
Pd/LA-catalyzed decarboxylation enabled exclusive [5 + 2] annulation toward N-aryl azepanes and DFT insights†
Teng
Liu‡
a,
Yuqi
Fang‡
b,
Linhong
Zuo
a,
Yulian
Yang
a,
Yin
Liu
a,
Weipeng
Chen
a,
Li
Dang
 b and
Wusheng
Guo
*a
b and
Wusheng
Guo
*a
aFrontier Institute of Science and Technology (FIST), Xi'an Jiaotong University, Yanxiang Road 99, Xi'an 710045, China. E-mail: wusheng.guo@mail.xjtu.edu.cn
bDepartment of Chemistry and Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University, Guangdong 515063, China
First published on 18th February 2021
Abstract
A practical and straightforward methodology towards the synthesis of non-fused N-aryl azepane derivatives with diversity is described. These Pd/LA-catalyzed reactions proceed smoothly under extremely mild conditions with ample reaction scope and CO2 as the byproduct. The resulting products can be easily converted into a range of highly functionalized azepanes. The synthetic value of this protocol is further proved in the formal synthesis of a pharmaceutically relevant Proheptazine derivative. DFT calculations revealed the details of the reaction pathway and the origin of this unusual exclusive [5 + 2] rather than empirical [3 + 2] annulation process.
Introduction
Azepane derivatives have been widely used as key intermediates in synthetic chemistry,1 and also found very important applications in biology as novel inhibitors, antidiabetics, anticancer and DNA binding reagents (Scheme 1a).2 For example, Proheptazine is an opioid analgesic and it produces similar effects to other opioids including analgesia, sedation and nausea.1d Appreciable progress has been made in the preparation of fused benzoazepanes3 while the diverse synthesis of the corresponding non-fused N-functionalized azepane derivatives is still underdeveloped,4,5 mainly due to the unfavourable enthalpic and entropic reasons.6 A handful of cyclization strategies4 were developed in the synthesis of non-fused N-alkyl/Ts azepanes, while the low nucleophilic character of the aryl versus alkyl amine moiety poses a huge challenge in the nucleophilic ring-closing stage toward the N-aryl azepanes. A few of approaches5 resulting in non-fused N-aryl azepanes generally suffered limitations on either reaction scope or functionality tolerance, thus restricting their further derivatization in synthetic chemistry.5 In this respect, Kleij's group recently developed a Pd-catalyzed one-pot, two-step cascade process in the presence of a dehydrating agent toward N-aryl azepane derivatives.7 To the best of our knowledge, a straightforward protocol toward the synthesis of non-fused N-aryl azepanes with diversity has not been established yet. Therefore, it is highly significant to explore such a process, especially in the development of new azepane-based pharmaceuticals.2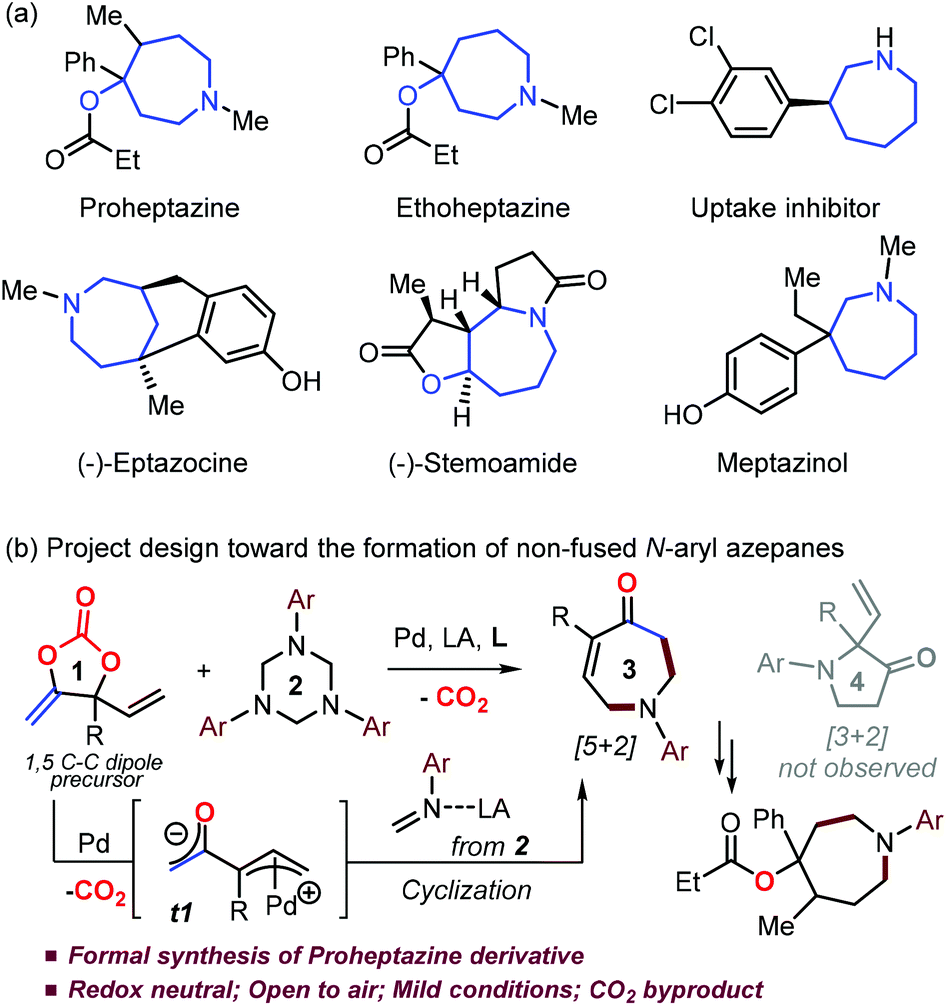 | ||
| Scheme 1 (a) Examples of azepanes as drugs or bio-active compounds and (b) Pd/LA-catalyzed decarboxylative formation of N-aryl azepanes. | ||
Decarboxylation of vinyl cyclic carbonates/carbamates has proved to be quite efficient in the preparation of a huge range of interesting molecules with CO2 as the sole byproduct under mild conditions.8,9 Very recently, our group designed and synthesized a class of new cyclic carbonates (1 in Scheme 1b) which are air-stable and could serve as reactive 1,3 or 1,5 C–C dipole precursors.9n Based on our knowledge of Pd-catalyzed transformations of cyclic carbonates,8,9h–k we envisaged that, in the presence of suitable palladium and Lewis acid (LA) catalysts with a ligand, the decarboxylation of carbonate 1 would generate a zwitterionic Pd-allyl intermediate t1; overcoming the challenge of the chemoselectivity of the reaction between t1 and an in situ formed N-aryl formaldimine10 would yield non-fused N-aryl azepane derivative 3via [5 + 2] annulation, although a formal [3 + 2] route resulting in pyrrolidinone 4 could be more favoured empirically (Scheme 1b).5,9f–g,9r Interestingly, we did not detect any pyrrolidinone product 4 throughout the whole condition screening process. With judicious choice of the catalytic system, the expected N-aryl azepanes could be obtained with decent yields. The carbonyl and alkenyl fractions in product 3 facilitate its further derivatization in synthetic chemistry. Therefore, the present methodology offers a general, practical and diverse protocol toward the formation of challenging non-fused N-aryl azepanes. We herein report our latest results on this topic and reveal the origin of this unusual exclusive [5 + 2] rather than empirical [3 + 2] annulation process via DFT calculations.
Results and discussion
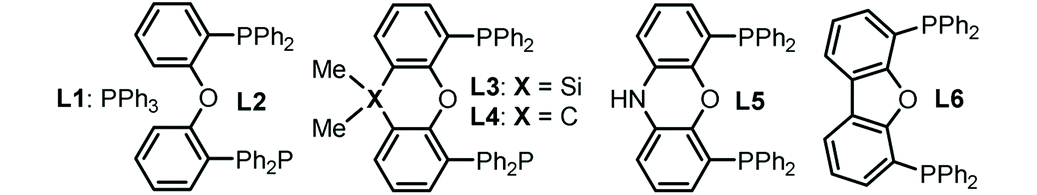
To start our research, the cyclic carbonate 1a and triazine 2a were submitted for the reaction in the presence of palladium and LA catalysts under different conditions at room temperature (Table 1). To our delight, the combination of Pd(TFA)2 and BF3·Et2O proved to be effective toward the formation of product 3aa with 19% NMR yield using DCM as the solvent (entry 1). Control experiments showed the key role of the palladium catalyst, ligand and Lewis acid additive to the successful reaction (entries 2 and 3). Keeping the other conditions unchanged, a range of biphosphane ligands (L2–L6) were then screened and ligand L5 showed more effective catalysis with a satisfactory NMR yield (entries 4–8). The category of Lewis acid additives affected the reaction outcome remarkably when using Pd(TFA)2 and L5 in the reaction (entries 9–13). The use of AgOTf gave rise to the best result with 91% yield of the desired product (entry 13), while the utilization of Sc(OTf)3 and Cu(OTf)2 proved to be ineffective (entries 9 and 10). Other palladium precatalyst were not active, suggesting the intrinsic reactivity of Pd(TFA)2 (entries 14–16). Further solvent screening resulted in an inferior reaction outcome (entries 17–19). No reaction was observed when N-benzylidenetosylamide was used as the substrate instead of trizaine 2a even at 60 °C,11 suggesting that the combination of carbonate 1a and 2a is compulsory for the desired conversions.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | L | LA | Solvent | Yieldb [%] |
| a The reactions were carried out with 1a (0.10 mmol), 2a (0.12 mmol), Pd catalyst (5 mol%), ligand (10 mol%), and Lewis acid (10 mol%) in solvent (0.5 mL) at room temperature for 8 h. b NMR yield using 2-methylnaphthalene as the internal standard. | |||||
| 1 | Pd(TFA)2 | L1 | BF3·Et2O | DCM | 19 |
| 2 | — | — | BF3·Et2O | DCM | — |
| 3 | Pd(TFA)2 | L1 | — | DCM | — |
| 4 | Pd(TFA)2 | L2 | BF3·Et2O | DCM | <1 |
| 5 | Pd(TFA)2 | L3 | BF3·Et2O | DCM | 33 |
| 6 | Pd(TFA)2 | L4 | BF3·Et2O | DCM | 29 |
| 7 | Pd(TFA)2 | L5 | BF3·Et2O | DCM | 56 |
| 8 | Pd(TFA)2 | L6 | BF3·Et2O | DCM | <1 |
| 9 | Pd(TFA)2 | L5 | Sc(OTf)3 | DCM | <1 |
| 10 | Pd(TFA)2 | L5 | Cu(OTf)2 | DCM | <1 |
| 11 | Pd(TFA)2 | L5 | AgTFA | DCM | 32 |
| 12 | Pd(TFA)2 | L5 | AgNTf2 | DCM | 81 |
| 13 | Pd(TFA) 2 | L5 | AgOTf | DCM | 91 |
| 14 | Pd(dba)2 | L5 | AgOTf | DCM | <1 |
| 15 | Pd(OAc)2 | L5 | AgOTf | DCM | <1 |
| 16 | White catalyst | L5 | AgOTf | DCM | <1 |
| 17 | Pd(TFA)2 | L5 | AgOTf | DCE | 78 |
| 18 | Pd(TFA)2 | L5 | AgOTf | THF | 12 |
| 19 | Pd(TFA)2 | L5 | AgOTf | ACN | 49 |
|
|||||
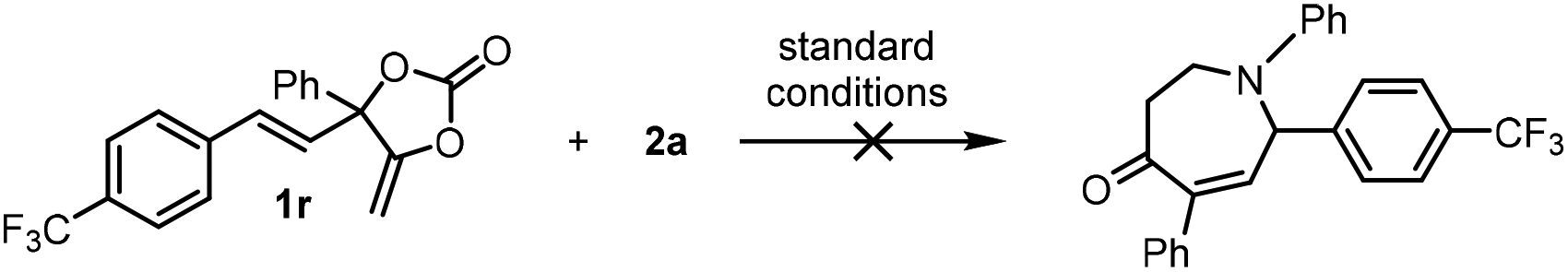
With the optimized reaction conditions in hand (Table 1, entry 13), the scope of the triazine partners (2a–2o) in the combination with carbonate 1a was first investigated (Fig. 1). The reaction proceeded smoothly in the presence of a wide range of triazines equipped with both electron-withdrawing (3ae–3ai, 3ak, 3al and 3ao) and electron-donating (3ab–3ad, 3aj, 3am and 3an) functionalized aryl substituents toward the formation of the desired products in good to excellent yields. In some cases, the use of small amounts of triazine (3ab and 3am) or elevated reaction temperature (3ak, 3ae, 3ah and 3an) was required for efficient catalysis. The triazine 2n bearing a heterocyclic group showed moderate reactivity toward the formation of product 3an at higher temperature with longer reaction time.12 The absolute configuration of product 3ao is unambiguously confirmed by X-ray analysis.13 Then, we focused on the investigation of the carbonate scope (3ba–3qa). Satisfactorily, a number of aryl- (3ba–3ma and 3pa–3qa) and alkyl-substituted carbonates (3na–3oa) equipped with different functional groups were tolerated under the optimized reaction conditions, giving access to the corresponding seven-membered azepanes in decent isolated yields. The introduction of naphthyl (3ja) or cyclohexyl (3na) in the carbonate substrates did not affect the efficiency of the catalysis. The present methodology allowed the installation of para-, meta- and otho-Ar substituents on the final products (Fig. 1; 3aa–3ao, 3ba–3ma, and 3pa–3qa), while the use of alkyl substituted triazines did not lead to the target products.14 It is noteworthy that the present protocol was highly efficient for the preparation of azepane derivatives equipped with F (3ag, 3al, 3fa and 3qa), OCF3 (3ae and 3ea) and CF3 (3ah, 3ga and 3ma) functional groups, which was otherwise synthetically challenging yet highly desired for biological applications.15 An attempt to synthesize α-functionalized azepane from a multisubstituted carbonate substrate 1r failed, indicating the synthetic limitations of the present methodology.16
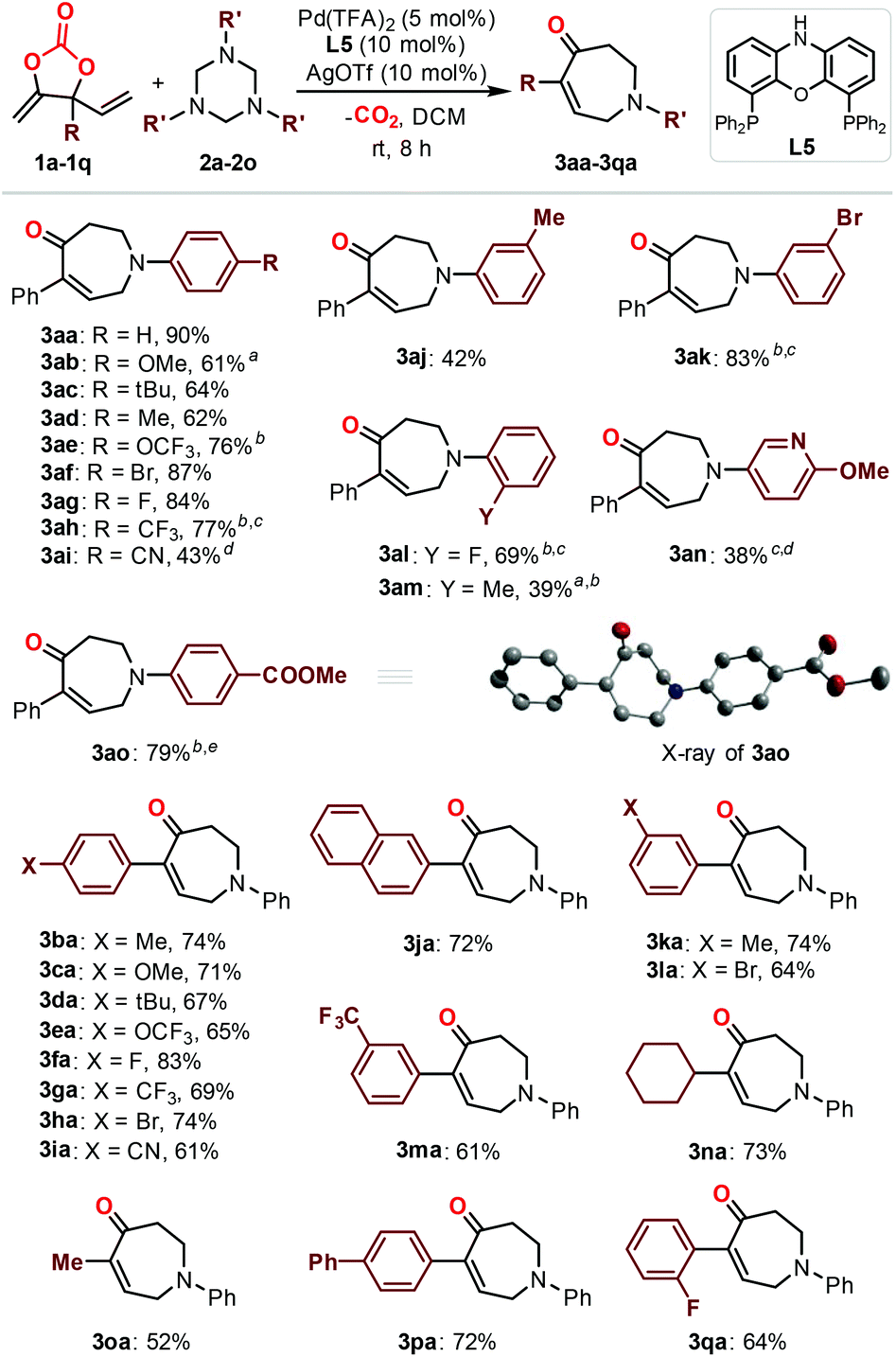 | ||
Fig. 1 Scope of 1,3,5-triazine and carbonate reaction partners. Reaction conditions: 1a–1q (0.1 mmol), 2a–2n (0.12 mmol), Pd(TFA)2 (5 mol%), L5 (10 mol%), AgOTf (10 mol%), DCM (0.5 mL), rt, 8 h. Isolated yields were obtained. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Using 0.8 equiv. of 1,3,5-triazine. b40 °C. c18 h. d60 °C. e42 h. The inset shows the solid state of 3ao and the hydrogen atoms are omitted for clarity. Using 0.8 equiv. of 1,3,5-triazine. b40 °C. c18 h. d60 °C. e42 h. The inset shows the solid state of 3ao and the hydrogen atoms are omitted for clarity. | ||
Notably, the reaction could be easily performed on a larger scale (8 mmol) without loss of efficiency, which was exemplified by the successful isolation of 1.68 g of compound 3aa (Fig. 2). In addition to the application potential of the resulting products reported previously,1,2 we further proved the synthetic utilization of these azepane derivatives using 3aa as a model substrate under different reaction conditions (Fig. 2a).17a–c,18,19 The double bond could be selectively reduced by H2 while retaining the carbonyl group (5a).17a Oxidation of the double bond gave rise to the corresponding epoxide 5b.17b The carbonyl group was selectively reduced with NaBH4 affording the alcohol 5c while retaining the double bond.17c In the presence of m-CPBA as the oxidant, a bicyclic isoxazolidine compound 5d was obtained with good isolated yield, and the solid state of the product was further characterized by single crystalline X-ray analysis.18 The reaction with trimethylsilylacetylene in the presence of n-BuLi allowed the introduction of a triple bond functionality in the azepane derivative (5e).19 The synthetic value of these azepane products was further proved in the formal synthesis of the N-phenyl Proheptazine derivative (Fig. 2b).17a,20 The azepane 3oa was reduced with dihydrogen17a and followed by a nucleophilic attack to afford the corresponding tertiary alcohol 7;20a the reaction of alcohol 7 and propionyl chloride20b gave rise to the pharmaceutically relevant N-phenyl Proheptazine 8 successfully.
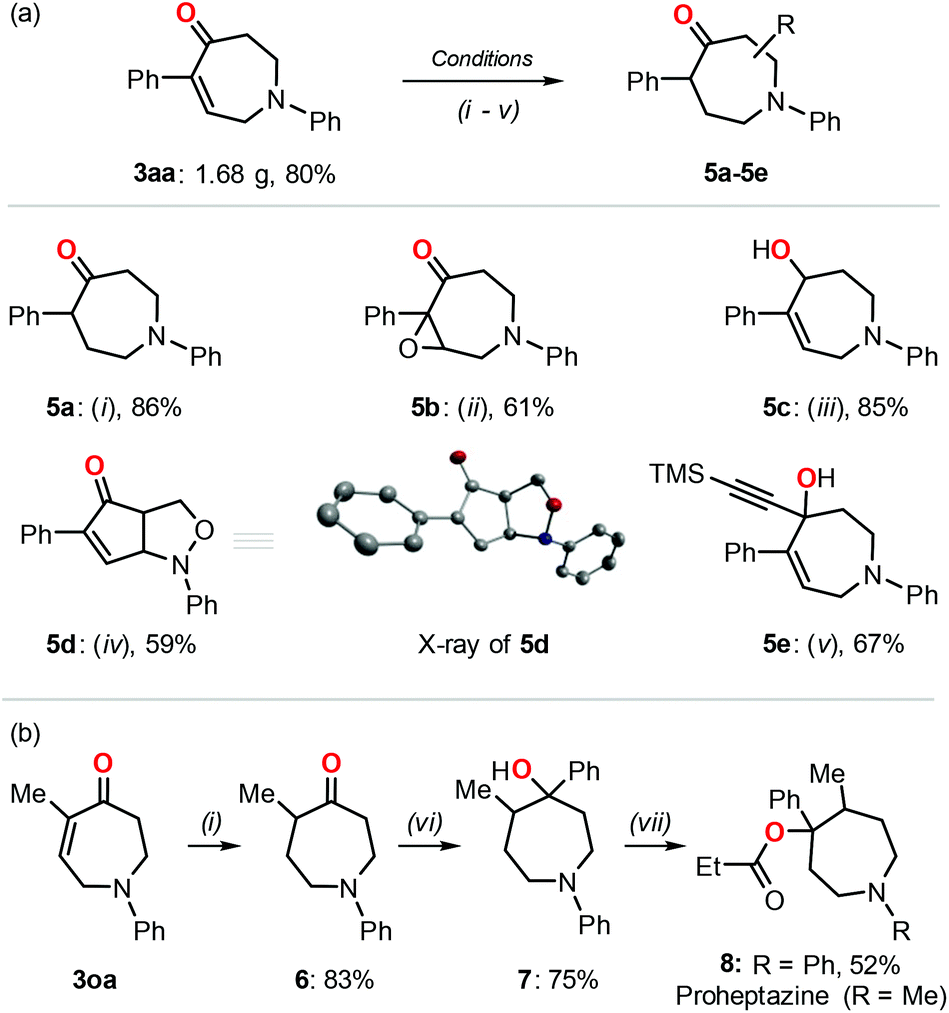 | ||
| Fig. 2 (a) Post-transformations of product 3aa under different reaction conditions: (i) H2 (1 bar), Pd/C (10%), MeOH, rt, 18 h; (ii) H2O2 (3 equiv.), aq. NaOH, MeOH, 0 °C to rt, 4 h; (iii) CeCl3 (1.2 equiv.), NaBH4 (1.2 equiv.), MeOH, 0 °C, 4 h; (iv) p-TsOH·H2O (10 mol%), m-CPBA (2.5 equiv.), benzene, rt, 6 h; (v) trimethylsilylacetylene (12 equiv.), n-BuLi (10 equiv.), anhydrous THF, under a N2 atmosphere, −78 °C to rt, 12 h. (b) Formal synthesis of Proheptazine derivative 8 from azepane 3oa; reaction conditions: (vi) PhLi (2.5 equiv.), THF, −78 °C, 12 h; (vii) propionyl chloride (4 equiv.), n-BuLi (4 equiv.), anhydrous THF, N2, −78 °C to 60 °C, 24 h. See the ESI† for details. The inset shows the X-ray image of 5d in which the hydrogen atoms are omitted for clarity. | ||
DFT calculations were performed using the reaction of 1a and 2a as a model in order to gain insights into the reaction pathways and the origin of the exclusive [5 + 2] rather than the [3 + 2] annulation process. All of the reported energies (in kcal mol−1) are relative free energies in solution. The species involved in the calculations are named with the prefix t (for theoretical) for clarity. The reaction process based on the calculations is shown in Scheme 2 as a simplified catalytic cycle, and the selected free energy profiles (in kcal mol−1) for the formation of relevant intermediates are shown in Scheme 3. The reaction started with the coordination of carbonate 1a with catalyst t1, followed by the oxidative C–O cleavage and CO2 extrusion toward the formation of palladacycle t4 (Scheme 2 and Scheme S1†);21 this process is similar to the one in our previous report on decarboxylative transformations of vinyl carbonates.9i The isomerization of t4 toward the formation of palladacycles t5, t5′ and t5′′ readily occurs within the 15 kcal mol−1 barrier (Scheme S1†).21 From palladacycles t5 and t5′′, the amine insertion and reductive elimination would give rise to AgOTf-bonded products t7 and t8 through either a six- or an eight-membered N-palladacycle t6 or t6′ (Scheme 3). The calculated results showed that the formation of t7 has much lower barriers than that of t8. Moreover, the calculations suggested that the isomerization from t7 to t8 commands a very high barrier (46.1 kcal mol−1), showing that it is impossible to generate t8 through the isomerization of t7. We further experimentally proved that the transformation of product 3aa into 4aa was impossible under the standard conditions or even at 60 °C (see the ESI† for details). Both the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N insertion and reductive elimination in the whole pathway toward the formation of AgOTf-bonded product t7 have lower than 16 kcal mol−1 barriers (Scheme 3 and Scheme S1†). These results are consistent with the experimental observation that the reaction occurred under mild conditions with 3aa (derived from t7) as the sole product.
N insertion and reductive elimination in the whole pathway toward the formation of AgOTf-bonded product t7 have lower than 16 kcal mol−1 barriers (Scheme 3 and Scheme S1†). These results are consistent with the experimental observation that the reaction occurred under mild conditions with 3aa (derived from t7) as the sole product.
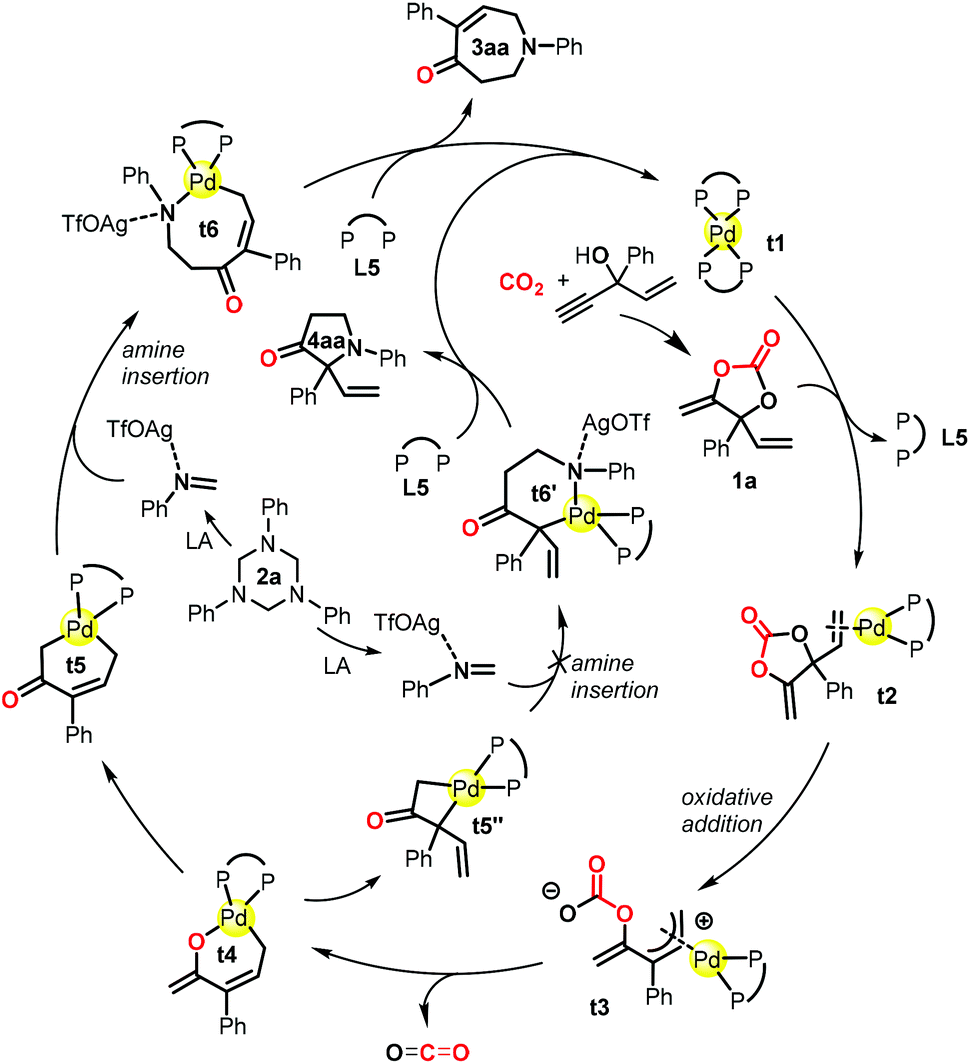 | ||
| Scheme 2 Proposed mechanism based on DFT calculations for the reaction of cyclic carbonate 1a and 1,3,5-triazine 2a toward the formation of compound 3aa and pyrrolidinone 4aa. | ||
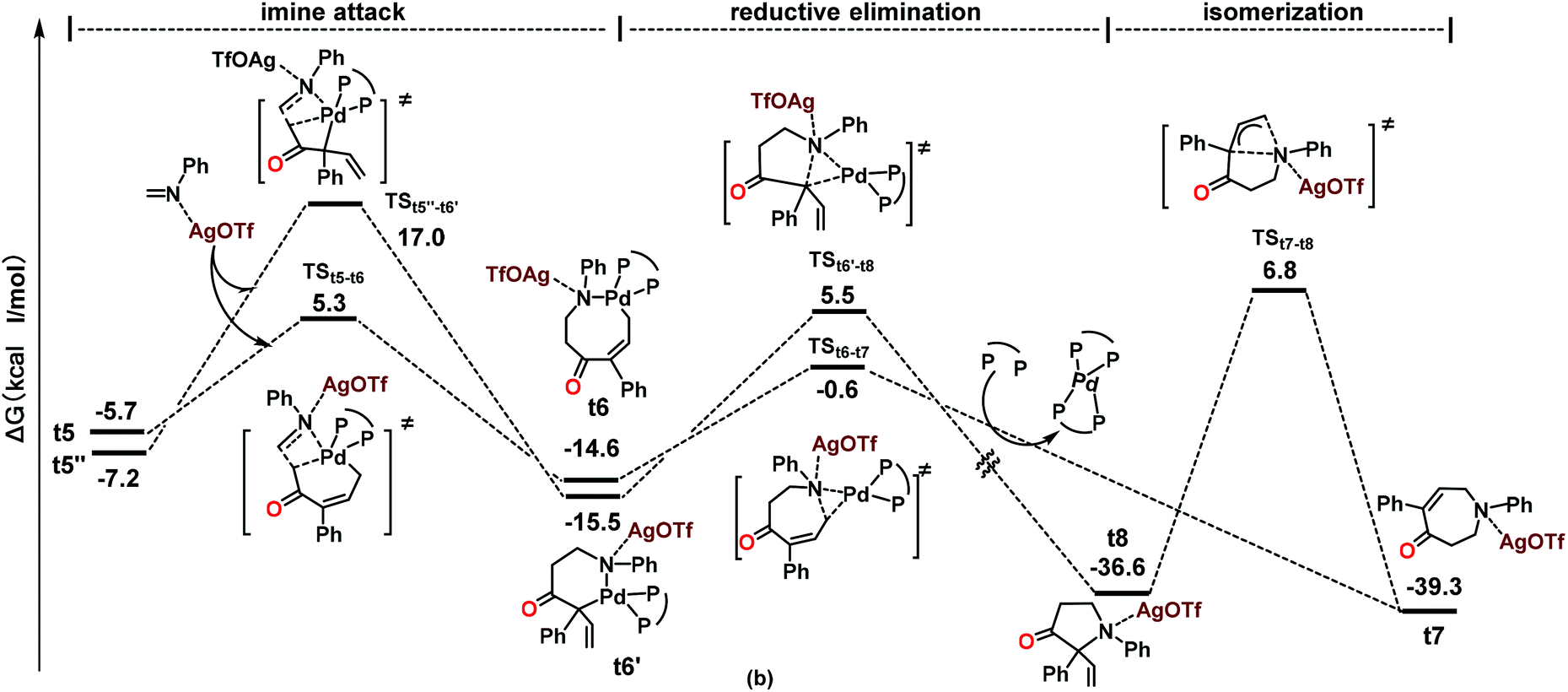 | ||
| Scheme 3 The free energy profiles (in kcal mol−1) for the formation of intermediates t7 and t8. | ||
Conclusions
To sum up, we herein report the first straightforward and diverse synthesis of challenging non-fused N-aryl azepane derivatives. This Pd/LA-catalyzed process could be performed open to air under extremely mild conditions with CO2 as the byproduct, thus offering a new and operationally friendly protocol for the preparation of N-aryl azepane derivatives. The resulting products could be easily transformed into a range of otherwise synthetically challenging and useful compounds. The synthetic value of this protocol is further proved in the formal synthesis of a pharmaceutically relevant N-phenyl Proheptazine derivative. The DFT calculations revealed the details of the reaction pathway and the origin of this unusual exclusive [5 + 2] rather than empirical [3 + 2] annulation process.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by the starting funding scheme of Xi'an Jiaotong University. We would also like to thank the Instrument Analysis Center of Xi'an Jiaotong University for the assistance with HRMS analysis.Notes and references
- For selected research articles, please refer to: (a) C. Draghici, T. Wang and D. A. Spiegel, Concise total synthesis of glucosepane, Science, 2015, 350, 294–298 CrossRef CAS; (b) W. Zawodny, S. L. Montgomery, J. R. Marshall, J. D. Finnigan, N. J. Turner and J. Clayden, Chemoenzymatic synthesis of substituted azepanes by sequential biocatalytic reduction and organolithium-mediated rearrangement, J. Am. Chem. Soc., 2018, 140, 17872–17877 CrossRef CAS; (c) T.-L. Shih, R.-Y. Yang, S.-T. Li, C.-F. Chiang and C.-H. Lin, Expeditious synthesis of tri-and tetrahydroxyazepanes from D-(–)-quinic acid as potent glycosidase inhibitors, J. Org. Chem., 2007, 72, 4258–4261 CrossRef CAS; (d) J. Diamond, W. F. Bruce and F. T. Tyson, Synthesis and properties of the analgesic DL-α-1,3-dimethyl-4-phenyl-4-propionoxyazacycloheptane (Proheptazine), J. Med. Chem., 1964, 7, 57–60 CrossRef CAS.
- For selected references, please see: (a) G.-F. Zha, K. P. Rakesh, H. M. Manukumar, C. S. Shantharam and S. Long, Pharmaceutical significance of azepane based motifs for drug discovery: A critical review, Eur. J. Med. Chem., 2019, 162, 465–494 CrossRef CAS; (b) D. S. Yamashita, R. W. Marquis, R. Xie, S. D. Nidamarthy, H.-J. Oh, J. U. Jeong, K. F. Erhard, K. W. Ward, T. J. Roethke, B. R. Smith, H.-Y. Cheng, X. Geng, F. Lin, P. H. Offen, B. Wang, N. Nevins, M. S. Head, R. C. Haltiwanger, A. A. N. Sarjeant, L. M. Liable-Sands, B. Zhao, W. W. Smith, C. A. Janson, E. Gao, T. Tomaszek, M. McQueney, I. E. James, C. J. Gress, D. L. Zembryki, M. W. Lark and D. F. Veber, Structure activity relationships of 5-, 6-, and 7-methyl-substituted azepan-3-one cathepsin K inhibitors, J. Med. Chem., 2006, 49, 1597–1612 CrossRef CAS.
- For selected articles on the synthesis of benzoazepanes, please refer to: (a) S. Cui, Y. Zhang, D. Wang and Q. Wu, Rh(III)-catalyzed C–H activation/[4 + 3] cycloaddition of benzamides and vinylcarbenoids: facile synthesis of azepinones, Chem. Sci., 2013, 4, 3912–3916 RSC; (b) J. Hao, Y. Xu, Z. Xu, Z. Zhang and W. Yang, Pd-catalyzed three-component domino reaction of vinyl benzoxazinanones for regioselective and stereoselective synthesis of allylic sulfone-containing amino acid derivatives, Org. Lett., 2018, 20, 7888–7892 CrossRef CAS; (c) H. He, W.-B. Liu, L.-X. Dai and S.-L. You, Enantioselective synthesis of 2,3-dihydro-1H-benzo[b]azepines: Iridium-catalyzed tandem allylic vinylation/amination reaction, Angew. Chem., Int. Ed., 2010, 49, 1496–1499 CrossRef CAS; (d) C. Guo, M. Fleige, D. Janssen-Müller, C. G. Daniliuc and F. Glorius, Cooperative N-heterocyclic carbene/palladium-catalyzed enantioselective umpolung annulations, J. Am. Chem. Soc., 2016, 138, 7840–7843 CrossRef CAS; (e) L.-Z. Yu, Q. Xu, X.-Y. Tang and M. Shi, Iron- or copper-catalyzed trifluoromethylation of acrylamide-tethered alkylidenecyclopropanes: facile synthesis of CF3-containing polycyclic benzazepine derivatives, ACS Catal., 2016, 6, 526–531 CrossRef CAS; (f) A. Claraz, F. Serpier and S. Darses, Organoboron initiated Rh-catalyzed asymmetric cascade reactions: a subtle switch in regioselectivity leading to chiral 3-benzazepine derivatives, ACS Catal., 2017, 7, 3410–3413 CrossRef CAS; (g) S. M. Wales, D. J. Rivinoja, M. G. Gardiner, M. J. Bird, A. G. Meyer, J. H. Ryan and C. J. T. Hyland, Benzoazepine-fused isoindolines via intramolecular (3 + 2)-cycloadditions of azomethine ylides with dinitroarenes, Org. Lett., 2019, 21, 4703–4708 CrossRef CAS; (h) D. Liu, M.-J. Jiao, X.-Z. Wang and P.-F. Xu, Metal-free visible-light-induced construction of difluoro-containing dibenzazepines, Org. Lett., 2019, 21, 4745–4749 CrossRef CAS; (i) Z. Zuo, J. Liu, J. Nan, L. Fan, W. Sun, Y. Wang and X. Luan, Highly stereoselective synthesis of imine-containing dibenzo[b,d]azepines by a palladium(II)-catalyzed [5 + 2] oxidative annulation of o-arylanilines with alkynes, Angew. Chem., Int. Ed., 2015, 54, 15385–15389 CrossRef CAS; (j) K.-Y. Ye, L.-X. Dai and S.-L. You, Enantioselective synthesis of 2,5-dihydrobenzo[b]azepine derivatives via iridium-catalyzed asymmetric allylic amination with 2-allylanilines and ring-closing-metathesis reaction, Org. Biomol. Chem., 2012, 10, 5932–5939 RSC; (k) Y. Yang, M.-B. Zhou, X.-H. Ouyang, R. Pi, R.-J. Song and J.-H. Li, Rhodium(III)-catalyzed [3 + 2]/[5 + 2] annulation of 4-aryl 1,2,3-triazoles with internal alkynes through dual C(sp2)-H functionalization, Angew. Chem., Int. Ed., 2015, 54, 6595–6599 CrossRef CAS; (l) L.-Z. Yu, Z.-Z. Zhu, X.-B. Hu, X.-Y. Tang and M. Shi, Palladium-catalyzed cascade cyclization of allylamine-tethered alkylidenecyclopropanes: facile access to iodine/difluoromethylene- and perfluoroalkyl-containing 1-benzazepine scaffolds, Chem. Commun., 2016, 52, 6581–6584 RSC; (m) G. Zhan, M.-L. Shi, Q. He, W. Du and Y.-C. Chen, [4 + 3] Cycloadditions with bromo-substituted Morita–Baylis–Hillman adducts of isatins and N-(ortho-chloromethyl)aryl amides, Org. Lett., 2015, 17, 4750–4753 CrossRef CAS; (n) Y. Zhao, J.-R. Chen and W.-J. Xiao, Visible-light photocatalytic decarboxylative alkyl radical addition cascade for synthesis of benzazepine derivatives, Org. Lett., 2018, 20, 224–227 CrossRef CAS; (o) M. Martínez-Mingo, N. Rodríguez and R. G. Arrayas, Access to benzazepinones by Pd-catalyzed remote C–H carbonylation of γ-arylpropylamine derivatives, Org. Lett., 2019, 21, 4345–4349 CrossRef; (p) C. Bressy, D. Alberico and M. Lautens, A route to annulated indoles via a palladium-catalyzed tandem alkylation/direct arylation reaction, J. Am. Chem. Soc., 2005, 127, 13148–13149 CrossRef CAS; (q) G.-W. Wang and J. F. Bower, Modular access to azepines by directed carbonylative C–C bond activation of aminocyclopropanes, J. Am. Chem. Soc., 2018, 140, 2743–2747 CrossRef CAS; (r) D. Tsvelikhovsky and S. L. Buchwald, Synthesis of heterocycles via Pd-ligand controlled cyclization of 2-chloro-N-(2-vinyl)aniline: preparation of carbazoles, indoles, dibenzazepines, and acridines, J. Am. Chem. Soc., 2010, 132, 14048–14051 CrossRef CAS; (s) K. Zhang, L. Cai, S. Hong and O. Kwon, Phosphine-catalyzed α-umpolung–aldol reaction for the synthesis of benzo[b]azapin-3-ones, Org. Lett., 2019, 21, 5143–5146 CrossRef CAS; (t) S. Undeela, G. Ravikumar, J. B. Nanubolu, K. K. Singarapuc and R. S. Menon, The facile synthesis of 1-benzoazepine derivatives via gold-catalyzed regioselective cycloisomerization reactions of N-(o-alkynylaryl)-N-vinyl sulfonamides, Chem. Commun., 2016, 52, 4824–4827 RSC.
- For representative articles, please refer to: (a) J.-J. Feng, T.-Y. Lin, H.-H. Wu and J. Zhang, Transfer of chirality in the rhodium-catalyzed intramolecular formal hetero-[5 + 2] cycloaddition of vinyl aziridines and alkynes: stereoselective synthesis of fused azepine derivatives, J. Am. Chem. Soc., 2015, 137, 3787–3790 CrossRef CAS; (b) J.-J. Feng, T.-Y. Lin, C.-Z. Zhu, H. Wang, H.-H. Wu and J. Zhang, The divergent synthesis of nitrogen heterocycles by rhodium(I)-catalyzed intermolecular cycloadditions of vinyl aziridines and alkynes, J. Am. Chem. Soc., 2016, 138, 2178–2181 CrossRef CAS; (c) A. Barbero, A. Diez-Varga, F. J. Pulido and A. González-Ortega, Synthesis of azepane derivatives by silyl-aza-Prins cyclization of allylsilyl amines: influence of the catalyst in the outcome of the reaction, Org. Lett., 2016, 18, 1972–1975 CrossRef CAS; (d) C.-Z. Zhu, J.-J. Feng and J. Zhang, Rhodium(I)-catalyzed intermolecular aza-[4 + 3] cycloaddition of vinyl aziridines and dienes: atom-economical synthesis of enantiomerically enriched functionalized azepines, Angew. Chem., Int. Ed., 2017, 56, 1351–1355 CrossRef CAS; (e) L. Cui, L. Ye and L. Zhang, Gold-catalyzed efficient synthesis of azepan-4-ones via a two-step [5+2] annulation, Chem. Commun., 2010, 46, 3351–3353 RSC Meyer's group recently reported a multi-step synthesis of azepanones; please see: (f) A. Dupas, P.-A. Lhotellier, G. Guillamot, C. Meyer and J. Cossy, Synthesis of highly substituted azepanones from 2H-azirines by a stepwise annulation/ring-opening sequence, Org. Lett., 2019, 21, 3589–3593 CrossRef CAS.
- (a) N. D. Shapiro and F. D. Toste, Synthesis of azepines by a gold-catalyzed intermolecular [4 + 3]-annulation, J. Am. Chem. Soc., 2008, 130, 9244–9245 CrossRef CAS; (b) K. Ramesh, S. N. Murthy and Y. V. D. Nageswar, Novel and efficient aqueous phase synthesis of N-substituted azepines via tandem Michael addition and cyclization in the presence of β-cyclodextrin, Tetrahedron Lett., 2011, 52, 2362–2366 CrossRef CAS; (c) D. Chaturvedi, A. K. Chaturvedi, N. Mishra and V. Mishra, Efficient, one-pot, BF3·OEt2-mediated synthesis of substituted N-aryl lactams, Synlett, 2012, 23, 2627–2630 CrossRef CAS; (d) Y. Shi, P. C. J. Kamer, D. J. Cole-Hamilton, M. Harvie, E. F. Baxter, K. J. C. Lim and P. Pogorzelec, A new route to N-aromatic heterocycles from the hydrogenation of diesters in the presence of anilines, Chem. Sci., 2017, 8, 6911–6917 RSC.
- (a) K. E. O. Ylijoki and J. M. Stryker, [5 + 2] Cycloaddition reactions in organic and natural product synthesis, Chem. Rev., 2013, 113, 2244–2266 CrossRef CAS; (b) M. Harmata, The (4 + 3)-cycloaddition reaction: heteroatom-substituted allylic cations as dienophiles, Chem. Commun., 2010, 46, 8904–8922 RSC; (c) M. Harmata, The (4 + 3)-cycloaddition reaction: simple allylic cations as dienophiles, Chem. Commun., 2010, 46, 8886–8903 RSC; (d) H. Butenschon, Seven-membered rings by cyclization at transition metals: [4 + 3], [3 + 2 + 2], [5 + 2], Angew. Chem., Int. Ed., 2008, 47, 5287–5290 CrossRef.
- J. Xie, X. Li and A. W. Kleij, Pd-catalyzed stereoselective tandem ring-opening amination/cyclization of vinyl γ-lactones: access to caprolactam diversity, Chem. Sci., 2020, 11, 8839–8845 RSC.
- For reviews, please refer to: (a) W. Guo, J. E. Gómez, À. Cristòfol, J. Xie and A. W. Kleij, Catalytic transformations of functionalized cyclic organic carbonates, Angew. Chem., Int. Ed., 2018, 57, 13735–13747 CrossRef CAS; (b) L. Zuo, T. Liu, X. Chang and W. Guo, An update of transition metal-catalyzed decarboxylative transformations of cyclic carbonates and carbamates, Molecules, 2019, 24, 3930 CrossRef CAS.
- For selected representative research articles, please refer to: (a) L.-C. Yang, Z. Y. Tan, Z.-Q. Rong, R. L. Liu, Y.-N. Wang and Y. Zhao, Palladium-titanium relay catalysis enables switch from alkoxide-π-allyl to dienolate reactivity for spiro-heterocycle synthesis, Angew. Chem., Int. Ed., 2018, 57, 7860–7864 CrossRef CAS; (b) Z.-Q. Rong, L.-C. Yang, S. Liu, Z. Yu, Y.-N. Wang, Z. Y. Tan, R.-Z. Huang, Y. Lan and Y. Zhao, Nine-membered benzofuran-fused heterocycles: enantioselective synthesis by Pd-catalysis and rearrangement via transannular bond formation, J. Am. Chem. Soc., 2017, 139, 15304–15307 CrossRef CAS; (c) S. Singha, T. Patra, C. G. Daniliuc and F. Glorius, Highly enantioselective [5 + 2] annulations through cooperative N-heterocyclic carbene (NHC) organocatalysis and palladium catalysis, J. Am. Chem. Soc., 2018, 140, 3551–3554 CrossRef CAS; (d) L.-C. Yang, Z.-Q. Rong, Y.-N. Wang, Z. Y. Tan, M. Wang and Y. Zhao, Construction of nine-membered heterocycles through palladium-catalyzed formal [5 + 4] cycloaddition, Angew. Chem., Int. Ed., 2017, 56, 2927–2931 CrossRef CAS; (e) Y. Wei, S. Liu, M.-M. Li, Y. Li, Y. Lan, L.-Q. Lu and W.-J. Xiao, Enantioselective trapping of Pd-containing 1,5-dipoles by photogenerated ketenes: access to 7-membered lactones bearing chiral quaternary stereocenters, J. Am. Chem. Soc., 2019, 141, 133–137 CrossRef CAS; (f) A. Khan, R. Zheng, Y. Kan, J. Ye, J. Xing and Y. J. Zhang, Palladium-catalyzed decarboxylative cycloaddition of vinylethylene carbonates with formaldehyde: enantioselective construction of tertiary vinylglycols, Angew. Chem., Int. Ed., 2014, 53, 6439–6442 CrossRef CAS; (g) A. Khan, L. Yang, J. Xu, L. Y. Jin and Y. J. Zhang, Palladium-catalyzed asymmetric decarboxylative cycloaddition of vinylethylene carbonates with Michael acceptors: construction of vicinal quaternary stereocenters, Angew. Chem., Int. Ed., 2014, 53, 11257–11260 CrossRef CAS; (h) A. Cai, W. Guo, L. Martínez-Rodríguez and A. W. Kleij, Palladium-catalyzed regio- and enantioselective synthesis of allylic amines featuring tetrasubstituted tertiary carbons, J. Am. Chem. Soc., 2016, 138, 14194–14197 CrossRef CAS; (i) W. Guo, L. Martínez-Rodríguez, R. Kuniyil, E. Martin, E. C. Escudero-Adán, F. Maseras and A. W. Kleij, Stereoselective and versatile preparation of tri- and tetrasubstituted allylic amine scaffolds under mild conditions, J. Am. Chem. Soc., 2016, 138, 11970–11978 CrossRef CAS; (j) J. E. Gómez, W. Guo and A. W. Kleij, Palladium-catalyzed stereoselective formation of substituted allylic thioethers and sulfones, Org. Lett., 2016, 18, 6042–6045 CrossRef; (k) W. Guo, L. Martínez-Rodríguez, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Highly efficient catalytic formation of (Z)-1,4-But-2-ene diols using water as a nucleophile, Angew. Chem., Int. Ed., 2016, 55, 11037–11040 CrossRef CAS; (l) A. Khan, S. Khan, I. Khan, C. Zhao, Y. Mao, Y. Chen and Y. J. Zhang, Enantioselective construction of tertiary C–O bond via allylic substitution of vinylethylene carbonates with water and alcohols, J. Am. Chem. Soc., 2017, 139, 10733–10741 CrossRef CAS; (m) L.-C. Yang, Y.-N. Wang, R. Liu, Y. Luo, X. Q. Ng, B. Yang, Z.-Q. Rong, Y. Lan, Z. Shao and Y. Zhao, Stereoselective access to [5.5.0] and [4.4.1] bicyclic compounds through Pd-catalysed divergent higher-order cycloadditions, Nat. Chem., 2020, 12, 860–868 CrossRef CAS; also see ref. 3d; (n) B. Yan, L. Zuo, X. Chang, T. Liu, M. Cui, Y. Liu, H. Sun, W. Chen and W. Guo, Kinetically controllable Pd-catalyzed decarboxylation enabled [5 + 2] and [3 + 2] cycloaddition toward carbocycles featuring quaternary carbons, Org. Lett., 2021, 23, 351–357 CrossRef CAS; (o) K. Komatsuki, Y. Sadamitsu, K. Sekine, K. Saito and T. Yamada, Stereospecific decarboxylative Nazarov cyclization mediated by carbon dioxide for the preparation of highly substituted 2-cyclopentenones, Angew. Chem., Int. Ed., 2017, 56, 11594–11598 CrossRef CAS; (p) K. Komatsuki, A. Kozuma, K. Saito and T. Yamada, Decarboxylative Nazarov cyclization-based chirality transfer for asymmetric synthesis of 2-cyclopentenones, Org. Lett., 2019, 21, 6628–6632 CrossRef CAS; (q) K. Kozuma, K. Komatsuki, K. Saito and T. Yamada, Thermal decarboxylative Nazarov cyclization of cyclic enol carbonates involving chirality transfer, Chem. Lett., 2020, 49, 60–63 CrossRef; (r) K. Ohmatsu, N. Imagawa and T. Ooi, Ligand-enabled multiple absolute stereocontrol in metal-catalysed cycloaddition for construction of contiguous all-carbon quaternary stereocentres, Nat. Chem., 2014, 6, 47–51 CrossRef CAS; (s) Y. Zheng, T. Qin and W. Zi, Enantioselective inverse electron demand [3 + 2] cycloaddition of palladium-oxyallyl enabled by a hydrogen-bond-donating ligand, J. Am. Chem. Soc., 2021, 143, 1038–1045 CrossRef CAS.
- For using 1,3,5-triazine as a precursor of N-aryl formaldimine, see examples: (a) C. Zhu, G. Xu and J. Sun, Gold-catalyzed formal [4 + 1]/[4 + 3] cycloadditions of diazo esters with triazines, Angew. Chem., Int. Ed., 2016, 55, 11867–11871 CrossRef CAS; (b) L. K. B. Garve, P. G. Jones and D. B. Werz, Ring-opening 1-amino-3-aminomethylation of donor–acceptor cyclopropanes via 1,3-diazepanes, Angew. Chem., Int. Ed., 2017, 56, 9226–9230 CrossRef CAS; (c) S. Oda, J. Franke and M. J. Krische, Diene hydroaminomethylation via ruthenium-catalyzed C–C bond forming transfer hydrogenation: beyond carbonylation, Chem. Sci., 2016, 7, 136–141 RSC; (d) S. Oda, B. Sam and M. J. Krische, Hydroaminomethylation beyond carbonylation: allene–imine reductive coupling by ruthenium-catalyzed transfer hydrogenation, Angew. Chem., Int. Ed., 2015, 54, 8525–8528 CrossRef CAS; (e) Y. Xu, L. Chen, Y. Yang, Z. Zhang and W. Yang, Vinylethylene carbonates as α,β-unsaturated aldehyde surrogates for regioselective [3 + 3] cycloaddition, Org. Lett., 2019, 21, 6674–6678 CrossRef CAS; (f) Y. Yang and W. Yang, Divergent synthesis of N-heterocycles by Pd-catalyzed controllable cyclization of vinylethylene carbonates, Chem. Commun., 2018, 54, 12182–12185 RSC.
- N-Benzylidenetosylamide is usually used as a substrate in cycloaddition reactions; see examples: (a) L. Yang, A. Khan, R. Zheng, L. Y. Jin and Y. J. Zhang, Pd-catalyzed asymmetric decarboxylative cycloaddition of vinylethylene carbonates with imines, Org. Lett., 2015, 17, 6230–6233 CrossRef CAS; (b) X.-F. Zhu, J. Lan and O. Kwon, An expedient phosphine-catalyzed [4 + 2] annulation: synthesis of highly functionalized tetrahydropyridines, J. Am. Chem. Soc., 2003, 125, 4716–4717 CrossRef CAS . See the ESI† for details of our reaction with N-benzylidenetosylamide as a substrate.
- The interaction of pyridine and the palladium catalyst may be the reason of the lower efficiency of the catalysis.
- For further details, please see CCDC 2038549.†.
- The utilization of N-benzyl or N-propyl triazines did not lead to the formation of the corresponding azepanes.
- (a) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, Applications of fluorine in medicinal chemistry, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS; (b) T. Furuya, A. S. Kamlet and T. Ritter, Catalysis for fluorination and trifluoromethylation, Nature, 2011, 473, 470–477 CrossRef CAS.
- The reaction of an internal alkene functionalized carbonate 1r and 2a under the standard conditions did not give the corresponding azepane product.
. - (a) P. Das, S. Gondo, P. Nagender, H. Uno, E. Tokunaga and N. Shibata, Access to benzo-fused nine-membered heterocyclic alkenes with a trifluoromethyl carbinol moiety via a double decarboxylative formal ring-expansion process under palladium catalysis, Chem. Sci., 2018, 9, 3276–3281 RSC; (b) M. Welker, S. Woodward and A. Alexakis, Tandem reactions with chiral enolates: preparation of allylic alcohols via trapping with vinyl oxiranes, Org. Lett., 2010, 12, 576–579 CrossRef CAS; (c) J. Nugent, E. Matoušová, M. G. Banwell and A. C. Willis, The palladium-catalyzed intramolecular Alder-ene reactions of O- and N-linked 1,6-enynes incorporating triethylsilyl capping groups, J. Org. Chem., 2017, 82, 12569–12589 CrossRef CAS.
- We found the formation of isoxazolidine 5d by accident; see CCDC 2038550† for details of the X-ray analysis. Isoxazolidines have found important applications in biology and their syntheses have gained much interest; for representative references, please refer to: (a) C.-H. Chen, Y.-C. Tsai and R.-S. Liu, Gold-catalyzed cyclization/oxidative [3 + 2] cycloadditions of 1,5-enynes with nitrosobenzenes without additional oxidants, Angew. Chem., Int. Ed., 2013, 52, 4599–4603 CrossRef CAS; (b) F. Xie, S. Yu, Z. Qi and X. Li, Nitrone directing groups in rhodium(III)-catalyzed C–H activation of arenes: 1,3-dipoles versus traceless directing groups, Angew. Chem., Int. Ed., 2016, 55, 15351–15355 CrossRef CAS; (c) A. N. Baumann, F. Reiners, T. Juli and D. Didier, Chemodivergent and stereoselective access to fused isoxazoline azetidines and thietanes through [3 + 2]-cycloadditions, Org. Lett., 2018, 20, 6736–6740 CrossRef CAS. The synthesis of bridged bicyclic isoxazolidines like 5d is underdeveloped. Compound 5d is supposed to be derived from the rearrangement of a nitrone intermediate; please refer to: (d) S.-I. Murahashi and Y. Imada, Synthesis and transformations of nitrones for organic synthesis, Chem. Rev., 2019, 119, 4684–4716 CrossRef CAS.
- S. Jansone-Popova and J. A. May, Synthesis of bridged polycyclic ring systems via carbene cascades terminating in C–H bond insertion, J. Am. Chem. Soc., 2012, 134, 17877–17880 CrossRef CAS.
- (a) A. M. Gilbert, G. P. Stack, R. Nilakantan, J. Kodah, M. Tran, R. Scerni, X. Shi, D. L. Smith and T. H. Andree, Modulation of selective serotonin reuptake inhibitor and 5-HT1A antagonist activity in 8-aza-bicyclo[3.2.1]octane derivatives of 2,3-dihydro-1,4-benzodioxane, Bioorg. Med. Chem. Lett., 2004, 14, 515–518 CrossRef CAS; (b) P. S. Salva, G. J. Hite, R. A. Heyman and G. Gianutsos, 3,7-Diazabicyclane. a new narcotic analgesic, J. Med. Chem., 1986, 29, 2111–2113 CrossRef CAS.
- See the ESI† for details.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data and crystallographic data for 3ao and 5d. CCDC 2038549 and 2038550. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo00070e |
| ‡ These authors contributed equally to this study. |
| This journal is © the Partner Organisations 2021 |