
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild reductive rearrangement of oximes and oxime ethers to secondary amines with hydrosilanes catalyzed by B(C6F5)3†
Huaquan
Fang‡
 a,
Guoqiang
Wang‡
ab and
Martin
Oestreich
*a
a,
Guoqiang
Wang‡
ab and
Martin
Oestreich
*a
aInstitut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: martin.oestreich@tu-berlin.de
bInstitute of Theoretical and Computational Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, P.R. China
First published on 1st April 2021
Abstract
The strong boron Lewis acid tris(pentafluorophenyl)borane, B(C6F5)3, has been found to catalyze the reductive rearrangement of oximes and their ether derivatives at room temperature with hydrosilanes as the reducing agents. Cyclic substrates undergo ring enlargement, and the secondary amine products are generally formed in good yields. Control experiments combined with a DFT computational analysis of the reaction mechanism suggest that there are three energetically accessible reaction pathways (paths A–C), either or not involving hydroxylamine derivatives. Paths A and B proceed through the intermediacy of a common N,O-bissilylated hydroxylamine, and the ring-expanding rearrangement yields an iminium ion. With no intermediate at the hydroxylamine oxidation level (path C), the reaction mechanism resembles that of the Beckmann rearrangement where an O-silylated oxime converts into a nitrilium ion. The reduction–rearrangement sequence (paths A and B) is slightly preferred over the rearrangement–reduction order of events (path C), especially at ambient temperature.
Introduction
A few years ago, our laboratory disclosed the previously elusive hydrogenation of various oxime ethers to the corresponding O-protected hydroxylamines by making use of FLP-type dihydrogen activation with B(C6F5)3 as the catalyst1 (Scheme 1, top).2 The goal of that project had been to avoid the otherwise typical cleavage of the weak N–O bond to afford primary amines.3 For this, the use of dihydrogen is critical because hydrosilanes as alternative reductants4 engage in deoxygenation.5 Treatment of those oxime ethers with hydrosilane/B(C6F5)3 combinations results in exactly that, and these reactions were not clean but there was evidence for a reductive rearrangement.2a Rearrangements of this sort that convert oximes and their ether derivatives into secondary amines can be achieved by the stoichiometric use of aluminum-6 and also boron-based7 reductants; the aforementioned reduction to primary amines is a common side reaction (Scheme 1, bottom). In the light of our earlier observations, we decided to investigate the B(C6F5)3-catalyzed reductive rearrangement of oximes with hydrosilanes to turn it into a useful chemoselective methodology.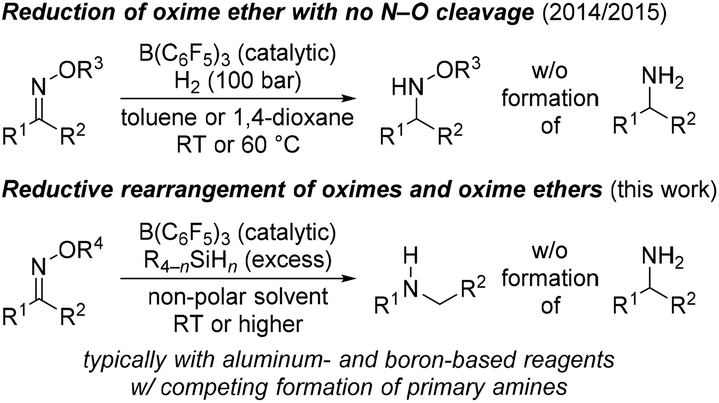 | ||
| Scheme 1 Reduction and reductive rearrangement of oximes and oxime ethers catalyzed by combinations of B(C6F5)3 and dihydrogen and hydrosilanes (n = 1–3), respectively. R1 and R2 = aryl, alkyl, and H; R3 = alkyl or silyl; R4 = alkyl, silyl, or H. | ||
Results and discussion
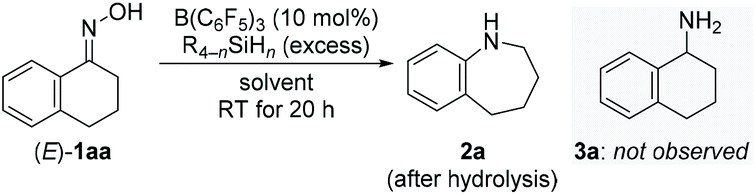
We began our investigations with optimizing the reductive rearrangement of α-tetralone-derived oxime into the corresponding 2,3,4,5-tetrahydro-1H-benzo[b]azepine [(E)-1aa → 2a; Table 1]. Treatment of (E)-1aa with 4.0 equiv. of PhSiH3 in the presence of 10 mol% of B(C6F5)3 in 1,2-C6H4F2 at room temperature afforded 2a in 40% yield after 20 h (entry 1). This reaction was highly chemoselective, favoring migration of the aryl group over simple reduction; no formation of any primary amine product 3a was detected (gray box). Among several solvents tested, toluene (toluene-d8 used) brought about the best result with 95% yield and 88% isolated yield (entries 2–6). In toluene-d8, excellent and good yields were also obtained with MePhSiH2 and Ph2SiH2, respectively (entries 7 and 8). Other hydrosilanes such as Et2SiH2 and Me2PhSiH were however completely unreactive even at higher temperature (entries 9 and 10, see the ESI† for details). Reactions conducted with a shorter reaction time (entry 11), less PhSiH3 (entry 12), or decreased catalyst loading (entry 13) resulted in lower yields. Incomplete conversion at reduced catalyst loadings could be compensated by longer reaction times.|
|
|||
|---|---|---|---|
| Entry | Hydrosilane (equiv.) | Solvent | Yieldb (%) |
| a All reactions were performed on a 0.10 mmol scale in a GC vial. b Yields determined by 1H NMR spectroscopy using mesitylene as an internal standard; isolated yield in parentheses. c 8 h. d 5.0 mol% B(C6F5)3. | |||
| 1 | PhSiH3 (4.0) | 1,2-C6H4F2 | 40 |
| 2 | PhSiH3 (4.0) | C6H5F | 64 |
| 3 | PhSiH3 (4.0) | C6H5Cl | 69 |
| 4 | PhSiH3 (4.0) | CH2Cl2 | 66 |
| 5 | PhSiH3 (4.0) | Benzene | 88 |
| 6 | PhSiH3 (4.0) | Toluene-d8 | 95 (88) |
| 7 | MePhSiH2 (4.0) | Toluene-d8 | 95 |
| 8 | Ph2SiH2 (4.0) | Toluene-d8 | 77 |
| 9 | Et2SiH2 (4.0) | Toluene-d8 | 0 |
| 10 | Me2PhSiH (4.0) | Toluene-d8 | 0 |
| 11c | PhSiH3 (4.0) | Toluene-d8 | 67 |
| 12 | PhSiH3 (2.0) | Toluene-d8 | 80 |
| 13d | PhSiH3 (4.0) | Toluene-d8 | 78 |
The yield of the above reductive rearrangement was also high on a 7.0 mmol scale, and we continued exploring the reaction scope under this optimized reaction setup (Scheme 2; cf. Table 1, entry 6). Related ring enlargements worked equally well [(E)-1ba,ca → 2b,c]. The functional-group tolerance was assessed with acetophenone-derived oxime derivatives (E)-1da–pa bearing a range of electron-donating and electron-withdrawing substituents on the aryl group. The four halo groups as in (E)-1ia–la were compatible with the reaction conditions but aryl iodide (E)-1la underwent partial hydrodeiodination to afford 2l in 57% yield along with 2d in 25% yield. A thioether as in (E)-1na and a free phenol as in (E)-1oa undergoing temporary silylation were tolerated as well. Exhaustive hydrodefluorination of the trifluoromethyl group in (E)-1ma occurred, furnishing 2h rather than 2m.8 The reaction of (E)-1pa with a dimethylamino group in the para position only gave a trace amount of 2p, and the deamination product 4-ethyl-N,N-dimethylaniline was formed in 64% yield.9 It is important to note that the configuration of the oxime does not affect the reaction outcome;6g both (E)-1ka and (Z)-1ka yielded 2k in 98% and 93%, respectively.
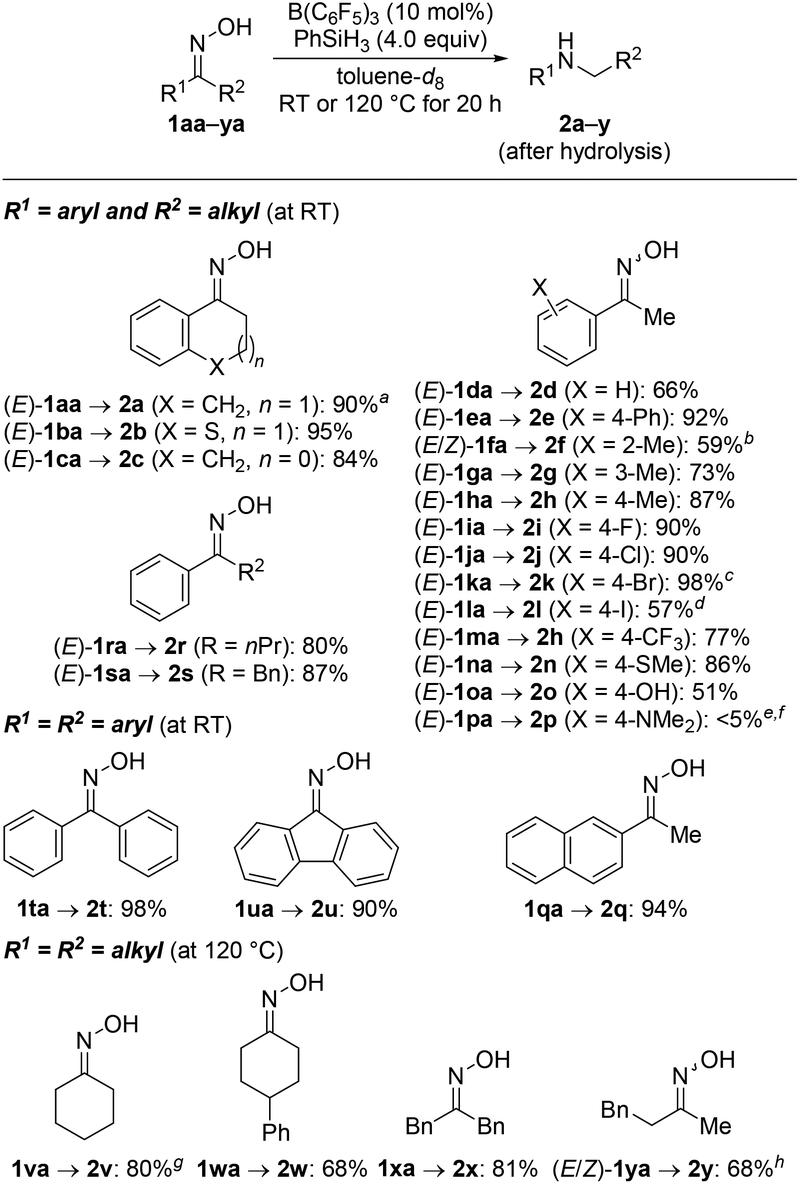 | ||
Scheme 2 Scope I: B(C6F5)3-catalyzed reductive rearrangement of ketone-derived oximes. Reaction conditions: 1 (0.20 mmol), B(C6F5)3 (10 mol%), PhSiH3 (4.0 equiv.), and toluene-d8 (0.20 mL) at RT for 20 h. Isolated yields after purification by flash chromatography on silica gel. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7.0 mmol scale. bE/Z = 75:25 for 1fa. c93% yield for (Z)-1ka. dFormed along with 2d in 25% yield. eRun at 120 °C. f4-Ethyl-N,N-dimethylaniline formed in 64% yield. gDetermined by 1H NMR spectroscopy using mesitylene as an internal standard. hE/Z = 71:29 for 1ya. 7.0 mmol scale. bE/Z = 75:25 for 1fa. c93% yield for (Z)-1ka. dFormed along with 2d in 25% yield. eRun at 120 °C. f4-Ethyl-N,N-dimethylaniline formed in 64% yield. gDetermined by 1H NMR spectroscopy using mesitylene as an internal standard. hE/Z = 71:29 for 1ya. | ||
We briefly investigated the reductive rearrangement of other alkyl-substituted benzylic oximes (Scheme 2). (E)-1ra and (E)-1sa with a 1° alkyl and a benzylic substituent converted selectively and in high yields into the corresponding secondary amines. However, substrates with 2° and 3° alkyl groups did lead to mixtures of rearranged products because the migrating ability of these groups (out)competes with that of the aryl group. This finding is different from previous reports.6g The results obtained with isopropyl, cyclohexyl, and tert-butyl are presented in the ESI.† Symmetric ketoximes such as diaryl-substituted 1ta and 1ua as well as dialkyl-substituted 1va–xa reacted cleanly but for the latter a reaction temperature of 120 °C was necessary to achieve high conversion. The rearrangement of unsymmetrically substituted (E/Z)-1ya furnished 2y selectively with migration of the phenethyl group. Additional substrates were subjected to the standard procedure (Scheme 3). Aldoxime (E)-1za rearranged to the aniline derivative 2z in a good yield. Moreover, diamine 5 was obtained from bisoxime (Z,Z)-4 by two-fold rearrangement in a moderate yield.
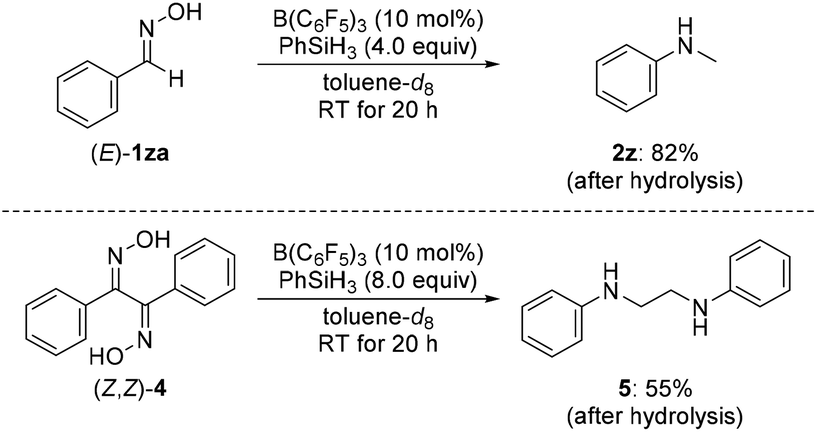 | ||
| Scheme 3 Scope II: B(C6F5)3-catalyzed reductive rearrangement of aldehyde- and benzil-derived oximes. Isolated yields after purification by flash chromatography on silica gel. | ||
We then turned our attention towards the reductive rearrangement of various oxime ethers (Scheme 4). After a slight reoptimization of the reaction conditions, we found that the reaction of O-silylated (E)-1db–de and O-alkylated ketoximes (E)-1df and (E)-1dg with 2.0 equiv. of PhSiH3 in the presence of 2.5 mol% of B(C6F5)3 afforded N-ethylaniline (2d) in moderate to near-quantitative yields. The scope also includes the successful reductive rearrangement of hydroxylamine 6aa and O-silylated hydroxylamine 7de (Scheme 5). Both yielded the desired secondary amines 2a and 2d in 61% and 81%, respectively.
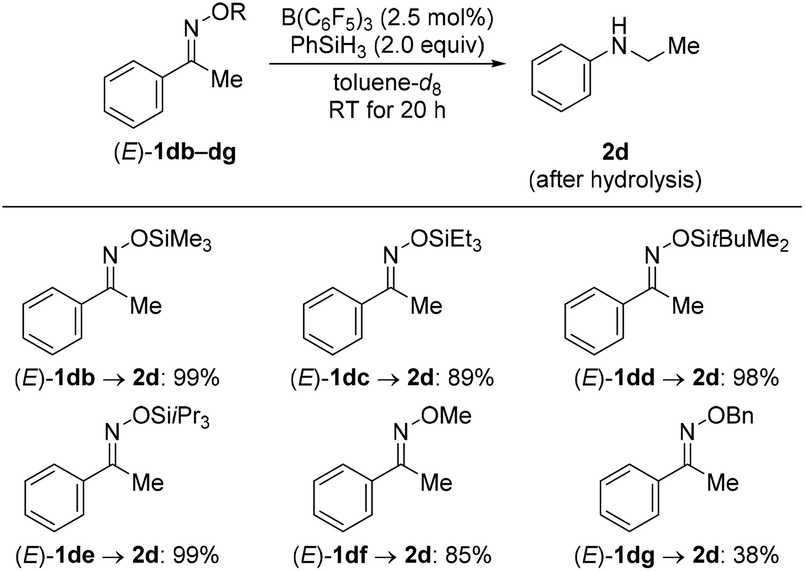 | ||
| Scheme 4 Scope III: B(C6F5)3-catalyzed reductive rearrangement of ketone-derived oxime ethers. Yields determined by 1H NMR spectroscopy using mesitylene as an internal standard. | ||
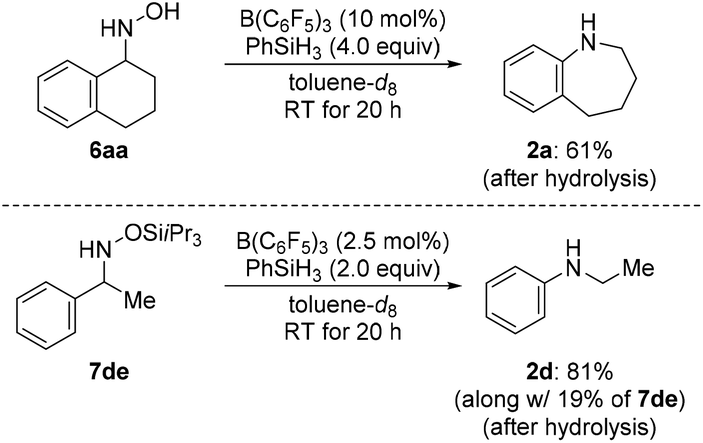 | ||
| Scheme 5 Scope IV: B(C6F5)3-catalyzed reductive rearrangement of a hydroxylamine and an O-silylated hydroxylamine. Yields determined by 1H NMR spectroscopy using mesitylene as an internal standard. | ||
The hydroxylamine derivatives were not only chosen as an expansion of the method but also for their potential intermediacy in the reductive rearrangement of oximes. The above results indicate that O-silylated oximes, free hydroxylamines, and O-silylated hydroxylamines may be involved. Further evidence came from deuterium labeling when employing PhSiD3 instead of PhSiH3 (Scheme 6). Bisdeuteration of the α-methylene group was obtained in the rearrangement of the oxime [(E)-1aa → 2a-d2] while monodeuteration was seen in the same position in the ring enlargement of the hydroxylamine (6aa → 2a-d1). This suggests that oximes and their congeners are reduced to the hydroxylamine oxidation level during the course of the reaction.2,10 Cho, Tokuyama, and co-workers had reported the same for the related DIBAL-H-mediated rearrangement, and their computed mechanism proceeds through a three-centered transition state for the concerted aryl migration followed by hydride reduction of the resulting iminium ion6g (silyliminium ion in the case of hydrosilane/B(C6F5)3 combinations).
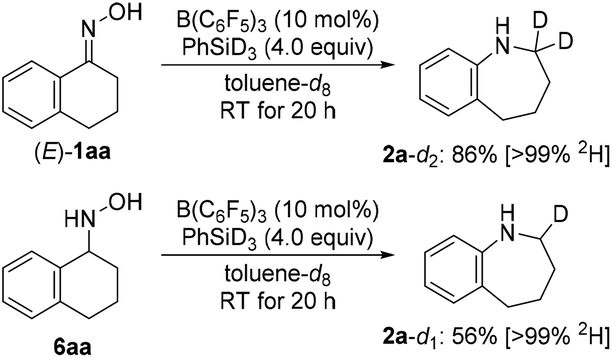 | ||
| Scheme 6 Deuterium-labeling experiments. | ||
We then conducted density functional calculations (DFT) to explore the mechanistic details of the reductive rearrangement of oximes to secondary amines. The calculations were performed at the M06-2X/cc-pVTZ//M06-2X/6-311G(d,p) level of theory11 using the Gaussian 16 software,12 with toluene solvation incorporated via a polarizable continuum model (PCM) at the same level of theory.13 The reaction between oxime (E)-1aa and PhSiH3 was chosen as a model system. Optimized geometries and free energy profiles (including the computational analysis on the relative stability of the B(C6F5)3·(E)-1aa Lewis pair and the B(C6F5)3·hydrosilane η1-adduct I114) involved in the catalytic cycle are provided in the ESI.†15
As shown in Scheme 7, the reaction begins with the activation of the Si–H bond in I1 by the nitrogen atom or oxygen atom in oxime (E)-1aa16,17 to afford N-silyliminium ion I2 (path A) or silyloxonium ion I8 (path B) with the borohydride [HB(C6F5)3]− as the counteranion. Our calculations indicate that the corresponding SN2-Si transition states do not exist in the case of PhSiH3.17,18 Instead, two stable trigonal bipyramidal transition complexes (rather than transition state) were located (I2′ or I8′, see Fig. S70 in the ESI†).19 These results are different from that found in a computational study on B(C6F5)3-catalyzed carbonyl hydrosilylation with a tertiary silane (Me3SiH).17 Subsequent hydride transfer from the borohydride to the silyliminium ion or dehydrogenation gives the N-silylated hydroxylamine (I2 → I3) and O-silylated oxime ether (I8 → I9), respectively. B(C6F5)3 is regenerated in either of these steps. The formation of O-silylated oxime ether I9 is favored over N-silylated hydroxylamine I3 because the dehydrogenative silylation of oxime (E)-1aa is calculated to be kinetically more favorable than the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N hydrosilylation reaction (ΔG‡ = 11.3 versus 21.0 kcal mol−1), probably due to the release of dihydrogen gas. The N-silylated hydroxylamine I3 or O-silylated oxime I9 then undergoes similar dehydrogenative silylation or CN hydrosilylation steps to form N,O-bissilylated hydroxylamine I5 through either I3 → I4 → I5 (path A) or I9 → I10 → I5 (path B). The hydrosilylation of O-silylated oxime ether I9 requires an activation barrier of 24.1 kcal mol−1, which is higher than that of the dehydrogenative silylation reaction of I3 (ΔG‡ = 13.3 kcal mol−1). These computational results suggest that the formation of N,O-bissilylated hydroxylamine I5 proceeding through CN hydrosilylation/Si–O dehydrogenative coupling (path A) is the dominant reaction sequence in the current system. I5 could further react with the B(C6F5)3·hydrosilane adduct I1 to form ion pair I6, which undergoes the rearrangement involving aryl migration and N–O bond cleavage to generate the iminium ion I7 along with the disiloxane byproduct (I6 → I7, ΔG‡ = 23.5 kcal mol−1).20 Finally, I7 further reacts through a barrierless hydride transfer process from the borohydride to the iminium ion to form the reductive rearrangement product 2a-Si (gray box). The overall reaction is exergonic by 98.1 kcal mol−1, and both path A and path B are likely related to the formation of the reductive rearrangement product because of the close activation barriers in the rate-determining steps involved (path A, I6 → I7, ΔG‡ = 23.5 kcal mol−1; path B, I10 → I5, ΔG‡ = 24.1 kcal mol−1). These computational results can account for the experimental observation that oxime ethers and several hydroxylamine derivatives are also applicable to this method.
N hydrosilylation reaction (ΔG‡ = 11.3 versus 21.0 kcal mol−1), probably due to the release of dihydrogen gas. The N-silylated hydroxylamine I3 or O-silylated oxime I9 then undergoes similar dehydrogenative silylation or CN hydrosilylation steps to form N,O-bissilylated hydroxylamine I5 through either I3 → I4 → I5 (path A) or I9 → I10 → I5 (path B). The hydrosilylation of O-silylated oxime ether I9 requires an activation barrier of 24.1 kcal mol−1, which is higher than that of the dehydrogenative silylation reaction of I3 (ΔG‡ = 13.3 kcal mol−1). These computational results suggest that the formation of N,O-bissilylated hydroxylamine I5 proceeding through CN hydrosilylation/Si–O dehydrogenative coupling (path A) is the dominant reaction sequence in the current system. I5 could further react with the B(C6F5)3·hydrosilane adduct I1 to form ion pair I6, which undergoes the rearrangement involving aryl migration and N–O bond cleavage to generate the iminium ion I7 along with the disiloxane byproduct (I6 → I7, ΔG‡ = 23.5 kcal mol−1).20 Finally, I7 further reacts through a barrierless hydride transfer process from the borohydride to the iminium ion to form the reductive rearrangement product 2a-Si (gray box). The overall reaction is exergonic by 98.1 kcal mol−1, and both path A and path B are likely related to the formation of the reductive rearrangement product because of the close activation barriers in the rate-determining steps involved (path A, I6 → I7, ΔG‡ = 23.5 kcal mol−1; path B, I10 → I5, ΔG‡ = 24.1 kcal mol−1). These computational results can account for the experimental observation that oxime ethers and several hydroxylamine derivatives are also applicable to this method.
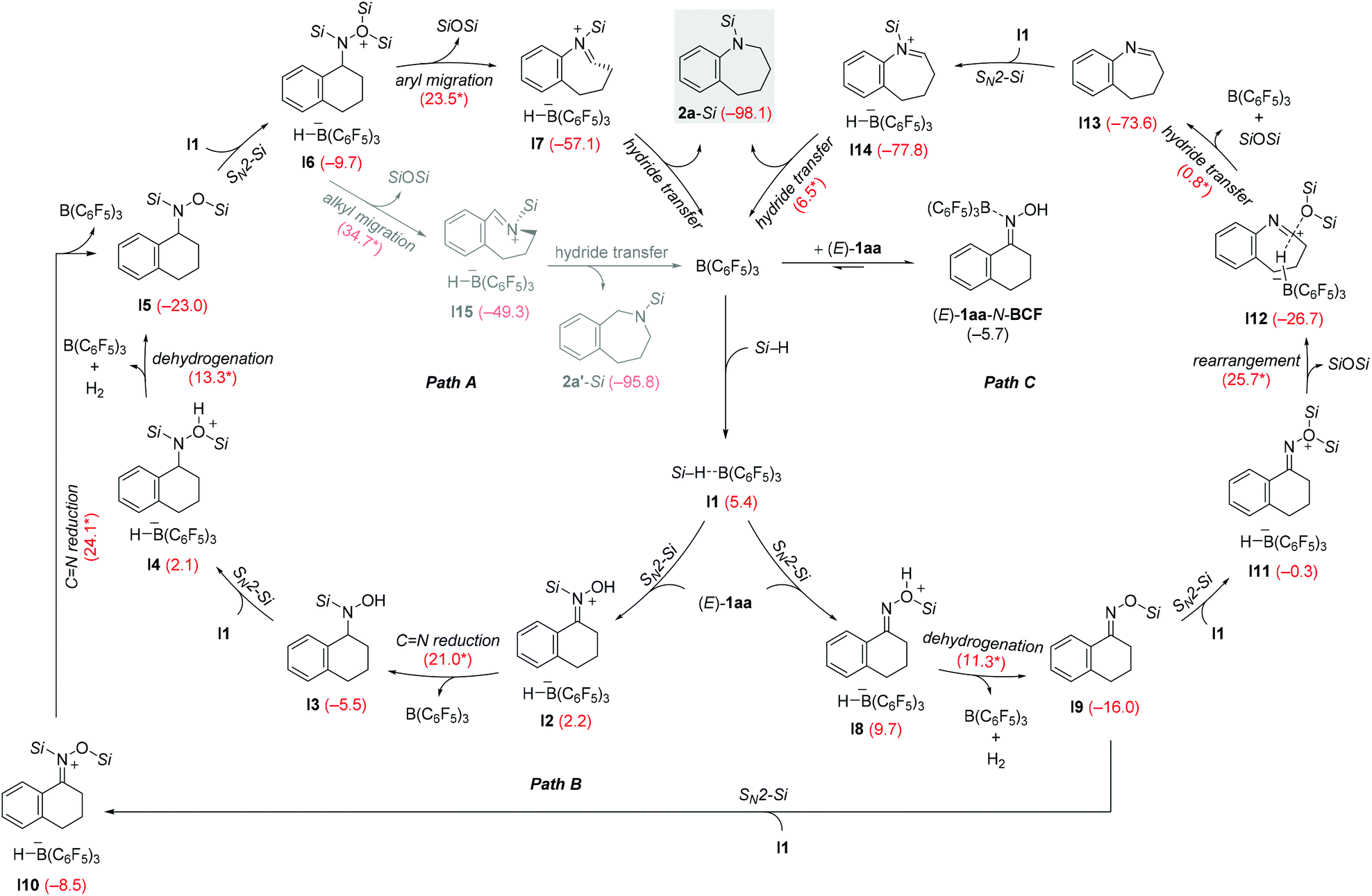 | ||
| Scheme 7 Reaction pathways for the reductive rearrangement of oximes to secondary amines calculated with the M06-2X functional. For each step, the Gibbs free reaction energies and barriers (labeled with an asterisk) are in kcal mol−1. (Si = SiH2Ph, SN2-Si = nucleophilic substitution at silicon). | ||
We also considered the possibility of the classical Beckmann rearrangement21 (path C, see Fig. S68 and S69 in the ESI† for details). This pathway proceeds through the reaction of I9 with the B(C6F5)3·hydrosilane adduct I1 to form the bissilyloxonium ion I11 with the oxygen atom as the nucleophilic center. Aryl migration and N–O bond cleavage of I11 further leads to the formation of ternary complex I12, in which both the borohydride and the disiloxane are loosely bound to the carbon atom of the nitrilium ion (see Fig. S69† for the structural details of I12). Subsequent hydride reduction accompanied by disiloxane displacement yields the ring-expanded imine I13 along with B(C6F5)3. A subsequent imine hydrosilylation10 through N-silyliminium ion I14 furnishes the reductive rearrangement product 2a-Si (gray box). The rate-determining step of this pathway is the aryl migration/N–O bond cleavage in intermediate I11 with an activation barrier of 25.7 kcal mol−1. Although this process is kinetically less favorable than path A or B, we cannot exclude whether this pathway contributes to the formation of the related reductive rearrangement product, especially in cases where higher reaction temperatures are applied.
The direct hydride transfer from the borohydride to the nitrogen atom of intermediate I6 or I11 to eventually form the primary amine 3a is kinetically disfavoured (cf. gray box in Table 1). The corresponding barriers are estimated to be at least 65.9 and 47.6 kcal mol−1, respectively (relative to separated reactants; see Fig. S64, S68, and S72†). These results can account for the high selectivity in favor of the secondary amine 2a under the experimental conditions.
Conclusion
In summary, we have developed an effective catalytic reductive rearrangement of oximes and their congeners to selectively give secondary amines. Aryl-substituted secondary and primary hydrosilanes work as stoichiometric reductants, and a catalyst loading of 10 or 2.5 mol% of B(C6F5)3 is sufficient to secure synthetically useful yields. Quantum-chemical calculations revealed that this reductive rearrangement can follow two distinct orders of events (Scheme 7): reduction–rearrangement involving hydroxylamine derivatives (paths A and B) or rearrangement–reduction (path C). The ring expansion yields an iminium ion (paths A and B) or a nitrilium ion (path C). All pathways are energetically accessible but paths A and B are kinetically favored at room temperature; path C becomes competitive in those cases where high reaction temperatures are required. Hence, a Beckmann rearrangement (path C) cannot be excluded but reduction followed by ring-expanding migration is more likely. The method indeed extends to hydroxylamine derivatives which is in agreement with the computed mechanism and control experiments. The new protocol avoids the previously used stoichiometric aluminum- and boron-based reductants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. F. gratefully acknowledges the Alexander von Humboldt Foundation for a postdoctoral fellowship (2018–2020). G. W. thanks the National Natural Science Foundation of China for financial support (Grant No. 21903043). All theoretical calculations were performed at the High-Performance Computing Center (HPCC) of Nanjing University. M. O. is indebted to the Einstein Foundation (Berlin) for an endowed professorship. We thank Dr Maria Schlangen and Dr Sebastian Kemper for expert advice with the MS and NMR measurements, respectively.Notes and references
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, FLP catalysis: Main group hydrogenations of organic unsaturated substrates, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC.
- (a) J. Mohr and M. Oestreich, B(C6F5)3-catalyzed hydrogenation of oxime ethers without cleavage of the N–O bond, Angew. Chem., Int. Ed., 2014, 53, 13278–13281 CrossRef CAS PubMed; (b) J. Mohr, D. Porwal, I. Chatterjee and M. Oestreich, Extending the scope of the B(C6F5)3-catalyzed C=N bond reduction: Hydrogenation of oxime ethers and hydrazones, Chem. – Eur. J., 2015, 21, 17583–17586 CrossRef CAS PubMed.
- Cramer and co-workers recently accomplished a transition-metal-catalyzed hydrogenation of oximes to hydroxyamines: J. Mas-Roselló, T. Smejkal and N. Cramer, Iridium-catalyzed acid-assisted asymmetric hydrogenation of oximes to hydroxylamines, Science, 2020, 368, 1098–1102 CrossRef PubMed.
- (a) D. Weber and M. R. Gagné, Si–H bond activation by main-group Lewis acids, in Organosilicon Chemistry: Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 33–85 Search PubMed; (b) M. Oestreich, J. Hermeke and J. Mohr, A unified survey of Si–H and H–H bond activation catalysed by electron-deficient boranes, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- H. Fang and M. Oestreich, Defunctionalisation catalysed by boron Lewis acids, Chem. Sci., 2020, 11, 12604–12615 RSC.
- For a review, see: (a) H. Cho, Rearrangement of oximes and hydroxylamines with aluminum reductants, Tetrahedron, 2014, 70, 3527–3544 CrossRef CAS. Selected examples involving LiAlH4: (b) D. R. Smith, M. Maienthal and J. Tipton, Reduction of oximes with lithium aluminum hydride, J. Org. Chem., 1952, 17, 294–297 CrossRef CAS; (c) R. E. Lyle and H. J. Troscianiec, Molecular rearrangements. VI. The rearrangement of oximes on reduction with lithium aluminum hydride, J. Org. Chem., 1955, 20, 1757–1760 CrossRef CAS; (d) M. N. Rerick, C. H. Trottier, R. A. Daignault and J. D. DeFoe, The lithium aluminum hydride - aluminum chloride reduction of oximes, Tetrahedron Lett., 1963, 4, 629–634 CrossRef. Selected examples involving DIBAL-H: (e) S. Sasatani, T. Miyazaki, K. Maruoka and H. Yamamoto, Diisobutylaluminum hydride a novel reagent for the reduction of oximes, Tetrahedron Lett., 1983, 24, 4711–4712 CrossRef CAS; (f) K. Maruoka, T. Miyazaki, M. Ando, Y. Matsumura, S. Sakane, K. Hattori and H. Yamamoto, Organoaluminum-promoted Beckmann rearrangement of oxime sulfonates, J. Am. Chem. Soc., 1983, 105, 2831–2843 CrossRef CAS; (g) H. Cho, Y. Iwama, K. Sugimoto, S. Mori and H. Tokuyama, Regioselective synthesis of heterocycles containing nitrogen neighboring an aromatic ring by reductive ring expansion using diisobutylaluminum hydride and studies on the reaction mechanism, J. Org. Chem., 2010, 75, 627–636 CrossRef CAS PubMed. An example with various aluminum reagents: (h) H. Cho, Y. Iwama, N. Mitsuhashi, K. Sugimoto, K. Okano and H. Tokuyama, Ring-expansion reaction of oximes with aluminum reductants, Molecules, 2012, 17, 7348–7355 CrossRef CAS PubMed.
- Selected examples involving BH3·SMe2: (a) M. Ortiz-Marciales, D. Figueroa, J. A. López, M. De Jesús and R. Vega, Steric and electronic effects on the reduction of O-silylated aromatic ketoximes with borane, Tetrahedron Lett., 2000, 41, 6567–6570 CrossRef CAS; (b) M. Ortiz-Marciales, L. D. Rivera, M. De Jesús, S. Espinosa, J. A. Benjamin, O. E. Casanova, I. G. Figueroa, S. Rodríguez and W. Correa, Facile Rearrangement of O-silylated oximes on reduction with boron trifluoride/borane, J. Org. Chem., 2005, 70, 10132–10134 CrossRef CAS PubMed.
- C. B. Caputo and D. W. Stephan, Activation of alkyl C–F bonds by B(C6F5)3: Stoichiometric and catalytic transformations, Organometallics, 2012, 31, 27–30 CrossRef CAS.
- H. Fang and M. Oestreich, Reductive deamination with hydrosilanes catalyzed by B(C6F5)3, Angew. Chem., 2020, 59, 11394–11398 CrossRef CAS PubMed.
- Selected examples of B(C6F5)3-catalyzed hydrosilylation of imines: (a) J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, B(C6F5)3-catalyzed hydrosilation of imines via silyliminium intermediates, Org. Lett., 2000, 2, 3921–3923 CrossRef CAS PubMed; (b) D. T. Hog and M. Oestreich, B(C6F5)3-catalyzed reduction of ketones and imines using silicon-stereogenic silanes: Stereoinduction by single-point binding, Eur. J. Org. Chem., 2009, 5047–5056 CrossRef CAS; (c) J. Hermeke, M. Mewald and M. Oestreich, Experimental analysis of the catalytic cycle of the borane-promoted imine reduction with hydrosilanes: Spectroscopic detection of unexpected intermediates and a refined mechanism, J. Am. Chem. Soc., 2013, 135, 17537–17546 CrossRef CAS PubMed.
- (a) Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed. The full citation of the Gaussian software is given in the ESI.†.
- J. Tomasi and M. Persico, Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Direct observation of a borane-silane complex involved in frustrated Lewis-pair-mediated hydrosilylations, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- 3D structures provided in the ESI† were prepared using CYL View. See: C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- (a) D. J. Parks, J. M. Blackwell and W. E. Piers, Studies on the mechanism of B(C6F5)3-catalyzed hydrosilation of carbonyl functions, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed; (b) S. Rendler and M. Oestreich, Conclusive evidence for an SN2-Si mechanism in the B(C6F5)3-catalyzed hydrosilylation of carbonyl compounds: Implications for the related hydrogenation, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed; (c) T. Fallon and M. Oestreich, A constellation of deuterium-labeled silanes as a simple mechanistic probe not requiring absolute configuration determination, Angew. Chem., Int. Ed., 2015, 54, 12488–12491 CrossRef CAS PubMed.
- K. Sakata and H. Fujimoto, Quantum chemical study of B(C6F5)3-catalyzed hydrosilylation of carbonyl group, J. Org. Chem., 2013, 78, 12505–12512 CrossRef CAS PubMed.
- For the reaction of (E)-1aa (or its reduced intermediates I3, I5, I9, and I13) with the B(C6F5)3·PhSiH3 complex I1, the corresponding SN2-Si transition state could not be located. After extensive scans for these processes, no electronic energy barrier was found. Some stable trigonal bipyramidal transition complexes (rather than transition states), such as I2′, I4′, I6′, I8′, I10′, I11′, and I14′, were located (see Fig. S70 in the ESI† for the optimized 3D structures). These results suggest that the activation barriers of these steps mainly come from the entropy loss for trimolecular processes.
- These transition complexes resemble the solid-state structure of the counteranion-stabilized H3Si+ cation described by us: Q. Wu, E. Irran, R. Müller, M. Kaupp, H. F. T. Klare and M. Oestreich, Characterization of hydrogen-substituted silylium ions in the condensed phase, Science, 2019, 365, 168–172 CrossRef CAS PubMed.
- We also calculated the rearrangement of bissilyloxonium ion I6 proceeding through an alkyl migration/N–O bond cleavage process (Scheme 7, I6 → I15, gray color). However, this process is kinetically unfavorable because of the involvement of a high-energy transition state (ΔG‡ = 34.7 kcal mol−1).
- For a recent review, see: (a) K. Kaur and S. Srivastava, Beckmann rearrangement catalysis: A review of recent advances, New J. Chem., 2020, 44, 18530–18572 RSC. Selected examples: (b) M. T. Nguyen, G. Raspoet and L. G. Vanquickenborne, A new look at the classical Beckmann rearrangement: A strong case of active solvent effect, J. Am. Chem. Soc., 1997, 119, 2552–2562 CrossRef CAS; (c) S. Yamabe, N. Tsuchida and S. Yamazaki, Is the Beckmann rearrangement a concerted or stepwise reaction? A computational study, J. Org. Chem., 2005, 70, 10638–10644 CrossRef CAS PubMed; (d) I. Lezcano-González, M. Boronat and T. Blasco, Investigation on the Beckmann rearrangement reaction catalyzed by porous solids: MAS NMR and theoretical calculations, Solid State Nucl. Magn. Reson., 2009, 35, 120–129 CrossRef PubMed; (e) N. An, B.-X. Tian, H.-J. Pi, L. A. Eriksson and W.-P. Deng, Mechanistic insight into self-propagation of organo-mediated Beckmann rearrangement: A combined experimental and computational study, J. Org. Chem., 2013, 78, 4297–4302 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and computational data. See DOI: 10.1039/d1qo00251a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2021 |