
Functionalisation of esters via 1,3-chelation using NaOtBu: mechanistic investigations and synthetic applications†
Hye Sung
Yang
a,
Lingamurthy
Macha
b,
Hyun-Joon
Ha
 *b and
Jung Woon
Yang
*a
*b and
Jung Woon
Yang
*a
aDepartment of Energy Science, Sungkyunkwan University, Suwon 16419, Republic of Korea. E-mail: jwyang@skku.edu
bDepartment of Chemistry, Hankuk University of Foreign Studies, Yongin 17035, Republic of Korea. E-mail: hjha@hufs.ac.kr
First published on 3rd November 2020
Abstract
For the first time, both 1,3-chelation and the formation of a tetrahedral intermediate were confirmed as the key factors for the unusual nucleophilic behavior of a metal t-butoxide in a transesterification reaction. NMR and real-time IR spectroscopies and deuterium-labeling experiments were used for the mechanistic investigation. Based on a pivotal point in the mechanistic understanding of the action of t-butoxide anion, several uncovering reactions such as direct access to value-added chiral α-amino acid t-butyl ester with almost complete retention of optical purity via elaborative transesterification, non-hydrolytic deesterification of esters, and selective bond cleavage of 3-benzoyloxazolidin-2-ones, were successfully achieved.
Introduction
Transesterification is one of the most important reactions1 that is widely used in biodiesel production2 and synthesis of pharmaceuticals3 and organic materials.4 Despite the versatility of this reaction, most of the methods reported for the synthesis of t-butyl esters via transesterification are inefficient. In 1986, however, the Meth-Cohn group successfully conducted the direct transesterification of methyl esters using lithium t-butoxide (generated in situ from t-BuOH and n-BuLi) to synthesize t-butyl esters.5 The widespread consideration of metal t-butoxide as non-nucleophilic base is highly prejudiced,6 because of which a possible role of t-butoxide anion as a nucleophile has been largely overlooked; Meth-Cohn's work and two other examples are exceptions.7Although there are a few examples of reactions using t-butoxide anion for transesterification, a comprehensive mechanistic understanding of the effect of t-butoxide anion on the reaction media is crucial to determine the important variables for developing new reactions. In this regard, we revisited the action of metal t-butoxide as a nucleophile8 and investigated the effect of environmental factors on the reaction mechanism using various spectroscopic techniques and deuterium-labeling experiments (Scheme 1). There are some distinctive features of our study: (i) the discovery of a new strategy for the generation of a t-butoxide nucleophile using sodium chelate of ester moiety (1,3-chelation)9 that facilitates a nucleophilic acyl substitution reaction via a tetrahedral intermediate.
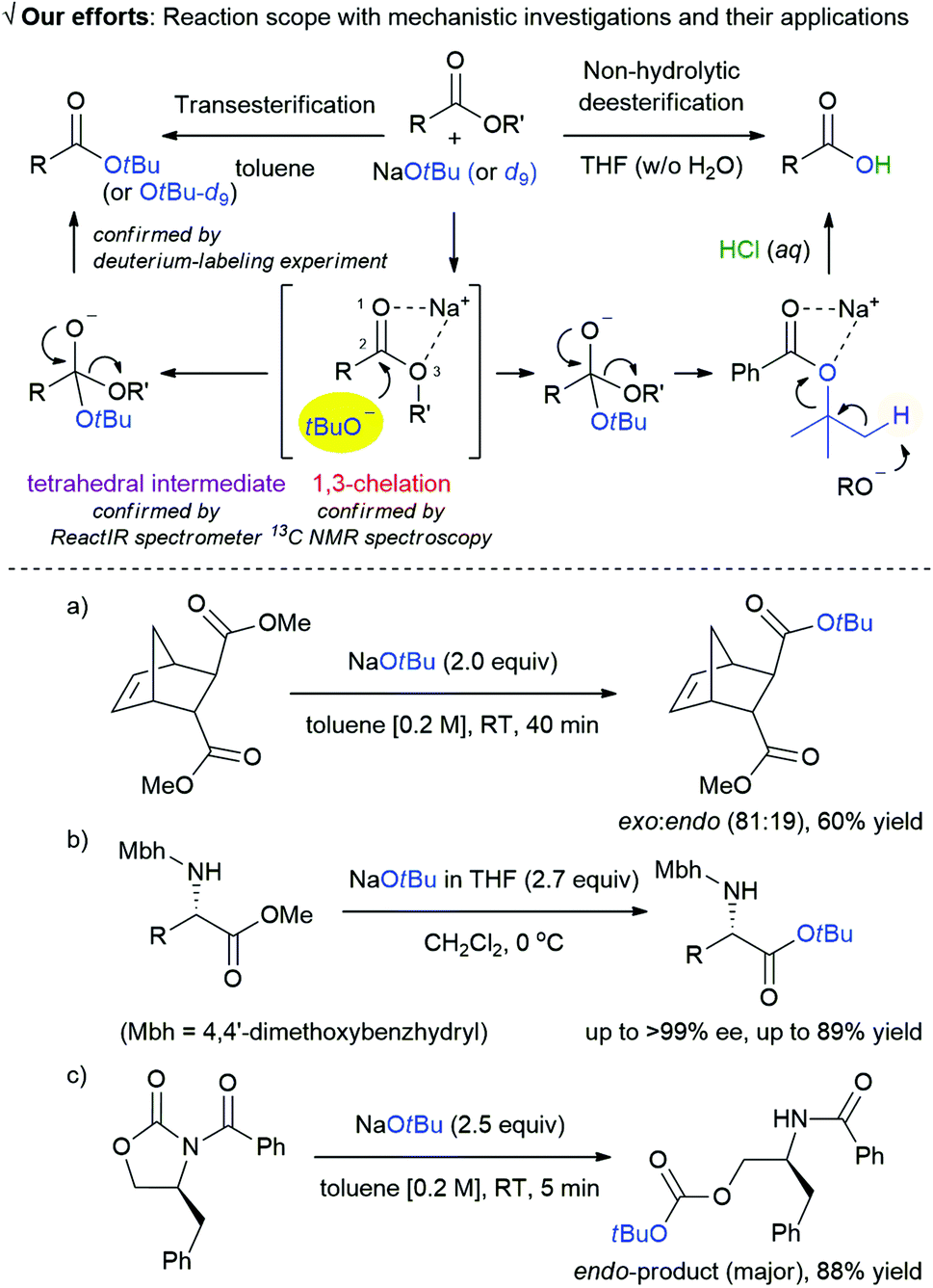 | ||
| Scheme 1 Transesterification and non-hydrolytic deesterification, along with mechanism determined using 13C NMR spectroscopy, ReactIR spectrometer, and deuterium-labeling experiment and general applications (a–c) based on our mechanistic analyses. | ||
The 1,3-chelation and formation of a tetrahedral intermediate are confirmed by NMR spectroscopy and ReactIR spectrometer, respectively. (ii) a new area for using NaOtBu as this nucleophile provides a platform for diverse array of applications including exo-selective transesterification of biomass-derived norbornene and transesterification of α-amino esters with a minimal loss of optical purity, and endo-selective cleavage reaction of oxazolidin-2-ones (a–c in Scheme 1).
Results and discussion
To test the nucleophilicity of NaOtBu, methyl benzoate 1a as a model substrate was reacted with NaOtBu (2.0 M solution in THF) in toluene at room temperature (Table 1). Surprisingly, t-butoxide acted as a nucleophile in a transesterification reaction to yield t-butyl benzoate 2a as the nucleophilic acyl substituted product. Analysis of the NMR spectra revealed that the yield of hydrolytic product, benzoic acid (3a), was slightly increased when the reaction time was gradually increased from 3 h to 24 h (Table 1, entries 1 and 2). Solid NaOtBu, which is easier to handle, was used for the transesterification of 1a to give t-butyl benzoate 2a as the sole product after 3 h (Table 1, entry 3). After identifying the existence of non-hydrolytic deesterified product, such as benzoic acid 3a, we focused on the development of non-hydrolytic deesterification reaction in various organic solvents. In contrast with toluene (Table 1, entry 4), the non-hydrolytic deesterified product 3a was produced exclusively in THF when the reaction was prolonged to 24 h (Table 1, entry 8). The optimal conditions for transesterification and non-hydrolytic deesterification are summarized as follows: for transesterification, NaOtBu (2.5 equiv.) in toluene (0.2 M) at room temperature for 3 h; for non-hydrolytic deesterification, NaOtBu (2.5 equiv.) in THF (0.2 M) at room temperature for 24 h.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time (h) | Conv.b (%) | Ratio of 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3ab :3ab |
| a Reaction conditions: 1a (0.5 mmol), NaOtBu (2.5 equiv.), solvent (0.2 M), rt. b Determined by 1H NMR spectroscopy. c Using NaOtBu (2.0 M solution in THF, 2.5 equiv.). | ||||
| 1c | Toluene | 3 | 96 | 92:8 |
| 2c | 24 | 97 | 72:28 |
|
| 3 | Toluene | 3 | 96 | 92:8 |
| 4 | 24 | 97 | 72:28 |
|
| 5 | Diethyl ether | 3 | 96 | 98:2 |
| 6 | 24 | 97 | 89:11 |
|
| 7 | THF | 3 | 92 | 70:30 |
| 8 | 24 | >99 | n.d:>99 |
|
| 9 | 1,4-Dioxane | 3 | 93 | 63:37 |
| 10 | 24 | 94 | 39:61 |
|
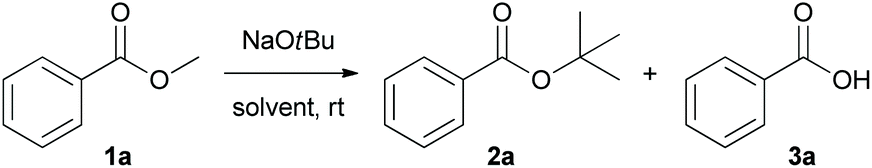
With the optimized conditions in hand, we explored the scope of NaOtBu-mediated transesterification (condition A) and non-hydrolytic deesterification (condition B). Various esters with different electronic properties and substitution patterns were examined, and the corresponding products were obtained in moderate to extremely high yields (Scheme 2).
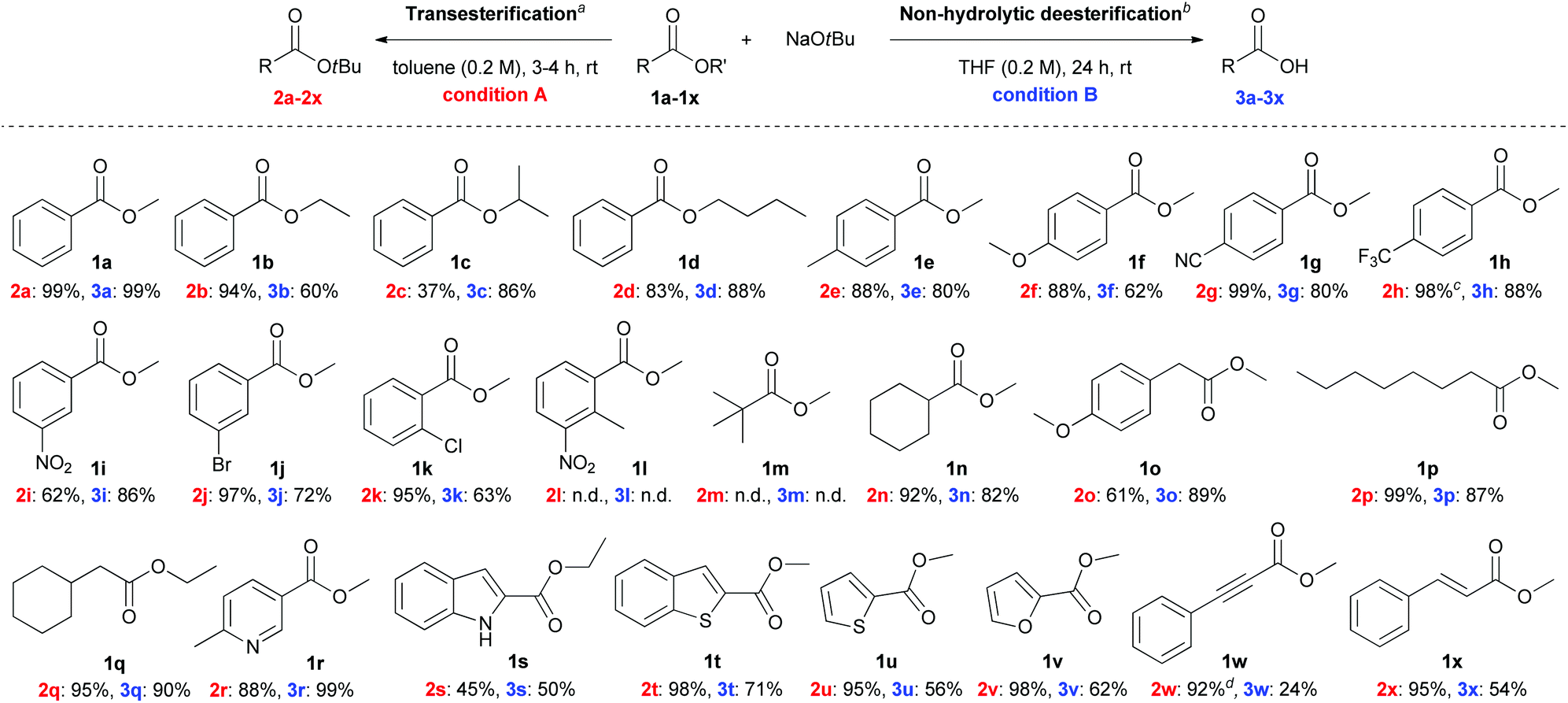 | ||
| Scheme 2 Substrate scope for the transesterification and non-hydrolytic deesterification reactions. aReaction conditions A: Ester 1 (0.5 mmol, 1.0 equiv.) and NaOtBu (2.5 equiv.) in toluene (0.2 M) at rt for 3–4 h. bReaction conditions B: Ester 1 (0.5 mmol, 1.0 equiv.) and NaOtBu (2.5 equiv.) in THF (0.2 M) at rt for 24 h. cFor 1 h. dFor 20 min. All yields are those of isolated products. | ||
The reaction with methyl and ethyl ester (1a and 1b, respectively) gave the expected t-butyl ester in 99% and 94% yields, respectively. As the alkyl moiety of the ester was changed to the relatively bulkier isopropyl (1c) group, the efficiency of transesterification decreased. This phenomenon is presumably attributed to the steric hindrance from the bulky t-butyl group, which impedes the approach of incoming nucleophile and prevents the reaction from proceeding further. Benzoates with diverse substituents at the phenyl ring and various positions have broad reaction scope and generate products in moderate to extremely high yields. Under the optimized reaction conditions, non-hydrolytic deesterification of esters also gives reasonably good yields.
This reaction has excellent compatibility with hydrolytically labile functional group (such as nitrile, 1g), which is easily hydrolysed with water in the presence of acid or base to yield carboxylic acid or carboxylate anion.10 In this regard, only a few examples of chemoselective transformation of ester bearing a cyano group to carboxylic acid were reported by using ion-exchange resin11 or using ferric sulphate with ionic liquid as a cocatalyst.12 When enolisable esters (1n–q) were employed for the transesterification and non-hydrolytic deesterification, the desired products were formed in good to high yields without the formation of Claisen condensation products, despite the conditions favouring the Claisen condensation reaction. A series of esters (1r–v) containing heterocyclic moieties, including pyridine, indole, thiophene, benzothiophene, and furan, were subjected to the individual reactions, and the corresponding products were obtained in moderate to high yields. In addition, esters (1w–x) bearing π-extended aromatic ring, for instance α,β-alkynyl ester and α,β-alkenyl ester, are compatible with these reactions.
We also investigated the utility of other alkoxides (e.g., ethoxide, isopropoxide, and t-amoxide) as potential nucleophiles and the effect of counterions (e.g., potassium and lithium ions) on the transesterification reaction (Table 2).
|
|
|||
|---|---|---|---|
| Entry | MOR′ | Conv.b (%) | Ratio of 2′ (or 2):3ab |
| a Reaction conditions: 1a (0.5 mmol), MOR′ (2.5 equiv.), toluene (0.2 M), rt. b Determined by 1H NMR spectroscopy. c Isolated yield. d In THF (0.2 M) at rt for 24 h. | |||
| 1 | NaOEt | 94 | 72:28 |
| 2 | NaOiPr | 97 | 80:20 |
| 3 | NaOtAm | 99c | >99:n.d.c |
| 4d | NaOtAm | 99c | n.d.:>99c |
| 5 | LiOtBu | 15 | 74:26 |
| 6 | KOtBu | >99 | n.d.:>99 |
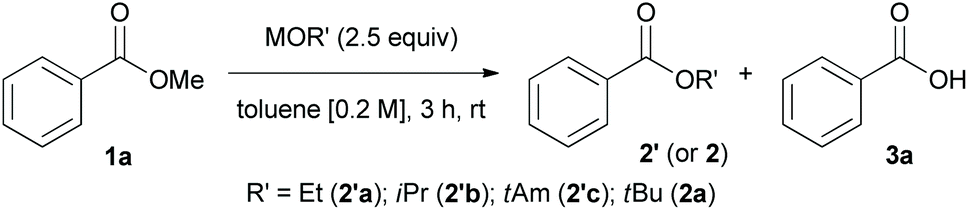
We realized other alkoxide could be used in the transesterification reaction to give corresponding product (2′a–c) in high yields. Interestingly, we found that counterions have a significant influence on the chemical reactivity and ratio of the products (2a or 3a). The percentage conversion using LiOtBu (product ratio of 2a:3a = 74:26) was extremely low (15%) compared to that using NaOtBu (96% conversion; product ratio of 2a:3a = 92:8). On the other hand, KOtBu was too reactive to stop the transesterification of methyl ester, and the reaction proceeded further to generate carboxylic acid 3a.
To demonstrate the practicality of transesterification, we performed gram-scale synthesis of 2a. The reaction of 1a was repeated on a 2.5 g (18.36 mmol) scale [36.7 times larger than the experiments described in Scheme 2] using NaOtBu as nucleophile, and the desired product 2a was isolated in 96% yield (Scheme 3).
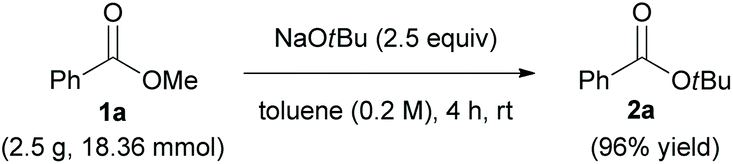 | ||
| Scheme 3 Gram-scale synthesis of 2a. | ||
Investigation of the mechanistic details of this transesterification and non-hydrolytic deesterification using the available analytical tools was challenging. NMR spectroscopy was used to clarify the interaction between the sodium ion and ester group (Fig. 1a). The reaction of methyl benzoate 1a (1 mmol) and NaOtBu (2.0 equiv.) was carried out in toluene-d8 (1.0 M) at room temperature for 1 min. As a result, peaks of both carbonyl carbon and the carbon of methoxy group show a small downfield shift in the crude 13C NMR spectra, indicating that the ester group is activated via the chelation between sodium ion and oxygen atoms in the ester moiety (4).
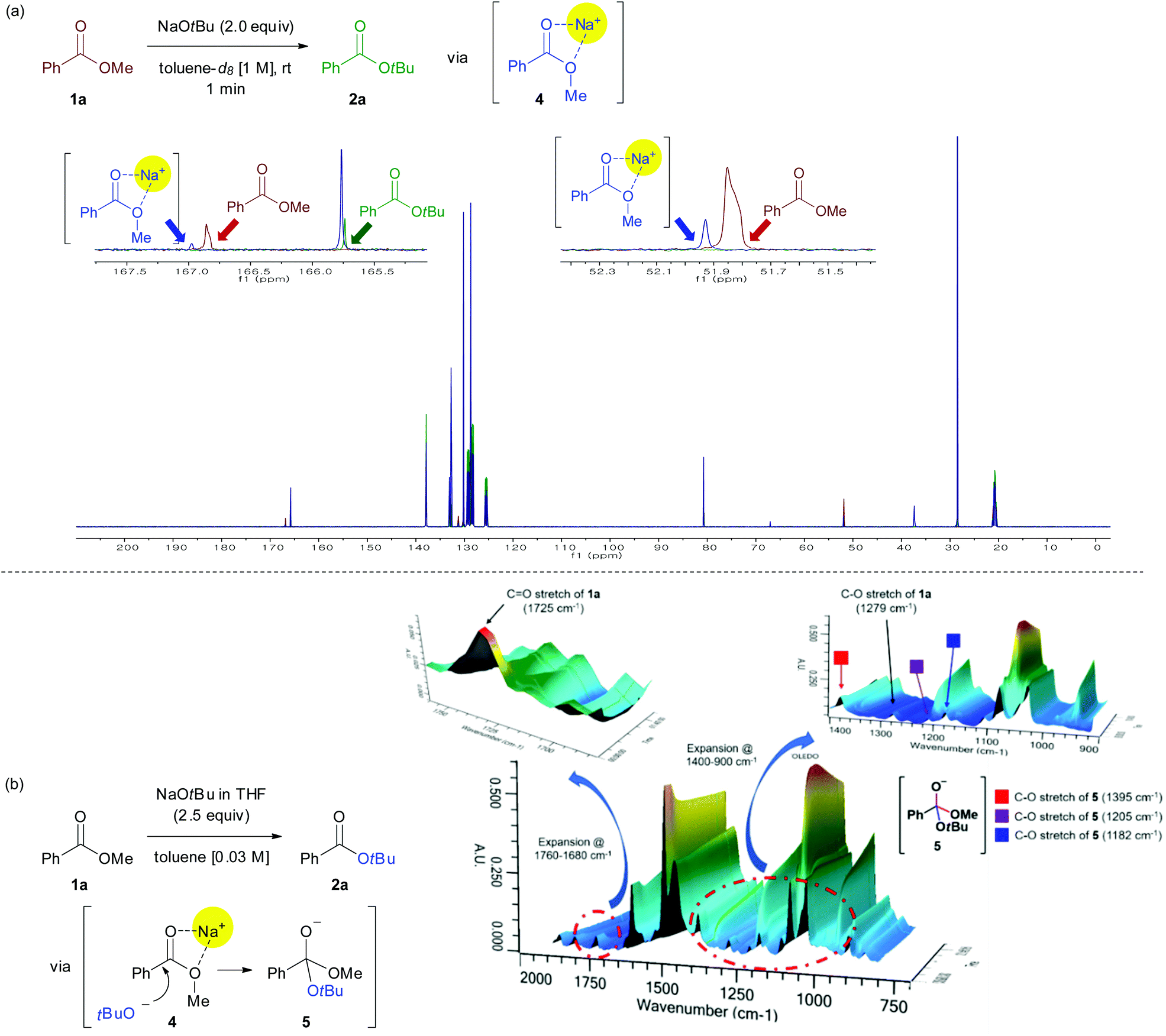 | ||
| Fig. 1 Mechanistic investigations through NMR spectroscopy and ReactIR spectrometer (a) Reactions conditions: Methyl benzoate 1a (1 mmol), NaOtBu (2.0 equiv.), toluene-d8 (1.0 M), rt. 13C NMR spectroscopy: brown line = methyl benzoate; green line = t-butyl benzoate; blue line = reaction mixture [reference peak: 137.86 ppm (toluene-d8)]. (b) Monitoring the formation of tetrahedral intermediate 5 by ReactIR during a nucleophilic acyl substitution reaction between 1a and NaOtBu and assignment of key absorption bands of tetrahedral intermediate 5. | ||
The existence of the tetrahedral intermediate 5, which resulted from the nucleophilic addition to an activated carbonyl group (4), was established by monitoring the reaction profiles in a ReactIR spectrometer (Fig. 1b).13 Upon the addition of NaOtBu to ester, the intensity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching absorption peak (1750–1725 cm−1) of the substrate 1a rapidly decreased. Simultaneously, new peaks appeared in the region 1400 to 1000 cm−1, which were presumably assigned to the C–O stretching vibrations of a tetrahedral intermediate 5. Individual wavenumber of the C–O stretching vibrations of 5 can be assigned according to the following principle in infrared spectroscopy: “many functional groups absorb infrared radiation at about the same wavenumber, regardless of the structure of the rest of the molecule”.14
O stretching absorption peak (1750–1725 cm−1) of the substrate 1a rapidly decreased. Simultaneously, new peaks appeared in the region 1400 to 1000 cm−1, which were presumably assigned to the C–O stretching vibrations of a tetrahedral intermediate 5. Individual wavenumber of the C–O stretching vibrations of 5 can be assigned according to the following principle in infrared spectroscopy: “many functional groups absorb infrared radiation at about the same wavenumber, regardless of the structure of the rest of the molecule”.14
Therefore, the wavenumbers of C–O stretching vibrations of 5 were assigned by comparing with wavenumbers of structurally similar molecules [e.g., (i) 1380 cm−1 for C–O stretch of Ph-CH2-OMe,15 (ii) 1197 cm−1 for C–O stretch of Ph-CH2-OtBu,16 (iii) 1203 cm−1 for C–O stretch of Ph-CH2-O−Na+].
A deuterium-labeling experiment was further conducted with methyl benzoate 1a and NaOtBu-d9 (generated in situ from the reaction of sodium hydride and tBuOH-d9 in THF) under standard conditions (Scheme 4). Deuterium-incorporated product 6 was obtained in 57% yield, and this was confirmed by high-resolution mass spectrometry [HRMS (EI) m/z [M]+ calcd for C11H5D9O2 187.1559, found 187.1534] and 1H and 13C NMR spectroscopies (see ESI†). These results indicated that −OtBu-d9 served as an excellent nucleophile in this transformation.
 | ||
| Scheme 4 Deuterium-labeling experiment. | ||
Additional experiments were performed for gaining further insights into the mechanism of non-hydrolytic deesterification reaction (Scheme 5).
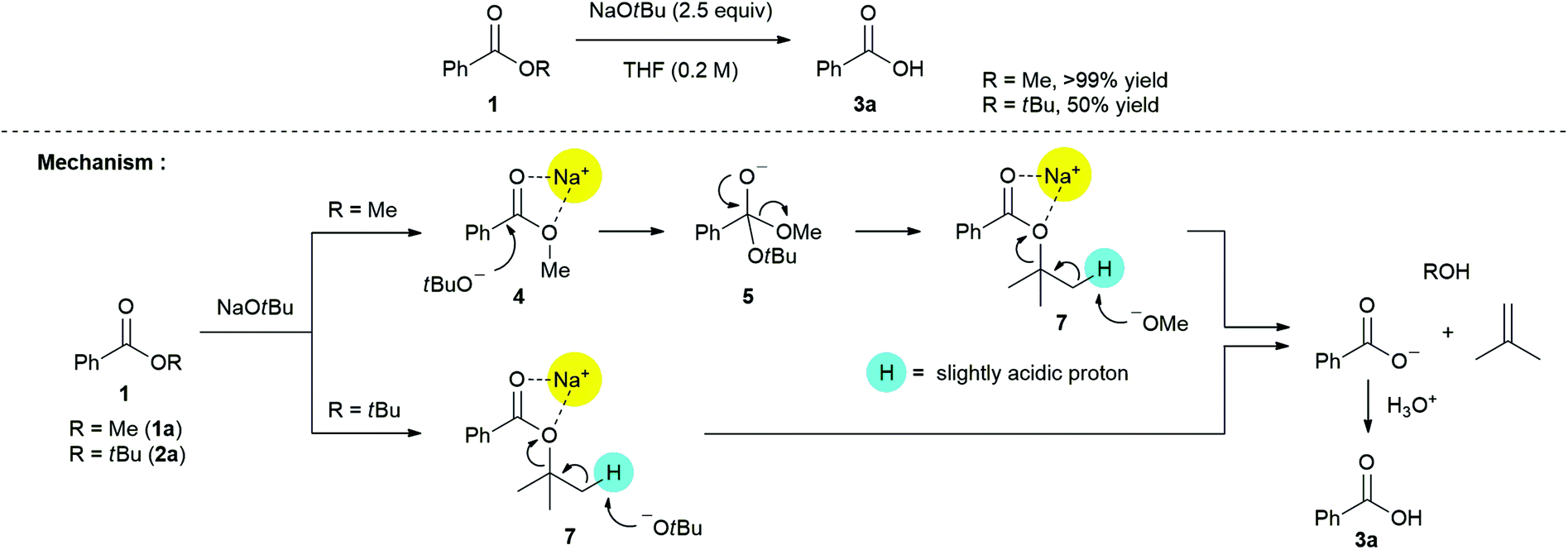 | ||
| Scheme 5 Two controlled experiments for elucidation of reaction mechanism. | ||
Based on the solid evidence of a tetrahedral intermediate 5, it is expected that the nucleophilic substitution reaction of 1a with NaOtBu is triggered naturally. The desired product 3a was obtained in >99% yield, followed by consecutive processes including 1,3-chelation between sodium ion and oxygen atoms in ester 7, abstraction of a slightly acidic proton by the liberated methoxide ion, and acidic work-up. Surprisingly, non-hydrolytic deesterification reaction of t-butyl benzoate 2a with NaOtBu also proceeded to give the desired product 3a in 50% yield. This process may be directly involved in 1,3-chelation between the sodium ion and oxygen atoms in ester 7, and this, rather than the formation of a tetrahedral intermediate (such as 5), is the so-called “activation step for carbonyl group”. This can be explained on the basis of the high energy barrier from the two t-butyl groups. The low yield of product 3a can be mainly attributed to steric hindrance between the t-butyl group on ester 7 and its bulkiness as a base in the proton abstraction step.
We further investigated the utility of the transesterification reaction with dimethyl bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate (8) for possible control of the endo and exo-selectivity (Scheme 6). In case of 8, the methyl ester at the sterically favoured exo-position was mostly substituted to t-butyl ester via nucleophilic acyl substitution reaction with high selectivity (exo:endo = 81:19). This result corroborates the behaviour of NaOtBu as an excellent nucleophile, rather than a base, that can react at a more easily accessible site.
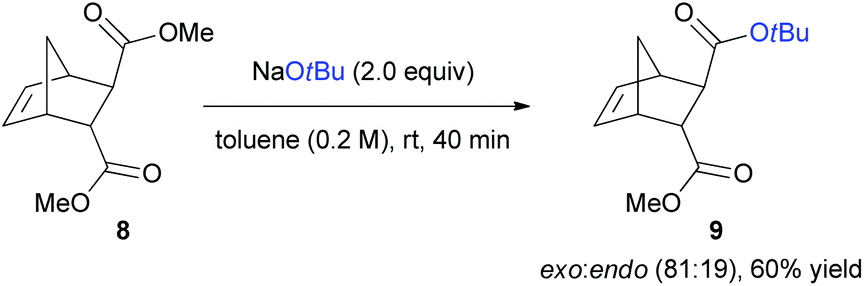 | ||
| Scheme 6 exo-Selective transesterification of norbornene with NaOtBu as a nucleophile. | ||
We next turned our attention to the spectacular elaborative transesterification reaction with amino acid esters. Amino acid esters are extensively used as building blocks in organic syntheses and peptide chemistry.17 In particular, t-butyl ester is one of the most promising functional groups, because it has the following relevant practical utilities over other alkyl esters:18 (i) t-butyl esters are resistant to saponification, whereas methyl, ethyl, and benzyl esters are prone to saponification.19 (ii) t-Butyl esters are easily removed to give carboxylic acids under acidic conditions.20 (iii) Sterically hindered t-butyl esters have superior resistance to nucleophilic attack over methyl or ethyl esters, and thus prevent the formation of unwanted 2,5-diketopiperazines (2,5-DKPs) as side-products of the self-condensation reaction of dipeptide amino acid esters.21 Most of the reported synthetic routes to amino acid t-butyl esters involve the addition of N-protected amino acid to isobutene or the transformation of carboxylic acid with t-butyl acetate in the presence of perchloric acid as catalyst (Scheme 7a).22 Given the importance of amino acid t-butyl esters in peptide synthesis, uncovering an approach to synthesize them is highly desirable.
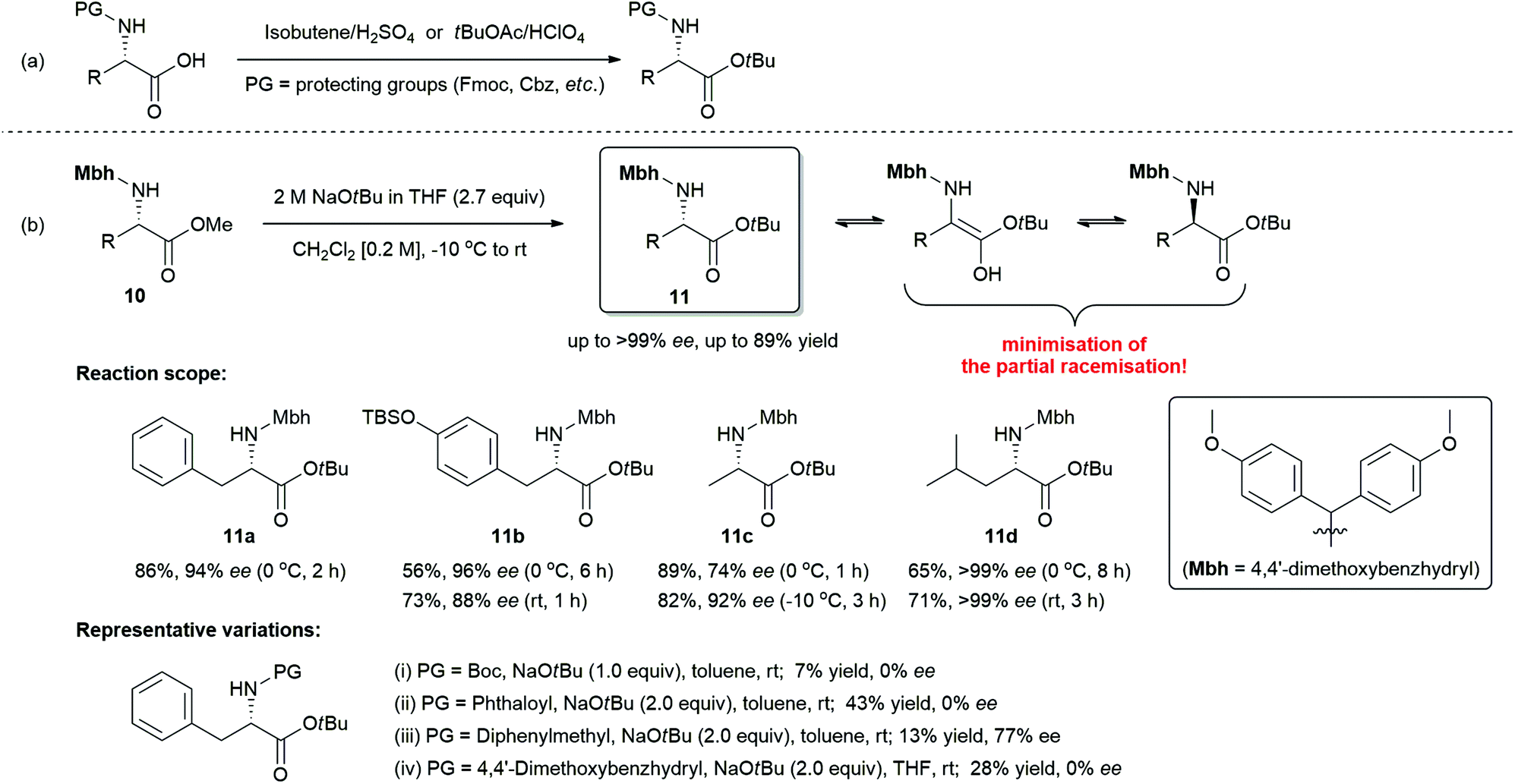 | ||
| Scheme 7 (a) Conventional syntheses of N-protected amino acid t-butyl esters using either isobutene or t-butyl acetate in the presence of strong acid catalysts (b) the current work with reaction scope using NaOtBu via transesterification reaction. | ||
With the newly discovered route for transesterification reaction utilizing NaOtBu, we further investigated N-Mbh-protected (S)-α-amino acid methyl ester (10)23 as a model substrate in this reaction (Scheme 7b).
However, there is an intrinsic problem in using NaOtBu as a strong base for the present reaction. In the presence of excess strong base, the racemisation reaction is proceeded by the removal of α-proton next to the activated carbonyl group. To circumvent or to retard an intrinsic racemisation pathway, we extensively examined various parameters such as solvents, concentrations, reaction temperatures, equivalents, type (i.e., solid or solution) of NaOtBu, and the nature of protecting group. All these parameters were found to have a significant influence in obstructing the intrinsic racemisation pathway (for detailed information, see ESI† and representative variations in Scheme 7b). The best results in terms of chemical yield and minimal loss of optical purity in the product were achieved under the following conditions: N-Mbh-protected α-amino acid esters (0.5 mmol) and 2 M NaOtBu in THF (2.7 equiv.) in dichloromethane (0.2 M) from −10 °C to rt.
Finally, we investigated the feasibility of a selective cleavage reaction of oxazolidin-2-ones (12a–c) with NaOtBu as a nucleophile. As depicted in Scheme 8a, most of the reported bond cleavage reactions with oxazolidin-2-ones occurred at either N(3)–C(1′) bond24 or C(5)–O(1) bond.25 As a result, the O-Boc-protected endocyclic cleaved products (13a–c) were obtained as major products in good yields (up to 88%) and high selectivity (up to 88:12 ratio of 13:2a), regardless of the substitution pattern of oxazolidin-2-ones at C4 position (12). The unique and unprecedented N(3)–C(2) bond cleavage pattern observed in these reactions is noteworthy and have not been reported so far (Scheme 8b).
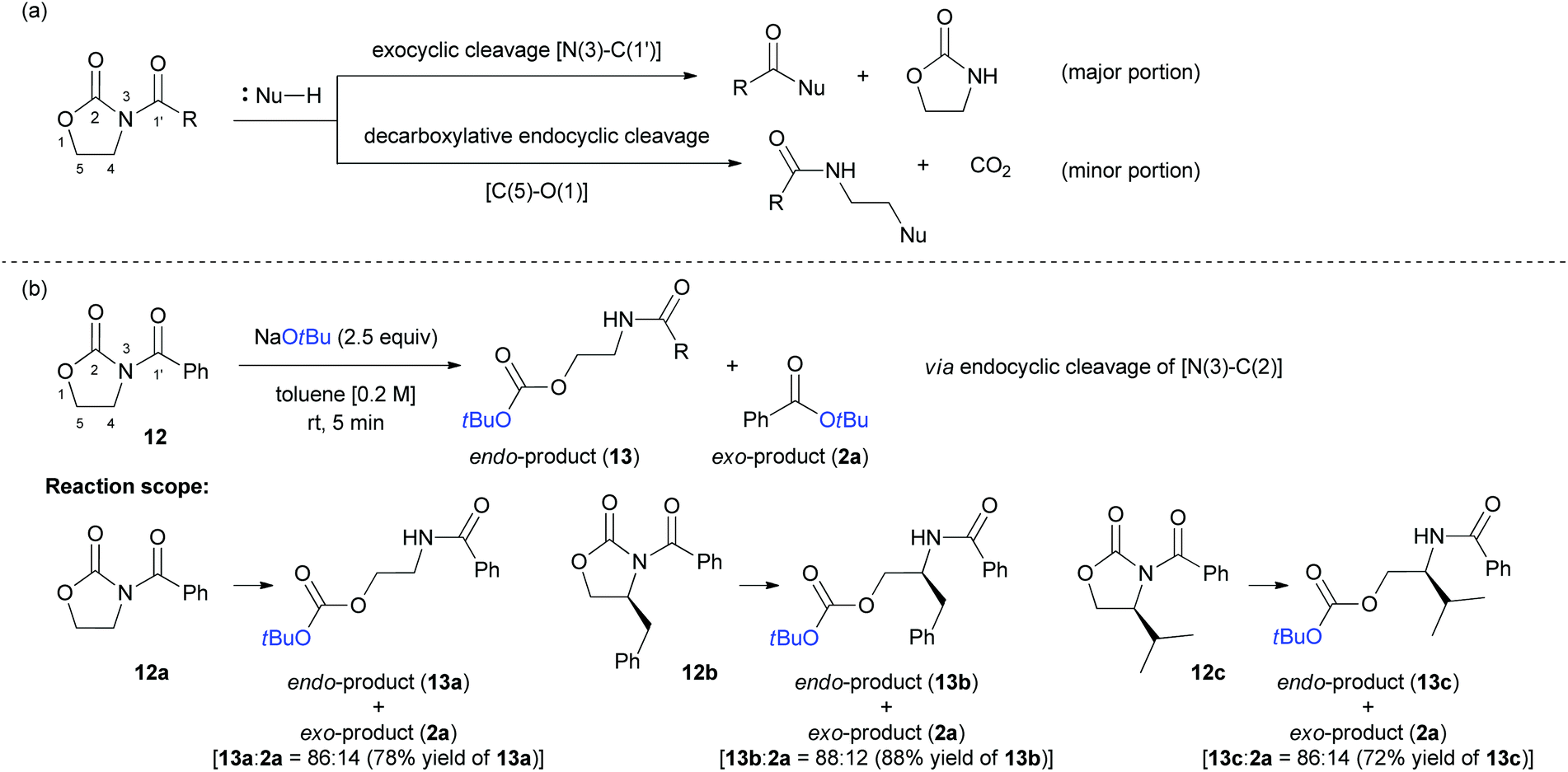 | ||
| Scheme 8 (a) Previous works on the bond cleavage reaction with oxazolidin-2-ones (b) the current work for endo-selective product formation and its reaction scope. | ||
Conclusions
In summary, we have demonstrated the use of NaOtBu as a nucleophile in the selective transesterification and non-hydrolytic deesterification of esters by suppressing its base-like character via 1,3-chelation between sodium ion and oxygen atoms in the ester. The reaction mechanism and the origin of high reactivity and selectivity was supported by extensive studies using NMR spectroscopy, ReactIR spectrometer, and deuterium-labeling experiment. Moreover, we also successfully conducted the transesterification reactions of methyl ester of chiral α-amino acids using NaOtBu, wherein NaOtBu acted as nucleophile to give the corresponding amino acid t-butyl esters and helped in almost complete retention of the optical purities by suppressing the racemisation as the main pathway. In addition, several uncovering reactions including non-hydrolytic deesterification of esters and selective bond cleavage of 3-benzoyloxazolidin-2-ones are successfully demonstrated for the first time. This yet unexploited strategy based on mechanistic investigations can provide general guidelines in multiple areas such as biodiesel production, peptide chemistry, and synthesis of pharmaceuticals and organic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by thanks the NRF Programs (2019R1A2C1003276 and 2019R1A4A2001440) for J. W. Yang. H.-J. Ha thanks the NRF Nano·Material Technology Development Program (2012M3A7B4049645) for financial support. We also thank Prof. Sukbok Chang (KAIST) for providing a real time IR spectrometer.Notes and references
- J. Otera, Transesterification, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS.
- For reviews of transesterification for biodiesel productions, see: M. E. Borges and L. Díaz, Recent developments on heterogeneous catalysts for biodiesel production by oil esterification and transesterification reactions: A review, Renewable Sustainable Energy Rev., 2012, 16, 2839–2849 CrossRef CAS.
- For representative examples of transesterification in pharmaceuticals, see: (a) H. Laurent, E. Gerhards and R. Wiechert, Synthesis of novel anti-inflammatory steroids, Angew. Chem., Int. Ed. Engl., 1975, 14, 65–69 CrossRef CAS; (b) T. Fujita, M. Tanaka, Y. Norimine, H. Suemune and K. Sakai, Enantioselective synthesis of (-)-curcumanolide a using enzymatic transesterification of meso-spirodiol, J. Org. Chem., 1997, 62, 3824–3830 CrossRef CAS.
- For representative examples of transesterification in organic materials, see: (a) J. Xiang, S. Toyoshima, A. Orita and J. Otera, A practical and green chemical process: fluoroalkyldistannoxane-catalyzed biphasic transesterification, Angew. Chem., Int. Ed., 2001, 40, 3670–3672 CrossRef CAS; (b) P. McKeown, M. Kamran, M. G. Davidson, M. D. Jones, L. A. Román-Ramírez and J. Wood, Organocatalysis for versatile polymer degradation, Green Chem., 2020, 22, 3721–3726 RSC.
- O. Meth-Cohn, A simple, powerful, and efficient method for transesterification, J. Chem. Soc., Chem. Commun., 1986, 695–697 RSC.
- (a) D. E. Pearson and C. A. Buehler, Potassium tert-butoxide in synthesis, Chem. Rev., 1974, 74, 45–86 CrossRef CAS; (b) T. M. Krülle and J. C. H. M. Wijkmans, Synthesis of novel 1,2,5-benzothiadiazepine 1,1-dioxides, Tetrahedron, 2001, 57, 7021–7026 CrossRef.
- (a) M. G. Stanton and M. R. Gagné, A mild protocol for the conversion of simple esters to tert-butyl esters, J. Org. Chem., 1997, 62, 8240–8242 CrossRef CAS; (b) V. A. Vasin and V. V. Razin, Mild and efficient method for preparation of tert-butyl esters, Synlett, 2001, 658–660 CrossRef CAS.
- Alkali metal t-butoxide as nucleophile, see: (a) S. A. DiBiase and G. W. Gokel, Crown-cation complex effects. 8. Reactions of crown ether activated tert-butoxide ion, J. Org. Chem., 1978, 43, 447–452 CrossRef CAS; (b) S. A. DiBiase, R. P. Wolak Jr., D. M. Dishong and G. W. Gokel, Crown cation complex effects. 10. Potassium tert-butoxide mediated penultimate oxidative hydrolysis of nitriles, J. Org. Chem., 1980, 45, 3630–3634 CrossRef CAS; (c) C. L. Cheong and B. J. Wakefield, Polyhalogenoaromatic compounds. Part 53. substitution in polyfluoroaromatic compounds by bulky nucleophiles, J. Chem. Soc., Perkin Trans. 1, 1988, 3301–3305 RSC; (d) S. M. Kim, H. S. Yang, H. Eum, H.-J. Ha and J. W. Yang, Mixed monosilyl acetals and catalyst-dependent chemoselective Mukaiyama aldol reactions, Chem. – Eur. J., 2017, 23, 16432–16437 CrossRef CAS.
- M. W. Drover, J. A. Love and L. L. Schafer, 1,3-N,O-Complexes of late transition metals. Ligands with flexible bonding modes and reaction profiles, Chem. Soc. Rev., 2017, 46, 2913–2940 RSC.
- (a) V. Y. Kukushkin and A. J. L. Pombeiro, Metal-mediated and metal-catalyzed hydrolysis of nitriles, Inorg. Chim. Acta, 2005, 358, 1–21 CrossRef CAS; (b) V. Theodorou, G. Paraskevopoulos and K. Skobridis, A mild alkaline hydrolysis of N- and N,N-substituted amides and nitriles, ARKIVOC, 2015, 101–112 Search PubMed.
- T. M. Morwick, High-throughput ester hydrolysis with catch-and-release isolation of carboxylic acids, J. Comb. Chem., 2006, 8, 649–651 CrossRef CAS.
- Y. L. Hu, H. Jiang, J. Zhu and M. Lu, Facile and efficient hydrolysis of organic halides, epoxides, and esters with water catalyzed by ferric sulfate in a PEG1000-DAIL[BF4]/toluene temperature-dependent biphasic system, New J. Chem., 2011, 35, 292–298 RSC.
- For elucidation of reaction mechanism by ReactIR spectrometer, see: (a) Z. Zhang, H. Y. Bae, J. Guin, C. Rabalakos, M. van Gemmeren, M. Leutzsch, M. Klussmann and B. List, Asymmetric counteranion-directed Lewis acid organocatalysis for the scalable cyanosilylation of aldehydes, Nat. Commun., 2016, 7, 12478 CrossRef CAS; (b) X.-P. Zeng, Z.-Y. Cao, X. Wang, L. Chen, F. Zhou, F. Zhu, C.-H. Wang and J. Zhou, Activation of chiral (Salen)AlCl complex by phosphorane for highly enantioselective cyanosilylation of ketones and enones, J. Am. Chem. Soc., 2016, 138, 416–425 CrossRef CAS; (c) J. M. V. Lauridsen, S. Y. Cho, H. Y. Bae and J.-W. Lee, CO2 (de)activation in carboxylation reaction: A case study using Grignard reagents and nucleophilic bases, Organometallics, 2020, 39, 1652–1657 CrossRef.
- (a) B. C. Smith, Fundamentals of Fourier Transform Infrared Spectroscopy, CRC Press, Boca Raton, 1995, p. 3 Search PubMed; (b) J. Coates, Interpretation of Infrared Spectra, A Practical Approach, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Inc., New York, 2006, pp. 1–23 Search PubMed.
- S. Torii, T. Inokuchi, S. Takagishi, H. Horike, H. Kuroda and K. Uneyama, Electrogenerated acid-catalyzed reactions of acetals, aldehydes, and ketones with organosilicon compounds, leading to aldol reactions, allylations, cyanations, and hydride additions, Bull. Chem. Soc. Jpn., 1987, 60, 2173–2188 CrossRef CAS.
- V. Lloret, M. Á. Rivero-Crespo, J. A. Vidal-Moya, S. Wild, A. Doménech-Carbó, B. S. J. Heller, S. Shin, H.-P. Steinrück, F. Maier, F. Hauke, M. Varela, A. Hirsch, A. Leyva-Pérez and G. Abellán, Few layer 2D pnictogens catalyze the alkylation of soft nucleophiles with esters, Nat. Commun., 2019, 10, 509 CrossRef.
- (a) M. Goodman and K. C. Stueben, Peptide syntheses via amino acid active esters, J. Am. Chem. Soc., 1959, 81, 3980–3983 CrossRef CAS; (b) Q. Wang, M. Leutzsch, M. van Gemmeren and B. List, Disulfonimide-catalyzed asymmetric synthesis of β3-amino esters directly from N-Boc-amino sulfones, J. Am. Chem. Soc., 2013, 135, 15334–15337 CrossRef CAS.
- G. W. Anderson and F. M. Callahan, t-Butyl esters of amino acids and peptides and their use in peptide synthesis, J. Am. Chem. Soc., 1960, 82, 3359–3363 CrossRef CAS.
- M. Saroja and T. N. B. Kaimal, A convenient method of esterification of fatty acids. Preparation of alkyl esters, sterol esters, wax esters and triacylglycerols, Synth. Commun., 1986, 16, 1423–1430 CrossRef CAS.
- (a) P. Strazzolini, N. Misuri and P. Polese, Efficient cleavage of carboxylic tert-butyl and 1-adamantyl esters, and N-Boc-amines using H2SO4 in CH2Cl2, Tetrahedron Lett., 2005, 46, 2075–2078 CrossRef CAS; (b) B. F. Lundt, N. L. Johansen, A. Vølund and J. Markussen, Removal of t-butyl and t-butoxycarbonyl protecting groups with trifluoroacetic acid, Int. J. Pept. Protein Res., 1978, 12, 258–268 CrossRef CAS.
- (a) A. Vollmar and M. S. Dunn, A convenient synthesis of t-alkyl esters of amino acids, J. Org. Chem., 1960, 25, 387–390 CrossRef CAS; (b) R. E. Shute and D. H. Rich, Prevention of diketopiperazine formation in peptide synthesis by a simultaneous deprotection-coupling procedure: entrapment of reactive nucleophilic species by in situ, acylation, J. Chem. Soc., Chem. Commun., 1987, 1155–1156 RSC; (c) N. Kise, H. Ozaki, H. Terui, K. Ohya and N. Ueda, A convenient synthesis of N-Boc-protected tert-butyl esters of phenylglycines from benzylamines, Tetrahedron Lett., 2001, 42, 7637–7639 CrossRef CAS; (d) M. Sedighi and M. A. Lipton, A convenient, general synthesis of 1,1-dimethylallyl esters as protecting groups for carboxylic acids, Org. Lett., 2005, 7, 1473–1475 CrossRef CAS.
- (a) S. B. Y. Shin and K. Kirshenbaum, Conformational rearrangements by water-soluble peptoid foldamers, Org. Lett., 2007, 9, 5003–5006 CrossRef CAS; (b) A. Odriozola, M. Oiarbide and C. Palomo, Enantioselective synthesis of quaternary Δ4- and Δ5-dehydroprolines based on a two-step formal [3+2] cycloaddition of α-aryl and α-alkyl isocyano(thio)acetates with vinyl ketones, Chem. – Eur. J., 2017, 23, 12758–12762 CrossRef CAS.
- W. König and R. Geiger, Eine neue amid-schutzgruppe, Chem. Ber., 1970, 103, 2041–2051 CrossRef.
- For representative examples of exocyclic cleavage with oxazolidin-2-ones, see: (a) P.-Q. Huang and H. Geng, Ni-catalyzed chemoselective alcoholysis of N-acyloxazolidinones, Green Chem., 2018, 20, 593–599 RSC; (b) C. Guissart, A. Barros, L. R. Barata and G. Evano, Broadly applicable ytterbium-catalyzed esterification, hydrolysis, and amidation of imides, Org. Lett., 2018, 20, 5098–5102 CrossRef CAS.
- For representative example of decarboxylative endocyclic cleavage with oxazolidin-2-ones, see: A. E. May, P. H. Willoughby and T. R. Hoye, Decarboxylative isomerization of N-acyl-2-oxazolidinones to 2-oxazolines, J. Org. Chem., 2008, 73, 3292–3294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data for compounds including 1H and 13C NMR spectra. See DOI: 10.1039/d0qo01135e |
| This journal is © the Partner Organisations 2021 |