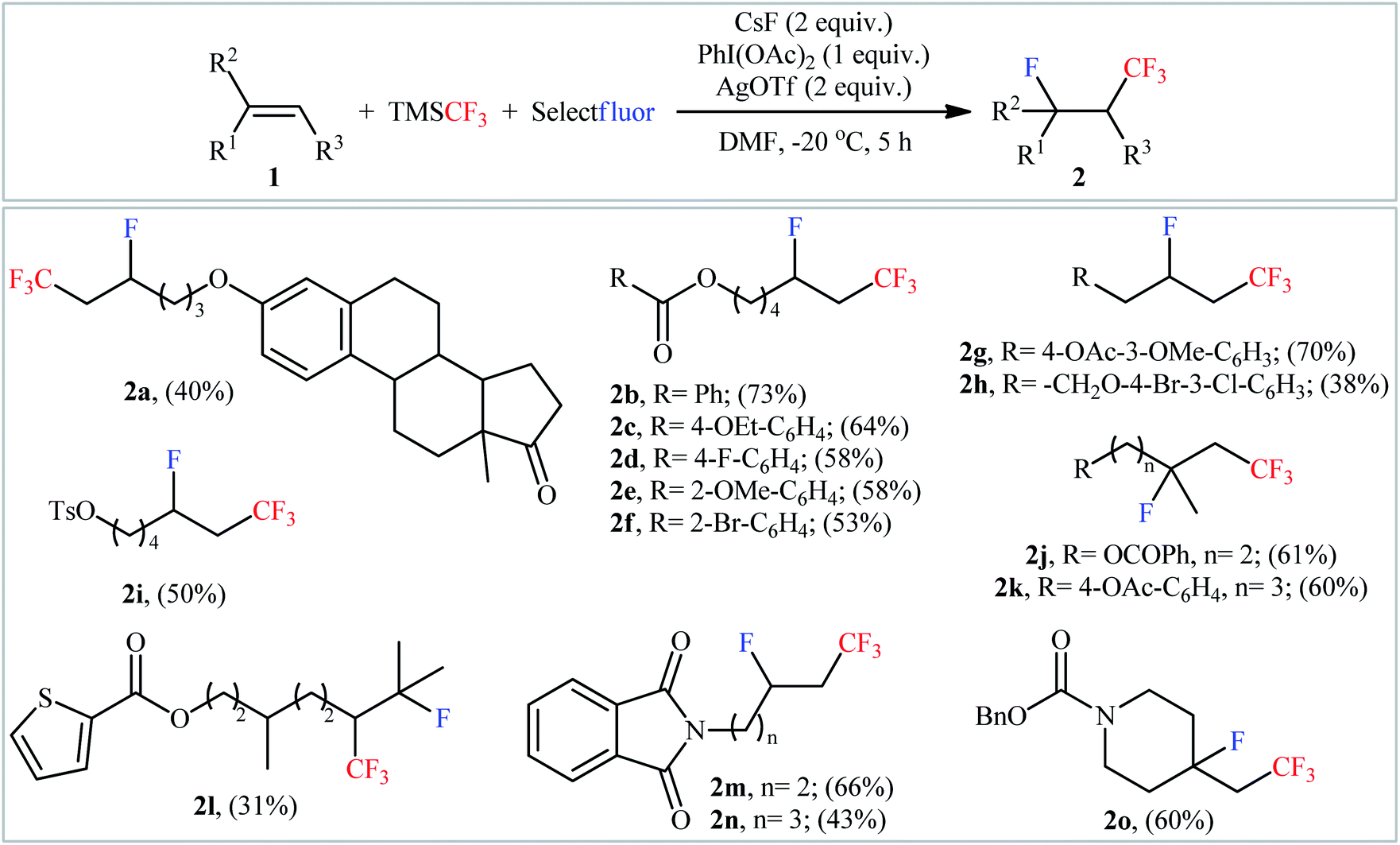
In 2015, Qing and co-workers disclosed for the first time the possibility of synthesizing β-CF3-substituted fluoroalkanes through the direct vicinal fluorotrifluoromethylation of the corresponding alkenes.15 To determine the optimum conditions, they screened the activities of different initiators (e.g., AgF, CsF, KF, NaOAc), oxidants (PhIO, PhI(OAc)2, PhI(OCOCF3)3, K2S2O8, O2, NCS, tBuOOtBu), and additives (e.g., AgNO3, AgOAc, AgOTf) in the fluorotrifluoromethylation of 5-hexen-1-yl benzoate using TMSCF3 and Selectfluor as the CF3 and F sources, respectively, as a model reaction. The optimal system was identified using CsF in combination with PhI(OAc)2 and AgOTf in DMF at −20 °C. A variety of terminal and internal unactivated aliphatic alkenes 1 were reacted well under the standard reaction conditions to afford the corresponding fluorotrifluoromethylated products 2 in modest to good yields, ranging from 31% to 73% (Scheme 2). However, no aromatic alkene was examined in this synthetic strategy. Notably, the reaction showed complete regioselectivity for unsymmetrical alkenes, in which CF3 group predominantly added to the less hindered carbon atom of the double bond. The radical trapping experiments with TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) revealed that the radical processes might be involved in this Ag-mediated oxidative fluorotrifluoromethylation reaction.
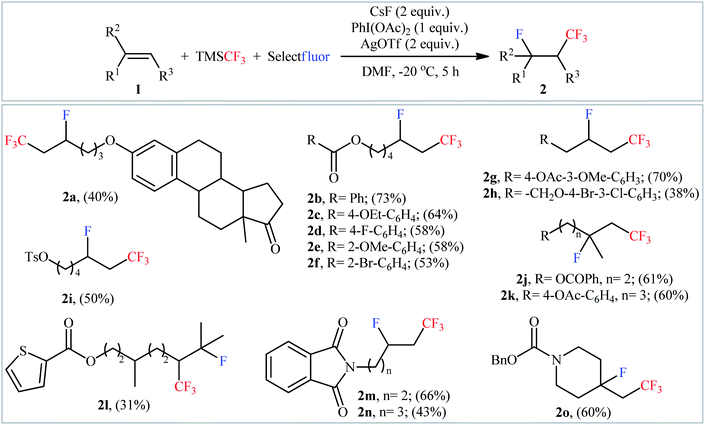 |
| | Scheme 2 Ag-mediated oxidative fluorotrifluoromethylation of alkenes 1 using TMSCF3 and Selectfluor, developed by Qing. | |
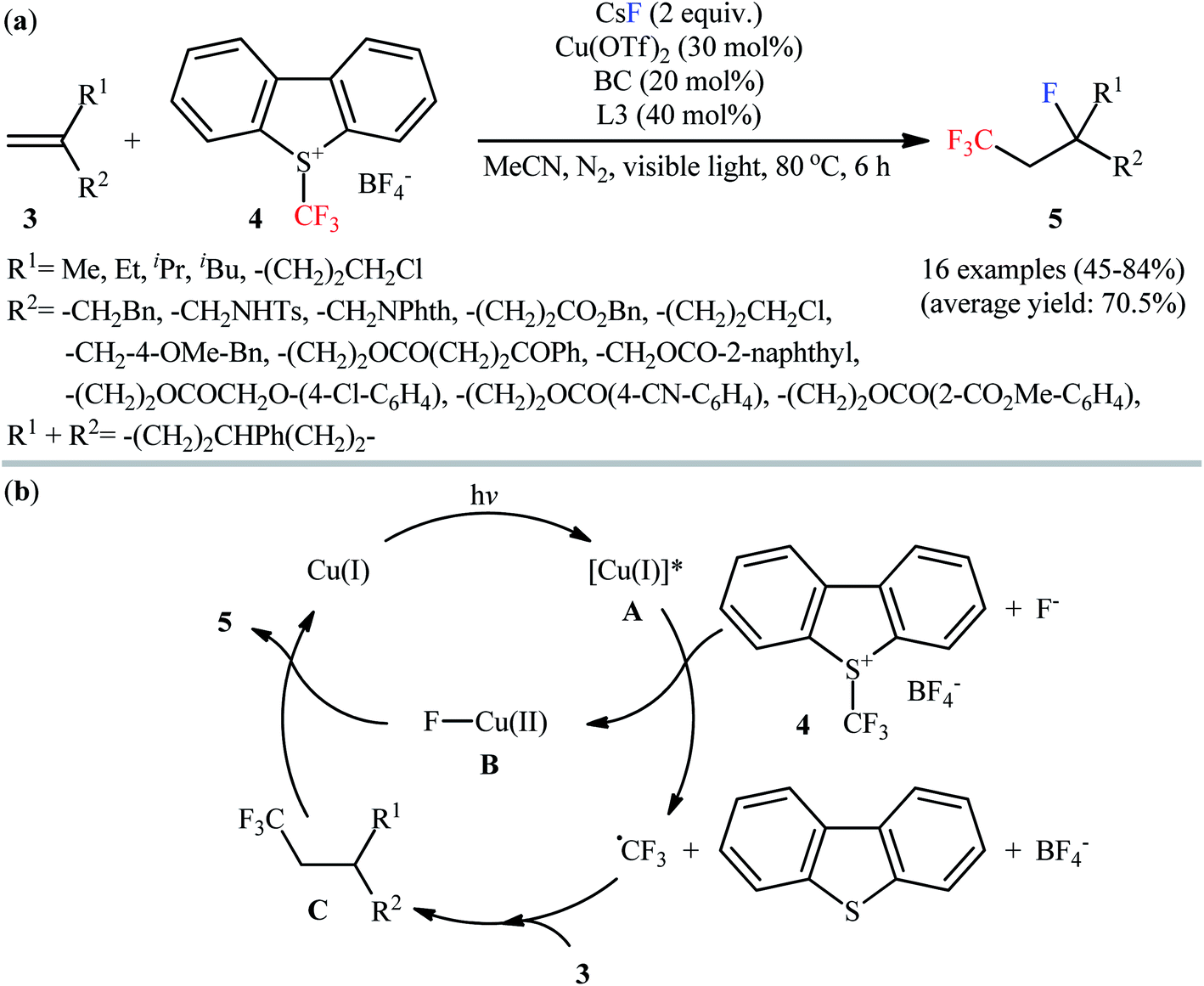
Following this work, Yu and Li along with their co-workers reported that Cu(OTf)2 enabled fluorotrifluoromethylation of 1,1-disubstituted aliphatic alkenes 3 with CsF (F source) and Umemoto's reagent 4 (CF3 source) in the presence of 40 mol% of 4,4′-bis(methoxycarbonyl)-2,2′-bipyridin (L3) and 20 mol% of bathocuproine (BC) as the ligands at 80 °C under an inert atmosphere.16 The reactions proceeded smoothly in MeCN under visible light irradiation (11 W), tolerated various functional groups (ketone, ether, ester, amide, sulfonamide, chloro, cyano), and generally provided moderate to high yields of fluorotrifluoromethylated products 5 with only cis-stereochemistry (Scheme 3a). 5-Hexen-1-yl benzoate gave the target product in only 18% yield and therefore no other monosubstituted alkenes were examined in the protocol. Internal alkenes could also undergo the fluorotrifluoromethylation but resulted in a mixture of cis–trans isomers. The proposed reaction mechanism involved the formation of trifluoromethyl radical (˙CF3) and a Cu(II)–F complex B via visible-light-promoted single-electron transfer between Umemoto's reagent and the excited Cu(I) complex A. The addition of electrophilic ˙CF3 to the alkene 3 produces the nucleophilic alkyl radical C, which abstracts a fluorine atom from the complex B to provide the desired product 5 and regenerate the Cu(I) complex (Scheme 3b).
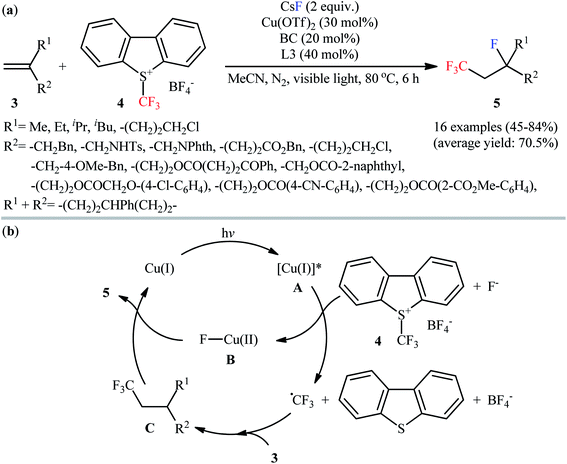 |
| | Scheme 3 (a) Visible light-induced Cu-catalyzed fluorotrifluoromethylation of 1,1-disubstituted alkenes 3 with CsF and Umemoto's reagent 4; (b) proposed mechanism for the formation of 5. | |
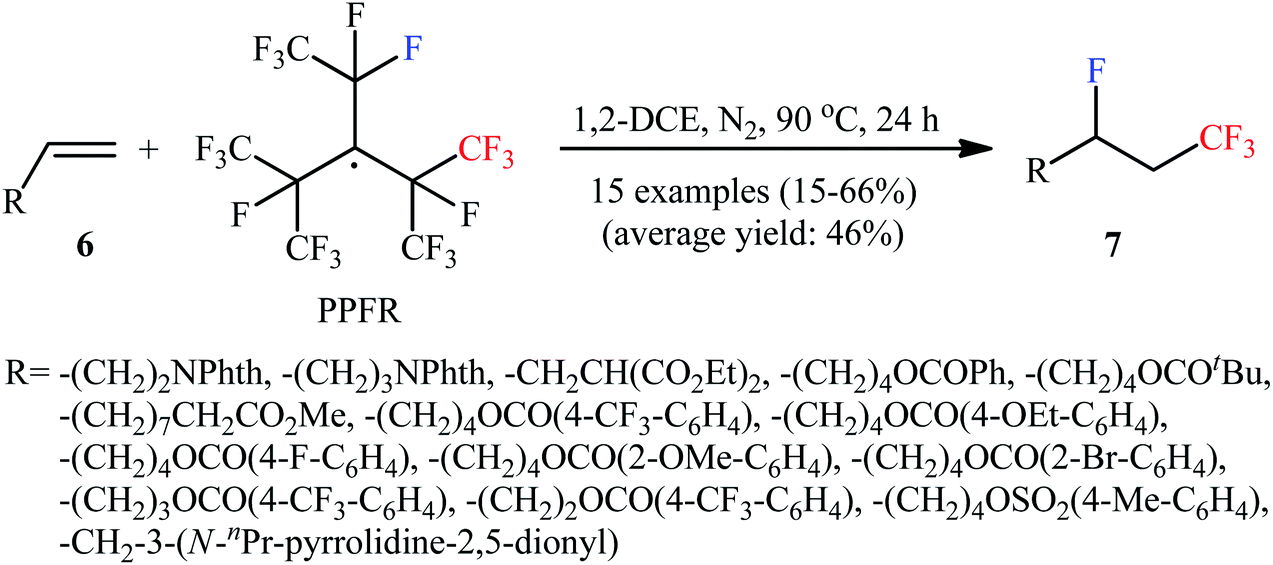
Very recently, Ponomarenko–Soloshonok's research team developed an efficient conversion of monosubstituted alkenes 6 to the corresponding 1-CF3-2-F-alkanes 7, using perfluoro-3-ethyl-2,4-dimethyl-3-pentyl radical (PPFR) as both trifluoromethylating and fluorinating agent under mediator- and additive-free conditions (Scheme 4).17 The reaction was experimentally simple, performed by simple heating of the substrates at 90 °C in DCE, and was applicable to various aliphatic monosubstituted alkenes with relatively wide functional group compatibility. However, the proposed system was unfruitful with aromatic monosubstituted alkenes (styrene derivatives), and lower yields were obtained with olefins bearing aromatic groups due to the side aromatic-trifluoromethylation.
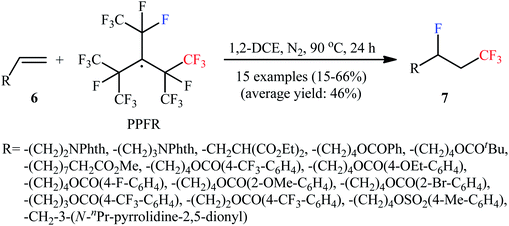 |
| | Scheme 4 Mediator and additive-free fluorotrifluoromethylation of terminal olefins 6 developed by Ponomarenko and Soloshonok. | |
3. Chloro-trifluoromethylation
Synthesis of vicinal chlorotrifluoromethylated alkanes though the direct chloro-trifluoromethylation of olefinic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds is the area that has experienced the most growth in the field of halotrifluoromethylation of unactivated alkenes over the past few years. In this section, we will summarize the available literature on this novel and appealing research arena with emphasize on the mechanistic proposals of the reactions. The section is organized by type of reagent used in two different sub-sections. The first describes chloro-trifluoromethylations using bifunctional reagents, while the second will cover examples that employ monofunctional reagents.
C bonds is the area that has experienced the most growth in the field of halotrifluoromethylation of unactivated alkenes over the past few years. In this section, we will summarize the available literature on this novel and appealing research arena with emphasize on the mechanistic proposals of the reactions. The section is organized by type of reagent used in two different sub-sections. The first describes chloro-trifluoromethylations using bifunctional reagents, while the second will cover examples that employ monofunctional reagents.
3.1. Bifunctional reagents
3.1.1. CF3SO2Cl. The first mention of the vicinal chlorotrifluoromethylation of unactivated alkenes using a bifunctional reagent can be found in a 1989 paper by Kamigata et al.,18 who showed that the treatment of monosubstituted alkenes 8 with commercially available inexpensive trifluoromethanesulfonyl chloride (CF3SO2Cl) in the presence of catalytic amounts of RuCl2(PPh3)2 in refluxing benzene in a sealed tube afforded the corresponding β-CF3 alkyl chlorides 9 in moderate to high yields along with a sulfur dioxide extrusion (Scheme 5). The reaction is interesting in that both aromatic and aliphatic alkenes were well-tolerated and electronic character of the peripheral substituents on the phenyl ring had no impact on the facility of reaction. This methodology was next efficiently extended to the chloroperfluoroalkylation of alkenes possessing an electron-withdrawing group using perfluoroalkanesulphonyl chlorides as the perfluoroalkyl radical and chloride ion sources.19
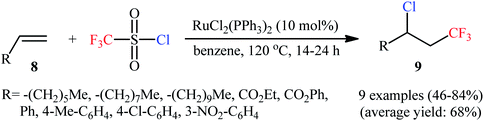 |
| | Scheme 5 Ru-catalyzed chlorotrifluoromethylation of monosubstituted alkenes 8 with CF3SO2Cl. | |
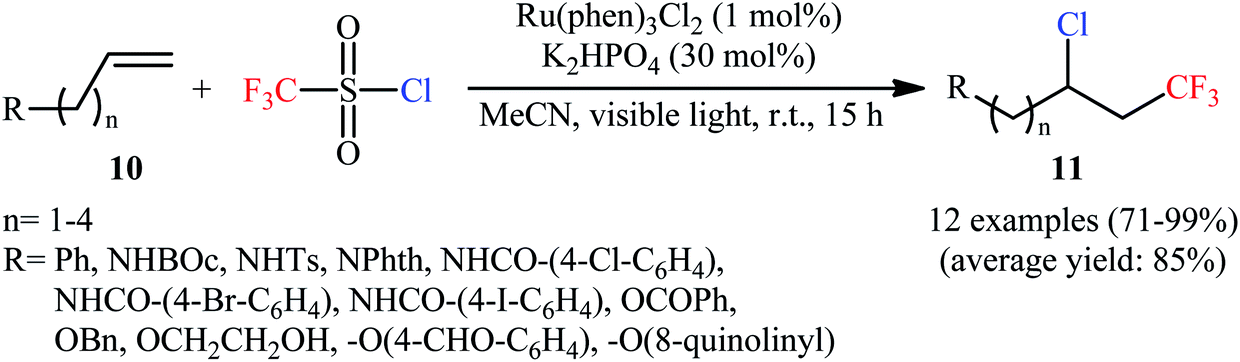
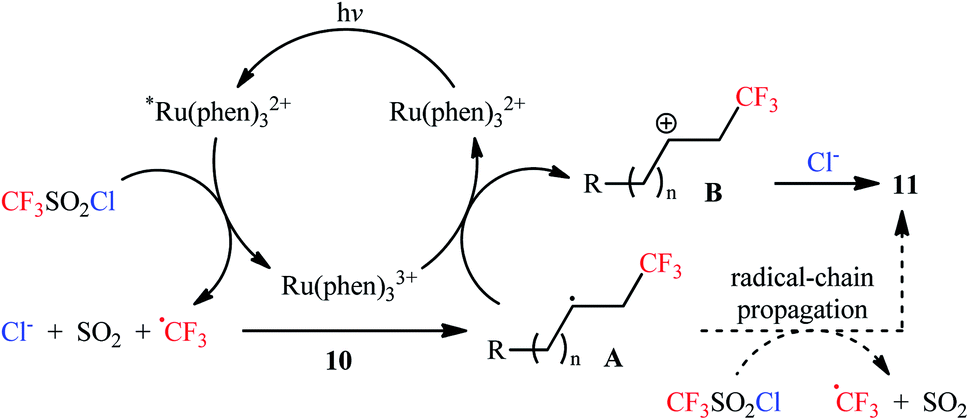
Along this line, Han et al. disclosed that, in the presence of visible light, Ru(Phen)3Cl2, and K2HPO4, monosubstituted aliphatic alkenes 10 reacted with CF3SO2Cl to deliver vicinal chlorotrifluoromethylated products 11 in good to almost quantitative yields and outstanding regioselectivity (Scheme 6).19 However, poor diastereoselectivities were observed in most cases. It is worthy of note that the process can be scaled up to provide multigram quantities of the desired chlorotrifluoromethylated products without sacrificing the yield or outcome of the methodology. Of note, Ru(bpy)3Cl2·6H2O was also found to promote this double functionalization reaction, albeit in lower yields. When the reaction was performed in the absence of either visible light, or photoredox catalyst, or additive, no product was obtained. This observation indicated the a photocatalyst, buffering agent and visible light irradiation are essential for the transformation. It should also be emphasized that this catalytic system was also effective for the chlorotrifluoromethylation of 1,1-disubstituted and internal alkenes. Furthermore, this procedure was successfully applied for difunctionalization of biologically active compounds such as rotenone and (+)-nootkatone. Mechanistic investigations indicated that this interesting transformation proceeds through the following key steps (Scheme 7): (i) photoexcitation of the ground state Ru(Phen)32+ by visible light to generate the excited state *Ru(Phen)32+; (ii) electron transfer from *Ru(Phen)32+ to CF3SO2Cl to produce Ru(Phen)33+ and CF3SO2Cl radical anion, which immediately converts into ˙CF3, SO2, and Cl− though intramolecular collapse; (iii) electrophilic addition of the ˙CF3 to alkene 10 to form the radical intermediate A; (iv) oxidation of intermediate A with Ru(Phen)33+ to produce the carbocation intermediate B and regenerate Ru(Phen)32+; and (v) nucleophilic addition of Cl− to cation B furnishes the final product 11.
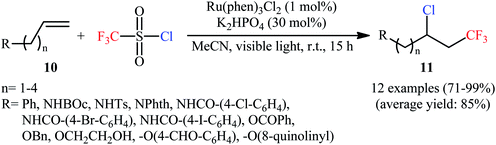 |
| | Scheme 6 Photoredox-catalyzed vicinal chlorotrifluoromethylation of alkenes 10 with CF3SO2Cl. | |
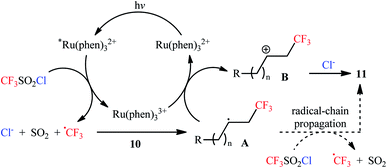 |
| | Scheme 7 The plausible reaction mechanism for the formation of chlorotrifluoromethylated products 11. | |
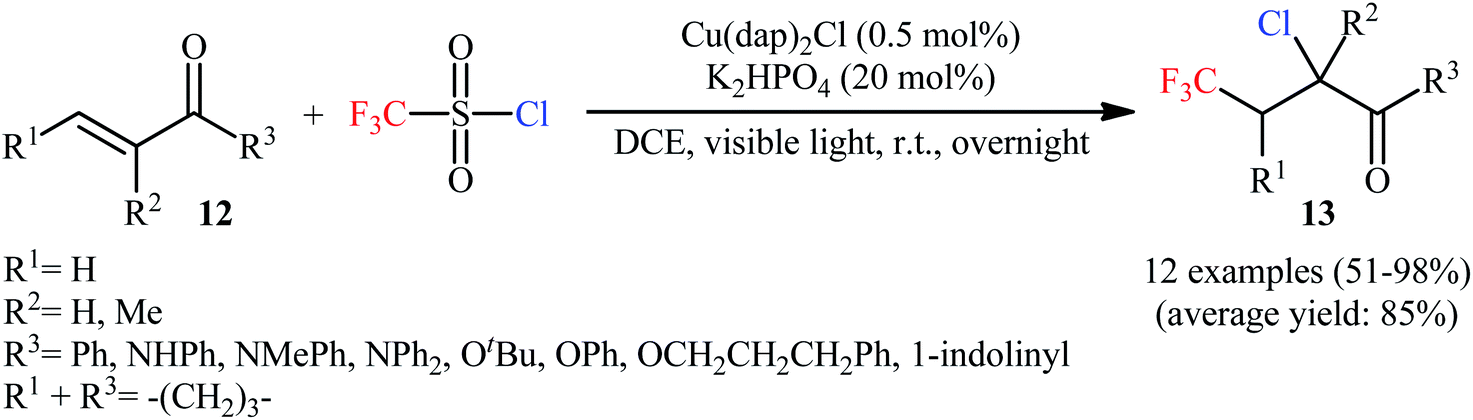
Subsequently, copper-catalyzed version of the titled difunctionalization reaction was developed by Tang and Dolbier.21 Thus, the reaction between electron-deficient unsaturated carbonyl compounds 12 and CF3SO2Cl in the presence of Cu(dap)2Cl/K2HPO4 combination as a catalytic system in DCE under visible light irradiation (r.t., overnight) afforded the corresponding α-chloro-β-trifluoromethylcarbonyl products 13 in moderate to excellent yields (Scheme 8). The reaction could also be conducted successfully on a gram scale (95% yield on a 1-gram scale). It should be mentioned that internal alkenes gave rise to mixtures of regio- and diastereoisomers unless symmetrical substrates. Besides CF3SO2Cl, other fluoroalkylsulfonyl chlorides (e.g., H2CFSO2Cl·HCF2SO2Cl, CF3CH2SO2Cl, and C4F9SO2Cl) have also been successfully used as chlorofluoroalkylating reagents in this transformation. In a related study, the group of Hu also reported an efficient Cu-based photoredox catalyst for the trifluoromethylchlorination of alkenes with CF3SO2Cl.22 The protocol is based on the use of only 0.5 mol% of [Cu(NN′)(Xantphos)][PF6]; (NN′ = 6-methyl-4-(2,4,5-trimethylphenyl)-2,2′-bipyridine) in the presence of over-stoichiometric amounts of K2CO3, in DCE under blue LEDs irradiation at 40 °C. Aryl and heteroaryl substituted terminal alkenes and terminal alkenes with adjacent electron-withdrawing groups (e.g., ester, amide) were compatible with the reaction conditions. However, the reactions of alkyl substituted terminal alkenes led to chlorosulfonation at the 2-position and trifluomethylation at the 1-position. Very recently, Xiao and co-workers improved the efficiency of this transformation in the terms of yield and reaction time by performing the process in the presence of CuCl2 (10 mol%) and pyridine (10 mol%) in 1,4-dioxane; however, elevated reaction temperature (100 °C) were necessary.23 Later, Matsubara's research group identified a novel system based on CoClTPP (TPP: 5,10,15,20-tetraphenylporphyrinato) and CF3SO2Cl as catalyst in MeCN.24 Although the process was efficient and provided high yields (51–100%) of products, the scope remains narrow as only a few terminal alkynes (8 examples) were evaluated.
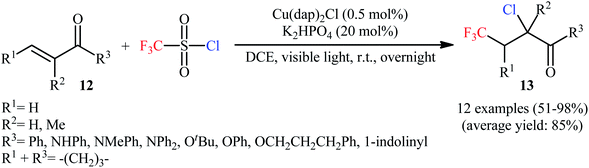 |
| | Scheme 8 Cu-catalyzed trifluoromethylchlorination of unsaturated carbonyl compounds 12 with CF3SO2Cl. | |
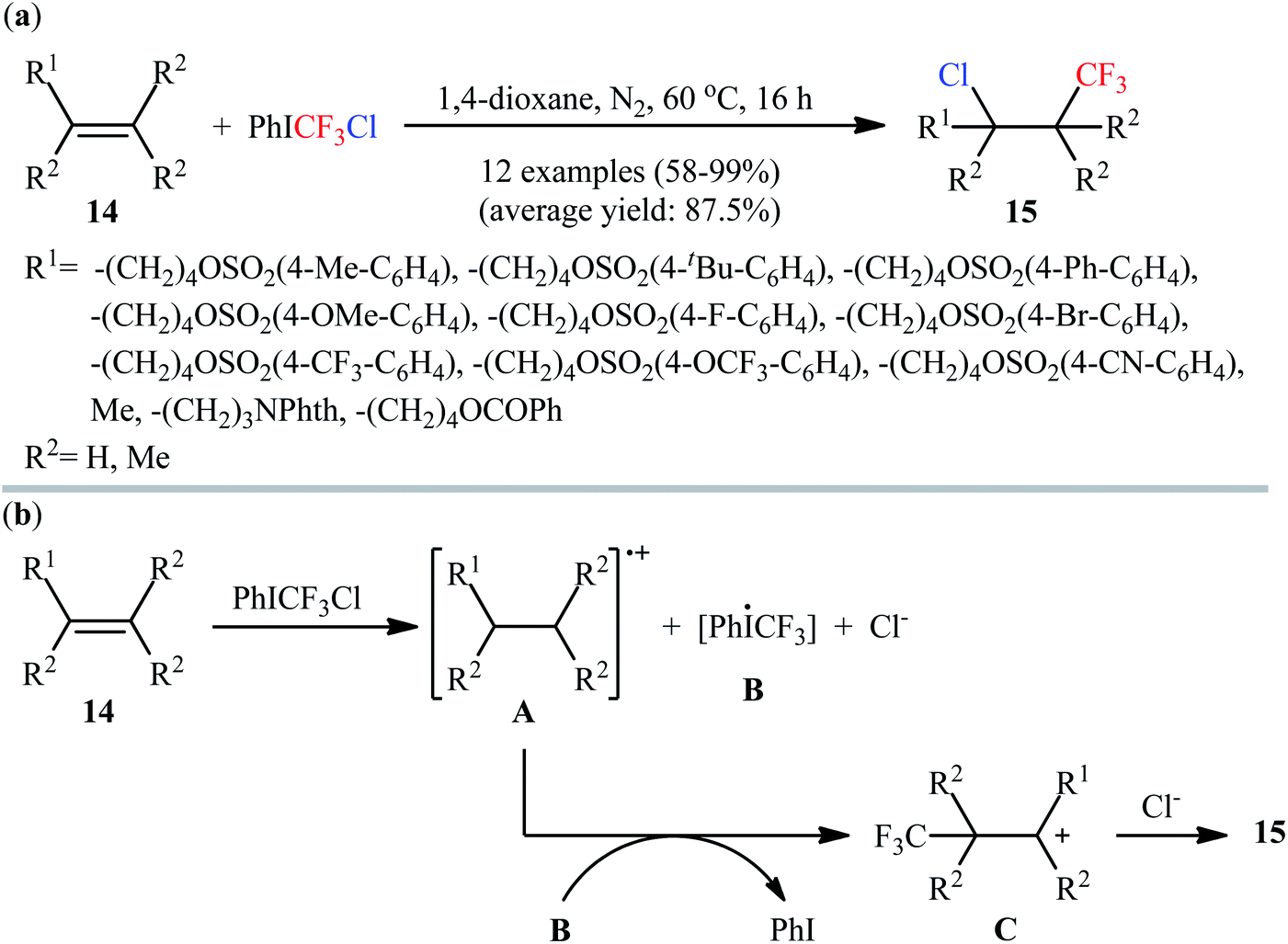
3.1.2. PhICF3Cl. Recently, the group of Wang prepared a super electrophilic trifluoromethylating reagent, chloro(phenyl)trifluoromethyliodane (PhICF3Cl), by simple ligand-exchange reaction between PhI(OCOCF3)2, Me3SiCF3, and NaCl.25 Comparison studies indicated that this CF3-containing λ3-iodane reagent is much more reactive than internally coordinated and neutral Togni's reagents in a wide range of electrophilic trifluoromethylation reactions.26 Interestingly, in the absence of any catalyst or additive in 1,4-dioxane under an inert atmosphere (N2), reaction of nonconjugated alkenes 14 with this reagent furnished the respective chlorotrifluoromethylated products 15 in good to excellent yields (Scheme 9a).27 The procedure was also applied to the high yielding chlorotrifluoromethylation of a series of pharmaceutically active substrates (e.g., oxaprozin, estrone, umbelliferone, ibuprofen, and indomethacin derivatives). However, except a few slightly electron-rich styrene derivatives, conjugated alkenes failed to participate in this reaction. A plausible mechanism is depicted in Scheme 9b. The transformation starts by a single electron transfer reaction between alkene 14 and PhICF3Cl, leading to the formation of radical cation A and hypervalent iodine radical B along with formation of Cl−. Subsequently, coupling of radical cation A with CF3 radical, generated from intermediate B with the concomitant release of PhI, produces carbocation C. Finally, nucleophilic attack of Cl− to carbocation C affords the observed chlorotrifluoromethylated products 15.
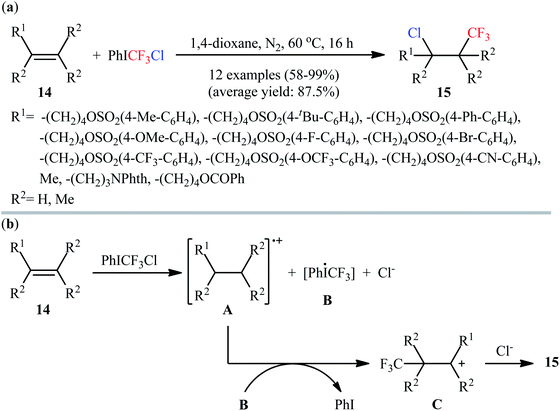 |
| | Scheme 9 (a) Catalyst-free chlorotrifluoromethylation of alkenes 14 with PhICF3Cl; (b) mechanistic proposal for the reaction in Scheme 8a. | |
3.2. Monofunctional reagents
In 2016, Qing and colleagues developed an interesting protocol for chlorotrifluoromethylation of olefinic double bonds through an appropriate combination of a trifluoromethylating agent and a metal chloride.28 They showed that treatment of various aromatic and aliphatic alkenes 16 with Langlois reagent (CF3SO2Na) and FeCl3 in the presence of K2S2O8 as an oxidant gave the corresponding chlorotrifluoromethylated products 17 in good to excellent yields within 10 h (Scheme 10). In this transformation, FeCl3 playing a dual role; the chlorine source and the mediator. Noteworthy, a diverse set of versatile functional groups including cyano, nitro, chloro, bromo, ester, ether, acid, and ketone functionalities were compatible by the reaction conditions employed and optically pure substrates preserved their optical purity. An improved access to vicinal chlorotrifluoromethylated alkanes under milder and transition-metal-free conditions has subsequently been described by Liu and co-workers, using N-chlorophthalimide as chlorine source and a catalytic amount of N-methyl-9-mesityl acridinium at room temperature.29
 |
| | Scheme 10 Iron-mediated chlorotrifluoromethylation of alkenes 16 with Langlois reagent. | |
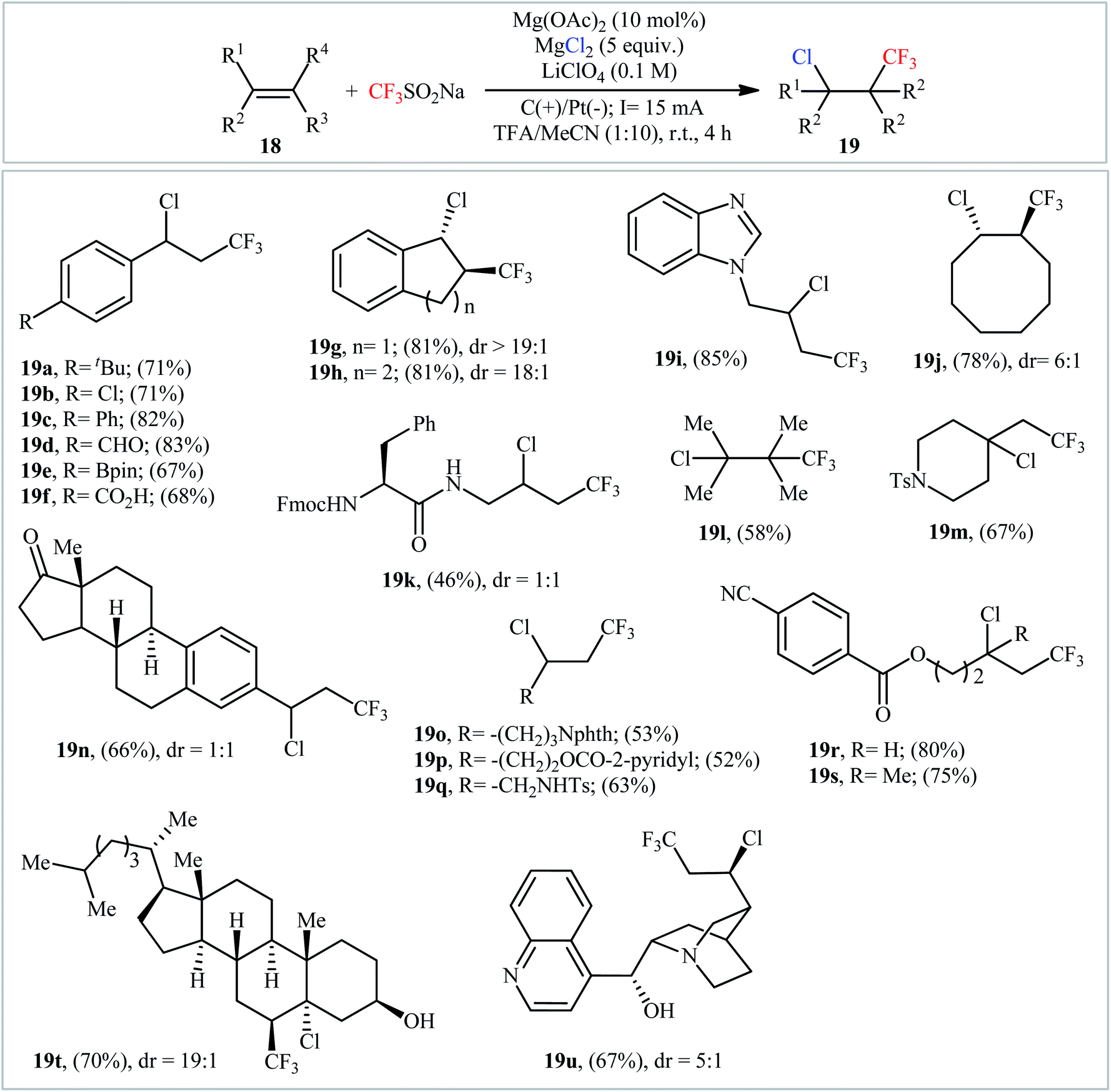
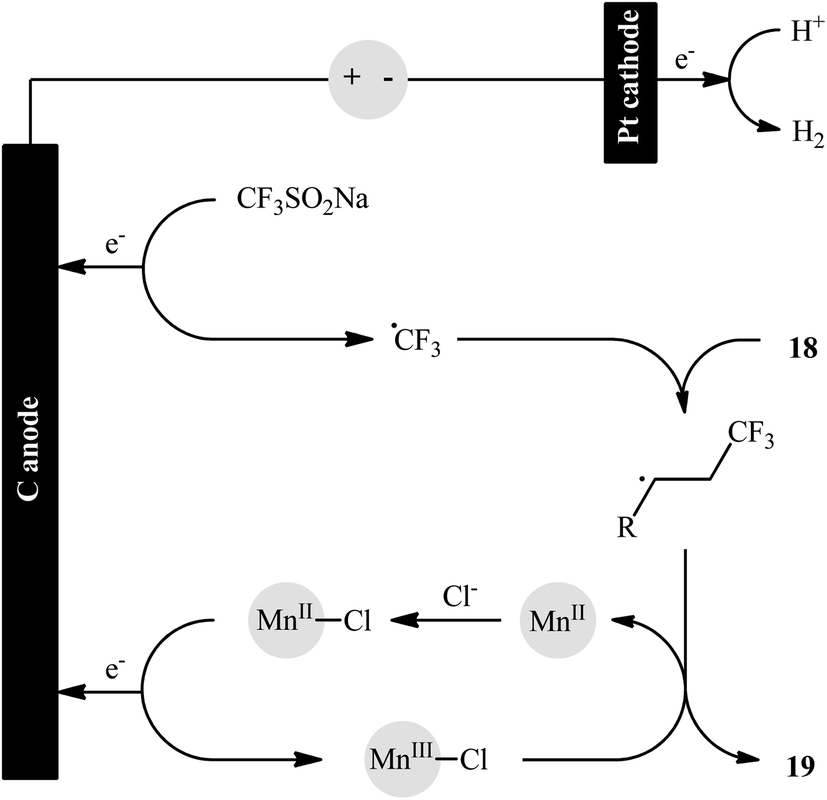
In an innovative study, the Lin laboratory demonstrated regioselective chlorotrifluoromethylation of alkenes 18 with CF3SO2Na and MgCl2 in an undivided carbon/Pt-cell under constant current conditions (15 mA).30 The optimal conditions consisted in using Mn(OAc)2 as the catalyst and LiClO4 as the electrolyte in the binary solvent CF3CO2H/MeCN with ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 at room temperature. The established electrochemical strategy efficiently provided the target chlorotrifluoromethylated products 19 in moderate to good yields within 4 h (Scheme 11). The mechanism suggested by the authors for this electrocatalytic chlorotrifluoromethylation is shown in Scheme 12.
:10 at room temperature. The established electrochemical strategy efficiently provided the target chlorotrifluoromethylated products 19 in moderate to good yields within 4 h (Scheme 11). The mechanism suggested by the authors for this electrocatalytic chlorotrifluoromethylation is shown in Scheme 12.
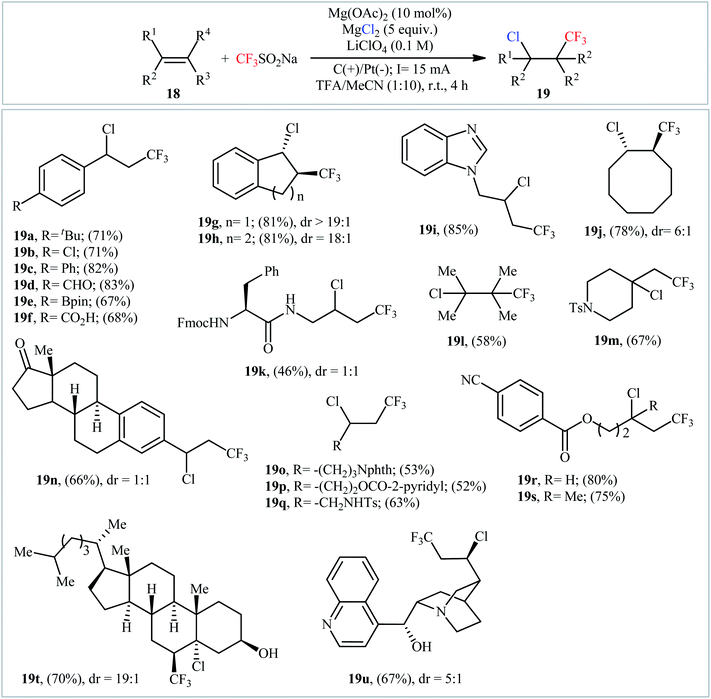 |
| | Scheme 11 Electrocatalytic chlorotrifluoromethylation of alkenes 18 with CF3SO2Na and MgCl2 developed by Lin. | |
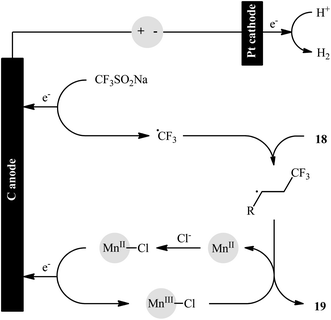 |
| | Scheme 12 Proposed mechanistic pathway for the formation of vicinal chlorotrifluoromethylated alkanes 19. | |
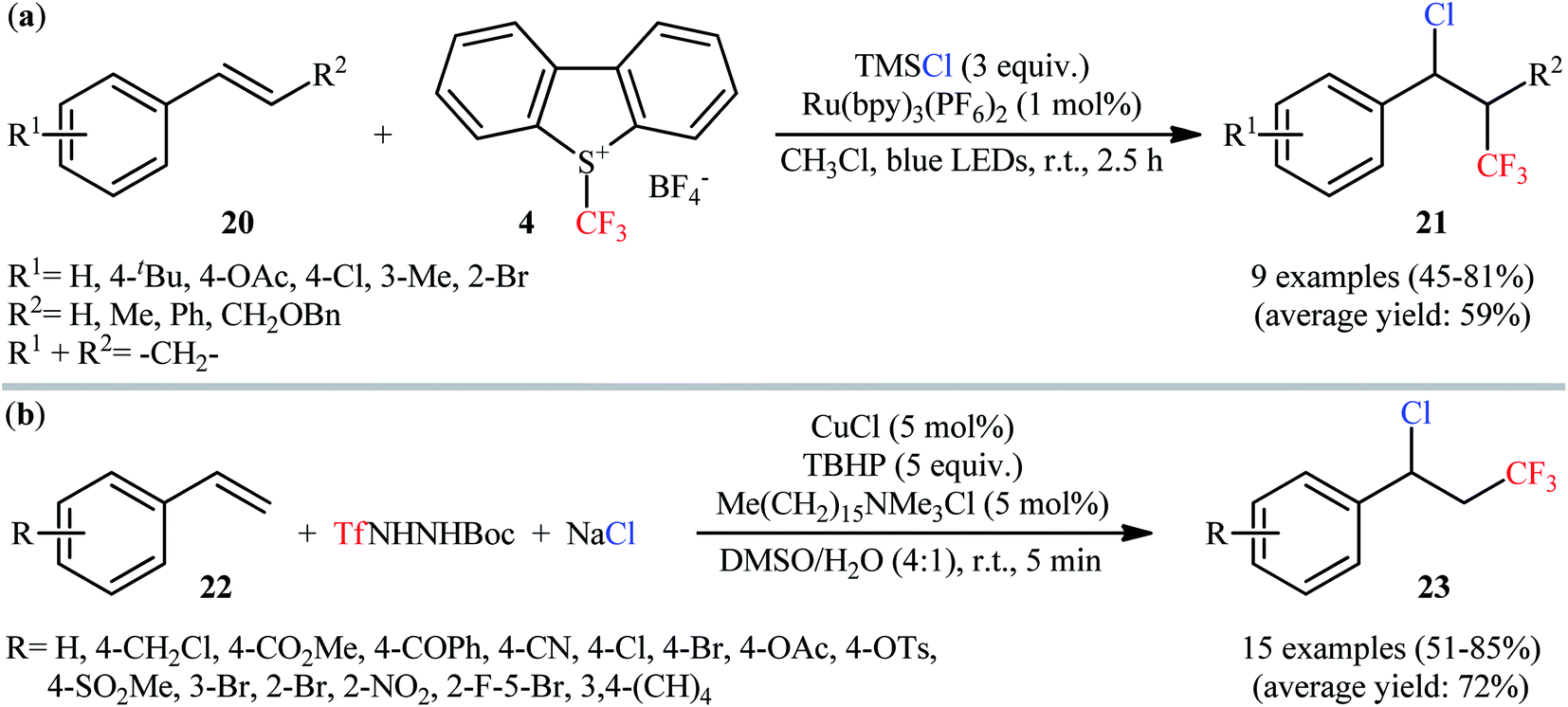
In this context, Masson and co-workers have described a three-component (1-chloro-3,3,3-trifluoropropyl)benzenes 21 synthesis by reaction between styrene derivatives 20, Umemoto's reagent 4, and TMSCl in the presence Ru(bpy)3(PF6)2 as the photocatalyst under visible light irradiation.31 This procedure efficiently provided the desired vicinal chlorotrifluoromethylated products in moderate to good yields at room temperature and without consuming any oxidant or additive (Scheme 13a). However, dibenzo[b,d]thiophene by-product generated from Umemoto's reagent was difficult to separate from the end products, especially when nonpolar styrenes were used as starting materials. The authors elegantly solved this problem by adding m-CPBA to the reaction mixture at the end of the reaction. In a closely related investigation, the group of Tian reported the use of TfNHNHBoc as the source of CF3 and NaCl as the source of Cl in this process.32 Thus, by employing CuCl as a redox catalyst, Me(CH2)15NMe3Cl as a phase-transfer catalyst, and TBHP as an oxidant in a 4:1 mixture of DMSO and H2O, the electron-poor styrenes 22 quickly afforded the respective (1-chloro-3,3,3-trifluoropropyl)benzene derivatives 23 in high yields (Scheme 13b). However, arylalkenes having strong electron-donating groups, alkylalkenes, electron-deficient terminal alkenes, and 1,2-disubstituted alkenes failed to enter into this reaction. Interestingly, by simply replacing NaCl and CuCl with NaBr and CuBr, respectively, the optimized reaction conditions were successfully applied to the vicinal bromotrifluoromethylation of an arylalkene. However, similar modification of the reaction conditions failed to execute the corresponding vicinal iodotrifluoromethylation of arylalkenes.
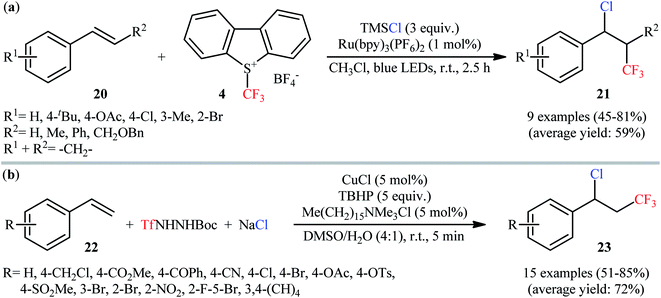 |
| | Scheme 13 (a) Photoredox-mediated synthesis of (1-chloro-3,3,3-trifluoropropyl)benzenes 21 by reaction between styrene derivatives 20, Umemoto's reagent 4, and TMSCl; (b) Cu-catalyzed synthesis of β-CF3 benzyl chlorides 23 via a three-component reaction between styrenes 22, TfNHNHBoc, and NaCl. | |
4. Bromotrifluoromethylation
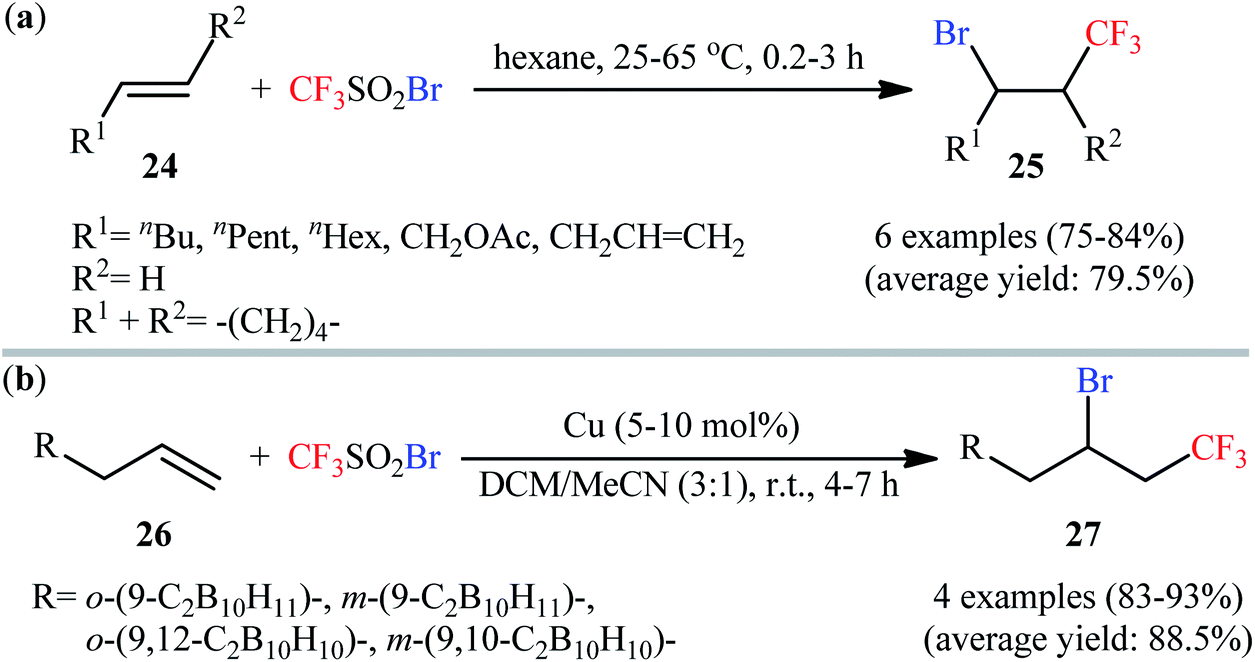
One of the earliest studies of the direct bromotrifluoromethylation of unactivated alkenes has been reported by Huang and Lü in 1992, when aliphatic alkenes underwent regioselective bromotrifluoromethylation with commercially available trifluoromethanesulfonyl bromide (CF3SO2Br) by simple heating in hexane.33 The reaction was conducted in the absence of any catalyst or additive, which tolerated both terminal and internal alkenes 24, affording the 1,2-difunctionalized products 25 in good to high yields (Scheme 14a). Furthermore, styrene also worked well in this reaction, albeit required a higher reaction temperature and a peroxide initiator. Beside alkenes, alkynes were also compatible with this halotrifluoromethylation reaction. Twenty-seven years later, this strategy associated with copper(0) catalysis applied by Ol'shevskaya and co-workers in the functionalization of allylcarboranes 26 and achieved the respective bromotrifluoromethylated carboranes 27 in high to excellent yields (Scheme 14b).34
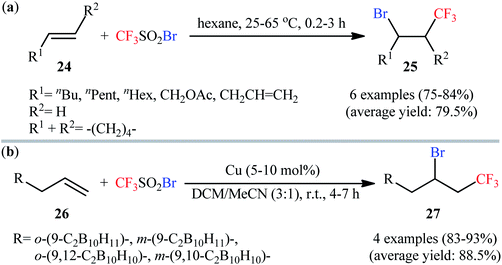 |
| | Scheme 14 (a) Huang's synthesis of β-CF3 alkyl bromides 25; (b) Cu-catalyzed bromotrifluoromethylation of allylcarboranes 27 with CF3SO2Br. | |
In 2015, Jung and Han demonstrated an efficient copper-mediated bromotrifluoromethylation of monosubstituted alkenes 28 using Umemoto's reagent 4 and CuBr, where CuBr acts as the source for both copper and bromine.35 This reaction employed bis(pinacolato)diboron (B2pin2) and K2HPO4 as additives and MeCN as the solvent, leading to the production of various 1-CF3-2-Br-alkanes 29 in good to excellent yields (Scheme 15a). 1,1-Disubstituted and internal alkenes were also effective in this reaction. However, the diastereoselectivities were poor in all cases tested. Noteworthy, by changing of CuBr with CuCl and CuI, the optimized reaction conditions were successfully applied to the chloro- and iodo-trifluoromethylation of the same set of alkenes. Concurrently, Masson found that CsBr could also be used as the source of bromine in the above-mentioned transformation.31 Thus, mediated by a catalytic amount of Ru(bpy)3(PF6)2 under constant irradiation by a blue LED, the reaction of a small series of terminal alkenes 30 with Umemoto's reagent 4 and CsBr took place readily, leading to the formation of corresponding bromotrifluoromethylated products 31 in moderate to good yields (Scheme 15b). The authors proposed mechanism for this reaction is analogous to the one depicted for Ru-catalyzed chlorotrifluoromethylation of alkenes with CF3SO2Cl (Scheme 7).
 |
| | Scheme 15 (a) Cu-mediated bromotrifluoromethylation of terminal alkenes 28 developed by Jung and Han; (b) photoredox-mediated bromotrifluoromethylation of unactivated alkenes 30 with Umemoto's reagent and CsBr. | |
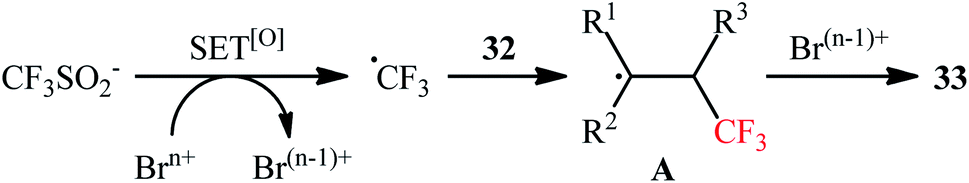
In 2017, Liu's research team developed an elegant metal-free method for the bromotrifluoromethylation of alkenes 32 on the use of NaSO2CF3 in the presence of more than stoichiometric amounts of sodium bromate (NaBrO3).36 The reactions were performed in DCM/H2O (4:1) at 110 °C in a sealed tube, tolerated various sensitive functional groups like OH, CHO, Cl, Br, NO2, SONR2, OSO2R, ester, ether, and ketone, and afforded the corresponding β-CF3 alkyl bromates 33 in fair to good yields (Scheme 16). Some important information of this synthetic procedure are listed below: (i) both terminal and nonterminal alkenes are compatible with this approach; (ii) optically pure substrates could be used without detected racemization; (iii) NaBrO3 acts not only as a bromine source, but also as an oxidant; and (iv) the process can be scaled up to provide multi-gram quantities of the target bromotrifluoromethylated products without sacrificing the yield and difficulty. The reaction mechanism involved single-electron oxidation of NaSO2CF3 by NaBrO3 to generate the ˙CF3 radical, which was then attacked to the double bond to furnish radical intermediate A. Finally, this intermediate underwent bromination to yield the observed product (Scheme 17). The exact bromination step remains not yet completely clear. One possible pathway is bromine atom abstraction by radical intermediate A from Br2, which is observed in the system. However, another process involving one-electron oxidation of this radical to give a carbocation followed by Br anion addition.
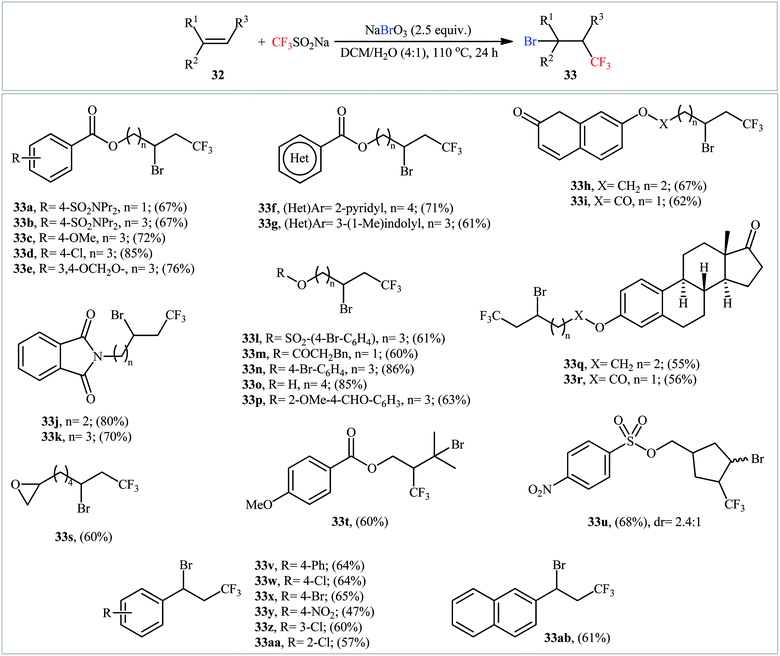 |
| | Scheme 16 Catalyst-free bromotrifluoromethylation of alkenes 32 by using NaSO2CF3 and NaBrO3. | |
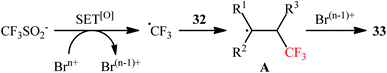 |
| | Scheme 17 Plausible mechanism for the reaction in Scheme 16. | |
5. Iodotrifluoromethylation
This section is divided into two parts according to iodotrifluoromethylating reagents. The first includes the iodotrifluoromethylation of alkenes using bifunctional reagents, while the second contains the three-component reactions.
5.1. Bifunctional reagents
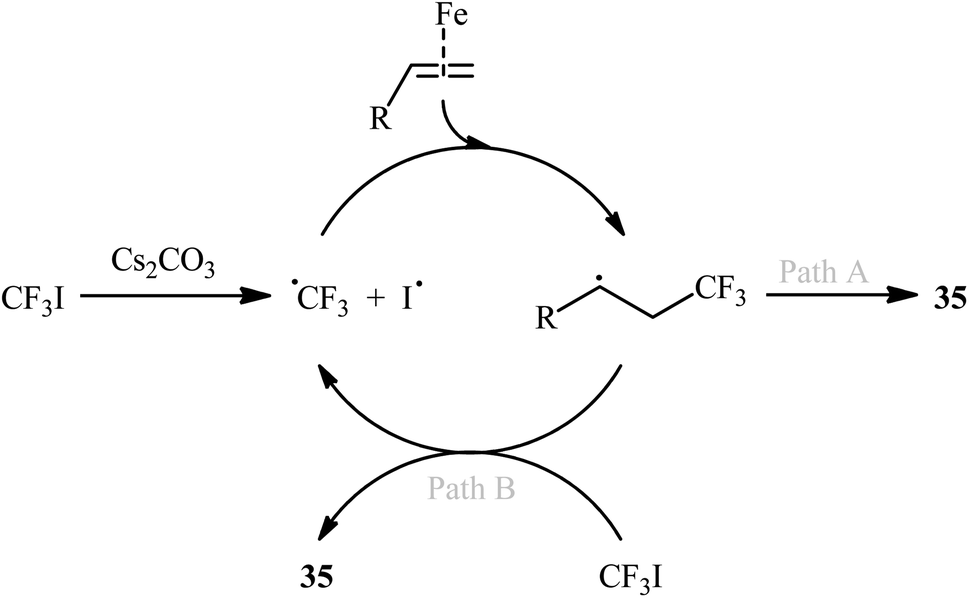
In 2004, Ogawa's research team reported a mild and fast photoinduced vicinal iodotrifluoromethylation of cyclohexylallene with trifluoroiodomethane (CF3I) upon irradiation with a xenon lamp under catalyst- and solvent-free conditions.37 Although only one poor yield example was provided, this paper represents the first example of the direct iodotrifluoromethylation of alkene CC double bonds. Three years later, Ignatowska and Dmowski provided two further examples of β-CF3 alkyl iodide preparation promoted by Na2S2O4/Na3PO4 in binary solvent MeCN/H2O with ratio 1:1.38 Concurrently, Yajima and Nagano described an interesting regioselective iodoperfluoroalkylation of acrylic acid derivatives bearing a chiral auxiliary with corresponding perfluoroalkyl iodides in the presence of an aqueous solution of Na2S2O3 under photoirradiation with a Hg lamp in a Pyrex tube.39 However, in this preliminary work, only one example of the iodotrifluoromethylation with CF3I was provided, without any substrate scope exploration. In 2014, with the objective of designing a more general protocol to β-CF3 alkyl iodides through iodotrifluoromethylation of the respective alkenes with CF3I, Hu and co-workers were able to showed that a small library of 1-CF3-2-I-alkanes 35 could be obtained in good to excellent yields from the reaction of terminal alkenes 34 with CF3I in 1,4-dioxane employing FeBr2/Cs2CO3 combination as the catalytic system (Scheme 18).40 Other perfluoroalkyl iodides, such as IC4F9, IC6F13, IC10H21, ICF2CO2Et, and IC6F12Cl were also compatible with these conditions, and furnished the corresponding iodoperfluoroalkylated products in high yields. Furthermore, this process also allowed the synthesis of diverse iodoperfluoroalkylated alkenes through the 1,2-addition of perfluoroalkyl iodides to the carbon–carbon triple bond of the corresponding alkynes. The putative mechanism for this alkene difunctionalization reaction is depicted in Scheme 19. Along this line, Reiser and co-workers have published the direct iodotrifluoromethylation of styrene with CF3I leading to (3,3,3-trifluoro-1-iodopropyl)benzene in yield of 45% via the combination of [Cu(dap)2]Cl catalyst and green LED irradiation.41
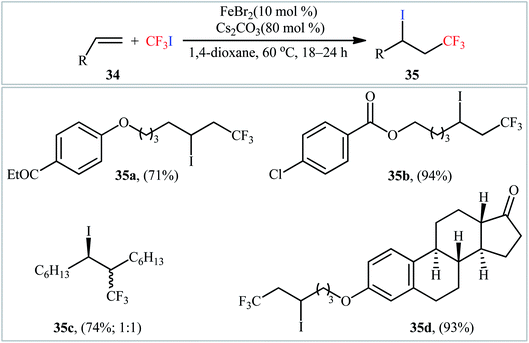 |
| | Scheme 18 Hu's synthesis of β-CF3 alkyl iodides 35. | |
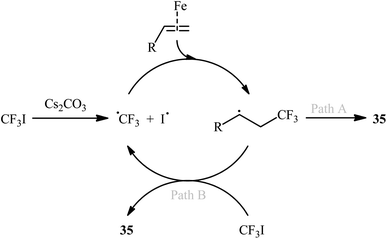 |
| | Scheme 19 Proposed reaction pathway for the formation of β-CF3 alkyl iodides 35. | |
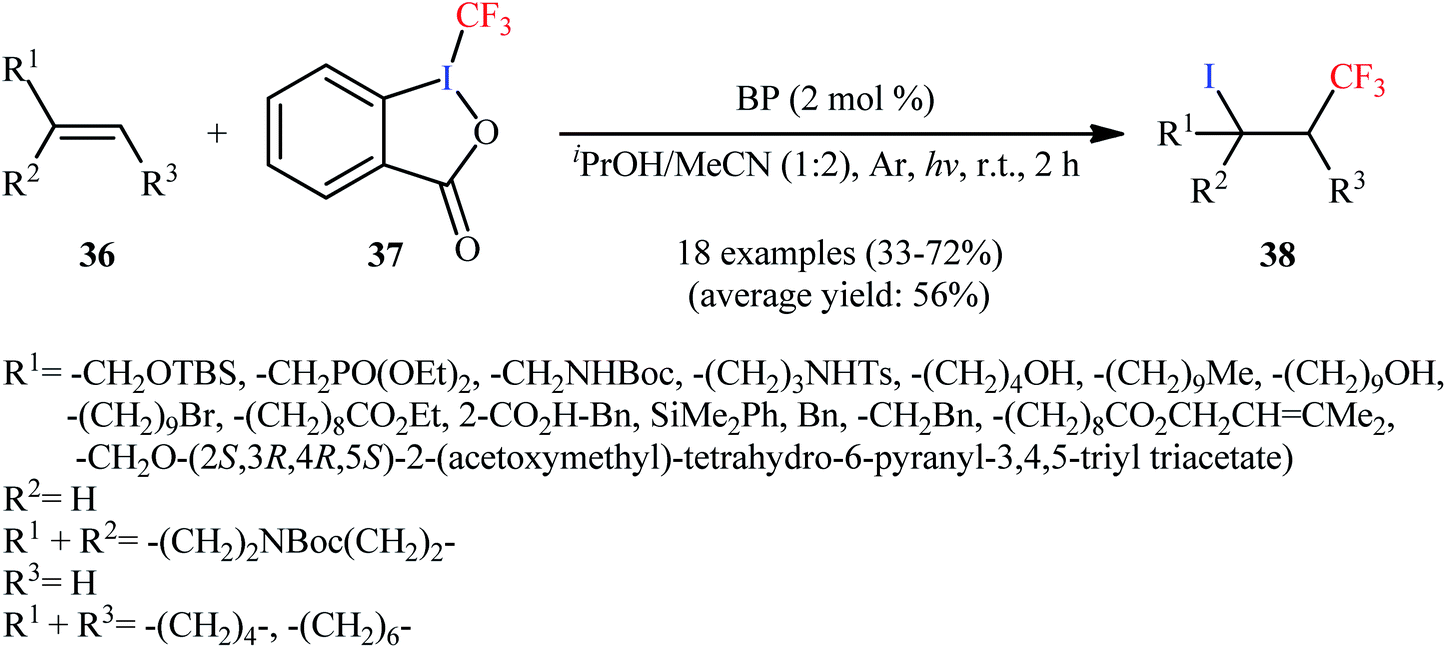
An improved version of this reaction was disclosed in 2017 by Vincent and colleagues, who developed a BP-sensitized (BP = benzophenone) light-promoted synthesis of β-CF3 alkyl iodides under mild conditions.42 A number of terminal and internal aliphatic alkenes 36 were rapidly converted to the corresponding iodotrifluoromethylated products 38 using 2 mol% of BP, employing Togni's reagent 37 as an effective iodotrifluoromethylating agent in a 1:2 mixture of iPrOH and MeCN (Scheme 20). Mechanistic studies suggested that this reaction proceeds via generation of isopropyl alcohol radical [MeC·(OH)Me] from the reaction of BP triplet state with iPrOH. Next, this radical reduces Togni's reagent to generate 2-iodobenzoic acid, acetone, and ˙CF3 radical. Subsequently, electrophilic addition of the latter to alkene forms the corresponding α-CF3 alkyl radical, which after abstraction a I˙ radical from 2-iodobenzoic acid affords the final product.
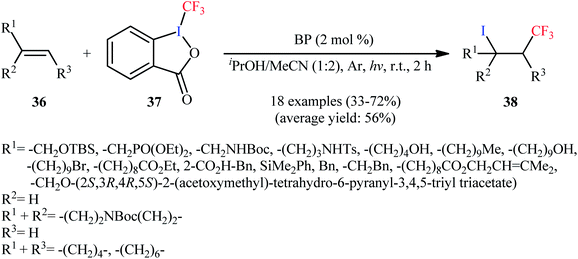 |
| | Scheme 20 Light-mediated iodotrifluoromethylation of alkenes 36 employing Togni's reagent 37 as source of CF3 and iodine. | |
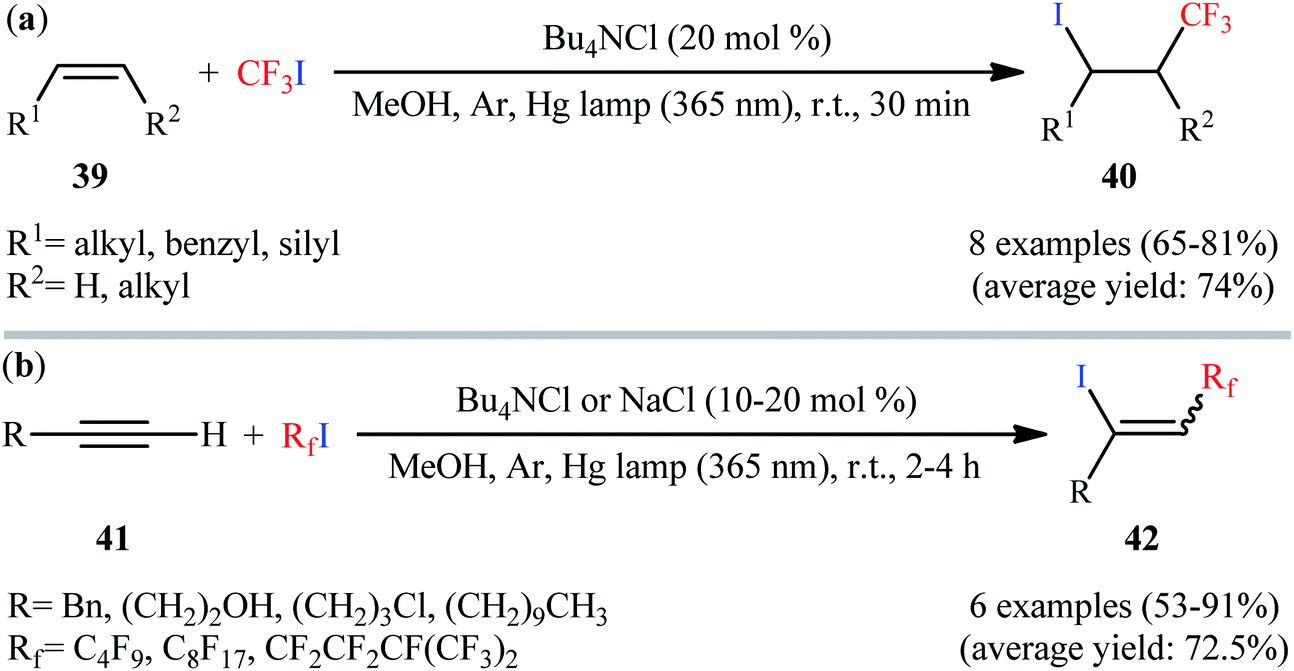
Very recently, the same group reported the direct iodotrifluoromethylation of alkenes using CF3I with the aid of 20 mol% of Bu4NCl under low intensity UVA irradiation (6 W) at room temperature (Scheme 21a).43 Various terminal and internal alkenes 39 were used to establish the general applicability of the procedure. Other than CF3I; IC4F9, IC5F11, and IC8F17 have also been used in this methodology. This procedure was also applicable to aliphatic terminal alkynes 41, giving the desired 1-perfluoroalkyl-2-iodoalkenes 42 in good yields and low to moderate stereoselectivity for the (E)-isomers (Scheme 21b). Noteworthy, NaCl was also found to effectively promote this iodoperfluoroalkylation reaction.
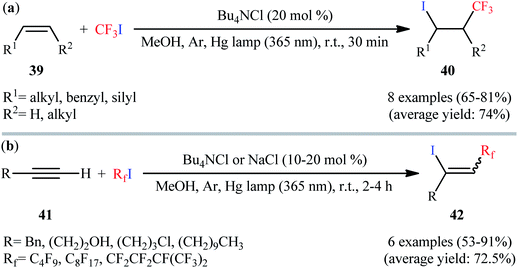 |
| | Scheme 21 (a) Light-mediated iodotrifluoromethylation of alkenes 39 with CF3I catalyzed by Bu4NCl; (b) light-mediated chloride ions-catalyzed iodoperfluoroalkylations of alkynes 41. | |
5.2. Monofunctional reagents
In 2014, Liu and co-workers disclosed that the iodotrifluoromethylation of various aromatic and aliphatic alkenes 43 was feasible with the employment of two simple solids, NaSO2CF3/I2O5, in aqueous medium.44 The reactions were performed in the absence of any catalyst or additive at 110 °C, tolerated a number of important functional groups (e.g., fluoro, chloro, bromo, nitro, hydroxyl, sulfonate, sulfamide, ketone, ester, ether), and provided the corresponding β-CF3 alkyl iodides 44 in moderate to excellent yields, ranging from 43% to 90% (Scheme 22). This facile synthetic procedure was also easily scaled up to the gram-level without harming the yield or outcome of the reaction. However, just like previous works, in the cases of internal alkenes, diastereoselectivity was modest at best. Mechanistic studies by electron spin resonance (ESR) spectroscopy suggested that the free-radical process might be involved in this reaction.
 |
| | Scheme 22 Iodotrifluoromethylation of alkenes 43 with NaSO2CF3 and I2O5 developed by Liu. | |
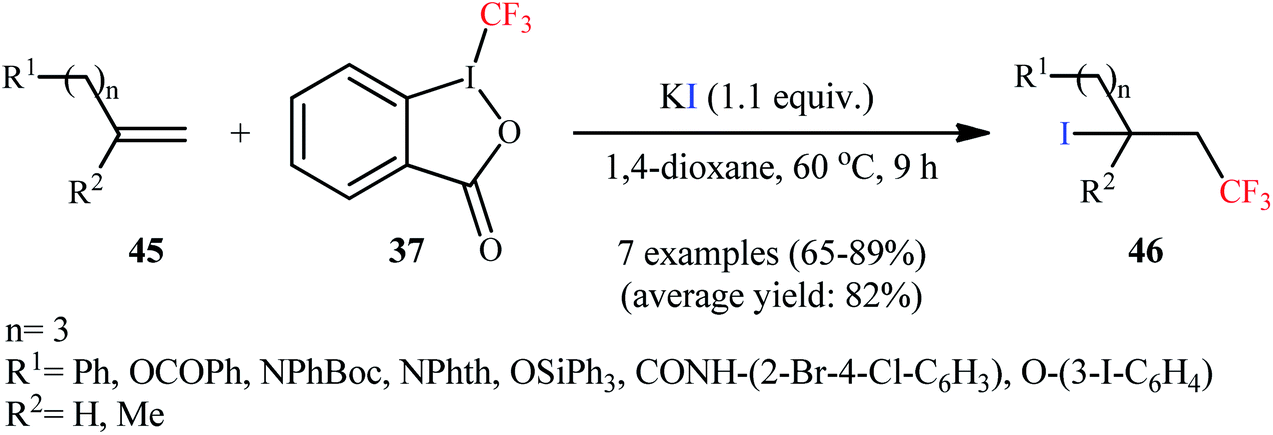
Subsequently, the Sodeoka group reported an efficient iodotrifluoromethylation of various of 1,1-disubstituted and monosubstituted alkenes 45 to afford the desired β-CF3 alkyl iodides 46 in good to high yields (65–89%), employing Togni reagent as CF3 source and KI as iodine source (Scheme 23).45 NaI and CsI were also found to be effective iodine source, and I2 and TBAI proved to be completely ineffective.
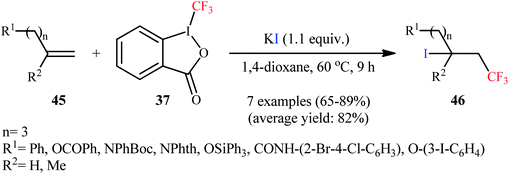 |
| | Scheme 23 Iodotrifluoromethylation of alkenes 45 using Togni's reagent and KI. | |
Finally, it should be noted that transition metal complexes play important role in halo-trifluoromethylation of alkenes and other compounds.46–65
6. Conclusion
Difunctionalizing trifluoromethylation of alkenes represents an extremely attractive, step- and atom-economical route to vicinal introduction of CF3 and another functional group across a carbon–carbon double bond via a single operation and in a selective fashion. Among these trifluoromethylation-involved 1,2-difunctionalization reactions, halotrifluoromethylation of unactivated alkenes, has attracted much attention because of versatility of the resulting alkyl halides as intermediates in more complex target syntheses. As illustrated, over the past few years, several interesting halotrifluoromethylation reactions of alkenes were designed that allow the high yielding construction of various β-CF3 alkyl fluorides, chlorides, bromides, and iodides (Table 1). Interestingly, most of these reactions were performed under catalyst-free conditions without consuming any additional chemical. Despite the extraordinary accomplishments during the last few years in this exciting research topic, several challenges still remain to be overcome: (i) the substrate scope, especially in fluoro-trifluoromethylations, is narrow and thus, expanding of the scope of these reactions are necessary; (ii) although regioselectivity of these reactions was generally high, the diastereoselectivities were poor and usually mixtures of isomers were achieved. Therefore, there is a further need for development of novel catalytic systems, which can allow for effective stereocontrolled halotrifluoromethylations for internal alkenes; and (iii) synthetic applicability of these reactions in the synthesis of natural products and biologically important compounds should be investigated.
Table 1 Comparison of results of direct halo-trifluoromethylation of alkenes
| Entry |
Halo-trifluoromethylating agent(s) |
Conditions |
Number of examples |
Yield (%) |
Ref. |
| Range |
Average |
| 1 |
TMSCF3, Selectfluor |
CsF, PhI(OAc)2, AgTf, DMF |
15 |
31–73 |
55 |
15 |
| 2 |
Umemoto's reagent, CsF |
Cu(OTf)2, BC, L3, MeCN |
16 |
45–84 |
70.5 |
16 |
| 3 |
PPFR |
Catalyst-free, 1,2-DCE |
15 |
15–66 |
46 |
17 |
| 4 |
CF3SO2Cl |
RuCl2(PPh3)2, benzene |
9 |
46–84 |
68 |
18 |
| 5 |
CF3SO2Cl |
Ru(Phen)3Cl2, K2HPO4, MeCN |
12 |
71–99 |
85 |
20 |
| 6 |
CF3SO2Cl |
Cu(dap)2Cl, K2HPO4, DCE |
12 |
51–98 |
85 |
21 |
| 7 |
CF3SO2Cl |
[Cu(NN′)(Xantphos)][PF6], K2CO3, DCM |
34 |
34–98 |
73.5 |
22 |
| 8 |
CF3SO2Cl |
CuCl2, pyridine, 1,4-dioxane |
26 |
33–93 |
68 |
23 |
| 9 |
CF3SO2Cl |
CoCITPP |
8 |
51–100 |
68 |
24 |
| 10 |
PhICF3Cl |
Catalyst-free, 1,4-dioxane |
12 |
58–99 |
87.5 |
27 |
| 11 |
CF3SO2Na, FeCl3 |
K2S2O8, MeCN |
17 |
51–93 |
77 |
28 |
| 12 |
CF3SO2Na, NCP |
Mes-Acr+, TFA, DCE |
19 |
41–88 |
67 |
29 |
| 13 |
CF3SO2Na, MgCl2 |
Mg(OAc)2, LiClO4, C(+)/Pr(−), THF, MeCN |
22 |
46–85 |
66 |
30 |
| 14 |
Umemoto's reagent, TMSCl |
Ru(bpy)3(PF6)2, CH3Cl |
9 |
45–81 |
59 |
31 |
| 15 |
TfNHNHBoc, NaCl |
CuCl, TBHP, Me(CH2)15NMe3Cl, DMSO, H2O |
15 |
51–85 |
72 |
32 |
| 16 |
CF3SO2Br |
Catalyst-free, hexane |
6 |
75–84 |
79.5 |
33 |
| 17 |
CF3SO2Br |
Cu, DCM, MeCN |
4 |
83–93 |
88.5 |
34 |
| 18 |
Umemoto's reagent, CuBr |
B2pin2, K2HPO4, MeCN |
10 |
70–93 |
81 |
35 |
| 19 |
Umemoto's reagent, CsBr |
Ru(bpy)3(PF6)2, MeCN |
6 |
42–77 |
59 |
31 |
| 20 |
CF3SO2Na, NaBrO3 |
Catalyst-free, DCM, H2O |
28 |
47–86 |
67.5 |
36 |
| 21 |
CF3I |
Catalyst-free, BTF |
1 |
81 |
81 |
37 |
| 22 |
CF3I |
Na2S2O4, Na3PO4, MeCN, H2O |
2 |
57–78 |
67.5 |
38 |
| 23 |
CF3I |
Na2S2O8, H2O |
1 |
63 |
63 |
39 |
| 24 |
CF3I |
FeBr2, Cs2CO3, 1,4-dioxane |
4 |
71–94 |
83 |
40 |
| 25 |
CF3I |
[Cu(dap)2]Cl, MeCN |
1 |
45 |
45 |
41 |
| 26 |
Togni's reagent |
BP, iPrOH, MeCN |
18 |
33–72 |
56 |
42 |
| 27 |
CF3I |
Bu4NCl, MeOH |
8 |
65–81 |
74 |
43 |
| 28 |
CF3SO2Na, I2O5 |
Catalyst-free, DCM, H2O |
40 |
43–90 |
70.5 |
44 |
| 29 |
Togni's reagent, KI |
Catalyst-free, 1,4-dioxane |
7 |
65–89 |
82 |
45 |
Conflicts of interest
There are no conflicts to declare.
References
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
-
(a) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS;
(b) S. Shafiei and S. Davaran, Chem. Rev. Lett., 2020, 3, 19–22 Search PubMed;
(c) S. Majedi and S. Majedi, J. Chem. Lett., 2020, 1, 2–8 Search PubMed;
(d) F. Nareetsile, J. T. P. Matshwele, S. Ndlovu and M. Ngaski, Chem. Rev. Lett., 2020, 3, 140–160 Search PubMed.
- V. Gouverneur and K. Seppelt, Chem. Rev., 2015, 115, 563–565 CrossRef CAS PubMed.
-
(a) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160–171 CrossRef CAS PubMed;
(b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- J. Sun, X. Zhen, H. Ge, G. Zhang, X. An and Y. Du, Beilstein J. Org. Chem., 2018, 14, 1452–1458 CrossRef CAS PubMed , and references therein..
-
(a) B. R. Langlois, T. Billard and S. Roussel, J. Fluorine Chem., 2005, 126, 173–179 CrossRef CAS;
(b) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 117, 218–234 CrossRef;
(c) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8954 CrossRef CAS PubMed.
- B. Gál, C. Bucher and N. Z. Burns, Mar. Drugs, 2016, 14, 206 CrossRef PubMed.
-
(a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed;
(b) A. D. Dilman and V. V. Levin, Eur. J. Org. Chem., 2011, 831–841 CrossRef CAS;
(c) P. Chen and G. Liu, Synthesis, 2013, 45, 2919–2939 CrossRef CAS;
(d) X. F. Wu, H. Neumann and M. Beller, Chem.–Asian J., 2012, 7, 1744–1754 CrossRef CAS PubMed;
(e) H. Liu, Z. Gu and X. Jiang, Adv. Synth. Catal., 2013, 355, 617–626 CrossRef CAS;
(f) C. Zhang, Adv. Synth. Catal., 2014, 356, 2895–2906 CrossRef CAS;
(g) C. Zhang, Org. Biomol. Chem., 2014, 12, 6580–6589 RSC;
(h) W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS;
(i) T. Koike and M. Akita, J. Fluorine Chem., 2014, 167, 30–36 CrossRef CAS;
(j) T. Koike and M. Akita, Top. Catal., 2014, 57, 967–974 CrossRef CAS;
(k) V. Bizet, T. Besset, J. A. Ma and D. Cahard, Curr. Top. Med. Chem., 2014, 14, 901–940 CrossRef CAS PubMed;
(l) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC;
(m) P. Gao, X. R. Song, X. Y. Liu and Y. M. Liang, Chem.–Eur. J., 2015, 21, 7648–7661 CrossRef CAS PubMed;
(n) X. Pan, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 1163–1185 RSC;
(o) X. Guo-Liang, J. Shang-Hua, S. Xiangdong and F. Behmagham, J. Fluorine Chem., 2020, 235, 109524 CrossRef.
- Q. Gu and E. Vessally, RSC Adv., 2020, 10, 16756–16768 RSC.
- C. Yang, A. Hassanpour, K. Ghorbanpour, S. Abdolmohammadi and E. Vessally, RSC Adv., 2019, 9, 27625–27639 RSC.
- M. Hamzeloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef.
- A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, J. Fluorine Chem., 2019, 220, 24–34 CrossRef CAS.
- A. Hosseinian, Y. J. Sadeghi, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 565–585 CrossRef CAS.
-
(a) M. Li, S. Abdolmohammadi, M. S. Hoseininezhad-Namin, F. Behmagham and E. Vessally, J. CO2 Util., 2020, 38, 220–231 CrossRef CAS;
(b) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 37 CrossRef CAS PubMed;
(c) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 20 CrossRef PubMed;
(d) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef PubMed;
(e) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef PubMed;
(f) J. Wang, P. Su, S. Abdolmohammadi and E. Vessally, RSC Adv., 2019, 9, 41684–41702 RSC;
(g) Y. Liu, A. G. Ebadi, L. Youseftabar-Miri, A. Hassanpour and E. Vessally, RSC Adv., 2019, 9, 25199–25215 RSC;
(h) S. Arshadi, A. Banaei, A. Monfared, S. Ebrahimiasl and A. Hosseinian, RSC Adv., 2019, 9, 17101–17118 RSC;
(i) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed;
(j) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed;
(k) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed;
(l) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed;
(m) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed;
(n) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed;
(o) S. Majedi, S. Majedi and F. Behmagham, Chem. Rev. Lett., 2019, 2, 187–192 Search PubMed;
(p) E. A. Mahmood, B. Azizi and S. Majedi, Chem. Rev. Lett., 2020, 3, 2–8 Search PubMed.
- Z. Liu, H. Chen, Y. Lv, X. Tan, H. Shen, H. Z. Yu and C. Li, J. Am. Chem. Soc., 2018, 140, 6169–6175 CrossRef CAS PubMed.
- A. Sato, M. V. Ponomarenko, T. Ono, G. V. Röschenthaler and V. A. Soloshonok, Eur. J. Org. Chem., 2019, 4417–4421 CrossRef CAS.
- N. Kamigata, T. Fukushima and M. Yoshida, J. Chem. Soc., Chem. Commun., 1989, 1559–1560 RSC.
- N. Kamigata, T. Fukushima, Y. Terakawa, M. Yoshida and H. Sawada, J. Chem. Soc., Perkin Trans. 1, 1991, 627–633 RSC.
- S. H. Oh, Y. R. Malpani, N. Ha, Y. S. Jung and S. B. Han, Org. Lett., 2014, 16, 1310–1313 CrossRef CAS PubMed.
- X. J. Tang and W. R. Dolbier Jr, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef CAS PubMed.
- M. Alkan-Zambada and X. Hu, Organometallics, 2018, 37, 3928–3935 CrossRef CAS.
- W. Zhang, J. H. Lin and J. C. Xiao, J. Fluorine Chem., 2018, 215, 25–31 CrossRef CAS.
- K. Maeda, T. Kurahashi and S. Matsubara, Eur. J. Org. Chem., 2019, 4613–4616 CrossRef CAS.
- C. Xu, X. Song, J. Guo, S. Chen, J. Gao, J. Jiang, F. Gao, Y. Li and M. Wang, Org. Lett., 2018, 20, 3933–3937 CrossRef CAS PubMed.
-
(a) J. Guo, C. Xu, X. Liu and M. Wang, Org. Biomol. Chem., 2019, 17, 2162–2168 RSC;
(b) J. Guo, C. Xu, L. Wang, W. Huang and M. Wang, Org. Biomol. Chem., 2019, 17, 4593–4599 RSC.
- C. Xu, W. Huang, R. Zhang, C. Gao, Y. Li and M. Wang, J. Org. Chem., 2019, 84, 14209–14216 CrossRef CAS PubMed.
- B. Yang, X. H. Xu and F. L. Qing, Chin. J. Chem., 2016, 34, 465–468 CrossRef CAS.
- J. Fang, Z. K. Wang, S. W. Wu, W. G. Shen, G. Z. Ao and F. Liu, Chem. Commun., 2017, 53, 7638–7641 RSC.
- K. Y. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, J. Am. Chem. Soc., 2018, 140, 2438–2441 CrossRef CAS PubMed.
- A. Carboni, G. Dagousset, E. Magnier and G. Masson, Synthesis, 2015, 47, 2439–2445 CrossRef CAS.
- J. Y. Guo, R. X. Wu, J. K. Jin and S. K. Tian, Org. Lett., 2016, 18, 3850–3853 CrossRef CAS PubMed.
- W. Y. Huang and L. Lü, Chin. J. Chem., 1992, 10, 268–273 CrossRef CAS.
- V. A. Ol'shevskaya, A. A. Tyutyunov, L. F. Ibragimova, E. G. Kononova and E. G. Rys, Polyhedron, 2019, 171, 508–514 CrossRef.
- W. An, N. Ha, H. M. Lee, Y. R. Malpani, D. H. Lee, Y. S. Jung and S. B. Han, Adv. Synth. Catal., 2015, 357, 3949–3960 CrossRef CAS.
- Z. Q. Liu and D. Liu, J. Org. Chem., 2017, 82, 1649–1656 CrossRef CAS PubMed.
- K. Tsuchii, M. Imura, N. Kamada, T. Hirao and A. Ogawa, J. Org. Chem., 2004, 69, 6658–6665 CrossRef CAS PubMed.
- J. Ignatowska and W. Dmowski, J. Fluorine Chem., 2007, 128, 997–1006 CrossRef CAS.
- T. Yajima and H. Nagano, Org. Lett., 2007, 9, 2513–2515 CrossRef CAS PubMed.
- T. Xu, C. W. Cheung and X. Hu, Angew. Chem., Int. Ed., 2014, 53, 4910–4914 CrossRef CAS PubMed.
- T. Rawner, E. Lutsker, C. A. Kaiser and O. Reiser, ACS Catal., 2018, 8, 3950–3956 CrossRef CAS.
- R. Beniazza, M. Douarre, D. Lastécouères and J. M. Vincent, Chem. Commun., 2017, 53, 3547–3550 RSC.
- R. Beniazza, L. Remisse, D. Jardel, D. Lastécouères and J. M. Vincent, Chem. Commun., 2018, 54, 7451–7454 RSC.
- Z. Hang, Z. Li and Z. Q. Liu, Org. Lett., 2014, 16, 3648–3651 CrossRef CAS PubMed.
- H. Egami, Y. Usui, S. Kawamura, S. Nagashima and M. Sodeoka, Chem.–Asian J., 2015, 10, 2190–2199 CrossRef CAS PubMed.
- X. Li, R. Zhang, X. Zhang, P. Zhu and T. Yao, Chem.–Asian J., 2015, 15, 1175–1179 CrossRef PubMed.
- M. Wang, M. Hu, Z. Li, L. He, Y. Song and Q. Jia, Biosens. Bioelectron., 2019, 142, 111536 CrossRef PubMed.
- H. Lu, Y. Zhu, Y. Yuan, L He, B. Zheng and X. Zheng, J. Mater. Sci.: Mater. Electron., 2021 DOI:10.1007/s10854-021-05310-0.
- H. Zhang, W. Guan, L. Zhang, X. Guan and S. Wang, ACS Omega, 2020, 29, 18007–18012 CrossRef PubMed.
- M. Wang, et al., Biosens. Bioelectron., 2018, 113, 16–24 CrossRef CAS PubMed.
- H. Zhang, M. Sun, L. Song, J. Guo and L. Zhang, Biochem. Eng. J., 2019, 147, 146–152 CrossRef CAS.
- M. Sun, L. Yan, L. Zhang, L. Song, J. Guo and H. Zhang, Process Biochem., 1991, 78, 108–113 CrossRef.
- F. Gharibzadeh, E. Vessally, L. Edjlali, M. Es haghi and R. Mohammadi, Iran. J. Chem. Chem. Eng., 2020, 39, 51–62 Search PubMed.
- W. Huang, et al., Chem, 2020, 6, 2300–2313 CAS.
- Y. Yang, L. Tao, H. Yang, S. Iglauer, X. Wang, R. Askari and H. Sun, J. Geophys. Res.: Solid Earth, 2020, 125, e2019JB018759 Search PubMed.
- Y. Yang, et al., Water Resour. Res., 2020, 56 DOI:10.1029/2019WR026112.
- Q. Wang, S. Sun, X. Zhang, H. Liu, B. Sun and S. Guo, Bioresour. Technol., 2021 DOI:10.1016/j.biortech.2021.125120.
- E. Vessally, S. Mohammadi, M. Abdoli, A. Hosseinian and P. Ojaghloo, Iran. J. Chem. Chem. Eng., 2020, 39, 11–19 CAS.
- J. Liu, M. Ren, X. Lai and G. Qiu, Chem. Commun., 2021 10.1039/D1CC00870F.
- Y. Duan, et al., Green Chem., 2020, 22, 44–53 RSC.
- N. N. Wang, X. Sun, Q. Zhao and P. Wang, Chem. Eng. J., 2021, 406, 126734 CrossRef CAS.
- Y. Yang, et al., ACS Appl. Mater. Interfaces, 2020, 12, 24845–24854 CrossRef CAS PubMed.
- H. Ahmadizadegan, Iran. J. Chem. Chem. Eng., 2020, 39, 33–47 Search PubMed.
- Y. Liu, Z. Wei, B. Zhong, H. Wang, L. Xia, T. Zhang and X. Huang, Energy Storage Mater., 2021, 35, 12–18 CrossRef.
- X. Yan, et al., Carbon, 2021, 174, 662–672 CrossRef CAS.
- M. Jamil, N. Sultana, M. Sarfraz, M. N. Tahir and M. I. Tariq, Iran. J. Chem. Chem. Eng., 2020, 39, 45–57 CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence e
e