
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluating the cytotoxicity of Ge–Sb–Se chalcogenide glass optical fibres on 3T3 mouse fibroblasts†
David Mabwa *ab,
Teo Kubienaab,
Harriet Parnellab,
Rong Suc,
David Furnissab,
Zhuoqi Tangab,
Richard Leachc,
Trevor M. Bensona,
Colin A. Scotchfordb and
Angela B. Seddon*ab
*ab,
Teo Kubienaab,
Harriet Parnellab,
Rong Suc,
David Furnissab,
Zhuoqi Tangab,
Richard Leachc,
Trevor M. Bensona,
Colin A. Scotchfordb and
Angela B. Seddon*ab
aMid-Infrared Photonics Group, George Green Institute for Electromagnetic Research Group, Faculty of Engineering, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: david.mabwa@nottingham.ac.uk; angela.seddon@nottinghan.ac.uk
bAdvanced Materials Research Group, Faculty of Engineering, University of Nottingham, Nottingham NG7 2RD, UK
cManufacturing Metrology Team, Faculty of Engineering, University of Nottingham, Nottingham NG7 2RD, UK
First published on 25th February 2021
Abstract
In vivo cancer detection based on the mid-infrared molecular fingerprint of tissue is promising for the fast diagnosis and treatment of suspected cancer patients. Few materials are mid-infrared transmissive, even fewer, which can be converted into functional, low-loss optical fibres for in vivo non-invasive testing. Chalcogenide-based glass optical fibres are, however, one of the few. These glasses are transmissive in the mid-infrared and are currently under development for use in molecular sensing devices. The cytotoxicity of these materials is however unknown. The cytotoxicity of Ge–Sb–Se chalcogenide optical glass fibres on 3T3 mouse fibroblast cells is here investigated. Fibres exposed to four different pre-treatment conditions are used: as-drawn (AD), propylamine-etched (PE), oxidised-and-washed (OW) and oxidised (Ox). To achieve the latter two conditions, fibres are treated with H2O2(aqueous (aq.)) and dried to produce a surface oxide layer; this is either washed off (OW) or left on the glass surface (Ox). Cellular response is investigated via 3 day elution and 14 day direct contact trials. The concentration of the metalloids (Ge, Sb and Se) in each leachate was measured via inductively coupled plasma mass spectrometry. Cell viability is assessed using the neutral red assay and scanning electron microscopy. The concentration of Ge, Sb and Se ions after a 3 day dissolution was as follows. In AD leachates, Ge: 0.40 mg L−1, Sb: 0.17 mg L−1, and Se: 0.06 mg L−1. In PE leachates, Ge: 0.22 mg L−1, Sb: 0.15 mg L−1, and Se: 0.02 mg L−1. In Ox leachates, Ge: 823.8 mg L−1, Sb: 2586.6 mg L−1, and Se: 3750 mg L−1. Direct contact trials show confluent cell layers on AD, PE and OW fibres after 14 days, while no cells are observed on the Ox surfaces. A >50% cell viability is observed in AD, PE and OW eluates after 3 days, when compared with Ox eluates (<10% cell viability). Toxicity in Ox is attributed to the notable pH change, from neutral pH 7.49 to acidic pH 2.44, that takes place on dissolution of the surface oxide layer in the growth media. We conclude, as-prepared Ge–Sb–Se glasses are cytocompatible and toxicity arises when an oxide layer is forced to develop on the glass surface.
1. Introduction
Over 100 different types of cancers occur in humans, and only four: breast, colon, prostate, and lung, are responsible for more than half of all diagnoses, with a comparable proportion of deaths. Cancer diagnosis has however improved in recent decades, with advancements occurring in for example, genetic profiling and diagnostic imaging. A relatively novel method is mid-infrared hyperspectral imaging.1 This is based on utilising mid-infrared (MIR) radiation to identify the presence and distribution of chemical species in biological tissue. When a tissue sample absorbs radiation in the MIR region (3.0 μm to 50 μm)2 covalently bonded bio-molecular species vibrate to give absorption bands whose absorption and characteristic wavelength, provide information pertaining to the quantity, and nature, of their bonding, respectively.3 The rapidity and high diagnostic accuracy of MIR hyperspectral imaging has been demonstrated by many, for instance: Old et al.4 and Pilling et al.5 Old et al. collected and rapidly processed 45 MIR hyperspectral images of oesophageal tissue (5 minutes per image), then accurately identified neoplastic Barrett's oesophagus with 95.6% sensitivity and 86.4% specificity.4 Similarly, Pilling et al. collected MIR spectra from 207-breast cancer patients using quantum cascade laser (QCL) imaging (5.55 μm–10.86 μm) and accurately differentiated between malignant and non-malignant stroma with 93.56% specificity.5Along with its numerous benefits, MIR hyperspectral imaging also comes with limitations. It is currently limited to only imaging excised tissue. This is due to traditionally weak MIR light blackbody sources and the lack of optical fibre that transmits sufficiently into the MIR spectral region.6 The latter limitation has since been resolved since the advent of chalcogenide-based optical fibres, derived from the chalcogen elements of group 16 in the periodic table (S, Se and Te – excluding O).7 With the aim of providing point-of-care testing, in previous work, we developed low optical loss Ge20Sb10Se70 atomic (at) % glass optical fibres capable of transmitting MIR light from 2.5 μm–13 μm.8 This glass system opens the possibility of utilising MIR hyperspectral imaging for the diagnosis of cancerous tissue, in vivo, through its incorporation into endoscopic probes.9
The metalloid elements used in formulating these optical fibres warrant an analysis of their cytotoxicity, prior to their application in point-of-care testing. Similar work was conducted by Wilhelm et al.,10 who investigated cell coverage by A549 human lung cells on glass preforms of Te2As3Se5 (TAS) at%. This study reported inconsistent and non-reproducible cell-attachment results, a problem attributed by the authors to the presence of arsenic (As) in the glass and a thin surface oxide layer. The toxic behaviour of the TAS glasses was then assessed by submerging the TAS fibres in cell cultures and analysing the cell metabolic activity via a WST-1 colorimetric assay. It was concluded that, after the TAS fibres were washed in water for 24 hours, they produced no toxic effect towards the cells (95% to 103% cell viability), whereas unwashed fibres produced a more toxic effect in comparison (65% to 69% cell viability).
As is included in many chalcogenide glass formulations due to its ability to stabilise the covalent glass network. However, the inclusion of this element into medical instruments (i.e., endoscopic probes) raises significant concerns. This is because, even in micro-quantities, As is highly toxic and possesses the potential to irreversibly damage cells and tissue.10 In previous work,11 we replaced As with antimony (Sb) to produce a glass composition more suitable for biological applications. The toxicity of these Sb-containing compositions has however, not previously been evaluated. This forms the aim of this paper. To produce an in-depth evaluation on the cytotoxicity of Sb-containing chalcogenide glasses and assess whether Sb-based compositions would be viable for their application into medical devices such as endoscopic probes.
Data related to the toxicity of antimony in humans come primarily from studies that investigate its connection with industrial-atmospheric exposure. Potkonjak and Pavlovich12 examined the respiratory condition of 51 workers in a Yugoslavian antimony smelter with chest X-ray analysis. X-ray changes indicated antimoniosis. The workers were exposed to 17 mg m−3 to 86 mg m−3 antimony dust for 9 to 31 years. Antimony trioxide (Sb2O3) made up 40% to 90% of the dust, antimony pentoxide (Sb2O5) 2% to 8% and free silica 1% to 5%. Positive X-ray findings characterised antimoniosis as diffuse, densely distributed punctate opacities, irregular in shape, and with a diameter less than 1 mm in the lung. In addition, chronic coughing was reported in 60.8% of workers, chronic bronchitis in 37.2%, chronic emphysema in 34.5%, inactive tuberculosis in 18.2% and pleural adhesions in 27.3%.
The chemo-toxicological characteristics of arsenic and antimony are very similar.13 In the trivalent state, both behave clastogenically in vivo and in vitro, are not directly mutagenic, and have a carcinogenic potential.14,15 Genotoxicity of antimony has been shown to be valence dependant but the underlying mechanism for its genotoxicity remains unclear.14,16 Kuroda et al.17 used the sister chromatid exchange (SCE) assay to show that Sb2O3 at a concentration of 0.34 μL mL−1 induced significant genotoxic activity, whereas Sb2O5 at a concentration of 40 μL mL−1 did not. A biomonitoring study by Cavallo et al.18 evaluated genotoxicity in 23 workers exposed to Sb2O3 (via skin contact and the respiratory pathway) using SCE assay, micronucleus tests and (Fpg)-modified comet assay. The results suggested that Sb(III) toxicity results from oxidative DNA damage. It has also been suggested by Grosskopf et al.16 that Sb(III) interferes with proteins involved in nucleotide excision repair, resulting in the partial retardation of this pathway and an indirect mechanism in the genotoxicity of Sb(III).
In view of this, we present a study that aims to evaluate the cytotoxicity of Ge20Sb10Se70 at% glass optical fibres on 3T3 mouse fibroblast cells. To observe the toxicity of the fibres, 3T3 fibroblast proliferation was investigated. This involved two trials. Firstly, cells were subjected to elution trials, in which the effect of any glass-derived leachates on the proliferation of the cells was assessed. The concentration of each metalloid species (leachates) was measured, by exposing glass fibres to deionised water over a 14 day period. This allowed for further evaluation of the cell viability outcome. This was followed by direct contact trials, which were performed to observe the adhesion/attachment behaviour of cells on the surface of the fibre. Cell viability was assessed using the neutral red assay; this chemo-sensitive assay was based on the ability of viable cells to incorporate and bind to the supravital neutral red dye. Since genotoxic activity is observed in literature as a response to Sb2O3, we generated oxides at the glass surface by exposing the Ge20Sb10Se70 at% glass optical fibres to aqueous H2O2, (H2O2(aq.)) and the potential toxicity of the oxide produced on 3T3 fibroblasts was evaluated.
2. Materials and methods
2.1. Ge20Sb10Se70 at% glass optical fibre pre-treatments and sample codes
Ge20Sb10Se70 at% bulk glass rod was synthesised in high purity, using 99.999% purity elements, in a pre-purified silica ampoule, under a 10−6 Pa vacuum at 900 °C for 12 h. The bulk glass preform was then drawn into a fibre with a diameter of 210 μm ± 20 μm. More details of the melting procedure can be found in the ESI.† The Ge20Sb10Se70 at% fibres were cleaved using a ruby-tipped scribe (S90R, Thorlabs) under ambient conditions into 5 mm and 10 mm lengths (for direct contact and elution trials respectively) and were subjected to one of three different post-fibre-drawing pre-treatments, as indicated below. All the pre-treatments were conducted under ambient conditions.To prepare propylamine etched (PE) fibres, as-drawn cleaved fibre pieces were collectively submerged in 5 mL of propylamine (Sigma-Aldrich, 99.0%), within a scintillation vial for 0.5 h, then washed in triplicate with acetone, and isopropanol. During each wash, the fibres were placed in a 10 mL scintillation vial and held in an ultrasonic cleaner for 5 min. Following this, the fibres were left in ambient conditions to allow the solvent to evaporate, then placed into a new scintillation vial, wrapped in lens tissue, and transferred into an MBraun glove box, with a nitrogen atmosphere, with 0.6 ppm H2O and 0.1 ppm O2 for storage up to 5 days before use.
After the pre-treatments described in Sections 2.1.1, 2.1.2 and 2.1.3, all fibre samples intended for direct contact and elution trials were disinfected via ultraviolet (UV) sterilisation in a class II microbiological safety cabinet (Nuaire biological safety cabinets, Triple Red Ltd) for 90 min, under ambient atmosphere and temperature, before the trials began.
In order to prepare the elution medium (eluate) for each condition, fibre samples from each condition were respectively submerged in culture medium (production method described in ESI†) for 24 h or 72 h, while being horizontally rotated in a sterile 20 mL disposable scintillation vial, at 50 rpm (revolutions per minute, Denley Spiramix 5). Oxidised samples (OW, O30 and O60) were only submerged for 24 h. Different submersion times were investigated to see what effect a longer fibre-matrix interaction time would have on cell viability. After 24 h or 72 h, the culture medium, now the elution medium (eluate) was decanted from the fibres and diluted. The following dilutions by volume of the eluate were then produced using the culture medium as the diluent: 100%, 75%, 50%, and 25%. The diluents were placed into 5 mL sterile universal tubes and stored in a chemical refrigerator (for a maximum of 3 days) at 4 °C until needed, after which the diluents were warmed to 37 °C in a water bath and used.
The control sample used in this work was tissue culture plastic (TCP), i.e. an untreated, fibreless well of a 96-well plate (material: polystyrene, ThermoScientific).
2.2. Cytotoxicity tests
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 (neutral red stock:culture media) dilution under sterile conditions, centrifuged at 1200 rpm for 4 minutes so that any crystals that may have formed, would settle at the base of the 25 mL sterile universal tube; care was taken not to shake the tube, so as not to re-suspend the crystals. The neutral red medium was not stored before/after an experiment. Any excess was discarded.
:100 (neutral red stock:culture media) dilution under sterile conditions, centrifuged at 1200 rpm for 4 minutes so that any crystals that may have formed, would settle at the base of the 25 mL sterile universal tube; care was taken not to shake the tube, so as not to re-suspend the crystals. The neutral red medium was not stored before/after an experiment. Any excess was discarded.At the indicated time points (day-1, day-3, day-7, and day-14 for direct contact trials and day-1 and day-3 for elution trials), the cells were submerged in neutral red medium and incubated for 2 hours at 37 °C with 5 vol% CO2. After incubation, the unincorporated neutral red was decanted and discarded. The cells were then washed in triplicate with phosphate buffer saline (PBS, Oxoid UK). Following the triplicate washes with PBS, a destaining solution was added to each well, to extract the incorporated dye from the viable cells. The destaining solution was prepared by volume in ambient temperature, by mixing pure ethanol (Scientific Laboratory Supplies, 99%), with deionised water and aqueous glacial acetic acid (99.74%, Fisher Scientific, UK)) to a ratio of 5:4.9:0.1, respectively. The 96-well plate was then placed on a plate shaker (Titramax 100, Heidolph) at 300 rpm for 10 min under ambient conditions to extract the incorporated neutral red dye from the cells. Finally, the 96-well plate was placed in the BioTek™ ELx800™ UV (Fisher Scientific) plate reader (primary wavelength 490 nm, reference wavelength 630 nm), to quantify the neutral red dye concentration. Cell viability was expressed as a percentage of the control of which, a cell viability under 70% of the viability expressed by the control cells, indicated material cytotoxicity, as stated by the ISO standards ISO 10993-5:2009.22
2.3. Materials and methods (ESI†)
See ESI for the following details:(1) Bulk Ge20Sb10Se70 at% preform fabrication and fibre preparation.
(2) A physical analysis of Ge20Sb10Se70 at% glass optical fibres using coherence scanning interferometry and scanning electron microscopy.
(3) A pH physical analysis of the following eluates: AD, PE and O60.
(4) The 3T3 fibroblast cell-line and method for producing the culture medium.
(5) Statistical analysis method performed on the results of this work.
3. Results
To recap, to simplify interpretation of the results, as described in Section 2.1, AD, PE, OW, O10, O30 and O60 fibre samples were prepared. Fibres were initially cleaved into 5 mm and 10 mm long lengths (for direct contact and elution trials, respectively), then subjected to one of three pre-treatment conditions. These were, submersion in propylamine for 0.5 h (PE); submersion in H2O2(aq.) for 10, 30 and 60 min and subsequent drying in ambient conditions for 12 h, to allow the build-up of a glass oxide layer (O10, O30, O60, respectively) and submersion in H2O2(aq.) for 60 min and subsequent drying as previously described and triplicate washing in both acetone and isopropanol to remove the oxide layer (OW) then drying in ambient conditions via evaporation. AD fibres were not subjected to any pre-treatment conditions.3.1. Chemical analysis: Ge, Sb and Se concentration in aqueous leachates
As in Fig. 1 (37 °C leaching temperature), O60 resulted in the highest Ge, Se, and Sb concentration, compared to AD and PE leachates. The concentration of Se and Sb in O60 dropped as the time for leaching increased, from 3858 mg L−1 and 2702 mg L−1, respectively in D1 to 1207 mg L−1 and 459 mg L−1, respectively, at D14. This drop is, however, more notable from D7 to D14, a 175.6% (Se) and 364.8% (Sb) drop, compared to the drop in concentration from D1 to D7 (Se: 16% and Sb: 26.7%). This time dependant relationship with concentration is also observed in AD and PE leachates. In AD, Ge and Sb concentrations increase by 222% and 159%, respectively, from D1 (Ge: 0.31 mg L−1, Sb: 0.19) to D14 (Ge: 1.02 mg L−1, Sb: 0.50 mg L−1), while Se is seen to decrease by 61.7% from D1 (Se: 0.12 mg L−1) to D14 (Se: 0.045 mg L−1). In PE, Ge and Sb concentrations increase by 1955% and 1757% from D1 (Ge: 0.13 mg L−1, Sb: 0.08 mg L−1) to D14 (Ge: 2.63 mg L−1, Sb: 1.48 mg L−1), however, this increase is more prominent from D7 to D14 (Ge: 832% and Sb: 663%), compared to D1 to D7 (Ge: 121% and Sb 143%). Se was observed to increase from 0.017 mg L−1, in D1, to 0.052 mg L−1 in D14 (a 196.5% increase).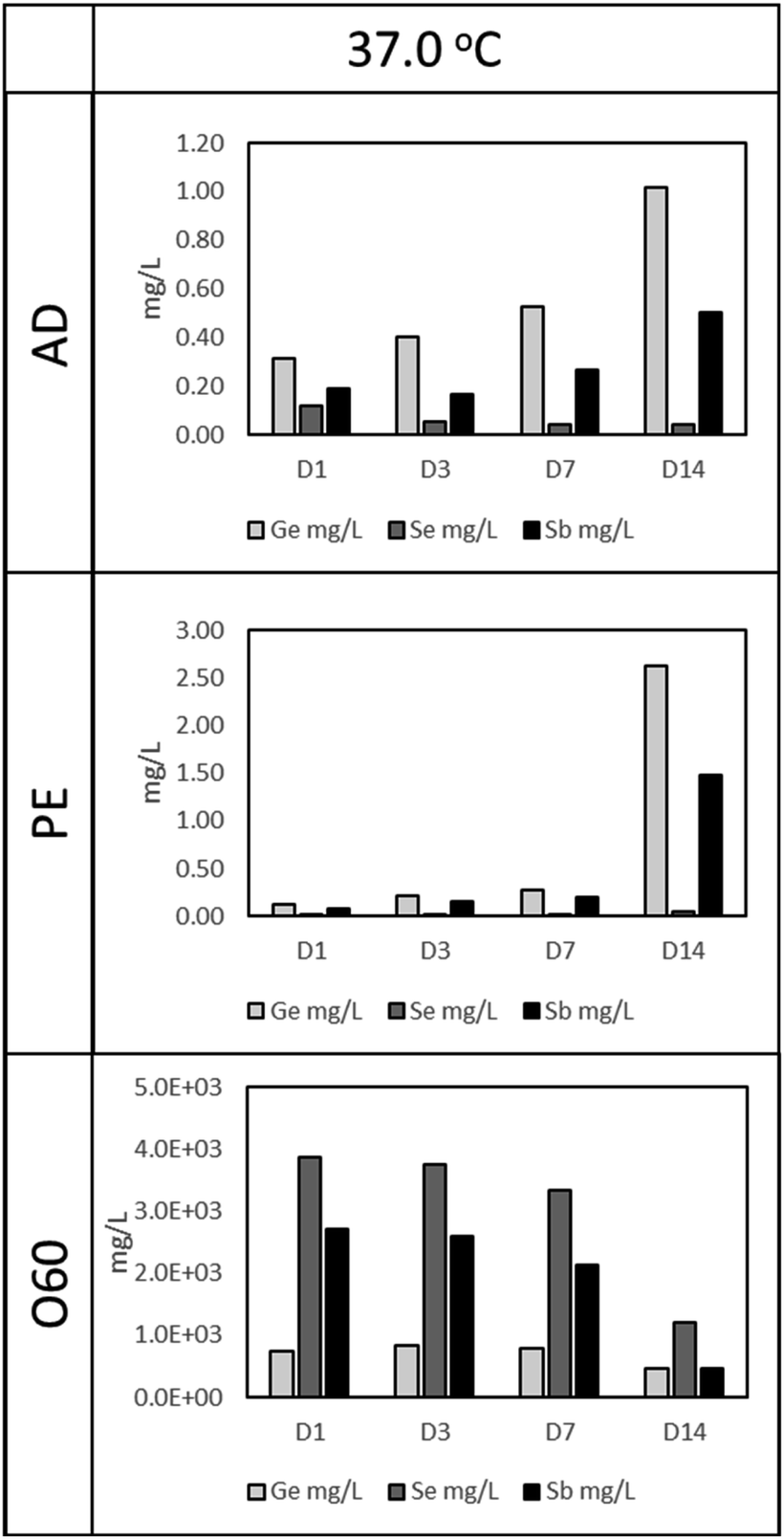 | ||
| Fig. 1 The concentration of Ge, Sb and Se in aqueous leachates after exposure of fibre (conditioned as AD, PE and O60) to deionised water at 37 °C for 1, 3, 7 and 14 days (D1, D3, D7 and D14, respectively). | ||
3.2. Cell response
After day-1 (from Fig. 2A), fibroblasts show a viability of >80%, in all eluates, excluding O30 and 060 which only show a viability of <20%. In Fig. 2A, a lower eluate concentration in PE-24 h and PE-72 h is seen to result in higher fibroblast viability. This correlation is also observed in AD-24 h, AD-72 h, and OW, up to an eluate concentration of 50%. At an eluate concentration of 25% in AD-24 h, AD-72 h and OW, the cell viability dropped by 10.4%, 6.8% and 4%, respectively. No statistically significant differences were found between the viabilities of cells grown in AD-24 h, AD-72 h, PE-24 h, PE-72 h and OW. Significant differences were found between the viabilities of cells grown in O60 and O30 and all other eluates, but not between O60 and O30. Fig. 2B shows that cell viability decreased in all eluates, excluding 50% and 25% concentrated PE-24 h, which increased by 8.2% and 10.7%, respectively, when compared to the cell viability observed in PE-24 h after day-1. Cell viability was highest in 25% concentrated PE-24 h (111.1% ± 3.1%) and lowest in 100% concentrated PE-72 h (53.9% ± 5.9%). A notable drop in the pH of O60 was also observed. The pH of the growth media was originally 7.42, and this dropped to 2.44 after 24 h interaction time with O60 fibres. It is inferred that this occurred when glass oxide from the surface of the O60 fibre samples dissolved into the growth media.
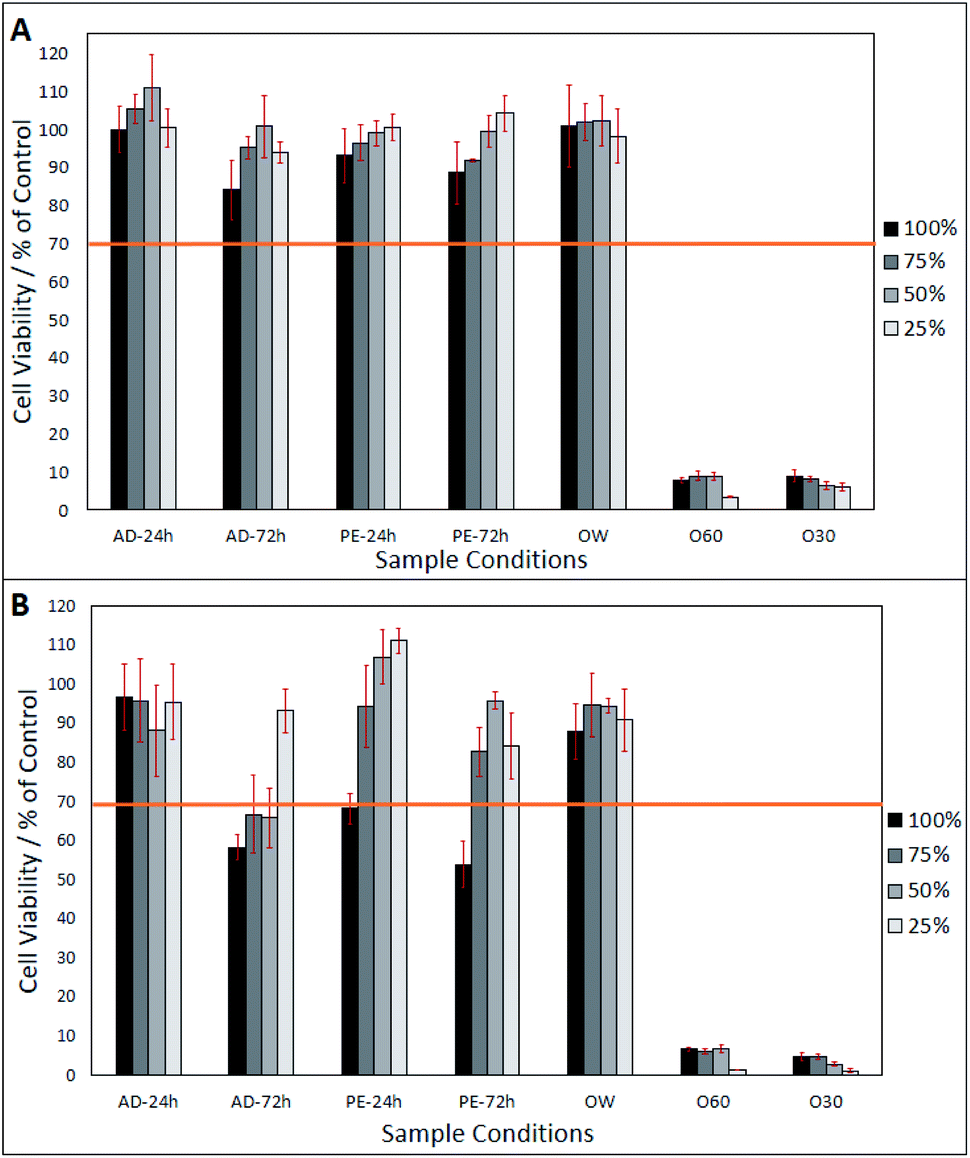 | ||
| Fig. 2 Neutral red assay of cells grown in eluates produced from AD-24 h, AD-72 h, PE-24 h, PE-72 h, OW, O60 and O30 concentrates. (A) Cell viability 24 hours after eluates were added to 3T3 fibroblasts. (B) Cell viability 72 hours after eluates were added. Sample number = n5. Significant differences determined using Tukey HSD, multiple comparisons, when p = <0.05. | ||
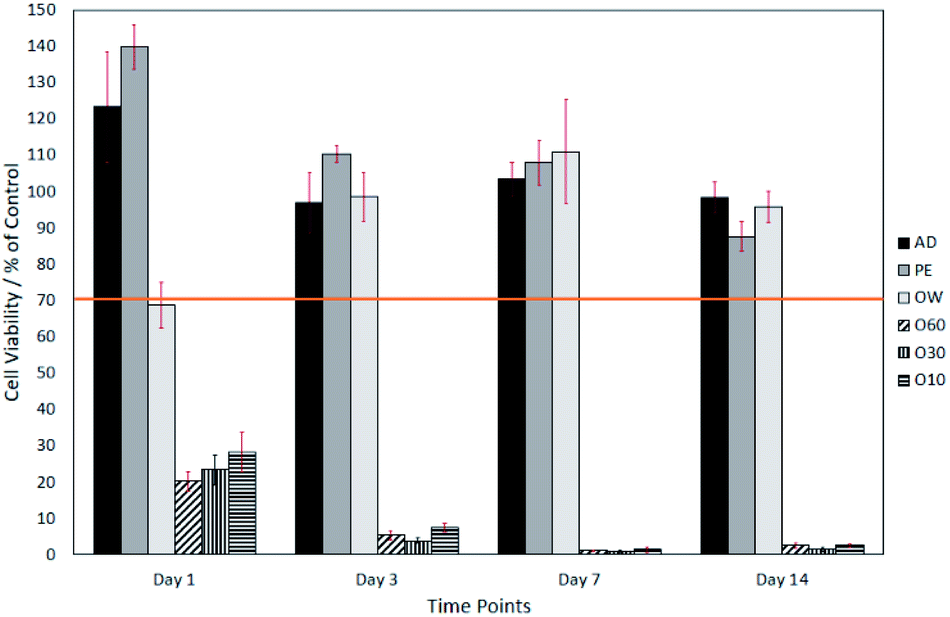 | ||
| Fig. 3 Neutral red assay of cells grown on AD, PE, OW, O60, O30 and O10 surfaces. Cell viability is displayed over a 14 day time period. Sample number = n5. Significant differences determined using Tukey HSD, multiple comparisons, when p = <0.05. | ||
3.3. SEM analysis
 | ||
| Fig. 4 Scanning electron micrographs of cell growth on Ge20Sb10Se70 fibres over a 14 day period. A1–A4 = AD fibre, B1–B4 = PE fibre, C1–C4 = OW fibre, D1–D4 = O10, E1–E4 = O30, F1–F4 = O60 fibre. | ||
(1) Surface roughness of the fibres obtained via coherence scanning interferometry.
(2) Surface texture obtained via SEM.
(3) pH measurements.
(4) Concentration of Ge, Sb, and Se obtained via intercoupled plasma mass spectrometry.
4. Discussion
In this study, a simple assessment of the cytotoxicity of Ge20Sb10Se70 at% chalcogenide glass optical fibre is presented. AD fibres were tested as they represent optical fibres that could be applied to MIR-based medical devices (i.e. endoscopic probes). When exposed to air, a passivating oxide layer begins to form on the surface of chalcogenide-based fibres. Fibres exposed to air for long periods of time (e.g. >1 year) can develop a surface oxide layer, with a thickness >30 Å.19 It has been previously shown10 that, when exposed to air for an extended period of time (3 years), fibres exert a significant toxic effect, however, when these fibred are washed, this toxic effect disappears. PE fibres are a representation of these “washed” fibres. When fibres were exposed to propylamine, this oxide layer was completely stripped off the fibre, and cells are therefore, exposed to ‘bare’ glass and not an oxide layer as in AD.The fibres used in this study were exposed to air for no longer than 90 days, and so, an oxide layer thick enough to influence cell response, it is suggested, would not have developed. To observe a similar glass-oxide based cytotoxicity as in ref. 10, a surface oxide layer was forced to develop via H2O2 aq. exposure. Finally, the OW treatment was selected to observe any remnant cytotoxicity in fibres, post H2O2 aq. treatment. In this condition, cells were exposed to fibres that were first treated with H2O2 aq. then washed to remove the surface oxide layer.
The surface oxide layer in O10–O60 was observed to be partially soluble in the cell growth medium. Once dissolved, this resulted in a notable drop in pH from the physiological 7.49 (ref. 23) to the acidic 2.44. This drop in pH was shown, through direct contact and elution trials, to have a toxic effect on cell viability. The elution trials show that a 1 day exposure to oxidised eluates was enough to result in a cell viability of <10% (see Fig. 2). Direct contact trials also show that a 1 day exposure to fibre surfaces with a glass oxide layer results in a cell viability of <30%, which dropped to <3% after day-3 (see Fig. 3).
This toxic effect on fibroblast cells may be explained as follows. Alterations to the cellular microenvironment can significantly affect cell physiology and induce pathology.24 An important aspect of the extracellular environment is its pH. This must be kept within strict boundaries (between 6 and 8 according to ref. 23) to facilitate correct cellular function and prevent cell death.25,26 Pathologies associated with an acidic extracellular environment include ischemia (pH <6.3 (ref. 27)), hypercapnia28 and metabolic acidosis (pH <7.35 (ref. 29)). Furthermore, mitochondrial dysfunction and the acidification of the extracellular environment have been linked to increased levels of reactive oxygen species (ROS) within cells.30,31 ROS (such as, superoxide (O2−), singlet oxygen (1O2), hydroxyl radical (OH.) and H2O2) are formed as products under physiological conditions as a result of the partial reduction of O2.32 Low concentrations of ROS are required to maintain normal physiological functions such as cellular proliferation, signal transduction, immunity, and genetic expression.32,33 However, an increased concentration of ROS results in the induction of oxidative stress.24
In a study by Teixeira et al.24 HEK293 cells were used to evaluate the effects of extracellular acidification by ambient temperature HCl aq. addition. They found that lowering the extracellular pH from 7.2 to 5.8, lowered cell viability by 70%, decreased cytosolic pH, hyperpolarised the mitochondrial membrane potential and increased the ROS levels. These results suggest that an acidic extracellular environment can induce cell death through an mPTP opening and ROS-mediated pathogenic pathway. This supports earlier work by Xue and Lucocq34 who showed that low extracellular pH results in the activation of the c-Jun N-terminal kinase (JNK) pathway, an important regulator of various cellular processes, including apoptosis, in Swiss 3T3 cells.35,36 In our work, the extracellular pH dropped from 7.49 to 2.44, and this was paralleled with a +70% drop in cell viability observed in both elution and direct contact trials after 24 h. Considering former work,24 the primary cause of cell death observed in our work is suggested to result from a potential increase in the concentration of ROS within the O10, O30 and O60 eluates.
A secondary potential cause of cytotoxicity involves the possible influence of Ge, Sb and Se metalloid oxides. Metal oxide nanoparticles (MO-NPs) are known to be highly toxic to biological tissue, so much so that some authors have demonstrated their potential applicability in cancer therapy.25–27,37–39 These oxide nanoparticles have shown toxic effect via pro-apoptotic activity, autophagy, proliferation inhibition, metal ion release and the increase of ROS.38,40 In a study by Wilhelm et al.10 on the biocompatibility of Te–As–Se glass fibres, they observed pronounced toxicity only for fibres exposed to air for 3 years. This toxicity was attributed to the formation of a soluble As2O3 layer on the fibre surface, which when exposed to aqueous solution, results in the release of arsenic (As) into solution as both As(III) and As(V). As then exert its toxicity by inactivating vital enzymes involved in ATP generation and DNA synthesis and repair.41 Similar results were observed in this study when fibre surface oxidation was chemically enforced over a shorter time. The proliferation rates of fibroblasts exposed to O10, O30 and O60 surfaces and eluates were significantly lower, when compared to the other glass surfaces. As mentioned above, an additional explanation of this is the release of metal ions into the growth media. The question to now address is, which metal oxide is the most plausible cause for this toxicity, GeO2, Sb2O3, Sb2O5, Se–O or SeO2?
It is generally accepted, that germanium metal nanomaterials have low toxicity,42 are non-carcinogenic and even inhibitory to the formation of tumour cells.43,44 Conversely, as discussed by Lin et al.,45 inorganic germanium compounds such as GeO2, can result in neuro- or nephrotoxicity, but only after very high doses (e.g. a minimum oral uptake of 5 g GeO2 per day for a 70 kg adult) and long-term exposures. In the same paper the authors showed that fabricated nano-Ge and nano-GeO2 are non-toxic to Chinese hamster ovary (CHO) K1 cells, displaying a relative viability greater than 70%. This shows that Ge and its products possess very low toxicity, such that it has found use as a health supplement, in the form of Ge-132.46 The levels of Ge observed in our work were considerably lower than the toxic levels indicated in literature (see above). While assessing the concentration of metalloid elements in our leachates via ICP-MS, we observed a maximum Ge concentration of 823.8 mg L−1, in O60 leachates after 3 days of dissolution, the toxicity of which, is attributed to the acidic extracellular microenvironment, generated by the drop in pH. We also observed a maximum Ge concentration of 1.02 mg L−1 and 2.63 mg L−1, in AD and PE leachates respectively, after 14 days of dissolution, and no toxicity was observed. GeO2 is therefore assumed not to contribute to cell death observed in our work.
Regarding antimony, a weak congruency exists amongst investigators on the toxicity of Sb2O3. Titma et al.47 have shown that, when human lung epithelial (A549) cells are exposed to 100 μg mL−1 of Sb2O3, cell viability falls from approximately (approx.) 170% after 12 hours, to approx. 30% after 36 hours. This agrees with the conclusions drawn by Mann et al.48 and Verdugo et al.49 that Sb2O3, Sb(III) and Sb(V), inhibit cell growth and induce apoptosis, by utilising similar signalling pathways to As2O3, As(III) and As(V), resulting in similar toxicities in acute promyelocytic leukaemia (NB4 APL) and human embryonic kidney (HEK-293) cell lines. On the contrary, Omura et al.50 showed a lack of toxicity resulting from Sb2O3 on the count, motility and morphology of rat and mouse sperm cells. Even at a repetitive administration of a 1.2 g kg−1 dose for 4 weeks, no toxicity was reported in mouse or rat testes. A possible explanation for this discrepancy is that the tissue distribution of Sb2O3 does not involve the testes, but primarily, the liver and kidneys. To add to the incongruity, Bregoli et al.,51 within the same study, showed that Sb2O3 nanoparticles (NPs) demonstrated toxicity towards primary cultures of human hematopoietic progenitor cells but not to human hematopoietic immortalised cell lines. The authors assigned this difference to the tumorigenic alterations of cells lines. In our studies of enforced surface oxidation of Ge–Sb–Se glass optical fibres, we cannot rule out the presence also of antimony pentoxide, however we find no toxicology reports about this particular oxide. As discussed in ref. 52, the human body is primarily exposed to antimony, via the inhalation route. Exposure via the gastrointestinal (GI) tract is low (<1%) and is limited by the emetic properties of antimony compounds. Finally, uptake through the skin makes no significant contribution to systemic exposure.52 The occupational aerosol exposure limit set by the American Conference of Governmental Industrial Hygienists (ACGIH) is 0.5 mg m−3.53 Cases where exposures are significantly in excess of this (4 mg m−3 to 86 mg m−3),12,14 have been associated with pulmonary toxicity.12,14,53 Pustular skin eruptions and antimony dermatosis have also been observed in people working with antimony and antimony salt.12,14,54
In our work, Sb ion concentration (see Fig. 1) in AD leachates after leaching in deionised water for 24 h and 72 h elution, at 37 °C (0.194 mg L−1 (i.e. 194.4 mg m−3) and 0.165 mg L−1 (i.e. 165.2 mg m−3), respectively) far exceeded the limits set by the ACGIH (0.5 mg m−3). Although elution was in aqueous media, and not in pure deionised water, it is surmised that the eluant Sb concentration would very likely have mirrored the aqueous leachate Sb concentration. It is notable that no cytotoxicity was observed in cells exposed to these eluates. These results seem to contradict Titma et al.,47 who showed that 100 μg mL−1 of Sb2O3 NPs result in a drop in cell viability from 100% to ∼45% in Balb/c 3T3 cells. These discrepancies are discussed later in this Discussion.
Selenium (Se) is an essential trace element incorporated into selenoproteins as selenocysteine. There are 25 selenium-based proteins in the human selenoproteome, with functions ranging from protection against oxidative damage to the regulation of cellular processes.14 Conversely, Se compounds have been shown to possess a clear cytotoxic activity against malignant cells.55,56 Forootanfar et al.57 investigated the cytotoxicity of SeO2 and Se NPs (spherical and ranging from 80 nm to 220 nm in size), using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. They found that the IC50 was 6.7 μg mL−1 ± 0.8 μg mL−1 in the MCF-7 cell-line treated with SeO2. Se NPs produced the same effect but at a significantly higher concentration of 41.5 μg mL−1 ± 0.9 μg mL−1. In previous work by Suzuki et al.,55 it was concluded that Se compounds induced apoptosis by causing endoplastic reticulum stress and activating the intrinsic apoptotic pathway. We however, observed no cytotoxic effect on cells exposed to AD eluates, containing 0.05 mg L−1 (after a 72 h elution time), see Fig. 1.
A tentative explanation for the discrepancies with the literature discussed here lie in the action of Se in high oxidative stress situations, and its interaction with As (and by extension, Sb). Firstly, it has previously been shown58 that Se-NPs activate the ATF4, SOD2 and Bcl-xL genes (the anti-oxidative stress pathway), to counter hydroquinone (HQ) induced oxidative stress and protect human dermal fibroblast cells. Additionally, internal ROS species were determined to be lower per cell, in cases where cells were exposed to Se-NPs (during HQ induced stress) in comparison to no exposure. Secondly, some authors have found that Se decreases the toxicity of As.59,60 As and Se have similar methylation pathways, and so, can mutually impede the secretion of their methylation metabolite.61 This led to the hypothesis that an As–Se compound is formed, that causes less damage on cells than As or Se alone.62 This compound was first discovered as seleno Bis(S-glutathionyl) arsinium ion [(GS)2AsSe]−.63 It was later found that simultaneous entry of As and Se into a cell, results in the formation of (GS)2AsOH first, which is then attacked by HSe−, displacing its –OH group, to form [(GS)2AsSe]−,64 which can later be excreted from the cell. Furthermore, Rossman and Uddin,65 proposed that Se can guard against As induced oxidative damage through the upregulation of selenoproteins, thioredoxin reductase and glutathione peroxidase. Due to the similar chemical and toxicological characteristics of As and Sb, we suggest that a similar interaction between Se and Sb, forming a seleno–antimony (Sb–Se) detoxication conjugate may have occurred in cells exposed to AD and PE eluates, and so, resulted in the mutual reduction of the toxicity of Sb and Se, and provided increased protection from Sb or Se based oxidative stress. This hypothesis is supported by Feng et al.,66 who showed that Se alleviated the toxicity of Sb in plants by inhibiting the uptake of Sb and relieving oxidative stress derived from Sb exposure.67
The level of Se generated in this study (discounting the levels produced in O60 leachates) is far lower than the permissible exposure limit of Se (400 μg day−1).68 The level of Sb, produced in this study (discounting levels produced in O60 leachates) is however, higher than the permissible exposure limit set by the ACGIH (0.5 mg m−3).53 We have however shown that, even when this is the case, there is a lack of cytotoxicity observed from AD fibres (via cell adhesion and cell propagation). This demonstrates the potential biocompatibility of medical devices developed using the Ge20Sb10Se70 at% glass composition.
Here, the surface texture of Ge20Sb10Se70 at% optical fibres was modified by etching with propylamine under ambient conditions or through oxidation by H2O2(aq.) (see Section 2.1). The results (shown in Fig. S1 and S2 of ESI†) have demonstrated that Ge20Sb10Se70 at% optical fibres are more susceptible to surface damage caused by oxidation by H2O2(aq.) for 60 min, than propylamine etching, as surfaces that underwent the former, generated the rougher surfaces (PE Sa = 169 nm ± 15 nm and O60 Sa = 236 nm ± 84 nm), results not shown here. This is not believed to have influenced cell viability, as cells would not have had the time to respond to the altered rougher surface due to the drastically altered chemistry of the cellular microenvironment after exposure to oxidised eluates and fibre surfaces.
5. Conclusions
Using a 3T3 fibroblast cell model, we provide evidence that unmodified Ge20Sb10Se70 at% optical fibres do not produce a cytotoxic response. A cytotoxic response was only observed after a glass oxide layer was forced to develop on unmodified, as-annealed, Ge20Sb10Se70 at% optical fibres, through submersion in H2O2(aq.). Such oxide growth does not occur under normal ambient conditions, nor on exposure to pH neutral water, but on exposure to a strong oxidising agent like H2O2(aq.). The primary cause of toxicity is attributed to the dramatic pH change that occurred in the extracellular environment, after interaction with the glass oxide. This drop in pH from the physiological 7.49 to the acidic 2.44 resulted in a +70% decrease in cell viability after 24 h in both elution and direct contact trials. A secondary negative influence on cell viability is attributed to the presence of Sb2O3 and SeO2 in solution. The lack of cytotoxicity in cells exposed to AD and PE samples is attributed to the mutual antagonistic effects of Sb and Se. The lack of cytotoxicity from the unmodified fibres in our work, demonstrates the potential biocompatibility of medical devices developed using the Ge20Sb10Se70 at% glass composition.Funding
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC) [via grant number EP/N50970X/1 and through the thematic programme: Wave Phenomena in Complex Media and EP/M008983/1].Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Saul Vazquez Reina and Dr Liz Bailey, from University Park, University of Nottingham, UK, for their technical help in measuring the concentrations of Ge, Sb and Se in our eluates.References
- M. Hermes, R. B. Morrish, L. Huot, L. Meng, S. Junaid, J. Tomko, G. R. Lloyd, W. T. Masselink, P. Tidemand-Lichtenberg, C. Pefersen, F. Palombo and N. Stone, Mid-IR hyperspectral imaging for label-free histopathology and cytology, J. Opt., 2018, 20, 21 CrossRef.
- BS-EN-ISO, Optics and photonics. Spectral bands. (BS ISO 20473:2007), British Standards Institution, 2007, p. 10 Search PubMed.
- M. C. Guidi, C. Mirri, E. Fratini, V. Licursi, R. Negri, A. Marcelli and R. Amendola, In vivo skin leptin modulation after 14 MeV neutron irradiation: a molecular and FT-IR spectroscopic study, Anal. Bioanal. Chem., 2012, 404(5), 1317–1326 CrossRef.
- O. J. Old, G. R. Lloyd, J. Nallal, M. Isabelle, L. M. Almond, N. A. Shepherd, C. A. Kendall, A. C. Shore, H. Barr and N. Stone, Rapid infrared mapping for highly accurate automated histology in Barrett's oesophagus, Analyst, 2017, 142(8), 1227–1234 RSC.
- M. J. Pilling, A. Henderson and P. Gardner, Quantum cascade laser spectral histopathology: breast cancer diagnostics using high throughput chemical imaging, Anal. Chem., 2017, 89(14), 7348–7355 CrossRef CAS.
- L. Shi and R. Alfano, Deep imaging in tissue and biomedical materials, using linear and nonlinear optical methods, Pan Stanford Publishing Pte. Ltd, Kent, 2017 Search PubMed.
- A. Seddon, Chalcogenide Glasses: A Review of their Preparation, Properties and Applications, J. Non-Cryst. Solids, 1995, 184, 44–50 CrossRef CAS.
- H. Parnell, Towards a Ge-Sb-Se/S hyperspectral imaging probe for early cancer diagnosis, University of Nottingham, Nottingham, 2018 Search PubMed.
- A. B. Seddon, Chalcogenide glass fibreoptics for new, mid-infrared medical endoscopy, Optical Fibers and Sensors for Medical Diagnostics and Treatment Applications XII, 2012, vol. proceedings, vol. 8218 Search PubMed.
- A. A. Wilhelm, P. Lucas, D. L. DeRosa and M. R. Riley, Biocompatibility of Te-As-Se glass fibres for cell-based bio-optic infrared sensors, J. Mater. Res. Technol., 2007, 22(4), 1098–1104 CrossRef CAS.
- H. Parnell, D. Furniss, Z. Tang, N. C. Neate, T. M. Benson and A. B. Seddon, Compositional dependence of crystallization in Ge–Sb–Se glasses relevant to optical fiber making, J. Am. Ceram. Soc., 2017, 101, 208–2019 CrossRef.
- V. Potkonjak and M. Pavlovich, Antimoniosis: A particular form of pneumoconiosis, Int. Arch. Occup. Environ. Health, 1983, 51, 199–207 CrossRef CAS.
- T. Gebel, Arsenic and antimony: comparative approach on mechanistic toxicology, Chem.-Biol. Interact., 1997, 107, 131–144 CrossRef CAS.
- G. E. Nordberg, B. A. Fowler and M. Nordberg, Handbook on the Toxicology of Metals, Elsevier, London, 2015 Search PubMed.
- M. De Boeck, M. Kirsch-Volders and D. Lison, Cobalt and antimony: genotoxicity and carcinogenicity, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 2003, vol. 533, pp. 135–152 Search PubMed.
- C. Grosskopf, T. Schwerdtle, L. H. F. Mullenders and A. Hartwig, Antimony impairs nucleotide excision repair: XPA and XPE as potential molecular targets, Chem. Res. Toxicol., 2010, 23(7), 1175–1183 Search PubMed.
- K. Kuroda, G. Endo, A. Okamoto, Y. S. Yoo and S. Horiguchi, Genotoxicity of beryllium, gallium and antimony in short-term assays, Mutat. Res. Lett., 1991, 264(4), 163–170 Search PubMed.
- D. Cavallo, I. Iavicoli, A. Setini, A. Marinaccio, B. Perniconi, G. Carelli and S. Iavicoli, Genotoxic risk and oxidative DNA damage in workers exposed to antimony trioxide, Environ. Mol. Mutagen., 2002, 40, 184–189 CrossRef CAS.
- P. Lucas, A. Wilhelm, M. Videa, C. Boussard-Pledel and B. Bureau, Chemical stability of chalcogenide infrared glass fibres, Corros. Sci., 2008, 50(7), 2047–2052 CrossRef CAS.
- BS-EN-ISO, Biological evaluation of medical devices: Part 12: Sample preparation and reference materials (10993-12:2012), British Standard, 2012 Search PubMed.
- G. Repetto, A. Peso and J. Zurita, Neutral red uptake assay for the estimation of cell viability/cytotoxicity, Nat. Protoc., 2008, 3(7), 1125–1131 CrossRef CAS.
- BS-EN-ISO, Biological evaluation of medical devices: Part 5: Tests for in vitro cytotoxicity, ISO 10993-5:2009), British Standard, 2009 Search PubMed.
- M. Nagy, D. Racz, Z. L. Nagy, P. P. Feher, S. L. Kovacs, C. Banko, Z. Basco, A. Kiss, M. Zsuga and S. Keki, Amino-isocyanoacridines: Novel, Tunable Solvatochromic Fluorophores as Physiological pH Probes, Sci. Rep., 2019, 9(8250), 1–12 CAS.
- J. Teixeira, F. Basit, H. G. Swarts, M. Forkink, P. J. Olivera, P. H. G. M. Willems and W. J. H. Koopman, Extracellular acidification induces ROS- and mPTP-mediated death in HEK293 cells, Redox Biol., 2018, 15, 394–404 CrossRef CAS.
- T. Nordstrom, K. Rajamaki, K. Murmi, K. E. O. Akerman, P. T. Kovanen, K. Oorni and K. K. Eklund, Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome, J. Biol. Chem., 2013, 288(19), 13410–13419 CrossRef.
- V. Sharma, R. Kaur, A. Bhatnagar and J. Kaur, Low-pH-induced apoptosis: role of endoplasmic reticulum stress-induced calcium permeability and mitochondria-dependent signaling, Cell Stress Chaperones, 2015, 20(3), 431–440 CrossRef CAS.
- M. L. Smith, R. v. Hanwehr and B. K. Siesjo, Changes in extra- and intracellular pH in the brain during and following Ischemia in hyperglycemic and moderately hypoglycemic rats, J. Cereb. Blood Flow Metab., 1986, 6(5), 574–583 CrossRef CAS.
- V. A. Ruffin, A. I. Salameh, W. F. Boron and M. D. Parker, Intracellular pH regulation by acid-base transporters in mammalian neurons, Front. Membr. Physiol. Membr. Biophys., 2014, 5(43), 1–11 Search PubMed.
- P. Ayers and L. Warrington, Diagnosis and treatment of simple acid-base disorders, Nutr. Clin. Pract., 2008, 23(2), 122–127 CrossRef.
- S. W. Caito and M. Aschner, Mitochondrial redox dysfunction and environmental exposures, Antioxid. Redox Signaling, 2015, 23(6), 578–595 CrossRef CAS.
- T. R. Figueira, M. H. Barros, A. A. Camargo, R. F. Castilho, J. C. B. Ferreira, A. J. Kowaltowski, F. E. Sluse, N. C. Souza-Pinto and A. E. Vercesi, Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health, Antioxid. Redox Signaling, 2013, 18(16), 2029–2074 CrossRef CAS.
- M. Nita and A. Grzybowski, The role of the reactive oxygen species and oxidative stress in pathomechanism of age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults, Oxid. Med. Cell. Longevity, 2015, 2016, 3164734 Search PubMed.
- W. Droge, Free radicals in the physiological control of cell function, Physiol. Rev., 2002, 82(1), 47–95 CrossRef CAS.
- L. Xue and J. M. Lucocq, Low extracellular pH induces activation of ERK 2, JNK, and p38 in A431 and Swiss 3T3 cells, Biochem. Biophys. Res. Commun., 1997, 241, 236–242 CrossRef CAS.
- A. Zeke, M. Misheva, A. Remenyi and M. A. Bogoyevitch, JNK signalling: Regulation and functions based on complex protein-protein partnerships, Microbiol. Mol. Biol. Rev., 2016, 80(3), 793–835 CrossRef CAS.
- J. Liu and A. Lin, Role of JNK activation in apoptosis: A double-edged sword, Cell Res., 2005, 15, 36–42 CrossRef.
- S. Jaworski, E. Sawosz, M. Kutwin, M. Wierzbicki, M. Hinzmann, M. Grodzik, A. Winnicka, L. Lipinska, K. Wlodyga and A. Chwalibog, In vitro and in vivo effects of graphene oxide and reduced oxide on glioblastoma, Int. J. Nanomed., 2014, 10(1), 1585–1596 Search PubMed.
- M. Vinardell and M. Mitjans, Metal/metal oxide nanoparticles for cancer therapy, Nanooncology, Springer, Cham, 2018, pp. 341–364 Search PubMed.
- M. P. Vinardell and M. Mitjans, Antitumor activities of metal oxide nanoparticles, Nanomaterials, 2015, 5(2), 1004–1021 CrossRef CAS.
- A. B. Djurisic, Y. H. Leung, A. M. C. Ng, X. Y. Xu, P. K. H. Lee, N. Degger and R. S. S. Wu, Toxicity of metal oxide nanoparticles: mechanisms, characterisation and avoiding experimental artefacts, Small, 2015, 11(1), 26–44 CrossRef CAS.
- R. N. Ratnaike, Acute and chronic arsenic toxicity, Postgrad. Med. J., 2003, 79(933), 391–396 CrossRef CAS.
- J. Hu, Q. Lu, C. Wu, M. Liu, H. Li, Y. Zhang and S. Yao, Germanium nanoparticles: Intrinsic peroxidase-like catalytic activity and its biosensing application, Talanta, 2019, 195, 407–413 CrossRef CAS.
- G. B. Gerber and A. Leonard, Mutagenicity, carcinogenicity and teratogenicity of germanium compounds, Mutat. Res., 1997, 387, 141–146 CAS.
- S. H. Tao and P. M. Bolger, Hazard assessment of germanium supplements, Regul. Toxicol. Pharmacol., 1997, 25(3), 211–219 CrossRef CAS.
- M. Lin, T. Hsu, P. Yang, M. Tsai, T. Perng and L. Lin, Comparison of organic and inorganic germanium compounds in cellular radiosensitivity and preparation of germanium nanoparticles as a radiosensitizer, Int. J. Radiat. Biol., 2009, 85(3), 214–226 CrossRef CAS.
- T. Nakamura, T. Nagura, K. Sato and M. Ohnishi, Evaluation of the effects of dietry organic germanium, Ge-132 and raffinose supplementation on caecal flora in rats, Biosci. Microbiota, Food Health, 2012, 31(2), 37–45 CrossRef.
- T. Titma, R. Shimmo, J. Siigur and A. Kahru, Toxicity of antimony, copper, cobalt, manganese, titanium and zinc oxide nanoparticles for the alveolar and intestinal epithelial barrier cells in vitro, Cytotechnology, 2016, 68, 2363–2377 CrossRef CAS.
- K. K. Mann, K. Davison, M. Colombo, A. L. Colosimo, Z. Diaz, A. M. S. Padovani, Q. Guo, P. J. Scrivens, W. Gao, S. Mader and W. H. Miller Jr, Antimony trioxide-induced apoptosis is dependent on SEK1/JNK signaling, Toxicol. Lett., 2006, 160, 158–170 CrossRef CAS.
- M. Verdugo, Y. Ogra and W. Quiroz, Mechanisms underlying the toxic effects of antimony species in human embryonic kidney cells (HEK-293) and their comparison with arsenic species, J. Toxicol. Sci., 2016, 41(6), 783–792 CrossRef CAS.
- M. Omura, A. Ranaka, M. Hirata and N. Inoue, Testicular toxicity evaluation of two antimony compounds, antimony trioxide and antimony potassium tartrate, in rats and mice, Environmental Health and Preventative Medicine, 2002, 7, 15–18 CrossRef CAS.
- L. Bregoli, F. Chiarini, A. Gambarelli, G. Sighinolfi, A. M. Gatti, P. Santi, A. M. Martelli and L. Cocco, Toxicity of antimony trioxide nanoparticles on human hematopoietic progenitor cells and comparison to cell lines, Toxicology, 2009, 262, 121–129 CrossRef CAS.
- ASTOR, Toxicological profile for antimony and compounds, Agency for Toxic Substances and Disease Registry, Division of Toxicology and Human Health Sciences, Atlanta, Georgia, 2019 Search PubMed.
- C. J. Boreiko and T. G. Rossman, Antimony and its compounds: Health impacts related to pulmonary toxicity, cancer, and genotoxicity, Toxicol. Appl. Pharmacol., 2020, 403, 1–17 CrossRef.
- G. P. White Jr, C. G. Mathias and J. S. Davin, Dermatitis in workers exposed to antimony in a melting process, J. Occup. Med., 1993, 35(4), 392–395 CAS.
- M. Suzuki, M. Endo, F. Shionhara, S. Echigo and H. Rikiishi, Differential apoptotic response of human cancer cells to organo selenium compounds, Cancer Chemother. Pharmacol., 2010, 66, 475–484 CrossRef CAS.
- M. Wallenberg, M. Sougat and M. Bjornstedt, Selenium cytotoxicity in cancer, Basic Clin. Pharmacol. Toxicol., 2014, 114, 377–386 CrossRef CAS.
- H. Forootanfar, M. Adeli-Sardou, M. Nikkhoo, M. Mehrabani, B. Amir-Heidari, A. R. Shahverdi and M. Shakibaie, Antioxidant and cytotoxic effect of biologically synthesised selenium nanoparticles in comparison to selenium dioxide, J. Trace Elem. Med. Biol., 2014, 28, 75–79 CrossRef CAS.
- S. Chung, A. K. Roy and T. J. Webster, Selenium nanoparticle protection of fibroblast stress: activation of ATF4 and Bcl-xL expression, Int. J. Nanomed., 2019, 14, 9995–10007 CrossRef CAS.
- R. E. Holmberg Jr and V. H. Ferm, Interrelationships of selenium, cadmium, and arsenic in mammalian teratogenesis, Arch. Environ. Health, 1969, 18(6), 873–877 CrossRef CAS.
- S. Biswas, G. Talukder and A. Sharma, Prevention of cytotoxic effects of arsenic by short-term dietary supplementation with selenium in mice, in vivo, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 1999, 441(1), 155–160 CrossRef CAS.
- O. A. Levander and L. C. Argrett, Effects of arsenic, mercury, thallium, and lead on selenium metabolism in rats, Toxicol. Appl. Pharmacol., 1969, 14(2), 308–314 CrossRef CAS.
- O. A. Levander and C. A. Baumann, Selenium metabolism: VI. Effect of arsenic on the excretion of selenium in the bile, Toxicol. Appl. Pharmacol., 1966, 9(1), 106–115 CrossRef CAS.
- J. Gailer, G. N. George, I. J. Pickering, R. C. Prince, S. C. Ringwald, J. E. Pemberton, R. S. Glass, H. S. Younis, D. W. DeYoung and H. V. Aposhian, A metabolic link between arsenite and selenite: The seleno-bis(S-glutathionyl) arsinium ion, J. Am. Chem. Soc., 2000, 122(19), 4637–4639 CrossRef CAS.
- J. Gailer, G. N. George, I. J. Pickering, G. A. Buttigieg, M. B. Denton and R. S. Glass, Synthesis, X-ray absorption spectroscopy and purification of the seleno-bis (S-glutathionyl) arsinium anion from selenide, arsenite and glutathione, J. Organomet. Chem., 2002, 650(1–2), 108–119 CrossRef CAS.
- T. G. Rossman and A. N. Uddin, Selenium prevents spontaneous and arsenite-induced mutagenesis, Int. Congr. Ser., 2004, 1275, 173–179 CrossRef.
- R. Feng, C. Wei, S. Tu and Z. Liu, Interactive effects of selenium and antimony on the uptake of selenium, antimony and essential elements in paddy rice, Plant Soil, 2013, 365, 375–386 CrossRef CAS.
- R. Feng, C. Wei, S. Tu, S. Tang and F. Wi, Detoxification of antimony by selenium and their interaction in paddy rice under hydroponic conditions, Microchem. J., 2011, 97(1), 57–61 CrossRef CAS.
- S. M. Hays, K. Macey, A. Nong and L. L. Aylward, Biomonitoring Equivalents for selenium, Regul. Toxicol. Pharmacol., 2014, 70, 333–339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00353k |
| This journal is © The Royal Society of Chemistry 2021 |