
Prolinamide plays a key role in promoting copper-catalyzed cycloaddition of azides and alkynes in aqueous media via unprecedented metallacycle intermediates†
Gargi
Chakraborti
,
Rajkumar
Jana
,
Tirtha
Mandal
,
Ayan
Datta
 * and
Jyotirmayee
Dash
*
* and
Jyotirmayee
Dash
*
School of Chemical Sciences, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700032, India. E-mail: spad@iacs.res.in; ocjd@iacs.res.in
First published on 17th March 2021
Abstract
We herein delineate cycloaddition of a wide range of azides with terminal alkynes using catalytic CuI and a prolinamide ligand in aqueous media under aerobic conditions. The catalytic system is used for a ‘one-pot’ synthesis of triazoles from aliphatic halides and alkynes. A detailed computational study predicts that prolinamide plays a unique role in facilitating the cycloaddition in water via the formation of remarkable metallacycle intermediates. The synthetic utility of the method is demonstrated by the synthesis of pharmacologically relevant triazolyl diaryl amines via a Cu(I) catalyzed relay cycloaddition and Ullmann coupling sequence.
Introduction
The Cu(I) catalyzed Huisgen1 1,3-dipolar cycloaddition2 of organic azides with alkynes is referred to as the prototypical ‘click reaction’ that has found diverse applications in fields ranging from medicinal chemistry to materials science.3,4 While the Huisgen reaction produces a mixture of 1,4- and 1,5-disubstituted triazole products in most cases,1 Sharpless5a and Meldal5b independently reported copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) to obtain 1,4-disubstituted 1,2,3-triazoles regiospecifically. Triazole derivatives are privileged scaffolds in medicinal chemistry, showing potent anti-cancer, anti-allergic, anti-bacterial, and anti-HIV activities.6,7 In our group, we have been using Cu(I) catalyzed and target directed azide–alkyne cycloaddition to prepare triazole ligands for DNA secondary structures with potent biological activities.7Numerous strategies based on Cu(I) based catalytic systems8 have been developed to efficiently synthesize triazole products.9,10 In some instances, high temperature or microwave conditions are required for copper(I) iodide catalyzed cycloadditions. However, the reaction can be performed at room temperature using Cu(I) in the presence of nitrogen, sulfur, NHC or polydentate ligands.11–14 Finn et al. reported that tris(2-benzimidazolylmethyl)amine ligands can accelerate copper(I)-catalyzed azide–alkyne cycloaddition with substantial improvements in the reaction rate as well as in yields of the triazole products.11a
Reiser et al. used a heterogeneous copper(I) isonitrile complex as an efficient catalyst for the 1,3-dipolar cycloaddition of azides and alkynes under mild conditions in water (Scheme 1).9h Díez-González et al. reported cycloaddition of azides and alkynes using the commercially available complex [CuBr(PPh3)3] under neat conditions.9i Although these methods have been widely used for the synthesis of triazoles from alkyl azides and alkynes, cycloaddition between substituted aromatic azides and alkynes in an aqueous environment under ambient conditions still remains challenging.
 | ||
| Scheme 1 Synthesis of triazole derivatives using CuAAC of azides and alkynes. | ||
In this context, we herein describe Cu(I) catalyzed azide–alkyne cycloaddition in aqueous media at room temperature using a triazolyl–prolinamide ligand, Pro-1 (Scheme 1). The reaction proceeds with low Cu(I) and Pro-1 loading (5 mol% CuI and 10 mol% Pro-1), providing highly pure triazole products in excellent yields, even obviating the need for column chromatographic purifications. Cu(I) salts have been directly used as catalysts in cycloaddition using polydentate nitrogen ligands that stabilize Cu(I) intermediates and accelerate the reactions.9,11–14 Inspired by this fact, we successfully employed the triazolyl–prolinamide Pro-1 as a ligand for CuAAC reactions (Scheme 1). The ligand contains two complementary hydrophobic and hydrophilic functional groups, which makes it ideal for assisting the reaction in aqueous media.15 It is further intriguing to find the role of Pro-1 in the catalytic cycle as well as in the transition state of CuAAC using DFT studies. Moreover, the synthetic utility has been demonstrated via a relay CuAAC–Ullmann coupling process.
Results and discussion
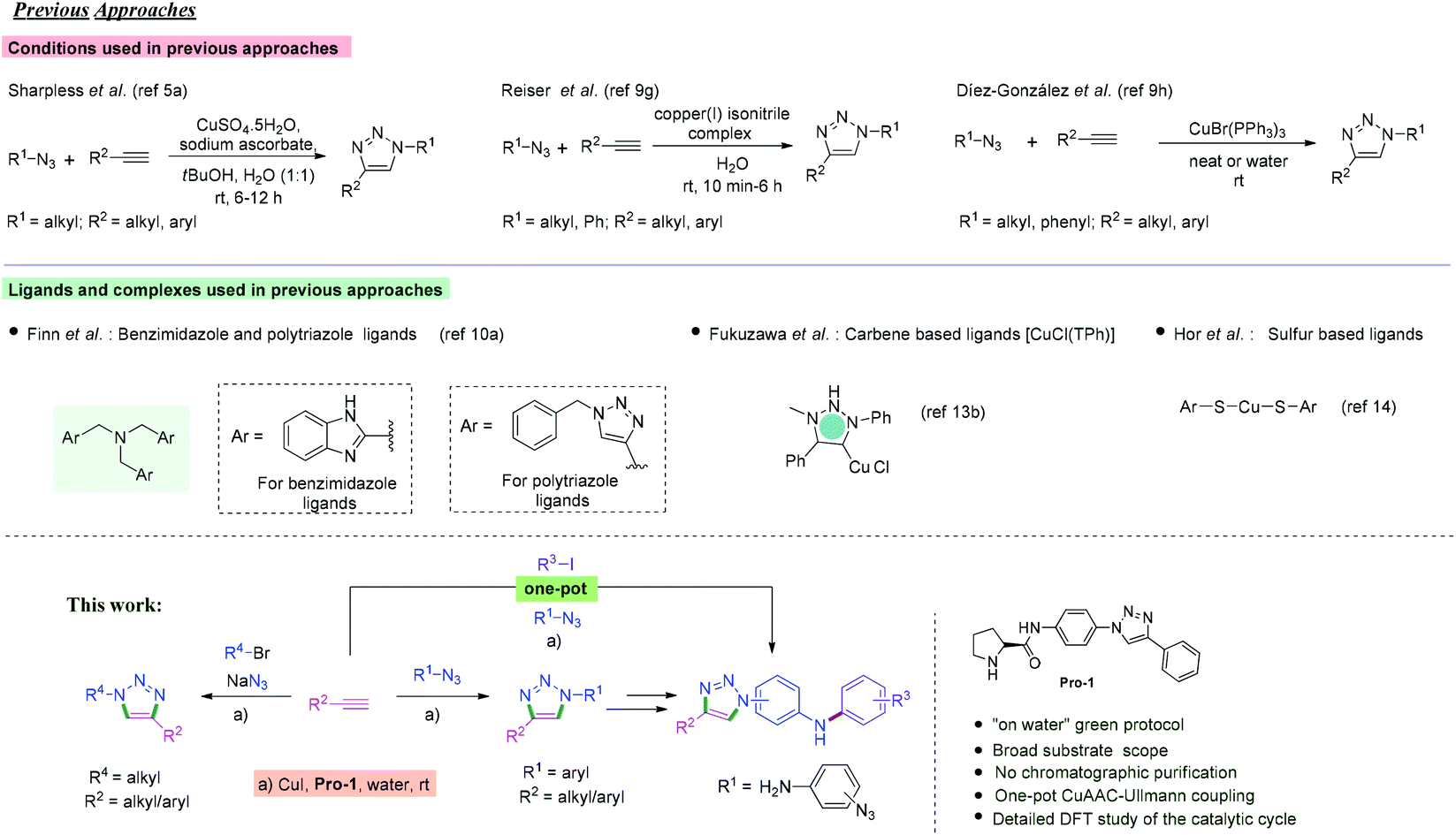
We commenced our studies with the CuAAC of p-azidoanisole (1a) with phenylacetylene (2a) as the model reaction at room temperature in aqueous media (Table 1; see Tables S1 and S2 in ESI,† for the detailed optimization study). To our delight, the reaction proceeded satisfactorily providing the desired product 3a in 95% yield (entry 1, Table 1). Later, DFT calculations revealed that water is an excellent solvent for the reaction to occur at room temperature (Fig. 1 and 2). Further, Pro-1 was found to be the optimal ligand for this transformation, indicating that the structure of prolinamide derivatives may be crucial for attaining high catalytic reactivity (entry 1).16 The synthetic details of prolinamide ligands (Pro-1, Pro-2, and Pro-3) are given in the ESI (Schemes S1–S5, ESI†). The reaction of p-azidoanisole (1a) with phenylacetylene (2a) proceeded satisfactorily in the presence of 5 mol% CuI and 10 mol% Pro-1. However, when the reaction was performed using half the loading of Pro-1, the yield of the triazole derivative 3a decreased (65%, Table S2†).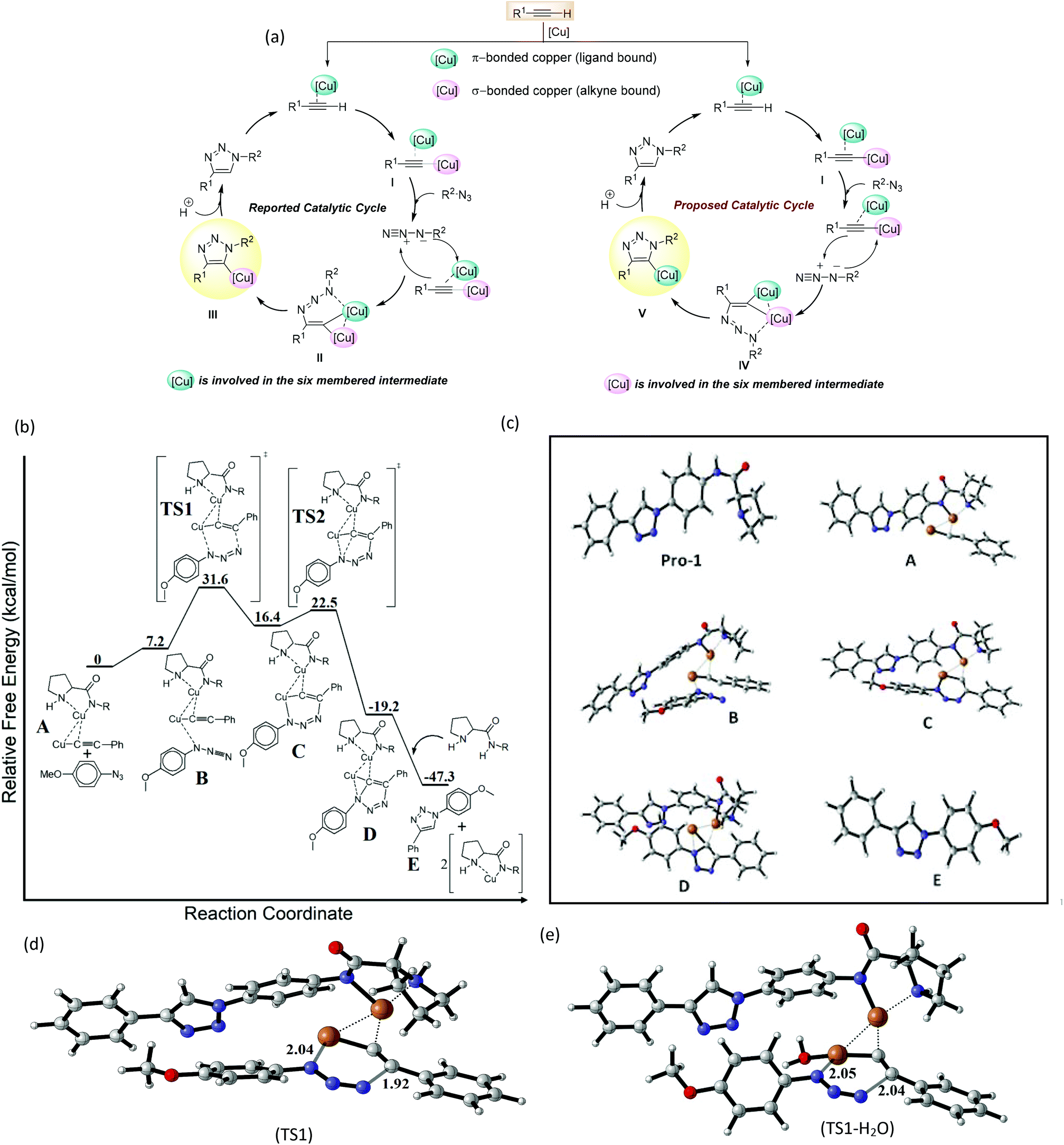 | ||
| Fig. 1 (a) Comparison between Fokin's mechanism and our proposed mechanistic model of CuAAC. (b) Free energy profile for the binuclear CuAAC reaction. (c) Optimized structures of all the reaction intermediates, reactants, products and catalysts for the CuAAC reaction. Optimized transition state (TS) structures with select bond lengths (in Å). (d) TS1 of the cycloaddition. (e) TS1 with the coordinated water molecule (explicit model). | ||
 | ||
| Fig. 2 Solvent dependent variation of activation energies for the rate-determining TS. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Catalyst | Solvent | Time (h) | Yieldb (%) |
a Reaction conditions: 1a (1.0 mmol), 2a (1.50 mmol), CuI (0.05 mmol), Pro-1 (0.1 mmol) in 2 mL water.
b Yield refers to the isolated yield without chromatographic purification. 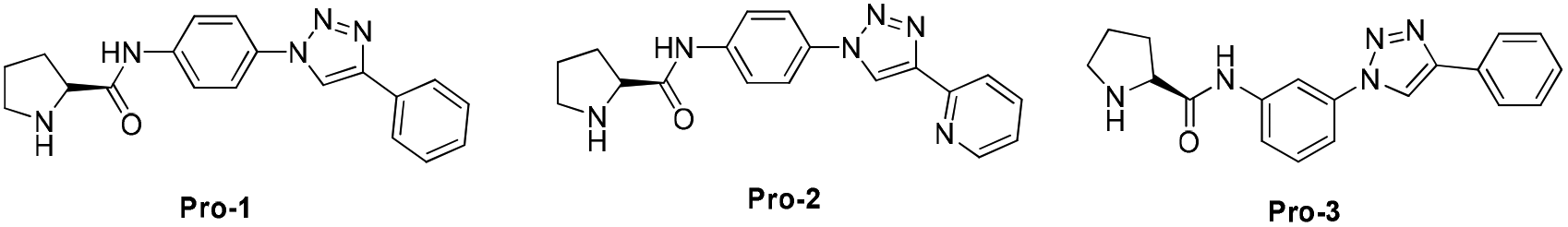
|
|||||
| 1 | Pro-1 | CuI | H2O | 8 | 95 |
| 2 | — | CuI | H2O | 48 | 20 |
| 3 | Pro-1 | — | H2O | 12 h | n.r. |
| 4 | DMEDA | CuI | H2O | 8 | 40 |
| 5 | Phenanthroline | CuI | H2O | 8 | 51 |
| 6 | L-Proline | CuI | H2O | 8 | 65 |
| 7 | Pro-2 | CuI | H2O | 8 | 68 |
| 8 | Pro-3 | CuI | H2O | 8 | 40 |
| 9 | Pro-1 | Cu(OAc)2 | H2O | 8 | 21 |
| 10 | Pro-1 | CuO | H2O | 8 | — |
| 11 | Pro-1 | CuBr | H2O | 8 | 57 |
| 12 | Pro-1 | Cu(0) | H2O | 8 | — |
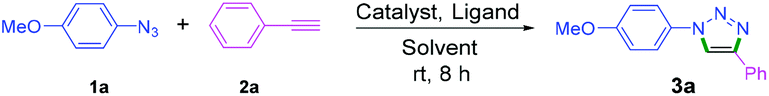
With the optimized reaction conditions (0.1 equiv. Pro-1, 0.05 eq. CuI, in 0.1 M water at room temperature; Table 1, entry 1), we next explored the scope of the reaction by varying azides and acetylenes (Schemes 2 and 3). It is worth noting that only a few studies on CuAAC of aromatic azides have previously been reported in aqueous media at room temperature.9,10
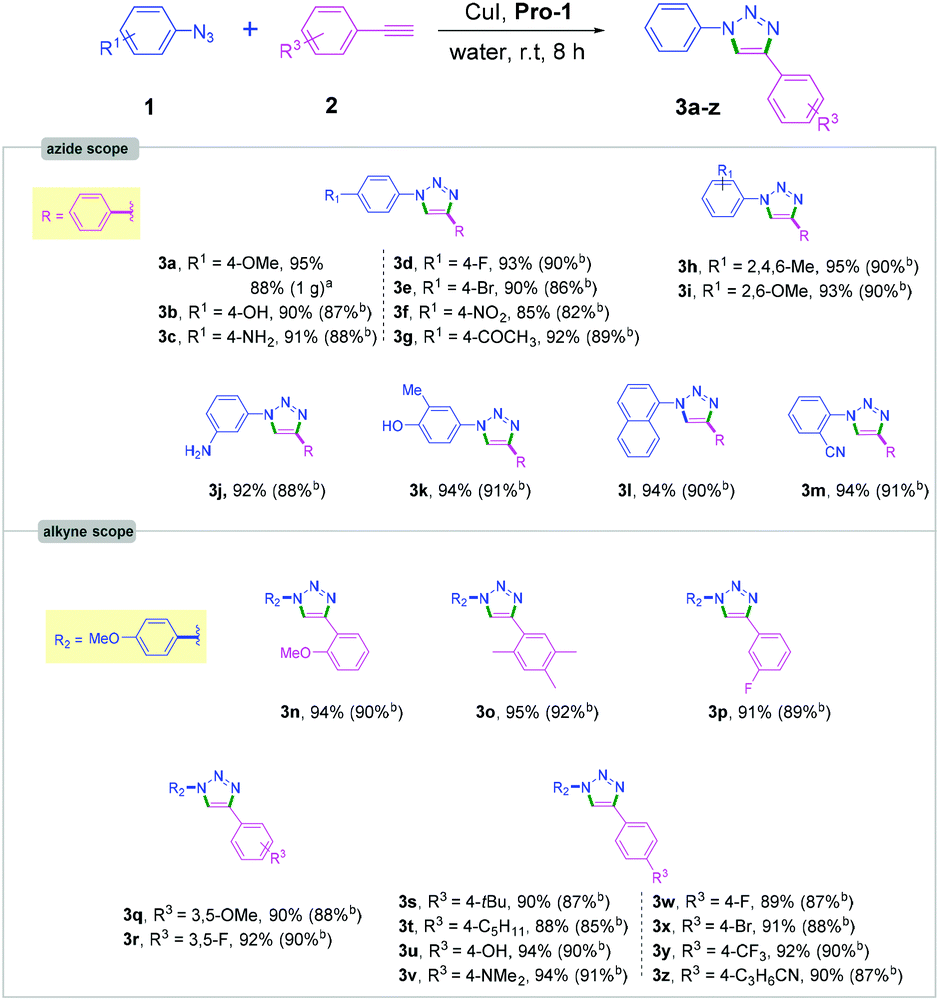 | ||
| Scheme 2 Cycloaddition of aromatic azides and alkynes: areaction performed with 1 g of 1a for 8 h, bisolated yields after chromatographic purification. | ||
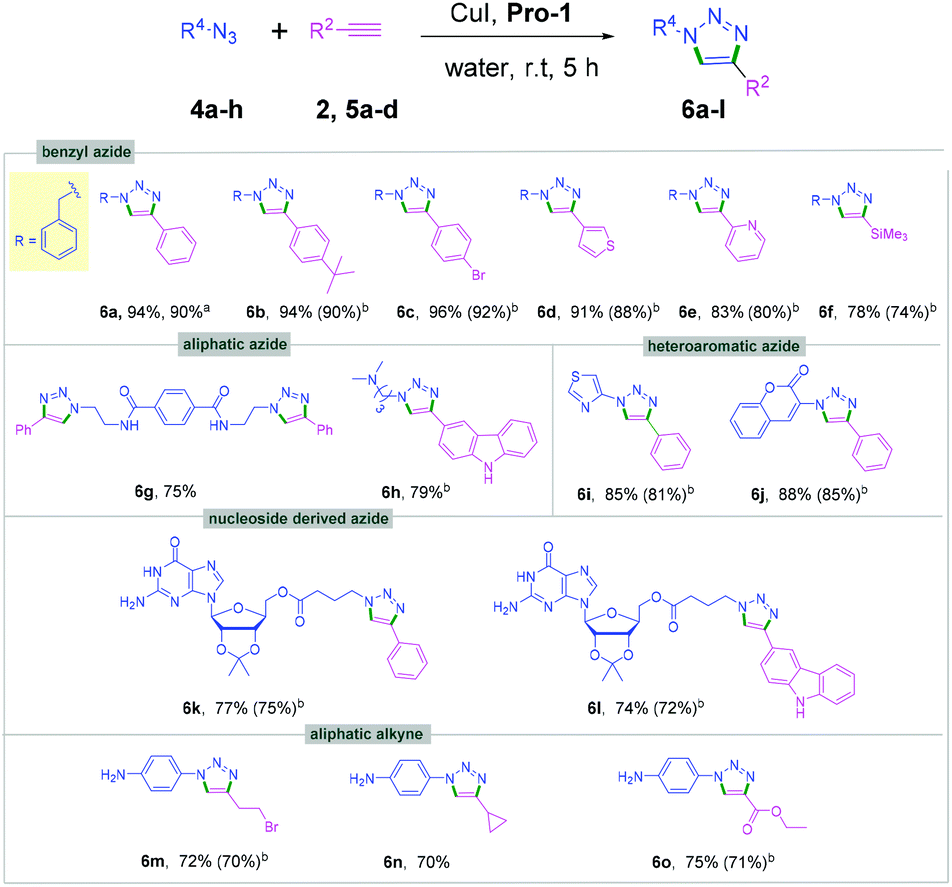 | ||
| Scheme 3 Reaction of benzyl, aliphatic, heteroaromatic and nucleobase derived azides with aromatic and aliphatic alkynes, areaction performed with 1 g of 1a for 5 h, bisolated yields after chromatographic purification. | ||
We were pleased to find that a variety of aromatic azides containing both electron-rich and electron-deficient functional groups (alkyl, methoxy, nitrile, nitro, ketone and halogen as substituents) reacted efficiently providing triazole products in excellent yields and high purity (3a–3z, Scheme 2). The ortho- and meta-substituted aromatic azides also provided the corresponding triazole products in high yields (92–95%, Scheme 2). Importantly, aryl azides containing hydroxy and amine substituents afforded the desired products (3b, 3c, 3u and 3v) in excellent yields (90–94%).
The viability of this catalytic protocol was next expanded using aliphatic, aromatic and heteroaromatic azides, nucleoside derived azides, and bis-azides as well as different aryl, alkyl and heteroaromatic alkynes 2a, 2s, 2x and 5a–d (Scheme 3). Aliphatic azides furnished the corresponding cycloaddition products (6a–6h) in 78%–96% yields (Scheme 3). The aromatic bis-azide 4b also produced the desired bis-triazole product 6g. Thus, the method can be used for the synthesis of both mono- and bis-triazole derivatives. Heteroaromatic azides such as thiazole azide 4d and coumarin azide 4e reacted with phenylacetylene 2a to provide the corresponding triazoles (6i and 6j) in high yields (Scheme 3).
Importantly, guanosine azide 4f could be coupled with alkynes 2a and 5d under the aqueous conditions to afford the products 6k and 6l in 74–77% yields (Scheme 3). Previously, the triazolylnucleobase derivatives were synthesized by CuAAC under a long reaction time.17 The present method offers an efficient approach for synthesizing nucleobase derived triazoles under mild reaction conditions in high yields. Aliphatic alkynes were also found to be compatible under the optimized conditions providing the desired triazoles 6f and 6m–o in good yields (Scheme 3). Gram-scale experiments with 1 g each of 1a and 4a were successfully performed affording the corresponding triazoles 3a and 6c in excellent yields (Schemes 2 and 3).
We next examined this protocol for a one-pot sequential synthesis of triazoles from benzyl/alkyl halides. Benzyl/alkyl halides 7a,b were converted to the corresponding azides in situ and then reacted with terminal alkynes 2a, 5a, 5b and 5d (Scheme 4 and Table S3, ESI†). Triazole derivatives 6 were obtained with excellent yields without any chromatographic purification (Scheme 4). However, we failed to isolate any triazole products when the one-pot reaction was performed using aromatic bromides.18
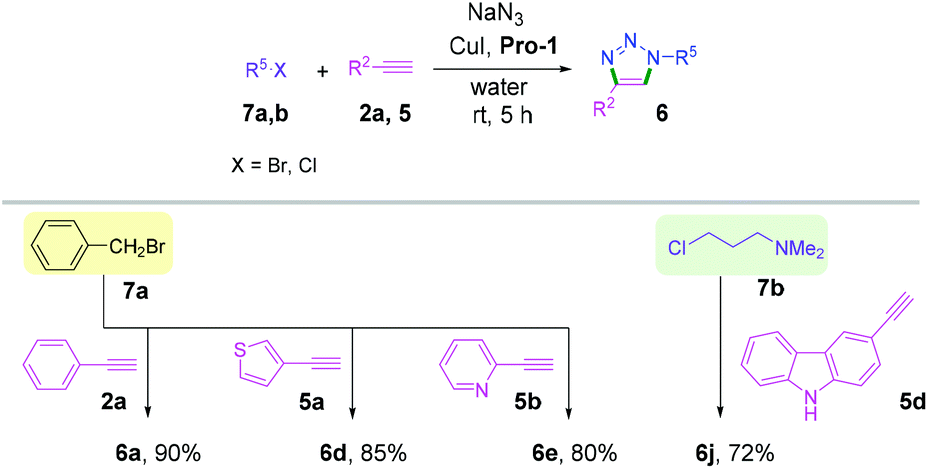 | ||
| Scheme 4 One-pot synthesis of triazoles from alkyl halides. | ||
The well-established catalytic model for CuAAC developed by Fokin et al. suggested that the σ-bound copper acetylide coordinates to an organic azide via the π-bound enriched copper atom to produce complex I (Fig. 1a).19 Next, the nucleophilic attack at N-3 of the azide by the β-carbon of the acetylide forms an unusual six-membered copper metallacycle II, containing the π-bound copper atom. Ring contraction of the intermediate II then provides triazole III containing the σ-bound copper atom.
Intriguingly, a bulky Cu-Pro-1 complex is generated similar to N-heterocyclic Cu catalysts reported by Chen et al.20,21 Owing to the bulky nature of the complex, the azide is stereochemically locked in a particular fashion, i.e. from below the molecular plane of the Cu-Pro-1 complex (Fig. 1a). The conformation of Pro-1 forces the orientation of the intermediate in such a way that the σ-bound copper atom is involved in the six membered metallacycle IV. Intermediate IV then leads to the formation of triazole V that contains the π-bound copper atom. Protonolysis of V provides the desired triazole derivative.
In order to gain insight into the reaction mechanism, density functional theory (DFT) studies were performed using the Gaussian 16 suite of programs22 (the details of the calculations are provided in the ESI† in the “General information on computational methods” section). The CuAAC was expected to be initiated through the activation of Pro-1 by CuI with the formation of the Cu-Pro-1 catalytic complex and deprotonation of phenylacetylene to form a Cu acetylide. Subsequently, the formation of an intermediate (A) occurred via Cu⋯Cu and Cu⋯C weak interactions between Cu-Pro-1 and Cu acetylide (Fig. 1b and c). The succeeding cycloaddition between p-azidoanisole and Cu-Pro-1–acetylide proceeded through the weak bonding interaction between the Cu atom of the acetylide and the nitrogen of the azide (Cu⋯N) (intermediate B). According to previous literature studies, the reaction barriers for a binuclear path of cycloaddition are lower than those for a mononuclear one.20,23–26 We have synthesized a Cu-acetylide using phenyl acetylene and used it to carry out the cycloaddition in the presence of Pro-1. It was observed that the kinetics of this mononuclear pathway proceeded slowly under the optimized reaction conditions (for details see ESI Scheme S8 and Table S5†). Therefore, the most accepted binuclear mechanism was used as a framework to investigate our method.
The cycloaddition step for the formation of six membered metallacycle is considered to be the rate-determining step for Cu-catalyzed alkyne–azide cycloaddition reaction.20,23,25 The estimated activation energy barrier for the cycloaddition was 24.4 kcal mol−1 (TS1, Fig. 1a and 2), which agrees well with the earlier studies.20,23 However, the activation energy for a mononuclear cycloaddition pathway was estimated to be 30.5 kcal mol−1 (Fig. S1†), which is relatively higher than that for a binuclear pathway. Therefore, even though the possibility of a mononuclear reaction pathway cannot be directly ruled out, cycloaddition preferably proceeds through a binuclear pathway. The structure of the optimized transition state (TS1, Fig. 1b) exhibits a bond forming interaction between the carbon atom of the acetylide and the nitrogen atom of the azide (C⋯N) with a bond distance of 1.92 Å. Again, a Cu⋯N interaction (with 2.04 Å bond distance) is also present in the TS forming a six-membered ring. The formation of the six-membered metallacycle (intermediate-C and intermediate IV) in a stereochemically locked configuration is the key difference between our proposed and the earlier reported catalytic cycle. The previously reported mechanism describes the formation of a six-membered metallacycle via the bonding interaction between the π-bound copper and azide N-3 atom while in this case, the interaction occurs between the σ-bonded Cu and azide N-3 atom. However, the resultant six-membered metallacycle is both kinetically and thermodynamically unstable. Hence, it rapidly undergoes ring contraction to form a more stable five-membered [Cu-Pro-1][(1-para-methoxyphenyl-4-phenyl-1,2,3-triazol-5-yl-Cu] complex (D) with a very low activation barrier of 6.1 kcal mol−1 (Fig. 1b and S2†). In the optimized transition state (TS2) leading to the formation of the five membered ring, the bonding interaction between the nitrogen of the azide and the carbon of the acetylide starts forming with a 1.98 Å bond distance (Fig. S2a†).
In order to mimic the experimental conditions, an SMD model was adopted with water, dimethyl sulfoxide and benzene as the continuum dielectrics (ε = 78.35, 46.83 and 2.27 respectively) and the corresponding energy activation barriers for transition states were determined. It was observed that the TS structures were stabilized in the presence of polar solvents, especially water, while for nonpolar solvents like benzene, there was a substantial increase in activation energy barriers. The activation barriers for TS1 are 24.4, 8.3, 14.1, and 20.5 kcal mol−1 respectively for the gas phase, water, DMSO and benzene (Fig. 2). A similar trend was also observed for TS2 with activation barriers of 6.1, 2.7, 3.6 and 4.8 kcal mol−1 respectively (Fig. S2†). Besides the implicit model, we have also considered the explicit solvation model to get a more accurate energy activation barrier as well as transition state structures. The coordination of solvent molecules (namely water, THF, and acetonitrile) with the σ-bonded copper has been reported to accelerate the reaction by stabilizing the transition state structures.23,25 In the explicit model, the effect of coordination of water molecules in the rate-determining transition state is studied (Fig. 1e). As in the implicit model, there is a substantial decrease in activation barrier (11.9 kcal mol−1) for the cycloaddition step. This reduction in energy activation barrier in the presence of a solvent can be attributed to the transition state stabilizing effect of the water molecule by coordinating to σ-bonded alkyne-Cu through the formation of a strong Cu–O bond. This observation nicely corroborates with the earlier studies.23,25 This study indicates that water, a polar protic solvent, not only is beneficial for the protonation of intermediates for the formation of the product but also substantially stabilizes the transition state through a strong Cu–O bonding interaction and hence water is an excellent solvent for this cycloaddition reaction. Even though the reaction might proceed in a polar aprotic solvent like DMSO, nonpolar solvents like benzene would retard the rate of reaction to a greater extent. This result very nicely corroborates with experimental studies. In the final step of cycloaddition, complex (D) is protonated by neighbouring Pro-1 to afford the corresponding triazole product (E) and two Cu-Pro-1 complexes were regenerated. This process was estimated to be more exoergic by 28.1 kcal mol−1. Thus, computational studies reveal in detail the mechanistic pathways for the cycloaddition of azides and alkynes and explain why water as a solvent facilitates this room temperature CuAAC process.
Next, we envisioned that the Ullmann type reaction15c,27 and cycloaddition be combined to develop a new relay process28 in a single reaction vessel, providing a class of triazolyldiaryl amine derivatives. Relay processes combine multiple steps in one-pot and significantly reduce reaction time, energy and waste of materials.29 The reactions of azidoanilines 1, alkyne 2 and aryl halides 10 were performed in a single pot in the presence of the CuI-Pro-1 catalytic system. The catalytic system efficiently promoted the relay process to provide triazolyldiaryl amines 11a–d in good to excellent yields (Scheme 5 and Table S4, ESI†).
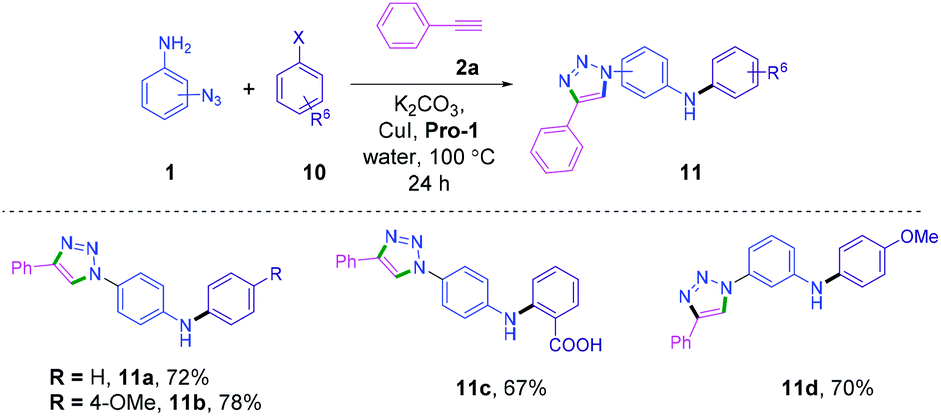 | ||
| Scheme 5 Relay Ullmann–CuAAC. | ||
Conclusions
In summary, we have developed azide–alkyne cycloaddition (AAC) in aqueous media at room temperature using CuI and a prolinamide ligand. The reaction is compatible with many functional groups, thereby enabling the synthesis of 1,4-disubstituted triazoles with diverse structural features. Importantly, the present catalytic system can be readily applied for the one-pot three-component preparation of triazoles using alkyl halides under relatively mild conditions. Furthermore, a Cu(I) catalyzed relay AAC and C–N bond formation is delineated providing an easy access to triazolyl diaryl amine derivatives. The reaction mechanism has been studied by detailed DFT analysis, which indicates the formation of unique metallacycles containing the ligand bound copper atom and further provides evidence that water is the best solvent. The robustness of the catalyst system, broad substrate scope, mild conditions, wide applicability and a different mechanistic approach render the method expeditious for the practical synthesis of triazole derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. D. thanks Wellcome Trust–DBT India Alliance [Grant Number, IA/S/18/2/503986] and CSIR for funding. G. C. thanks Wellcome Trust–DBT India Alliance for the research fellowship. A. D. thanks SERB vide the DIA/2018/000013 project for partial support. We thank Technical Research Centre (TRC) of IACS for funding. R. J. and T. M. thank DST and IACS, Kolkata, for the research fellowship.Notes and references
- (a) R. Huisgen, 1,3-Dipolar Cycloadditions. Past and Future, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef; (b) R. Huisgen and A. Padwa, in 1,3-Dipolar Cycloaddition Chemistry, ed. Wiley, New York, 1984, pp. 1–176 Search PubMed.
- (a) B. M. Trost and I. Fleming, in Comprehensive Organic Synthesis, ed. Pergamon, Oxford, 1991, vol. 4, pp. 1069–1109 Search PubMed; (b) T. Hashimoto and K. Maruoka, Recent Advances of Catalytic Asymmetric 1,3-Dipolar Cycloadditions, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed; (c) L. Maiuolo, V. Algieri, F. Olivito and A. De Nino, Recent Developments on 1,3-Dipolar Cycloaddition Reactions by Catalysis in Green Solvents, Catalysts, 2020, 10, 65–92 CrossRef; (d) M. Breugst and H.-U. Reissig, The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition, Angew. Chem., 2020, 59, 12293–12307 CrossRef CAS PubMed and the references therein.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Click Chemistry : Diverse Chemical Function from a Few Good Reactions, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- For reviews, see: (a) S. K. Mamidyala and M. G. Finn, In situ click chemistry: probing the binding landscapes of biological molecules, Chem. Soc. Rev., 2010, 39, 1252–1261 RSC; (b) K. D. Hanni and D. A. Leigh, The application of CuAAC ‘click’ chemistry to catenane and rotaxane synthesis, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC; (c) Y. Hua and A. H. Flood, Click chemistry generates privileged CH hydrogen-bonding triazoles: the latest addition to anion supramolecular chemistry, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC; (d) C. Le Droumaguet, C. Wang and Q. Wang, Fluorogenic click reaction, Chem. Soc. Rev., 2010, 39, 1233–1239 RSC; (e) J. M. Holub and K. Kirshenbaum, Tricks with clicks: modification of peptidomimetic oligomers via copper-catalyzed azide-alkyne [3+2] cycloaddition, Chem. Soc. Rev., 2010, 39, 1325–1337 RSC; For selected examples, see: (f) K. B. Sharpless and R. Manetsch, In situ click chemistry: a powerful means for lead discovery, Expert Opin. Drug Discovery, 2006, 1, 525–538 CrossRef CAS PubMed; (g) D. D. Diaz, K. Rajagopal, E. Strable, J. Schneider and M. G. Finn, “Click” Chemistry in a Supramolecular Environment: Stabilization of Organogels by Copper(I)-Catalyzed Azide–Alkyne [3+2] Cycloaddition, J. Am. Chem. Soc., 2006, 128, 6056–6057 CrossRef CAS PubMed; (h) J. F. Lutz, Copper-Free Azide–Alkyne Cycloadditions: New Insights and Perspectives, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. Tornoe, C. Christensen and M. Meldal, Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- (a) R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, E. De Clercq, C.-F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, 1,2,3-Triazole-[2,5-Bis-O-(tert-butyldimethylsilyl)-.beta.-D-ribofuranosyl]-3′-spiro-5′′-(4′′-amino-1′′,2′′-oxathiole 2′′,2′′-dioxide) (TSAO) Analogs: Synthesis and Anti-HIV-1 Activity, J. Med. Chem., 1994, 37, 4185–4194 CrossRef CAS PubMed; (b) M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester, D. K. Hutchinson, J. Morris, R. J. Reischer, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaadt, D. Stapert and B. H. Yagi, Substituent Effects on the Antibacterial Activity of Nitrogen−Carbon-Linked (Azolylphenyl)oxazolidinones with Expanded Activity Against the Fastidious Gram-Negative Organisms Haemophilus influenzae and Moraxella catarrhali, J. Med. Chem., 2000, 43, 953–970 CrossRef CAS PubMed.
- For a review, see: (a) P. Saha, D. Panda and J. Dash, The application of click chemistry for targeting quadruplex nucleic acids, Chem. Commun., 2019, 55, 731–750 RSC; For selected examples, see: (b) D. Dutta, M. Debnath, D. Müller, R. Paul, T. Das, I. Bessi, H. Schwalbe and J. Dash, Cell penetrating thiazole peptides inhibit c-MYC expression via site-specific targeting of c-MYC G-quadruplex, Nucleic Acids Res., 2018, 46, 5355–5365 CrossRef CAS PubMed; (c) R. Paul, D. Dutta, R. Paul and J. Dash, Target–Directed Azide–Alkyne Cycloaddition for Assembling HIV–1 TAR RNA Binding Ligands, Angew. Chem., 2020, 59, 12407–12411 CrossRef CAS PubMed; (d) P. Saha, D. Panda, D. Müller, A. Maity, H. Schwalbe and J. Dash, In situ formation of transcriptional modulators using non-canonical DNA i-motifs, Chem. Sci., 2020, 11, 2058–2067 RSC.
- (a) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Copper-Catalyzed C−H Functionalization Reactions: Efficient Synthesis of Heterocycles, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed and the references therein; (b) R. Bohmann and C. Bolm, Copper-Catalyzed C–N Cross-Coupling of Sulfondiimines with Boronic Acids, Org. Lett., 2013, 15, 4277–4279 CrossRef CAS PubMed; (c) Z.-B. Dong, X. Liu and C. Bolm, Copper-Catalyzed C(sp2)–S Coupling Reactions for the Synthesis of Aryl Dithiocarbamates with Thiuram Disulfide Reagents, Org. Lett., 2017, 19, 5916–5919 CrossRef CAS PubMed.
- For reviews, see: (a) M. Meldal and C. W. Tornøe, Cu-Catalyzed Azide−Alkyne Cycloaddition, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (b) J. E. Hein and V. V. Fokin, Copper-catalyzed azide–alkynecycloaddition (CuAAC) and beyond: new reactivity of copper(I) acetylides, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC; (c) V. K. Tiwari, B. B. Mishra, N. Mishra, K. B. Mishra, A. S. Singh and X. Chen, Cu-Catalyzed Click Reaction in Carbohydrate Chemistry, Chem. Rev., 2016, 5, 3086–3240 CrossRef PubMed; For selected examples, see: (d) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed; (e) V. O. Rodionov, S. I. Presolsky, S. Gardinier, Y.-H. Lim and M. G. Finn, Benzimidazole and Related Ligands for Cu-Catalyzed Azide–Alkyne Cycloaddition, J. Am. Chem. Soc., 2007, 129, 12696–12704 CrossRef CAS PubMed; (f) C. Nolte, P. Mayer and B. F. Straub, Isolation of a Copper(I) Triazolide: A “Click” Intermediate, Angew. Chem., Int. Ed., 2007, 46, 2101–2103 CrossRef CAS PubMed; (g) S. Őzçubukçu, E. Ozkal, C. Jimeno and M. A. Pericàs, A Highly Active Catalyst for Huisgen 1,3-Dipolar Cycloadditions Based on the Tris(triazolyl)methanol−Cu(I) Structure, Org. Lett., 2009, 11, 4680–4683 CrossRef PubMed; (h) M. Liu and O. Reiser, A Copper(I) Isonitrile Complex as a Heterogeneous Catalyst for Azide–Alkyne Cycloaddition in Water, Org. Lett., 2011, 3, 1102–1105 CrossRef PubMed; (i) S. Lal and S. Díez-González, [CuBr(PPh3)3] for Azide−Alkyne Cycloaddition Reactions under Strict Click Conditions, J. Org. Chem., 2011, 76, 2367–2373 CrossRef CAS; (j) J.-A. Shin, Y.-G. Lim and K.-H. Lee, Copper-Catalyzed Azide–Alkyne Cycloaddition Reaction in Water Using Cyclodextrin as a Phase Transfer Catalyst, J. Org. Chem., 2012, 77, 4117–4122 CrossRef CAS PubMed; (k) R. Berg and B. F. Straub, Advancements in the mechanistic understanding of the copper-catalyzed azide–alkyne cycloaddition, Beilstein J. Org. Chem., 2013, 9, 2715–2750 CrossRef PubMed; (l) E. Tasca, G. L. Sorella, L. Sperni, G. Strukul and A. Scarso, Micellar promoted multi-component synthesis of 1,2,3-triazoles in water at room temperature, Green Chem., 2015, 17, 1414 RSC; (m) J.-A. Shin, S.-J. Oh, H.-Y. Lee and Y.-G. Lim, An efficient Cu-catalyzed azide–alkyne cycloaddition (CuAAC) reaction in aqueous medium with a zwitterionic ligand, betaine, Catal. Sci. Technol., 2017, 7, 2450–2456 RSC.
- (a) Y. M. A. Yamada, S. M. Sarkar and Y. Uozumi, Self-Assembled Polymeric Copper Catalyst to Parts per Million Levels: Click Chemistry, J. Am. Chem. Soc., 2012, 134, 9285–9286 CrossRef CAS PubMed; (b) W. Wang, F. Wei, Y. Ma, C.-H. Tung and Z. Xu, Copper(I)-Catalyzed Three-Component Click/Alkynylation: One-Pot Synthesis of 5-Alkynyl-1,2,3-triazoles, Org. Lett., 2016, 18, 4158–4161 CrossRef CAS PubMed; (c) B. G. Pasupuleti and G. Bez, CuI/L-proline catalyzed click reaction in glycerol for the synthesis of 1,2,3-triazoles, Tetrahedron Lett., 2019, 60, 142–146 CrossRef CAS; (d) F. Krauskopf, K.-N. Truong, K. Rissanen and C. Bolm, [3+2]-Cycloadditions of N-Cyano Sulfoximines with 1,3-Dipoles, Eur. J. Org. Chem., 2020, 2761–2765 CrossRef CAS.
- (a) W. G. Lewis, F. G. Magallon, V. V. Fokin and M. G. Finn, Discovery and Characterization of Catalysts for Azide–Alkyne Cycloaddition by Fluorescence Quenching, J. Am. Chem. Soc., 2004, 126, 9152–9153 CrossRef CAS PubMed; (b) V. O. Rodionov, S. I. Presolski, S. Gardinier, Y. H. Lim and M. G. Finn, Benzimidazole and Related Ligands for Cu-Catalyzed Azide–Alkyne Cycloaddition, J. Am. Chem. Soc., 2007, 129, 12696–12704 CrossRef CAS PubMed; (c) V. O. Rodionov, S. I. Presolski, D. D. Diaz, V. V. Fokin and M. G. Finn, Ligand-Accelerated Cu-Catalyzed Azide−Alkyne Cycloaddition: A Mechanistic Report, J. Am. Chem. Soc., 2007, 129, 12705–12712 CrossRef CAS PubMed.
- W. Wang, F. Hua, Y. Y. Jiang and Y. F. Zhao, Quick and highly efficient copper-catalyzed cycloaddition of aliphatic and aryl azides with terminal alkynes “on water”, Green Chem., 2008, 10, 452–456 RSC.
- (a) S. Díez-González and S. P. Nolan, [(NHC)2Cu]X Complexes as Efficient Catalysts for Azide–Alkyne Click Chemistry at Low Catalyst Loadings, Angew. Chem., Int. Ed., 2008, 47, 8881–8884 CrossRef PubMed; (b) T. Nakamura, T. Terashima, K. Ogata and S. Fukuzawa, Copper(I) 1,2,3-Triazol-5-ylidene Complexes as Efficient Catalysts for Click Reactions of Azides with Alkynes, Org. Lett., 2011, 13, 620–623 CrossRef CAS PubMed.
- F. W. Li and T. S. A. Hor, Facile Synthesis of Nitrogen Tetradentate Ligands and Their Applications in CuI–Catalyzed N–Arylation and Azide–Alkyne Cycloaddition, Chem. – Eur. J., 2009, 15, 10585–10592 CrossRef CAS PubMed.
- (a) S. Paladhi, J. Das, M. Samanta and J. Dash, Asymmetric Aldol Reaction of Thiazole–Carbaldehydes: Regio– and Stereoselective Synthesis of Tubuvalin Analogues, Adv. Synth. Catal., 2014, 356, 3370–3376 CrossRef CAS; (b) S. Paladhi, M. Bhati, D. Panda and J. Dash, Thiazolidinedione-Isatin Conjugates via an Uncatalyzed Diastereoselective Aldol Reaction on Water, J. Org. Chem., 2014, 79, 1473–1480 CrossRef CAS PubMed; (c) G. Chakraborti, S. Paladhi, T. Mandal and J. Dash, “On Water” Promoted Ullmann-Type C–N Bond-Forming Reactions: Application to Carbazole Alkaloids by Selective N-Arylation of Aminophenols, J. Org. Chem., 2018, 83, 7347–7359 CrossRef CAS PubMed.
- As a chiral ligand is not necessary for this reaction, the reactions were carried out using D,L-proline and an achiral prolinamide Pro-1 (prepared from D,L-proline) and the same results were obtained (Table S1, ESI†). However, chiral Pro-1, prepared from L-proline was used in this reaction as L-proline is easily available and commercially cheap compared to D,L-proline.
- (a) Y. P. Kumar, S. Bhowmik, R. N. Das, I. Bessi, S. Paladhi, R. Ghosh, H. Schwalbe and J. Dash, A Fluorescent Guanosine Dinucleoside as a Selective Switch–On Sensor for c-myc G–Quadruplex DNA with Potent Anticancer Activities, Chem. – Eur. J., 2013, 19, 11502–11506 CrossRef CAS PubMed; (b) Y. P. Kumar, R. N. Das, S. Kumar, O. M. Schtte, C. Steinem and J. Dash, Triazole–Tailored Guanosine Dinucleosides as Biomimetic Ion Channels to Modulate Transmembrane Potential, Chem. – Eur. J., 2014, 20, 3023–3028 CrossRef CAS PubMed.
- (a) J. Andersen, U. Madsen, F. Björkling and X. Liang, Rapid Synthesis of Aryl Azides from Aryl Halides under Mild Conditions, Synlett, 2005, 2209–2213 CAS; (b) W. Zhua and D. Ma, Synthesis of aryl azides and vinyl azides via proline-promoted CuI-catalyzed coupling reactions, Chem. Commun., 2004, 888–889 RSC.
- B. T. Worell, J. A. Malik and V. V. Fokin, Direct Evidence of a Dinuclear Copper Intermediate in Cu(I)-Catalyzed Azide-Alkyne Cycloadditions, Science, 2013, 340, 457–460 CrossRef PubMed.
- Y. C. Lin, Y. J. Chen, T. Y. Shih, Y. H. Chen, Y. C. Lai, M. Y. Chiang, G. C. Senadi, H. Y. Chen and H. Y. Chen, Mechanistic Study in Click Reactions by Using (N-Heterocyclic carbene)Copper(I) Complexes: Anionic Effects, Organometallics, 2019, 38, 223–230 CrossRef CAS.
- H. B. El Ayouchia, L. Bahsis, H. Anane, L. R. Domingo and S. E. Stiriba, Understanding the mechanism and regioselectivity of the copper(I) catalyzed [3+2] cycloaddition reaction between azide and alkyne: a systematic DFT study, RSC Adv., 2018, 8, 7670–7678 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- Y. özkılıc and N. Ş. Tüzün, A DFT Study on the Binuclear CuAAC Reaction: Mechanism in Light of New Experiments, Organometallics, 2016, 35, 2589–2599 CrossRef.
- M. Ahlquist and V. Fokin, Enhanced Reactivity of Dinuclear Copper(I) Acetylides in Dipolar Cycloadditions, Organometallics, 2007, 26, 4389–4391 CrossRef CAS.
- B. F. Straub, μ-Acetylide and μ-alkenylidene ligands in “click” triazole syntheses, Chem. Commun., 2007, 3868–3870 RSC.
- E. Boz and N. Ş. Tüzün, Ag-catalyzed azide alkyne cycloaddition: a DFT approach, Dalton Trans., 2016, 45, 5752–5764 RSC.
- (a) D. Maiti and S. L. Buchwald, Orthogonal Cu- and Pd-Based Catalyst Systems for the O- and N-Arylation of Aminophenols, J. Am. Chem. Soc., 2009, 131, 17423–17429 CrossRef CAS PubMed; (b) D. Maiti and S. L. Buchwald, Cu-Catalyzed Arylation of Phenols: Synthesis of Sterically Hindered and Heteroaryl Diaryl Ethers, J. Org. Chem., 2010, 75, 1791–1794 CrossRef CAS PubMed; (c) D. Maiti, Chemoselectivity in the Cu-catalyzed O-arylation of phenols and aliphatic alcohols, Chem. Commun., 2011, 47, 8340–8342 RSC.
- (a) A. K. Ganai, P. Shinde, B. B. Dhar, S. S. Gupta and B. L. V. Prasad, Development of a multifunctional catalyst for a “relay” reaction, RSC Adv., 2013, 3, 2186–2191 RSC; (b) J. E. Camp, Auto–Tandem Catalysis: Activation of Multiple, Mechanistically Distinct Process by a Single Catalyst, Eur. J. Org. Chem., 2017, 425–433 CrossRef CAS; (c) Q.-J. Liang, Y.-H. Xu and T.-P. Loh, Multi-catalyst promoted asymmetric relay reactions, Org. Chem. Front., 2018, 5, 2765–2768 RSC; (d) H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Catalytic cascade reactions by radical relay, Chem. Soc. Rev., 2019, 48, 4626–4638 RSC.
- (a) L. F. Tietze and N. Rackelmann, Domino reactions in the synthesis of heterocyclic natural products and analogs, Pure Appl. Chem., 2004, 76, 1967–1983 CAS; (b) D. E. Fogg and E. N. dos Santos, Tandem catalysis: a taxonomy and illustrative review, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS; (c) J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Concurrent Tandem Catalysis, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qo01150a |
| This journal is © the Partner Organisations 2021 |