
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-stimuli pseudorotaxane switches under kinetic control†‡
Marius
Gaedke
 a,
Henrik
Hupatz
a,
Hendrik V.
Schröder§
a,
Simon
Suhr
b,
Kurt F.
Hoffmann
c,
Arto
Valkonen
d,
Biprajit
Sarkar
b,
Sebastian
Riedel
c,
Kari
Rissanen
d and
Christoph A.
Schalley
*a
a,
Henrik
Hupatz
a,
Hendrik V.
Schröder§
a,
Simon
Suhr
b,
Kurt F.
Hoffmann
c,
Arto
Valkonen
d,
Biprajit
Sarkar
b,
Sebastian
Riedel
c,
Kari
Rissanen
d and
Christoph A.
Schalley
*a
aInstitut für Chemie und Biochemie der Freien Universität Berlin, Arnimallee 20, 14195 Berlin, Germany. E-mail: c.schalley@fu-berlin.de
bLehrstuhl für Anorganische Koordinationschemie, Institut für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
cInstitut für Chemie und Biochemie der Freien Universität Berlin, Fabeckstr. 34/36 14195, Berlin, Germany
dDepartment of Chemistry P.O. Box 35, 40014 Jyväskylä, Finland
First published on 29th April 2021
Abstract
A series of dumbbell-shaped sec-ammonium salts with bulky (pseudo)stoppers (‘speed bumps’) were tested for their ability to form pseudorotaxanes with a redox-switchable, tetrathiafulvalene (TTF)-decorated [24]crown-8 ether. Depending on the size of the pseudostoppers, fast (less than ten minutes), slow (hours to days) and very slow (no pseudorotaxanes observed) threading has been observed. NMR spectroscopy as well as tandem mass spectrometry indicate the formation of non-threaded face-to-face complexes prior to pseudorotaxanes formation. Both isomers can be distinguished by their substantially different stability in collision-induced dissociation (CID) experiments. Two external stimuli affect the stability of the pseudorotaxanes: Deprotonation of the ammonium ion results in fast dethreading, while dethreading is much slower when induced by the charge repulsion upon chemical oxidation of the TTF moiety. Remarkably, the same steric bulk of the pseudostopper thus leads to different dethreading rates depending on the stimulus applied. Based on these findings, two redox-switchable rotaxanes containing a 1-naphthyl and a phenyl moiety as sterically different ‘speed bumps’ in the axle centre were synthesised. Bulk electrolysis of the rotaxanes did not result in the expected macrocycle translocation on the axle independent of the ‘speed bump’ as a remarkable consequence of the mechanical bond.
Introduction
Artificial molecular switches1 are controlled by external chemical,2,3 light,4,5 or electrochemical6 stimuli. In mechanically interlocked molecules (MIMs) such as rotaxanes and catenanes, stimuli-induced switching manipulates the interactions of the wheels with their binding sites on the other component in a controlled manner.7,8 Pseudorotaxanes are non-interlocked, but threaded axle-wheel complexes able to dissociate without breaking a covalent bond. Depending on the bulkiness of the axle end groups, they encompass a broad range of dethreading rates with the rotaxane at the very end, where no wheel exchange takes place (Fig. 1a).9 Thus, (pseudo)rotaxanes allow us to study stimuli-induced switching with and without wheel exchange. In rotaxanes, the wheel's translational freedom is limited to the track provided by the axle10 and switching can be converted into directed co-conformational changes, which can propel artificial molecular motors and machines.7,8,11 The impact of the wheel exchange rate on dual-stimuli-induced switching has not been studied for kinetically hindered pseudorotaxanes, but rather for pseudorotaxanes with fast exchange or for rotaxanes (no exchange). This raises an interesting question: Can different stimuli applied to a dual-stimuli-responsive and kinetically hindered pseudorotaxane result in different timescales for dethreading over a ‘speed bump’ pseudostopper, even when the steric size of the ‘speed bump’ remains unchanged?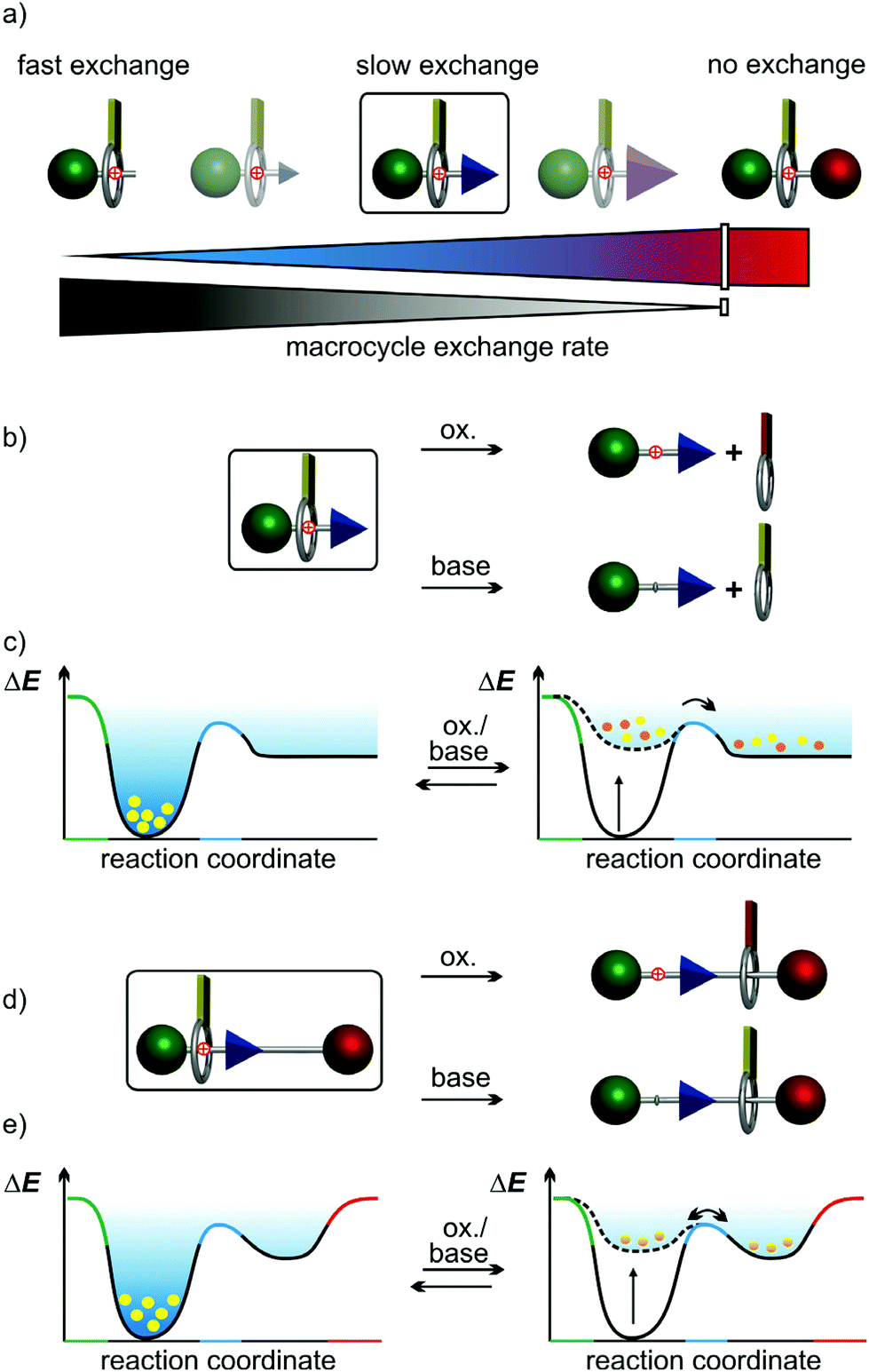 | ||
| Fig. 1 (a) Range of macrocycle exchange rates from a quickly reversible pseudorotaxane formation (light blue, small ‘speed bump’) to a fully interlocked rotaxane (red stopper). (b) Stimuli-induced shuttling or unthreading and (c) the corresponding changes of the energy landscapes of a pseudorotaxane as well as (d and e) of a rotaxane (stoppers: green/red, steric ‘speed bump’ in the axle: blue, TTF oxidation states in yellow (reduced)/orange (oxidized to TTF2+). | ||
The energy landscapes of switchable (pseudo)[2]rotaxanes change, when stimuli are applied. In Fig. 1, which exemplarily depicts an acid/base- and redox-switchable pseudorotaxane (Fig. 1b and c) and its ‘speed bump’ containing rotaxane analogue (Fig. 1d and e), the yellow dots represent the (pseudo)rotaxane populations. Initially, the wheels reside on the binding site. Upon applying one of the two stimuli, the former binding site becomes disfavoured and the macrocycles migrate over the blue ‘speed bump’. For the pseudo[2]rotaxane, the wheel is released from the axle over the barrier caused by the pseudostopper, while the wheel cannot escape from the axle in the [2]rotaxane. It will nevertheless move across the ‘speed bump’ back and forth and a new equilibrium distribution of positional isomers is finally reached on a time scale depending on the ‘speed bump’ size.
From the broad variety of available binding motifs,12,13sec-ammonium/crown ether (pseudo)rotaxanes provide an easy access to dual-stimuli-responsive compounds that allow us to investigate the above question in detail. In this motif, a crown ether encircles the ammonium ion with binding constants typically in the range of 102 to 106 M−1 in non-protic organic solvents. In thermodynamic terms, ammonium ions form threaded complexes depending on the number of hydrogen bonds (including C–H⋯O hydrogen bonds involving the methylene groups next to the ammonium ion), charge distribution and ion pair separation energies of the ammonium salt.14,15 Only face-to-face interactions are possible,16 when the axle end groups are sterically too bulky. A large number of pseudo[2]rotaxanes with different crown ether sizes and different end groups have been reported to form in a broad variety of solvents.17–19 In marked contrast, only few examples of rotaxanes with ‘speed bumps’ along their axles have been studied to unravel the influence of the mechanical bond.20–25 The studies on shuttling in crown ether rotaxanes mostly focus on degenerate shuttles with two or more equivalent binding sites.20,24 However, non-degenerate shuttles are more suitable for directional movement,26 among which only very few are responsive to both acid/base and redox stimuli.27
Here, we make use of the sec-ammonium/crown ether binding motif and describe a series of pseudorotaxanes responsive to both stimuli with differently sized and differently shaped pseudostoppers. The ammonium/crown ether interaction can be disrupted by deprotonation of the ammonium group with a strong base.28 Introducing a tetrathiafulvalene (TTF) into the crown ether (TTFC8, Fig. 2) implements also the second stimulus: Two-electron oxidation of the TTF unit to its dication induces coulombic repulsion between the wheel and the ammonium station and is expected to lead to dethreading as well.29,30 Hence, these dual-stimuli-responsive pseudorotaxanes allow comparing the dethreading rates after deprotonation with those after oxidation, while maintaining the same steric bulk of the ‘speed bump’ pseudostopper. After identifying a suitable pseudostopper in pseudorotaxane dethreading experiments, this ‘speed bump’ can be integrated into a rotaxane axle to investigate co-conformational changes in the dual-stimuli-responsive [2]rotaxane, in which the mechanical bond prevents dethreading.
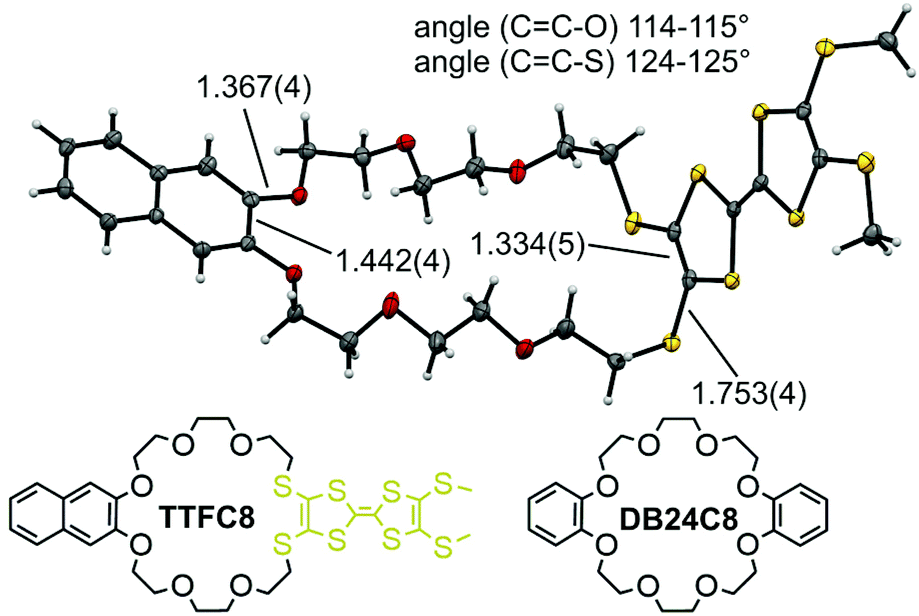 | ||
| Fig. 2 Solid-state structure of TTFC8 with selected bond lengths (Å) and angles. | ||
Results and discussion
The wheel: crystal structure of TTFC8
The redox-active wheel TTFC8 used in this study was synthesized as reported earlier.29 This macrocycle and the commercially available and widely used dibenzo[24]crown-8 ether (DB24C8) have the same number of 24 atoms along their circumference. Nevertheless, the substitution of two catechol oxygen by sulfur atoms affects both the structure and the conformational properties of the ring,31 as becomes clear, when comparing the new solid state structure of TTFC8 (Fig. 2, also see ESI section 8‡) with the known structure of DB24C8.31The naphthalene-substituted side of TTFC8 only shows a minor elongation in the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (1.44 Å compared to 1.41 Å for DB24C8), the C–O bond lengths and CC–O angles are virtually the same (1.37 Å and 115°). Although a shorter central CC bond (1.33 Å) can be found on the TTF side, the C–S bond lengths (1.75 Å vs. 1.37 Å) and CC–S bond angles (125° vs. 115°) notably enlarge the macrocycle cavity on the TTF-substituted side consistent with other published thiacrown derivatives.32–34 The S atoms are exo to the ring rather than endo, like the oxygens on the other side. The effect of the structure on binding ammonium guests was investigated in our previous paper.30
C bond (1.44 Å compared to 1.41 Å for DB24C8), the C–O bond lengths and CC–O angles are virtually the same (1.37 Å and 115°). Although a shorter central CC bond (1.33 Å) can be found on the TTF side, the C–S bond lengths (1.75 Å vs. 1.37 Å) and CC–S bond angles (125° vs. 115°) notably enlarge the macrocycle cavity on the TTF-substituted side consistent with other published thiacrown derivatives.32–34 The S atoms are exo to the ring rather than endo, like the oxygens on the other side. The effect of the structure on binding ammonium guests was investigated in our previous paper.30
Threading experiments
To test whether these differences in ring annulus lower the barrier to overcome bulky substituents, various sec-ammonium axles were synthesised as tetrakis(3,5-bis(trifluoromethyl)phenyl) borate (BArF24−) salts to compensate for the reduced binding constants of the thiacrown (see ESI section 5‡ for details).29 Using BArF24− with respect to PF6− results in a 10 to 20-fold increase of the binding constant.30 The axles only differ in their terminal groups on one side (shown in blue in Fig. 3). As a guideline for the pseudostopper size and shape literature data available for DB24C8-based (pseudo–)rotaxanes with PF6− counterions was used.16,19,35 | ||
| Fig. 3 The synthesised axles PAn. | ||
The axles were mixed with either DB24C8 or TTFC8 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in CD2Cl2. 1H NMR spectra were taken at regular intervals to compare the half-lives (t1/2) of the threading reactions for 4 mM solutions at room temperature by following the changing integrals of the C
:1 ratio in CD2Cl2. 1H NMR spectra were taken at regular intervals to compare the half-lives (t1/2) of the threading reactions for 4 mM solutions at room temperature by following the changing integrals of the C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–NH2–C2 signals (see Table 1 and ESI section 2 Fig. S27–41‡ for the NMR spectra). Upon threading, the two methylene group signals shift downfield (Δδ ≥ +0.2 ppm). Furthermore, diastereotopic splitting of the crown ether methylene groups is observed for the pseudorotaxanes due to the directionality of the axle.16,36
2–NH2–C2 signals (see Table 1 and ESI section 2 Fig. S27–41‡ for the NMR spectra). Upon threading, the two methylene group signals shift downfield (Δδ ≥ +0.2 ppm). Furthermore, diastereotopic splitting of the crown ether methylene groups is observed for the pseudorotaxanes due to the directionality of the axle.16,36
| Entrya | PA1 | PA2 | PA3 | PA4 | PA5 | PA6 | PA7 | PA8 | PA9 | PA10 | PA11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Axles denoted with “≫14 days” were considered to be not suitable for this study. It does not necessarily exclude a threaded complex to be achievable over time with suitable temperature and concentration. | |||||||||||
| DB24C8 | <10 min | <10 min | ≫14 days | <10 min | ≫14 days | ≫14 days | ≫14 days | <10 min | <10 min | <10 min | 10 days |
| TTFC8 | <10 min | <10 min | ≫14 days | <10 min | 50 h | 14 days | 22 h | <10 min | <10 min | <10 min | 2 h |
An initial kinetic screening categorizes the axles into three groups. The first group (PA1, PA2, PA4, PA8, PA9 and PA10) shows complete conversion into the threaded complex with TTFC8 and DB24C8 within less than 10 min. The second group (PA5, PA6, PA7 and PA11) reveals half-lives in the range of hours to days. In addition, PA5, PA6 and PA7 form pseudorotaxanes with TTFC8, but not with DB24C8 (even within 14 days). Both crown ethers can, however, pass the 1-naphthalene pseudostopper of PA11 to form threaded complexes. Here, the slightly larger TTFC8 threaded onto the axle approximately 120 times more quickly. Finally, no sign of threading is observed for PA3 neither with DB24C8 nor TTFC8 within 14 days. These results are in good agreement with a recent study by Credi et al.19 which also showed the 3,5-dimethyl phenyl moiety to be an efficient stopper in DB24C8 containing rotaxanes37–40 and identified the 2,6-dimethyl phenyl moiety as kinetically inert to threading of DB24C8.19 Consequently, out of these candidates we chose PA11 (1-naphthyl) as the most suitable pseudostopper for dual-stimuli-induced switching experiments, because threading occurs at time intervals that are conveniently observable by NMR spectroscopy.
In summary, the slight differences in size and shape apparent from the comparison of the crystal structures clearly affect the threading kinetics even though TTFC8 and DB24C8 share the same number of atoms in the wheel circumference.
Face-to-face complexes vs. pseudorotaxanes
In order to obtain some orienting insight into the thermodynamic binding properties of PA11, axle PA12 has been synthesized, which allows fast threading of TTFC8 over the phenyl group. The association constant obtained by isothermal titration calorimetry amounts to Ka = (3.9 ± 0.7) 105 M−1 (ΔG° = (−31.9 ± 0.4) kJ mol−1, ΔH° = (−44.1 ± 1.2) kJ mol−1, TΔS° = (−12.2 ± 1.6) kJ mol−1; also, see ESI section 5 and Table S2†). This binding data is very similar to that of the symmetrical dibenzylammonium axle (DBA) (Ka = (7.7 ± 1.0) 105 M−1 (ΔG° = (−33.6 ± 0.3) kJ mol−1, ΔH° = (−39.8 ± 1.0) kJ mol−1, TΔS° = (−6.2 ± 1.3) kJ mol−1). We therefore assume the binding data for PA11 to be in a similar range.An ITC experiment with PA11 was also attempted with titration intervals of 20 min to account for the slow threading. Remarkably, very sharp heat signals were observed. The quick heat evolution is not consistent with the slow pseudorotaxane formation as observed in 1H NMR experiments. Obviously, the axle and the crown ether quickly form a non-threaded complex (Fig. 4a),16 for which an upper limit for Ka of ca. 104 M−1 is estimated (see ESI section 5 and Fig. S53‡). 1H NMR experiments confirm this face-to-face complex formation that is expressed in a minor shift for the methylene protons next to the ammonium moiety. These signals vanish slowly over time with the appearance of pseudorotaxane signals (Fig. S41‡).
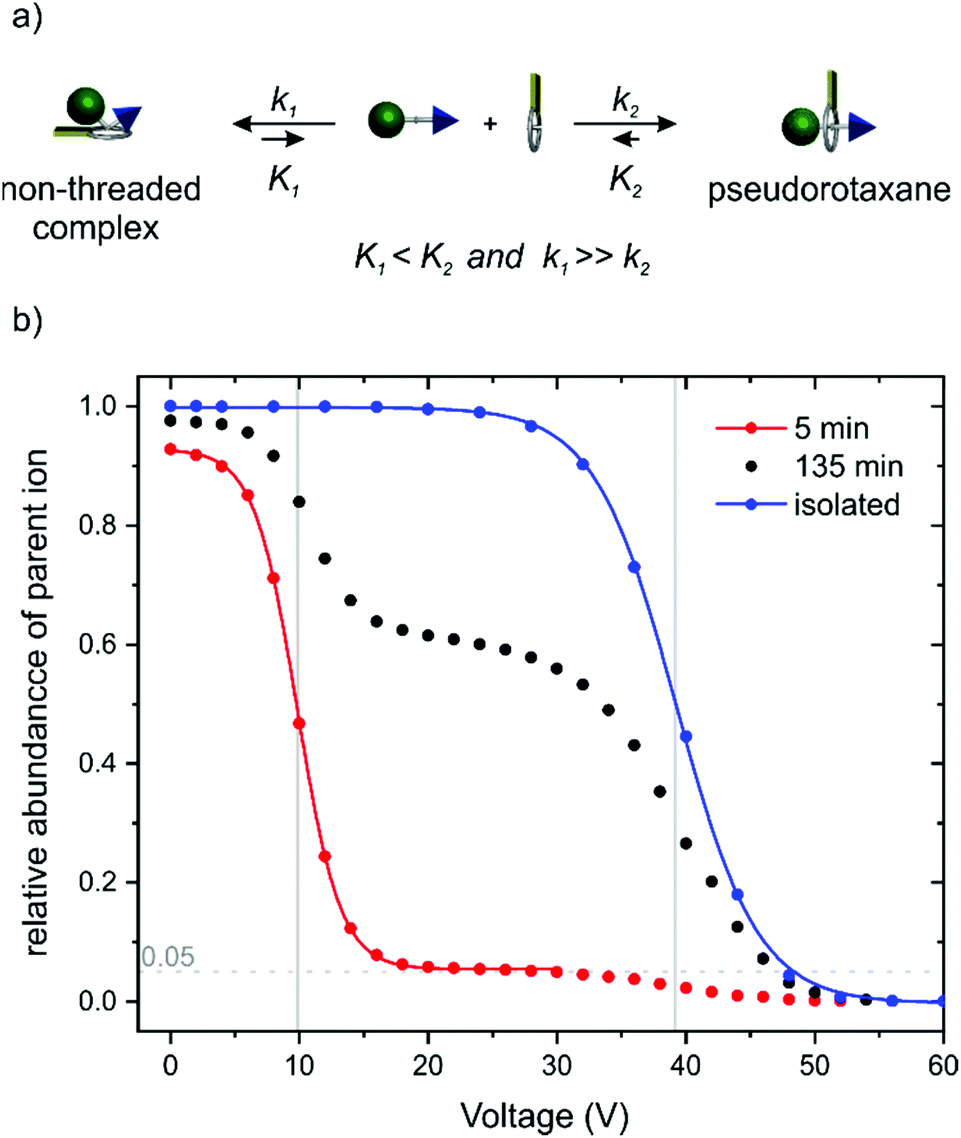 | ||
| Fig. 4 (a) Schematic representation of the equilibrium for face-to-face complexation which competes with the formation of PA11@TTFC8. (b) Survival yield curves obtained for mass-selected [PA11·TTFC8]+ ions (m/z 1108) at increasing collision voltages. Red curve: sample 5 min; black curve: sample 135 min after mixing of axle and wheel. Blue curve: pre-formed pseudorotaxane isolated by column chromatography before the experiment. The solid lines represent a sigmoidal fitting to determine 50% survival yield voltages E1/2: red: E1/2 = (9.8 ± 0.2) V; blue: E1/2 = (39.2 ± 0.4) V (for experimental details, see ESI section 3‡). | ||
Besides the evidence from ITC and NMR experiments, tandem mass spectrometry complements the picture. As one can expect the face-to-face complexes to easily dissociate in the gas phase upon collision-induced dissociation (CID), the dethreading of the pseudorotaxanes should require higher collision energies due to the steric barrier caused by the pseudostopper. Fig. 4b shows the survival yield curves for PA11@TTFC8 separately prepared and isolated by column chromatography (blue) in comparison to a mixture of PA11 and TTFC8 subjected to the collision experiment 5 (red) and 135 minutes (black) after sample preparation. The red curve is indicative of a complex, which fragments at rather low collision voltages (E1/2 = 9.8 ± 0.2 V). A small amount of 5% of the complex remains intact and half of it is fragmented at ca. 39 V. The blue curve shows only one, more stable complex (E1/2 = 39.2 ± 0.4 V). For the sample taken after 135 min roughly 40% of the complex fragment at the lower collision voltage, while 60% fragment at the higher one. Clearly, two differently stable complexes form, i.e. the instantly forming face-to-face complex and the pseudorotaxane, which increases in abundance over time. This nicely shows the conversion of the instantly formed, but weaker non-threaded complex (lower equilibrium constant K1, but higher rate constant k1) into the pseudorotaxane (higher equilibrium constant K2, but lower rate constant k2).
Dual-stimuli responsiveness
When pseudo[2]rotaxane PA11@TTFC8 is deprotonated with an excess of polystyrene-immobilized P2 base, quick dethreading is observed (Fig. 5b and ESI section 2 Fig. S32, S34, S36 and S42‡). No indication for hydrogen bonding of neutral TTFC8 to the neutral axle was observed in the 1H NMR spectra in contrast to earlier reports on complexes of neutral primary amines.41 Dethreading is indicated by an emerging set of methylene proton signals (Hd, He), upfield shifted by Δδ ≈ 0.8 ppm upon deprotonation and dethreading. The pseudo[2]rotaxanes PA5@TTFC8 (2,3-dimethylbenzyl end group), PA6@TTFC8 (2-ethylbenzyl) and PA7@TTFC8 (2-methoxybenzyl) were also subjected to deprotonation. Again, dethreading via deprotonation occurred immediately (see ESI Fig. S42 and Table S1‡).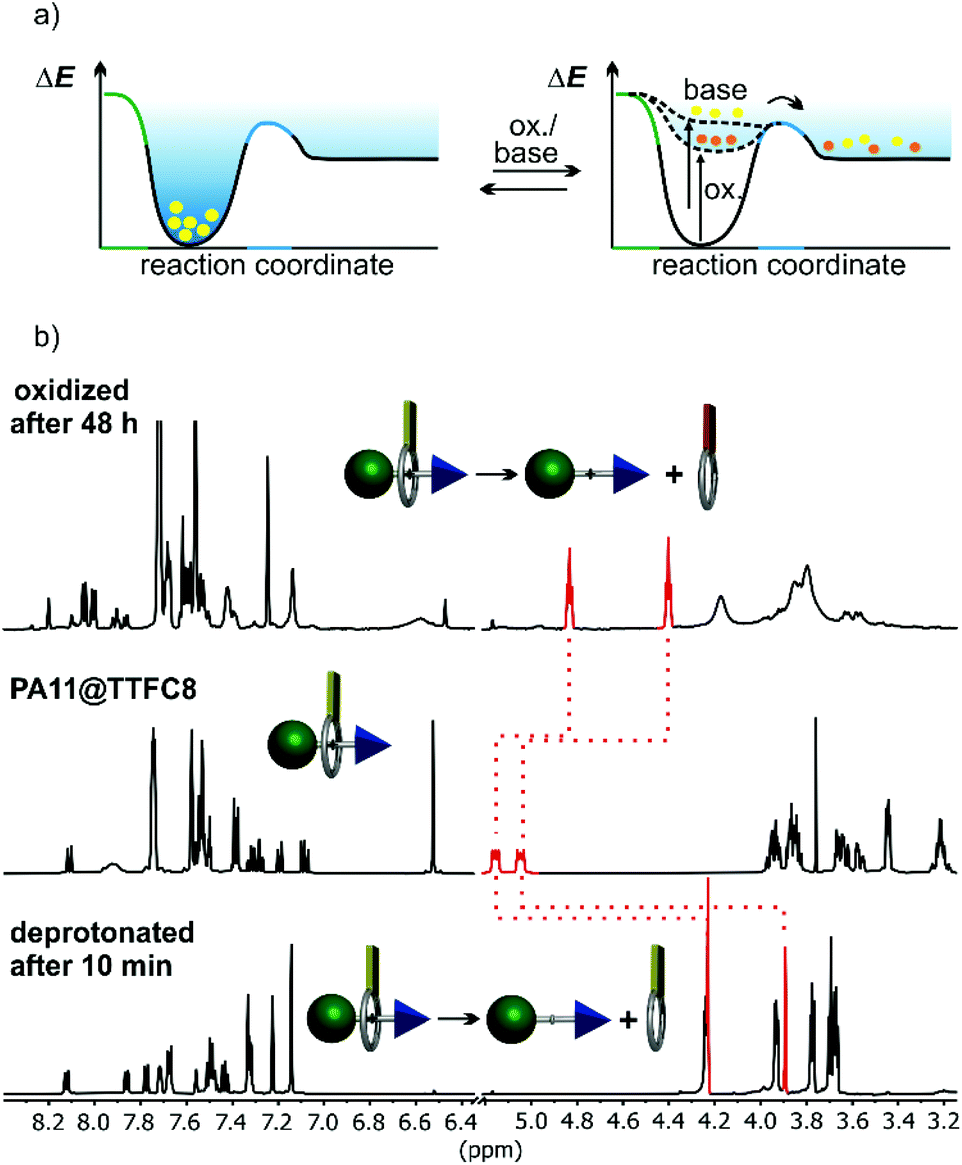 | ||
| Fig. 5 (a) Dual-stimuli switching expressed in terms of the potential energy surface of the pseudorotaxane PA11@TTFC8. (b) 1H NMR spectra (600 MHz, dry CD2Cl2, 298 K) of PA11@TTFC8 (center), 48 hours after oxidation with excess NOSbF6 (top), and 10 minutes after deprotonation with an excess of Schwesinger's P2 base immobilized on polystyrene (bottom). | ||
The pseudorotaxane PA11@TTFC8 can also successfully be oxidized with excess NOSbF6 in dry CD2Cl2 under argon atmosphere. Two subsequent one-electron-oxidation steps lead to the TTF dication PA11@TTFC82+ easily recognizable from its instantaneously appearing deep blue colour. The oxidized macrocycle unthreads with a t1/2 of 11 h (Fig. 5b, ESI section 2 Fig. S42‡). As the dication is again diamagnetic, unthreading can also be followed by integrating of a new set of methylene proton signals (Hd, He), upfield shifted by Δδ ≈ 0.3 and 0.6 ppm. This is confirmed by monitoring the dethreading of pseudo[2]rotaxanes PA5@TTFC8, PA6@TTFC8 and PA7@TTFC8 after oxidation. Complete dethreading occurred only for PA7@TTFC8 and required a substantially longer time (t1/2 = 22 h, see ESI section 2 Fig. S36‡).
These experiments clearly reveal that the dethreading rate does not only depend on the steric bulk of the axles’ terminal groups, but also change significantly with the nature of the stimulus applied. Earlier, we reported attractive forces to still occur even between the doubly oxidized TTFC8 and secondary ammonium axles.29 Assuming that the ring circumference and conformational flexibility of TTFC8 is not significantly altered upon oxidation42 and taking into account that the oxidation itself occurs almost instantaneously, we therefore hypothesize that these residual interactions are the cause for the slower dethreading after oxidation. Fig. 5a qualitatively visualizes this in the framework of the energy landscapes after oxidation and after deprotonation: The oxidized pseudorotaxane resides in a somewhat lower local minimum on its potential energy surface as compared to the deprotonated pseudorotaxane.
Synthesis of ‘speed bump’ rotaxanes
Axle 6 (Fig. 6) features the bulky 1-naphthalene moiety and can easily be equipped with a stopper. After stirring 6 with TTFC8 for three days at 40 °C (for R1, Fig. 6) or S4 (axle of rotaxane R2) with TTFC8 for 10 minutes at room temperature (for R2), nitrile-oxide stopper St143 was added and the mixtures were stirred at 35 °C overnight. Two rotaxanes (R1 and R2) were isolated only differing by the size of the connecting blue ‘speed bump’. | ||
| Fig. 6 Synthesis of rotaxane R1. Rotaxane R2 and pseudo[2]rotaxane PA11@TTFC8 are shown for comparison. For details of the synthesis of R2, see ESI, Scheme S3.‡ | ||
In the 1H NMR spectra, successful rotaxane formation is evidenced by a downfield shift (Δδ = +0.5 ppm) of the methylene protons next to the ammonium, as well as the strong downfield shift (Δδ = +4.0 ppm) of the former alkyne proton that is incorporated in the newly formed isoxazole (Hp for R1 and Hj for R2). Additionally, the methylene protons next to the axle ether oxygen atom (Hl for R1 and Hf for R2) shows an upfield shift (Δδ = 1.0 ppm) due to the close proximity to the naphthalene and/or TTF moiety of TTFC8 (see ESI Fig. S19 and S26‡). The interlocked structure was further supported by tandem mass spectrometry. Collision-induced dissociation of mass-selected rotaxane parent ions occurred only at high collision energies. As the intact axle is not observed among the fragments, it is clear that dissociation of the two components is only possible after covalent bond cleavage within the axle, which excludes a non-threaded complex. Also, the collision energies required for dissociation as well as the observed fragment ions are comparable to other similar rotaxanes.29
Deprotonation of the rotaxanes
Deprotonation of R1 with polystyrene-immobilized P2 base yields a neutral amine (Fig. 7d) which is indicated by a strong highfield shift of methylene protons Hd,e (Δδ = 1 ppm solid line). The positions of these signals are similar to those of the deprotonated free axle (Fig. 7e). Other significant shifts include the isoxazole proton Hp (Δδ = −0.5 ppm), and alkyl chain methylene protons Hl (Δδ = +0.7 ppm solid line). Together with the downfield shift of Hl, this clearly indicates a deprotonation-induced shuttling away from the amine and towards the alkyl chain. No such shift of Hl is observed when free axle 7 (6+St1) is deprotonated (Fig. 7a/e). Rotaxane R2 behaves similarly (Fig. S43 bottom‡). Variable temperature 1H NMR experiments (see ESI, Fig. S51 and S52‡) performed with deprotonated R1 and R2 to test whether the two positional isomers of the wheel would be distinguishable unfortunately remained inconclusive.24,44–50 Nevertheless, the NMR data is in agreement with a shift of the wheel across the ‘speed bump’ in the axle centre upon deprotonation.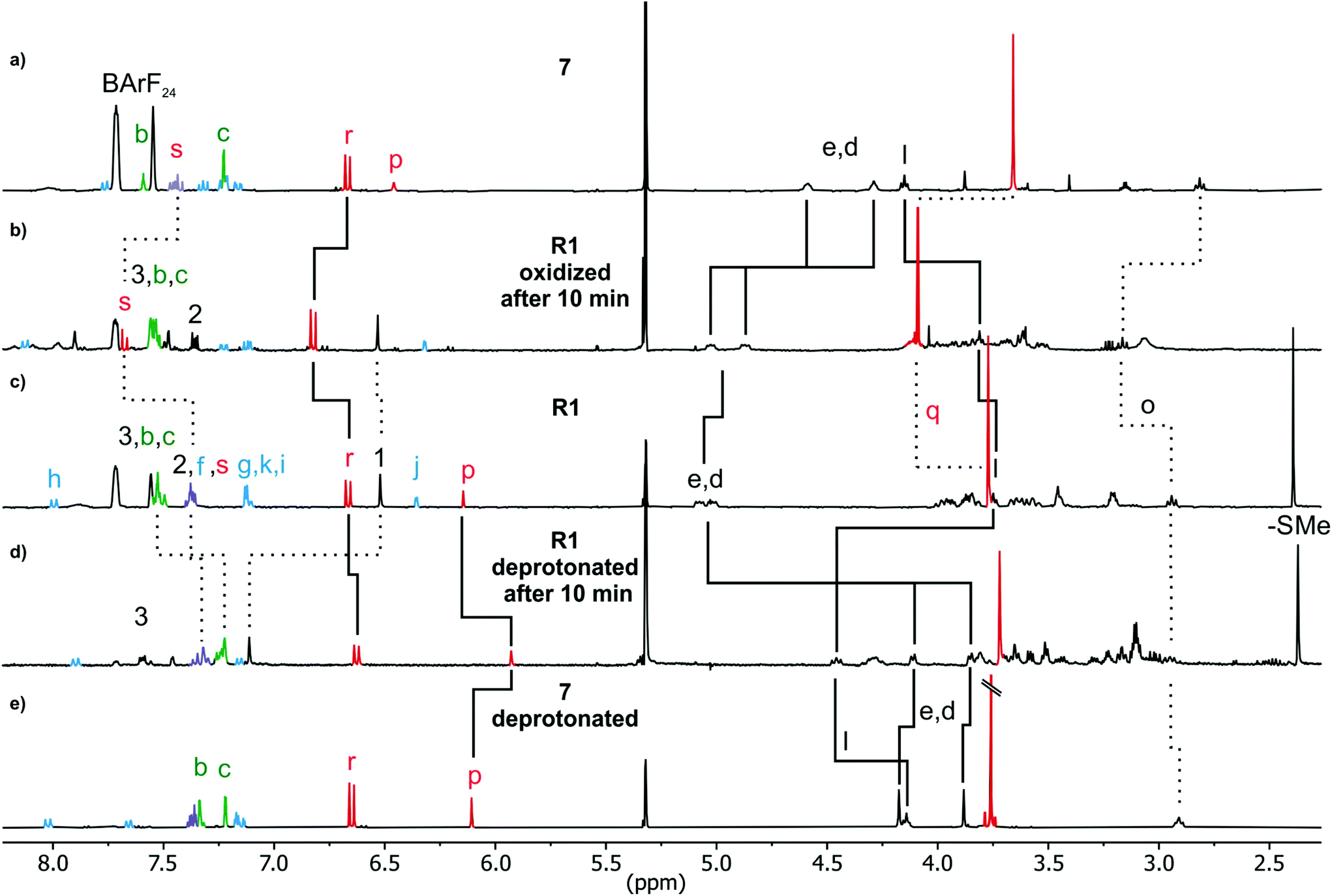 | ||
| Fig. 7 1H NMR spectra (400 MHz, 298 K, CD2Cl2) of (a) stoppered axle 7 (b) R1 10 min after two-electron-oxidation (c) R1 (d) R1 10 minutes after deprotonation and (e) deprotonated 7 with selected shifts highlighted. | ||
Oxidation of the rotaxanes
The 1H NMR spectrum of R1 recorded 10 minutes after oxidation with NOSbF6 to R12+ is shown in Fig. 7b and did not change over the following 48 hours. The methylene protons Hd,e next to the ammonium ion undergo only a small upfield shift (Δδ = −0.1 ppm). Also, proton Hl is shifted only by a small amount. As the small shifts can easily be rationalized by the additional charge induced through oxidation, these findings indicate that the wheel still occupies the ammonium station. However, protons Hr and Hs of the dimethoxyphenyl stopper at the opposite end of the axle shift downfield to some extent (Δδ = 0.2 to 0.25 ppm). Although this observation may at first glance seem inconsistent with the interpretation that the wheel remains on the ammonium side of the axle, these shift differences can be easily explained by invoking p-donor–p-acceptor complex formation between the strongly electron-deficient TTF2+ dication and the rather electron-rich dimethoxyphenyl stopper. The 1H NMR spectrum of the oxidized rotaxane thus indicates that the macrocycle still encircles the ammonium ion, while refolding of the more remote axle half occurs.More evidence can be obtained from cyclovoltammetric (CV) and differential pulse voltammetric (DPV) experiments with which the half-wave potentials of the two reversible TTF redox waves can be obtained. These potentials characteristically shift depending on the interactions between axle and wheel, as shown earlier for TTFC8-containing rotaxanes.29,51,52 The correlation diagram in Fig. 8a contains the peak potentials obtained by DPV measurements in CH2Cl2 with n-Bu4NBArF24 as the supporting electrolyte. Compared to the free wheel, the redox potentials E11/2(TTF0/+) and E21/2(TTF+/2+) are anodically shifted for all species bound to an axle. PA12@TTFC8 is a diagnostic control compound, as two sets of TTF redox processes corresponding to the threaded (E11/2, E21/2) and unthreaded wheel (E11/2,  ) are observed (see ESI Fig. S55‡). The low steric bulk of the phenyl group allows fast interconversion between threaded and unthreaded on the CV timescale (in the DPV only one peak for TTF0/+ redox couple is visible). In contrast, the isolated pseudorotaxane PA11@TTFC8 only shows one redox couple for TTF0/+ and TTF+/2+ due to the sterically hindered unthreading. The mechanically interlocked structures R1 and R2 show no third feature (
) are observed (see ESI Fig. S55‡). The low steric bulk of the phenyl group allows fast interconversion between threaded and unthreaded on the CV timescale (in the DPV only one peak for TTF0/+ redox couple is visible). In contrast, the isolated pseudorotaxane PA11@TTFC8 only shows one redox couple for TTF0/+ and TTF+/2+ due to the sterically hindered unthreading. The mechanically interlocked structures R1 and R2 show no third feature (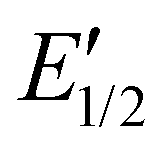 ), meaning that the voltammograms indicate no significant wheel shuttling away from the ammonium station on the timescale of the experiment. Bulk electrolysis (Fig. 8b–d) of the (pseudo)rotaxanes for 2 h did not show a change in the DPV for R1 and R2. However, for PA11@TTFC8 signal broadening and lowered amperage in the course of electrolysis was observed. Bulk electrolysis gave rise to a new signal, which corresponds to the second oxidation of unbound TTFC8. The bulk electrolysis experiments suggest, that electrochemically–induced migration over the naphthalene moiety does take place for pseudorotaxane PA11@TTFC8. On the contrary, no such behavior was observed for the rotaxanes R1 and R2. These results clearly support the 1H NMR data, that no oxidation-induced shuttling occurs in the mechanically interlocked species.
), meaning that the voltammograms indicate no significant wheel shuttling away from the ammonium station on the timescale of the experiment. Bulk electrolysis (Fig. 8b–d) of the (pseudo)rotaxanes for 2 h did not show a change in the DPV for R1 and R2. However, for PA11@TTFC8 signal broadening and lowered amperage in the course of electrolysis was observed. Bulk electrolysis gave rise to a new signal, which corresponds to the second oxidation of unbound TTFC8. The bulk electrolysis experiments suggest, that electrochemically–induced migration over the naphthalene moiety does take place for pseudorotaxane PA11@TTFC8. On the contrary, no such behavior was observed for the rotaxanes R1 and R2. These results clearly support the 1H NMR data, that no oxidation-induced shuttling occurs in the mechanically interlocked species.
 | ||
| Fig. 8 Electrochemical data: (a) Correlation diagram of DPV peak potentials of all relevant (pseudo)rotaxane species, DPVs before and after electrolysis of (b) R1, (c) R2, (d) PA11@TTFC8 (CH2Cl2, with n-Bu4NBArF24 as the electrolyte (0.1 M), 298 K, 1 mM analyte, 25 mV modulation amplitude, 50 ms modulation time, 5 mV step potential, 0.5 s interval time). | ||
Consequently, the behaviour of the rotaxanes parallels that of the pseudorotaxanes in that the two different stimuli cause significant differences in dethreading/shuttling rates despite of the steric barrier remaining unchanged. Also, the reason – a binding interaction of the crown ether wheel to the ammonium ion that is stronger than the effects of charge repulsion – is likely the same. Nevertheless, there are also differences: While the pseudorotaxane finally dethreads over 48 hours, no change is observed on a similar time scale for the rotaxane. This is an effect of the mechanical bond. While dethreading of the pseudorotaxane is entropically favoured by an increase in particle number, such wheel shuttling in the rotaxanes does not benefit from such entropic benefit.
Conclusions
A set of BArF24− salts of sec-ammonium ions bearing different bulky end groups have been investigated with respect to their ability to form crown ether/ammonium pseudorotaxanes. The small differences in bond lengths between the two crown ethers DB24C8 and TTFC8 change the threading barrier significantly and threading occurs significantly faster for TTFC8. Three groups of quickly threading, kinetically hindered slowly threading, and non-threading axles can be distinguished. For the slowly and non-threading axle/wheel pairs, the formation of face-to-face crown ether/ammonium complexes occurs, which has been exemplarily studied in more detail for PA11 by 1H NMR, tandem MS and ITC experiments. To our knowledge this is the first time that an equilibrium of a threaded and non-threaded complex was quantitatively studied for the same axle macrocycle combination.Deslipping of TTFC8 from different axles could be achieved by deprotonation of the axle or by chemical oxidation of the macrocycle. Upon deprotonation, the pseudorotaxanes dissociate quickly. Chemical oxidation to the TTF dication lead to much slower dethreading. The difference in dethreading rates for the two stimuli can be explained by residual attractive interaction of the oxidized TTFC82+ and the axle. It is quite remarkable that two different stimuli lead to so clearly different dethreading rates, even though the steric barrier remains unchanged. Clearly, an interplay between the steric barrier and the changes in the interactions between wheel and its binding stations is observed here which complicates the analysis of such steric ‘speed bumps’ in molecular interlocked switches and machines.
The shuttling of the wheel in the rotaxanes on one hand behaves similarly in that the two stimuli cause different effects on the shuttling. But the behaviour is not fully analogous. The comparison of rotaxanes and pseudorotaxanes clearly reveals a significant and non-negligible influence of the mechanical bond. At the end, the shuttling is affected by changing interactions between the wheel and the available binding stations as well as steric barriers on its way along the axle and the particular effects of the mechanical bond.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) – Projektnummer 387284271 – SFB 1349 – und Projektnummer 434455294 (funded by the Deutsche Forschungsgemeinschaft – project-ID 387284271 – SFB 1349 and project-ID 434455294). A.V. kindly acknowledges the Academy of Finland (grant no. 314343) for financial support. We also thank Jennifer Anders for help with the synthesis.Notes and references
- J. D. Harris, M. J. Moran and I. Aprahamian, New molecular switch architectures, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9414–9422 Search PubMed.
- J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, A molecular elevator, Science, 2004, 303, 1845–1849 Search PubMed.
- D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Coumarin-Based Small-Molecule Fluorescent Chemosensors, Chem. Rev., 2019, 119, 10403–10519 Search PubMed.
- D. Bleger and S. Hecht, Visible-Light-Activated Molecular Switches, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 Search PubMed.
- D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Design of Collective Motions from Synthetic Molecular Switches, Rotors, and Motors, Chem. Rev., 2020, 120, 310–433 Search PubMed.
- A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, High hopes: can molecular electronics realise its potential?, Chem. Soc. Rev., 2012, 41, 4827–4859 Search PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Artificial Molecular Machines, Chem. Rev., 2015, 115, 10081–10206 Search PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Synthetic molecular motors and mechanical machines, Angew. Chem., Int. Ed., 2007, 46, 72–191 Search PubMed.
- T. Felder and C. A. Schalley, Secondary isotope effects on the deslipping reaction of rotaxanes: high-precision measurement of steric size, Angew. Chem., Int. Ed., 2003, 42, 2258–2260 Search PubMed.
- E. A. Neal and S. M. Goldup, Chemical consequences of mechanical bonding in catenanes and rotaxanes: isomerism, modification, catalysis and molecular machines for synthesis, Chem. Commun., 2014, 50, 5128–5142 Search PubMed.
- M. Baroncini, S. Silvi and A. Credi, Photo- and Redox-Driven Artificial Molecular Motors, Chem. Rev., 2020, 120, 200–268 Search PubMed.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Surveying macrocyclic chemistry: from flexible crown ethers to rigid cyclophanes, Chem. Soc. Rev., 2017, 46, 2459–2478 Search PubMed.
- A. Carrasco-Ruiz and J. Tiburcio, Electrostatic kinetic barriers in the threading/dethreading motion of a rotaxane-like complex, Org. Lett., 2015, 17, 1858–1861 Search PubMed.
- P. R. Ashton, P. J. Campbell, P. T. Glink, D. Philp, N. Spencer, J. F. Stoddart, E. J. T. Chrystal, S. Menzer, D. J. Williams and P. A. Tasker, Dialkylammonium Ion/Crown Ether Complexes: The Forerunners of a New Family of Interlocked Molecules, Angew. Chem., Int. Ed. Engl., 1995, 34, 1865–1869 Search PubMed.
- R. M. Izatt, J. D. Lamb, N. E. Izatt, B. E. Rossiter, J. J. Christensen and B. L. Haymore, A calorimetric titration study of the reaction of several organic ammonium cations with 18-crown-6 in methanol, J. Am. Chem. Soc., 1979, 101, 6273–6276 Search PubMed.
- P. R. Ashton, I. Baxter, M. C. T. Fyfe, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, Rotaxane or Pseudorotaxane? That Is the Question!†, J. Am. Chem. Soc., 1998, 120, 2297–2307 Search PubMed.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, John Wiley & Sons, Inc., 2016 Search PubMed.
- T. Clifford, A. Abushamleh and D. H. Busch, Factors affecting the threading of axle molecules through macrocycles: binding constants for semirotaxane formation, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4830–4836 Search PubMed.
- J. Groppi, L. Casimiro, M. Canton, S. Corra, M. Jafari-Nasab, G. Tabacchi, L. Cavallo, M. Baroncini, S. Silvi, E. Fois and A. Credi, Precision Molecular Threading/Dethreading, Angew. Chem., Int. Ed. Engl., 2020, 59, 14825–14834 Search PubMed.
- P. G. Young, K. Hirose and Y. Tobe, Axle length does not affect switching dynamics in degenerate molecular shuttles with rigid spacers, J. Am. Chem. Soc., 2014, 136, 7899–7906 Search PubMed.
- M. Asakawa, P. R. Ashton, R. Ballardini, V. Balzani, M. Bělohradský, M. T. Gandolfi, O. Kocian, L. Prodi, F. M. Raymo, J. F. Stoddart and M. Venturi, The Slipping Approach to Self-Assembling [n]Rotaxanes†, J. Am. Chem. Soc., 1997, 119, 302–310 Search PubMed.
- B. Riss-Yaw, C. Clavel, P. Laurent and F. Coutrot, The relationship between the conformational degree of freedom of template-containing threads and slippage in the formation of [2]rotaxane building blocks, Chem. Commun., 2017, 53, 10874–10877 Search PubMed.
- Y. Yamashita, Y. Saito, S. Kikkawa, Y. Mutoh, S. Hosoya, I. Azumaya and S. Saito, Evaluation of the Steric Bulk of Substituents Utilizing the Shuttling Behavior of [2]Rotaxanes with N-Arylpyrrole Moieties, Eur. J. Org. Chem., 2019, 2019, 3412–3420 Search PubMed.
- G. Gholami, K. Zhu, G. Baggi, E. Schott, X. Zarate and S. J. Loeb, Influence of axle length on the rate and mechanism of shuttling in rigid H-shaped [2]rotaxanes, Chem. Sci., 2017, 8, 7718–7723 Search PubMed.
- A. C. Catalan and J. Tiburcio, Self-assembly of pseudo-rotaxane and rotaxane complexes using an electrostatic slippage approach, Chem. Commun., 2016, 52, 9526–9529 Search PubMed.
- C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Mastering the non-equilibrium assembly and operation of molecular machines, Chem. Soc. Rev., 2017, 46, 5491–5507 Search PubMed.
- G. Ragazzon, C. Schafer, P. Franchi, S. Silvi, B. Colasson, M. Lucarini and A. Credi, Remote electrochemical modulation of pKa in a rotaxane by co-conformational allostery, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9385–9390 Search PubMed.
- K. Nakazono and T. Takata, Neutralization of a sec-ammonium group unusually stabilized by the “rotaxane effect”: synthesis, structure, and dynamic nature of a “free” sec-amine/crown ether-type rotaxane, Chem. – Eur. J., 2010, 16, 13783–13794 Search PubMed.
- H. V. Schröder, S. Sobottka, M. Nössler, H. Hupatz, M. Gaedke, B. Sarkar and C. A. Schalley, Impact of mechanical bonding on the redox-switching of tetrathiafulvalene in crown ether-ammonium [2]rotaxanes, Chem. Sci., 2017, 8, 6300–6306 Search PubMed.
- H. Hupatz, M. Gaedke, H. V. Schröder, J. Beerhues, A. Valkonen, F. Klautzsch, S. Müller, F. Witte, K. Rissanen, B. Sarkar and C. A. Schalley, Thermodynamic and electrochemical study of tailor-made crown ethers for redox-switchable (pseudo)rotaxanes, Beilstein J. Org. Chem., 2020, 16, 2576–2588 Search PubMed.
- A. Crochet, E. Kottelat, A. Fleury, M. Neuburger and K. M. Fromm, Polymorph of Dibenzo-24-Crown-8 and its Mercury Complex, Z. Anorg. Allg. Chem., 2011, 637, 672–675 Search PubMed.
- H. Nagai, Y. Suzaki and K. Osakada, Thiacrown Ethers with Oxygen and Sulfur for Coordination: Formation of the Pd and Pt Complexes and Pseudorotaxane with Dialkylammonium, Eur. J. Inorg. Chem., 2014, 2014, 4376–4384 Search PubMed.
- F. Le Derf, M. Mazari, N. Mercier, E. Levillain, P. Richomme, J. Becher, J. Garin, J. Orduna, A. Gorgues and M. Salle, Electroregulated Metal-Binding with a Crown Ether Tetrathiafulvalene Derivative: Toward Electrochemically Addressed Metal Cation Sponges, Inorg. Chem., 1999, 38, 6096–6100 Search PubMed.
- H. Nagai, Y. Suzaki and K. Osakada, Chemical Modification of a [2]Rotaxane Composed of Dithiacrown Ether and Dialkylammonium with Organic and Inorganic Compounds, Chem. Lett., 2016, 45, 834–836 Search PubMed.
- A. R. Williams, B. H. Northrop, K. N. Houk, J. F. Stoddart and D. J. Williams, The influence of constitutional isomerism and change on molecular recognition processes, Chem. – Eur. J., 2004, 10, 5406–5421 Search PubMed.
- S. J. Cantrill, D. A. Fulton, A. M. Heiss, A. R. Pease, J. F. Stoddart, A. J. P. White and D. J. Williams, The Influence of Macrocyclic Polyether Constitution upon Ammonium Ion/Crown Ether Recognition Processes, Chem. – Eur. J., 2000, 6, 2274–2287 Search PubMed.
- T. Takata, Stimuli-Responsive Molecular and Macromolecular Systems Controlled by Rotaxane Molecular Switches, Bull. Chem. Soc. Jpn., 2019, 92, 409–426 Search PubMed.
- S. Miyagawa, M. Kimura, S. Kagami, T. Kawasaki and Y. Tokunaga, Utilization of a Crown Ether/Amine-Type Rotaxane as a Probe for the Versatile Detection of Anions and Acids by Thin-Layer Chromatography, Chem. – Asian J., 2020, 15, 3044–3049 Search PubMed.
- J. Nishiyama, Y. Makita and N. Kihara, Rapid and Efficient Acylative Active Transport on a Rotaxane, Asian J. Org. Chem., 2015, 4, 1056–1064 Search PubMed.
- H. Sato, D. Aoki and T. Takata, Synthesis and Star/Linear Topology Transformation of a Mechanically Linked ABC Terpolymer, ACS Macro Lett., 2016, 5, 699–703 Search PubMed.
- S. D. P. Fielden, D. A. Leigh, C. T. McTernan, B. Perez-Saavedra and I. J. Vitorica-Yrezabal, Spontaneous Assembly of Rotaxanes from a Primary Amine, Crown Ether and Electrophile, J. Am. Chem. Soc., 2018, 140, 6049–6052 Search PubMed.
- H. V. Schröder, F. Witte, M. Gaedke, S. Sobottka, L. Suntrup, H. Hupatz, A. Valkonen, B. Paulus, K. Rissanen, B. Sarkar and C. A. Schalley, An aryl-fused redox-active tetrathiafulvalene with enhanced mixed-valence and radical-cation dimer stabilities, Org. Biomol. Chem., 2018, 16, 2741–2747 Search PubMed.
- T. Matsumura, F. Ishiwari, Y. Koyama and T. Takata, C-C bond-forming click synthesis of rotaxanes exploiting nitrile N-oxide, Org. Lett., 2010, 12, 3828–3831 Search PubMed.
- V. N. Vukotic, K. Zhu, G. Baggi and S. J. Loeb, Optical Distinction between “Slow” and “Fast” Translational Motion in Degenerate Molecular Shuttles, Angew. Chem., Int. Ed., 2017, 56, 6136–6141 Search PubMed.
- K. Hirose, Molecular brake systems controlled by light and heat, J. Inclusion Phenom. Macrocyclic Chem., 2010, 68, 1–24 Search PubMed.
- J. Dale and P. O. Kristiansen, Cyclic oligo-ethers related to ethylene oxide, J. Chem. Soc., Chem. Commun., 1971, 13, 670 Search PubMed.
- C. I. Ratcliffe, J. A. Ripmeester, G. W. Buchanan and J. K. Denike, A molecular merry-go-round: motion of the large macrocyclic molecule 18-crown-6 in its solid complexes studied by deuterium NMR, J. Am. Chem. Soc., 1992, 114, 3294–3299 Search PubMed.
- V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Metal-organic frameworks with dynamic interlocked components, Nat. Chem., 2012, 4, 456–460 Search PubMed.
- K. Zhu, V. N. Vukotic, C. A. O'Keefe, R. W. Schurko and S. J. Loeb, Metal-organic frameworks with mechanically interlocked pillars: controlling ring dynamics in the solid-state via a reversible phase change, J. Am. Chem. Soc., 2014, 136, 7403–7409 Search PubMed.
- N. D. Suhan, L. Allen, M. T. Gharib, E. Viljoen, S. J. Vella and S. J. Loeb, Colour coding the co-conformations of a [2]rotaxane flip-switch, Chem. Commun., 2011, 47, 5991–5993 Search PubMed.
- M. Gaedke, F. Witte, J. Anhäuser, H. Hupatz, H. V. Schröder, A. Valkonen, K. Rissanen, A. Lützen, B. Paulus and C. A. Schalley, Chiroptical inversion of a planar chiral redox-switchable rotaxane, Chem. Sci., 2019, 10, 10003–10009 Search PubMed.
- H. V. Schröder, F. Stein, J. M. Wollschläger, S. Sobottka, M. Gaedke, B. Sarkar and C. A. Schalley, Accordion-Like Motion in Electrochemically Switchable Crown Ether/Ammonium Oligorotaxanes, Angew. Chem., Int. Ed., 2019, 58, 3496–3500 Search PubMed.
Footnotes |
| † Dedicated to Prof. Placido Neri upon the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2073308. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo00503k |
| § Present address: Dr H. V. Schröder Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ08544, USA |
| This journal is © the Partner Organisations 2021 |