
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The sol–gel autocombustion as a route towards highly CO2-selective, active and long-term stable Cu/ZrO2 methanol steam reforming catalysts†
Kevin
Ploner
a,
Parastoo
Delir Kheyrollahi Nezhad
a,
Albert
Gili
 bc,
Franz
Kamutzki
b,
Aleksander
Gurlo
b,
Andrew
Doran
d,
Pengfei
Cao
e,
Marc
Heggen
e,
Nicolas
Köwitsch
f,
Marc
Armbrüster
f,
Maximilian
Watschinger
a,
Bernhard
Klötzer
a and
Simon
Penner
*a
bc,
Franz
Kamutzki
b,
Aleksander
Gurlo
b,
Andrew
Doran
d,
Pengfei
Cao
e,
Marc
Heggen
e,
Nicolas
Köwitsch
f,
Marc
Armbrüster
f,
Maximilian
Watschinger
a,
Bernhard
Klötzer
a and
Simon
Penner
*a
aDepartment of Physical Chemistry, University of Innsbruck, Innrain 52c, A-6020 Innsbruck, Austria. E-mail: simon.penner@uibk.ac.at; Tel: +43 51250758003
bChair of Advanced Ceramic Materials, Institut für Werkstoffwissenschaften und -technologien, Technical University Berlin, Hardenbergstr. 40, D-10623 Berlin, Germany
cInstitute of Chemistry, Technical University Berlin, Sekretariat TC 8, Straße des 17. Juni 124, D-10623 Berlin, Germany
dAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
eErnst Ruska-Centre for Microscopy and Spectroscopy with Electrons, Forschungszentrum Jülich GmbH, Leo-Brandt-Str. 1, D-52428 Jülich, Germany
fFaculty of Natural Sciences, Institute of Chemistry, Materials for Innovative Energy Concepts, Technical University Chemnitz, Straße der Nationen 62, D-09111 Chemnitz, Germany
First published on 20th May 2021
Abstract
The adaption of the sol–gel autocombustion method to the Cu/ZrO2 system opens new pathways for the specific optimisation of the activity, long-term stability and CO2 selectivity of methanol steam reforming (MSR) catalysts. Calcination of the same post-combustion precursor at 400 °C, 600 °C or 800 °C allows accessing Cu/ZrO2 interfaces of metallic Cu with either amorphous, tetragonal or monoclinic ZrO2, influencing the CO2 selectivity and the MSR activity distinctly different. While the CO2 selectivity is less affected, the impact of the post-combustion calcination temperature on the Cu and ZrO2 catalyst morphology is more pronounced. A porous and largely amorphous ZrO2 structure in the sample, characteristic for sol–gel autocombustion processes, is obtained at 400 °C. This directly translates into superior activity and long-term stability in MSR compared to Cu/tetragonal ZrO2 and Cu/monoclinic ZrO2 obtained by calcination at 600 °C and 800 °C. The morphology of the latter Cu/ZrO2 catalysts consists of much larger, agglomerated and non-porous crystalline particles. Based on aberration-corrected electron microscopy, we attribute the beneficial catalytic properties of the Cu/amorphous ZrO2 material partially to the enhanced sintering resistance of copper particles provided by the porous support morphology.
1. Introduction
Concerning renewable energy storage, methanol is a promising candidate due to its beneficial properties with respect to stocking and distribution.1 It is liquid at ambient conditions, has a high hydrogen-to-carbon ratio and the absence of a C–C bond considerably decreases the necessary temperature for selective reforming (e.g. CO2-selective steam reforming of methanol at 200–300 °C as opposed to ethanol steam reforming with inherently reduced CO2 selectivity at temperatures above 400 °C).1–3 Due to its high volumetric energy density, it is especially suitable for automotive applications, where the H2/CO2 reformate can be used in a polymer electrolyte membrane fuel cell (PEMFC).1,4 For this application, the concentration of CO has to be kept in the low ppm regime, as even small traces of CO can deteriorate the performance of PEMFC electrodes.5The methanol conversion reaction exhibiting the highest hydrogen yield is stoichiometric methanol steam reforming6 (MSR):
| CH3OH(g) + H2O(g) ⇄ 3H2,(g) + CO2,(g) ΔH0r = 49.6 kJ mol−1 | (1) |
| CH3OH(g) ⇄ 2H2,(g) + CO(g) ΔH0r = 90.6 kJ mol−1 | (2) |
| CO2,(g) + H2,(g) ⇄ H2O(g) + CO(g) ΔH0r = 41.1 kJ mol−1 | (3) |
| CO(g) + 3H2,(g) ⇄ CH4,(g) + H2O(g) ΔH0r = −206 kJ mol−1 | (4) |
| CO2,(g) + 4H2,(g) ⇄ CH4,(g) + 2H2O(g) ΔH0r = −165 kJ mol−1 | (5) |
The archetypical MSR catalyst is commercially available Cu/ZnO/Al2O3.2,7,9,10 It suffers from various drawbacks, including significant deactivation by copper particle sintering, and features too high CO levels for direct use in a PEMFC.5,7 Therefore, alternative systems have to be developed fulfilling all requirements of an efficient MSR catalyst to render its use economically feasible.1
ZrO2 is a promising candidate as a synergistically active support for highly CO2-selective copper-based MSR catalysts.2,11,12 ZrO2 exhibits three crystal structures: monoclinic under ambient conditions (m-ZrO2, room temperature to 1170 °C, group P21/c)13 and the high-temperature polymorphs tetragonal (t-)ZrO2 (1170–2370 °C, space group P42/nmc)14 and cubic (c-)ZrO2 (2370–2680 °C, space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m),15 respectively. The latter two can be preserved as metastable structures at room temperature via particle size control or doping.16–18 Both Cu/m-ZrO2, as well as Cu/t-ZrO2 systems, were tested with conflicting reports in MSR. Examples of both well and undesirably performing catalysts with respect to CO2 selectivity in MSR are reported for Cu/m-ZrO219,20 as well as Cu/t-ZrO211,12,19 materials.
m),15 respectively. The latter two can be preserved as metastable structures at room temperature via particle size control or doping.16–18 Both Cu/m-ZrO2, as well as Cu/t-ZrO2 systems, were tested with conflicting reports in MSR. Examples of both well and undesirably performing catalysts with respect to CO2 selectivity in MSR are reported for Cu/m-ZrO219,20 as well as Cu/t-ZrO211,12,19 materials.
A comprehensive study on Cu/ZrO2 catalysts in MSR following dedicated synthesis routines revealed that especially the synthesis approach for the ZrO2 phase (determining its surface-chemical properties as the key parameter for a high CO2 selectivity) is crucial for the performance of Cu/ZrO2 catalysts in MSR. The influence of the type of copper precursor is limited.19 The surface-chemical (defect) behaviour of a specific ZrO2 polymorph could be steered by the synthesis pathway and the preparation history. Its bulk crystallographic structure is of minor importance for the MSR performance.19
Various synthesis approaches for MSR catalysts are reported in literature, aiming at the optimisation of the activity, CO2 selectivity and long-term stability.2 Among the most prominent ones are wet impregnation (Cu/ZrO2,19 Cu/Zn + Cu/Cr + CuZr on Al2O3,21 Cu/Al2O3 + Zn and Ce,22 CeO2- + ZrO2-promoted Cu/ZnO on Al2O323) and co-precipitation (Cu/ZrO2,19 CuO/CeO2,24 ZrO2- and Al2O3-promoted Cu/ZnO,25 Cu/CeO226), but also other less commonly utilised methods like hydrothermal synthesis (Cu/Zn/Al27), soft reactive grinding (Cu1.5Mn1.5O4 spinel28), oxalate gel co-precipitation (Cu/MnOx,28 Cu/ZrO2,29 Cu/ZrO230), the polymer template sol–gel method (Cu/ZrO212) and urea nitrate combustion (Cu/CeO2,31 Cu/CeO2 doped with Sm, Zn, La, Zr, Mg, Gd, Y, Ca32) were employed. Each of these syntheses offers certain advantages, but alternative methods satisfying all prerequisites of an ideal MSR catalyst are still to be developed.
One promising candidate is the sol–gel autocombustion method – the urea nitrate combustion is a subgroup of this method – that grants access to homogeneous oxide powders by the use of complexing agents like glycine or urea to prevent selective precipitation of metal ions during the removal of water.33 At the same time, they act as the fuel for the autocombustion of the correspondingly employed metal nitrates, yielding finely dispersed oxidised powders. Additionally, the sol–gel autocombustion method offers the advantages of simplicity as a one-step approach, a high versatility concerning the educts and a reasonably high surface area.33
The sol–gel autocombustion technique can be utilised for the synthesis of many material classes and is primarily employed for perovskite or spinel phases with various applications (CoFe2O434 and Li0.5Fe2.5O435 with special magnetic properties, (CeO2)0.9(SmO1.5)0.136 as electrolyte in a solid oxide fuel cell, LaMnO337 for the oxygen reduction reaction, LaFeO338 as well as doped La(Cu0.7Mn0.3)O339 for the reduction of NO by CO), but also for other catalysts, e.g. NiO/ZrO240 for chemical-looping combustion. For the preparation of catalysts for MSR, this method was mainly used for Cu/ZnO/Al2O3 systems and different variants obtained by substitution/addition of selected components.41–45 All of these systems display a specific morphology featuring so-called combustion pores that are formed by the expansion of the gases produced during the reaction of the fuel and the oxidizing agent.43,44 One particular CuO/ZnO/Al2O3 system prepared with ethylene glycol as the fuel displayed a high activity and long-term stability in MSR, which is explained by a high number of combustion pores and a small particle size.44 Yu et al.46 were successful in the preparation of a highly active and stable CuFeO2–CeO2 MSR catalyst via an analogous sol–gel autocombustion routine.
For binary Cu/ZrO2 MSR catalysts, specific wet impregnation,19,29 co-precipitation,19,29 oxalate gel co-precipitation,29,30 the polymer template sol–gel method12 and conventional sol–gel methods47 have been employed. Wet impregnation is a comparably easy synthesis route, but the resulting Cu/ZrO2 catalysts can suffer from elevated deactivation in MSR.19 Similar observations concerning long-term stability have been made for aqueous co-precipitation,29 while more sophisticated techniques like oxalate gel co-precipitation29 and the polymer template sol–gel method12 alleviate these stability issues. Conventional sol–gel methods provide exceptionally high surface areas and copper dispersion,47 but the latter three approaches all consist of multiple synthesis steps, making the process more complex and costly.
In the present work, the sol–gel autocombustion method is adapted to obtain binary Cu/ZrO2 catalysts for methanol steam reforming, making use of a number of advantages, which potentially lead to highly active and CO2-selective catalyst materials. Apart from the rather simple one-step process and the generally high versatility of educts, the high dispersion of metal ions and the characteristic morphology featuring combustion pores lead to a high number of catalytically active sites. Characterisation of the catalysts by in situ X-ray diffraction studies during the post-combustion calcination in combination with aberration-corrected electron microscopy allows us to directly follow the development of the bulk and particle structures of the active Cu/ZrO2 phase and to directly relate the obtained morphology to activity and CO2 selectivity under MSR operation. For the latter, we correlate batch reactor studies, detailing the development of different trace products as a function of pre-MSR calcination temperature, with long-term stability tests in a flow reactor.
2. Experimental
2.1. Sample synthesis
The catalysts consisting of ≈10 mol% Cu (exact loading is difficult to reach because of unknown water content of ZrO(NO3)2·xH2O; 10 mol% Cu correspond to 5.4 wt% Cu to ensure comparability with other systems from our workgroup19) supported on ZrO2 were prepared using the sol–gel autocombustion method.33 Cu(NO3)2·3H2O (≈0.03 mol l−1 in water, Merck, Darmstadt, ≥99.5%) and ZrO(NO3)2·xH2O (≈0.25 mol l−1 in water, Alfa Aesar, Puratronic, 99.994% metals basis) were separately dissolved in deionised water and subsequently added to an aqueous solution of glycine (≈0.56 mol l−1 in water, Sigma Aldrich, ≥99%). This yields a NH2:NO3− ratio being slightly larger than equimolar and final concentrations of 0.01 mol l−1 Cu(NO3)2·3H2O, 0.08 mol l−1 ZrO(NO3)2·xH2O and 0.19 mol l−1 glycine. The blueish solution was stirred at room temperature for 30 min and subsequently, the temperature was increased to 90–95 °C to evaporate the water. Over the course of 3 h, the solution transformed into a viscous blue gel. This gel was heated in a beaker to 250 °C in air, where the metal oxides, as well as carbon residues, were formed in a self-ignited combustion. Calcination at 400 °C, 600 °C and 800 °C for 2 h removed the remaining carbon, indicated by the change of the initially black colour, with simultaneous formation of either a mostly amorphous, a tetragonal or a monoclinic polymorph of ZrO2 (cf. ESI,† Fig. S7). The nominal Cu loading has been verified by dedicated TGA oxidation–reduction experiments. The latter have been proven to yield very accurate values of the Cu content, especially for Cu/ZrO2 catalysts.192.2. Catalytic characterisation
The MSR mixture is provided as a liquid solution of methanol and water in a ratio to yield an equilibrium gas phase composition of methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water = 1:2 at room temperature. The mixture and the gas phase are initially purified with three freeze–pump–thaw cycles before the gas phase is expanded into the pre-evacuated reactor. As standard pre-treatments for MSR catalysts, oxidative cleaning in pure O2 at 400 °C for 1 h (termed O400) and pre-reduction in pure H2 at 300 °C for 1 h (termed H300) are conducted before the methanol steam reforming experiment. For MSR, the reaction mixture (≈28 mbar) is introduced into the reactor at 100 °C (to prevent condensation on the sample) and Ar is added to correct the mass traces of the reactants and products with respect to thermal expansion during heating and the slow gas withdrawal to the QMS vacuum chamber through the capillary. Helium is added as a carrier gas up to atmospheric pressure, which enhances the heat transfer to and in the catalyst bed, as well as the recirculation efficiency. After an equilibration period of 40 min, the temperature program is started and the gas-phase composition is quantified via the QMS. Baseline and Ar intensity correction in combination with external calibration of the relevant gases including their relative fragmentation patterns (e.g. for correction of the m/z = 28 fragment of CO2) yields the evolution of the effective temperature- and total pressure drop-corrected partial pressures of the gaseous species.
:water = 1:2 at room temperature. The mixture and the gas phase are initially purified with three freeze–pump–thaw cycles before the gas phase is expanded into the pre-evacuated reactor. As standard pre-treatments for MSR catalysts, oxidative cleaning in pure O2 at 400 °C for 1 h (termed O400) and pre-reduction in pure H2 at 300 °C for 1 h (termed H300) are conducted before the methanol steam reforming experiment. For MSR, the reaction mixture (≈28 mbar) is introduced into the reactor at 100 °C (to prevent condensation on the sample) and Ar is added to correct the mass traces of the reactants and products with respect to thermal expansion during heating and the slow gas withdrawal to the QMS vacuum chamber through the capillary. Helium is added as a carrier gas up to atmospheric pressure, which enhances the heat transfer to and in the catalyst bed, as well as the recirculation efficiency. After an equilibration period of 40 min, the temperature program is started and the gas-phase composition is quantified via the QMS. Baseline and Ar intensity correction in combination with external calibration of the relevant gases including their relative fragmentation patterns (e.g. for correction of the m/z = 28 fragment of CO2) yields the evolution of the effective temperature- and total pressure drop-corrected partial pressures of the gaseous species.
To obtain the formation rates in terms of specific activities, the partial pressures are differentiated with respect to the time and converted to molar amounts utilizing the ideal gas law. Normalisation to the copper mass yields the specific activity in μmol gCu−1 s−1. The methanol conversion (xMeOH) is obtained as a relative value by relating the m/z = 31 signal at each time to the value at the start of the temperature ramp (eqn (6)). The accuracy of the methanol conversion was estimated utilizing the standard deviation of the constant m/z = 31 signal at its highest point in a reference measurement, as the noise scales with the total signal height. This standard deviation was multiplied by 3 and propagated to the accuracy of the methanol conversion according to eqn (6), yielding a maximum accuracy of ≈0.01 (≈1%) at low methanol conversions, which becomes smaller at higher conversions.
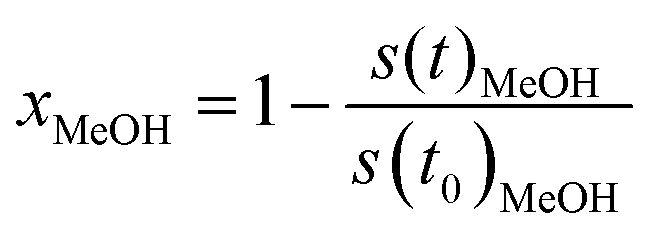 | (6) |
The integral CO2 selectivity is obtained by division of the partial pressure of CO2 by the sum of the values of CO, CO2 and CH4. Values larger than 1 caused by slight deviations in the baseline (when the sum of the partial pressures is close to 0, division by it causes huge artefacts in the integral CO2 selectivity) were manually set to 1.
The apparent activation energy of CO2 formation EA(CO2) was calculated by fitting an Arrhenius function to the specific activity plotted vs. the absolute temperature at the beginning of the rate increase at a methanol conversion below 10%. Simultaneous variation of both EA and the pre-exponential factor A yields values ranging from 1 × 107 to 5 × 109 μmol gCu−1 s−1 for the latter. Hence, a fixed average value of 1 × 108 μmol gCu−1 s−1 is employed in the fits to enhance the relative comparability of the related activation energies.
:1 H2O/MeOH vapour mixture (via simultaneous evaporation of 0.01 ml min−1 H2O(l), 0.0225 ml min−1 MeOH(l), Fisher Scientific, HPLC grade). The unconverted fraction of the reactant vapours was condensed downstream in a cooling trap and the gas stream was further dried by a Nafion® membrane, which was dried in counter flow by a N2-flow of 100 ml min−1. Finally, the dry gas stream was analysed by online gas chromatography to determine the specific activity using the ideal gas law and normalisation to the copper mass. The CO2 selectivity is obtained by division of the specific activity of CO2 by the sum of the values of CO, CO2 and CH4.
2.3. In situ X-ray diffraction (in situ XRD)
The bulk structural changes of the post-combustion powders of ZrO2 and Cu/ZrO2 were investigated with temperature-resolved in situ synchrotron XRD measurements at the beamline 12.2.2 at the Advanced Light Source (ALS) at the Lawrence Berkeley National Laboratory (LBNL), California, in a setup previously described by Doran et al.48 and Schlicker et al.49 A monochromatic beam with an energy of 25 keV and a spot size of 30 μm was utilised in transmission mode and the diffraction pattern was recorded with a PerkinElmer XRD 1621 image plate detector, collecting one pattern per 60 s. A LaB6 NIST standard was measured for the calibration of the sample-to-detector distance, the detector tilt and the exact wavelength (0.4959 Å) using the Dioptas software,50 which was also utilised for the conversion of the 2D detector images to diffraction patterns by radial integration.The sample powder (≈1 mg) was placed in a quartz capillary with a diameter of 700 μm located inside a SiC sleeve that was heated with two infrared lights.48 A flow of 2 ml min−1 O2 and 8 ml min−1 Ar was supplied by Alicat mass flow controllers and a sequence of heating with a rate of 5 °C min−1 from 25–800 °C, an isothermal period of 20 min and cooling to room temperature with 20 °C min−1 was executed.49
2.4. Scanning transmission electron microscopy (STEM)
Scanning transmission electron microscopy (STEM) was performed using a FEI Titan 80–200 (ChemiSTEM) electron microscope with a Cs-probe corrector (CEOS GmbH) and a high-angle annular dark field (HAADF) detector. The microscope was operated at 200 kV. To achieve “Z-Contrast” conditions, a probe semi-angle of 24.7 mrad and an inner collection semi-angle of the detector of 88 mrad were used. Compositional maps were obtained with energy-dispersive X-ray (EDX) spectroscopy using four large-solid-angle symmetrical Si drift detectors. For EDX elemental mapping, Cu K and Zr K peaks were used.2.5. N2 adsorption according to the BET method
The specific surface area was measured using a NOVA 2000e Surface Area & Pore Size Analyzer (Quantachrome Instruments) and the software Quantachrome NovaWin. Prior to the adsorption of N2 at −196 °C, the samples were heated in vacuo under active pumping at 200 °C for 1 h to remove any adsorbed water. The adsorption isotherm was measured at five points from 0.05 to 0.30 p/p0 and evaluated according to the BET model.512.6. Dissociative N2O adsorption
The determination of the accessible specific copper surface area is based on the selective oxidation of metallic Cu to Cu2O at the surface using N2O.52–54 The measurements were conducted in a quartz-glass reactor with a volume (including the sample) that is precisely calibrated by expansion of He (5.0, Messer) from a manifold with a defined volume. To account for the temperature gradient in the reactor, this calibration is performed at a sample temperature of 300 °C. To prevent the contribution of water formed by the reduction to the total pressure, a degassed zeolite trap is placed in the cold zone of the reactor. Since only metallic copper can be oxidised by N2O, the calcined samples are pre-reduced in pure flowing H2 (5.0, Messer) at 300 °C for 30 min. Then, the reactor is evacuated, purged with He and evacuated once more, before the sample is cooled down to 70 °C and subjected to a defined pressure of N2O (2.0, Messer) measured by a Baratron pressure transducer (MKS Instruments). A quadrupole mass spectrometer (QMS, Balzers QMA125 + QME 125-9) was employed for monitoring the formation of N2 and the consumption of N2O for 90 min. Following evacuation and flushing with He, the temperature was increased to 300 °C in vacuo. Thereafter, a defined pressure of H2 was provided in the manifold, which can be converted to a molar amount utilizing the ideal gas law. After opening the valve to the sample, the pressure decrease caused by the reduction of the N2O-induced surface Cu2O layer was monitored for 35 min. Based on the preceding He calibration and the thereby calculated effective volume at 300 °C, the molar amount nH2 of consumed H2 can be calculated via the ideal gas law. Therefore, the remaining molar amount of H2 in the effective volume Veff at 300 °C is subtracted from the supplied molar amount of H2 in the manifold at room temperature before expansion (eqn (7)). | (7) |
Since the reduction of 1 mol surface Cu2O with H2 yields 2 mol Cu0, the consumed molar amount of H2 is equal to twice the amount of accessible Cu surface atoms. Hence, the specific copper surface area SACu and the dispersion DCu can be determined utilizing eqn (8) and (9), respectively.54
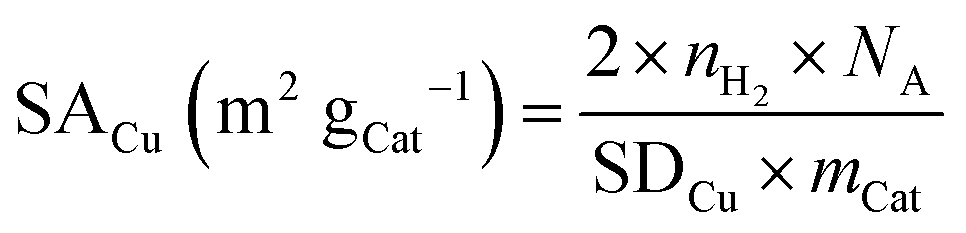 | (8) |
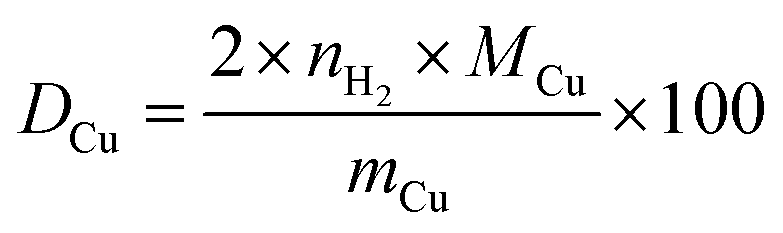 | (9) |
The average particle diameter of Cu dCu can be estimated according to eqn (10).55 This model evaluation is based on the assumption of spherical particles embedded in the support. Hence, only half of their surface area is accessible.
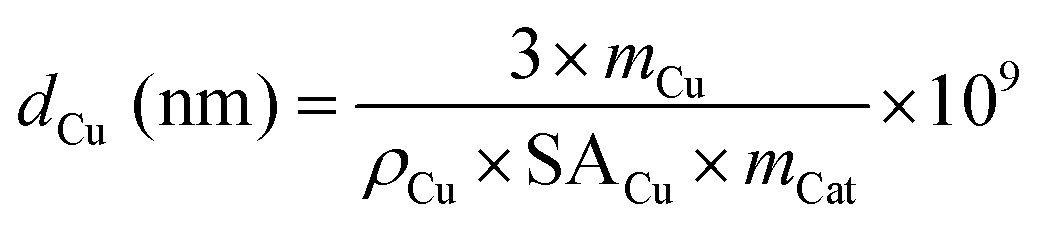 | (10) |
2.7. Thermogravimetric analysis (TGA)
The copper loading of the catalysts was determined via oxidation–reduction–oxidation–reduction treatments in a NETZSCH STA 449F3 Jupiter TGA/DSC apparatus. To ensure complete oxidation, the samples were subjected to a temperature program consisting of heating with 5 °C min−1 from 25–400 °C, an isothermal period of 30 min and subsequent cooling to 25 °C with 5 °C min−1 in a gas atmosphere of 10 vol% O2 (99.999%, Linde) in Ar (99.999%, Linde) with a total flow rate of 100 ml min−1. The same temperature program was applied for reduction in a gas mixture of 10 vol% H2 (99.999%, Linde) in Ar with a total flow rate of 100 ml min−1. The reduction of the catalysts corresponds to the transformation of CuO to Cu0, where the weight loss of oxygen is quantified and can be utilised for the calculation of the copper loading. The additional oxidation and reduction steps were conducted to ensure the reproducibility of the measurements.3. Results and discussion
A schematic representation of the sol–gel autocombustion approach is depicted in Fig. 1. The educts are dissolved separately in deionised water and then combined in one solution (A), where glycine acts as a complexing agent for copper and zirconium, ensuring a high dispersion in the dissolved state even upon removal of water.33 After ageing (B), the solvent is removed by evaporation at 90 °C, leading to the formation of a gel (C). The autocombustion is triggered by increasing the temperature to approximately 250 °C, causing a spontaneous strongly exothermic reaction of glycine and nitrate.33 The resulting powder (D) is still amorphous and can be calcined at selected temperatures (E), yielding the samples (F) in this study. This procedure can be conducted without copper as well, only utilizing ZrO(NO3)2 and glycine. The obtained Zr precursor exhibits a similar tunability of its structure by application of different calcination treatments, whereas the temperature regions of stability are distinct from the copper-containing samples.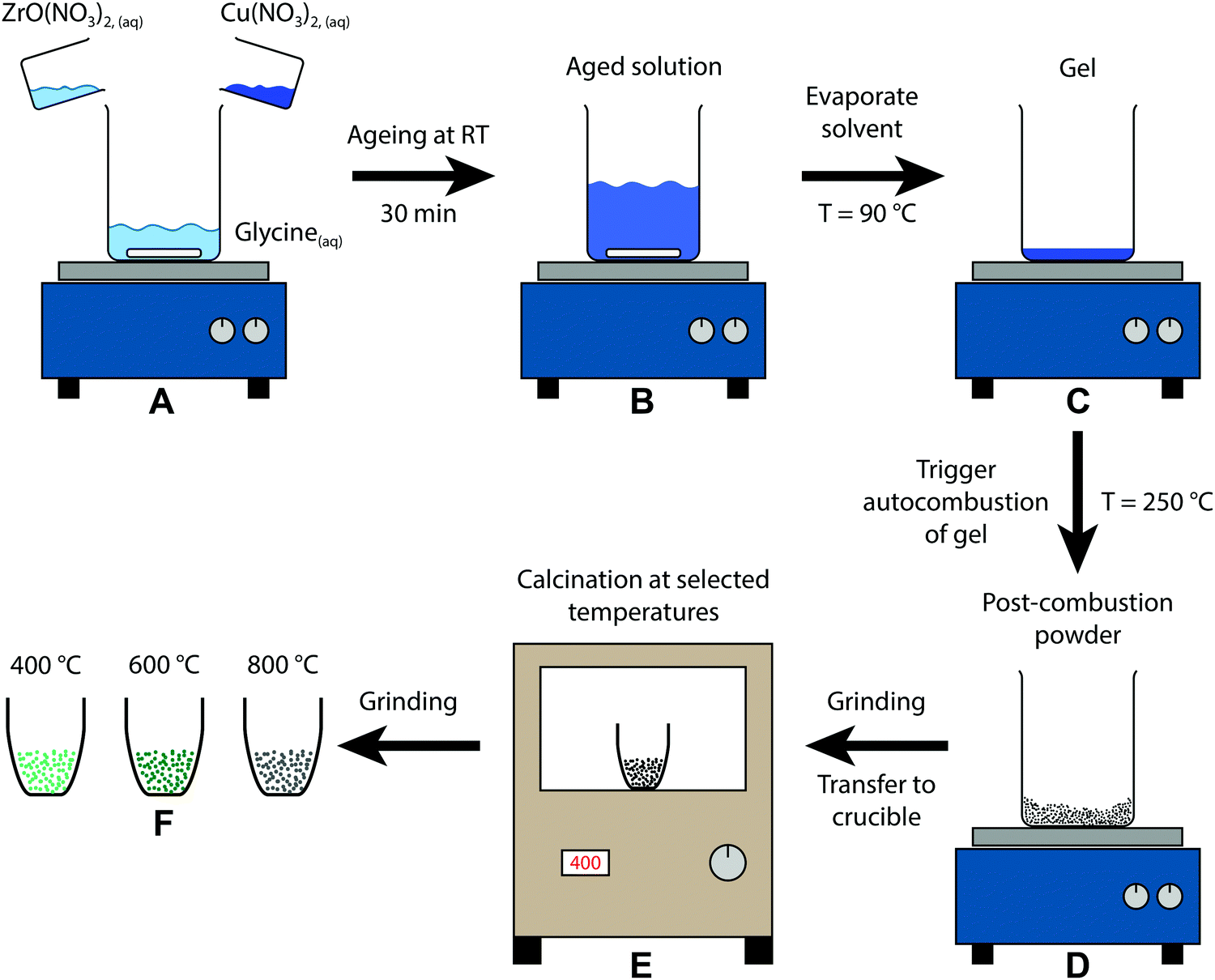 | ||
| Fig. 1 Schematic depiction of the sol–gel autocombustion yielding the three samples of this study by calcination of the post-combustion precursor at different temperatures. | ||
Variation of the calcination temperature from 400–900 °C for the pure amorphous Zr precursor material reveals a dependency of the colour on the calcination temperature (see ESI,† Fig. S1). The colour changes from dark brown at 400 °C, over brown to white at 800 °C. A tentative explanation for the darker colour at lower calcination temperatures can be given by an elevated number of defects in ZrO2 acting as colour centres.57,58 At lower calcination temperatures (400–600 °C), more oxygen vacancies, which can be created by the reaction of carbonaceous species from the synthesis with lattice oxygen,59 can be retained. Stabilisation of the tetragonal structure at lower calcination temperatures (cf. Fig. S5, ESI†) is mainly ascribed to the presence of an elevated number of oxygen vacancies (which are themselves stabilised by the nanoparticle size effect).59 Since the calcination was conducted in air, the oxygen vacancies can be quenched at higher temperatures, resulting in monoclinic zirconia as the predominant polymorph. This is a result of the missing stabilisation of the tetragonal phase due to a decreased number of oxygen vacancies, which in turn could explain the white colour, as less colour centres in the form of oxygen vacancies are present. We have tried to assess the influence of surface-near defects by evaluating the Zr 3d spectra (Fig. S4, ESI†), but while we did observe a high number of sub-stoichiometric Zr oxides for all samples, a clear trend with respect to the annealing temperature was not obtained. We hence conclude that mostly bulk-related vacancies contribute to the colour. Note that the different sample colour after calcination can be masked by both carbon residues from the synthesis60 and the addition of copper. Additionally, copper may affect the stability of vacancies in ZrO2, altering the prerequisites altogether. One example of the effect of Cu addition in binary Cu/ZrO2 systems was provided by doping ZrO2 with copper in a co-precipitation synthesis, which leads to increasing stabilisation of the tetragonal and cubic ZrO2 polymorphs and a reduction of the crystallite size of the zirconia phase with increasing copper content.61 Another study of three binary Cu/ZrO2 catalysts for methanol synthesis prepared by impregnation and co-precipitation techniques reported that the stabilisation of the tetragonal polymorph at lower temperature is caused by the presence of oxygen vacancies. Furthermore, incorporation of Cu+ or Cu2+ into the ZrO2 lattice compensates for the negative charge of vacancies in ZrO2, further contributing to the stabilisation of the tetragonal polymorph.62
The three Cu/ZrO2 samples in this study exhibit a distinct colour after calcination, which is clearly different from the colour of the black post-combustion powder (see ESI,† Fig. S2). The sample calcined at 400 °C for 2 h in air (termed CZ400) is between green and turquoise, treatment at 600 °C for 2 h (sample termed CtZ600) leads to a darker green colour and calcination at 800 °C for 2 h (sample termed CmZ800) yields a grey powder. A list of the investigated Cu/ZrO2 samples is provided in Table 1. The green colour of CZ400 indicates the incorporation of Cu into ZrO2, which was also observed by Tada et al.63,64 upon impregnating amorphous zirconia with copper nitrate, identifying the resulting phase with X-ray absorption studies as an amorphous ternary CuxZryOz compound. The grey colour of CmZ800 can be explained as a mixture of the purely white m-ZrO2 and the black CuO, whereas the dark green colour of CtZ600 might be interpreted as an intermediate state, consisting of partially remaining CuxZryOz as well as already present CuO and t-ZrO2.
| Acronym | Calcination temperature/°C | ZrO2 polymorph after calcination | Copper loading from TGA (reduced state)/wt% |
|---|---|---|---|
| CZ400 | 400 | Amorphous/tetragonal | 6 |
| CtZ600 | 600 | Tetragonal | 6 |
| CmZ800 | 800 | Monoclinic | 6 |
The copper loading of the samples was determined via an oxidation–reduction–oxidation–reduction cycle in a thermogravimetric analysis (TGA) setup as described in Section 2.7. The mass decrease in the reduction with H2 up to 400 °C corresponds to the Cu loading, which is identical for all three samples. This confirms the expectations, as all catalysts were obtained by calcination of the same post-combustion material.
The effect of the different calcination treatments on the structure of the pure ZrO2 samples is visualised in ex situ XRD measurements depicted in the ESI† (Fig. S5). At 400 °C, the material remains amorphous, whereas at 500 °C, pure t-ZrO2 is formed. From 600 °C to 900 °C, the amount of m-ZrO2 increases until only small contributions of t-ZrO2 prevail. Similar trends can be observed in an in situ XRD experiment with the post-combustion powder of ZrO2 (ESI,† Fig. S6). Heating this precursor in 20 vol% O2 in Ar from 25–800 °C reveals an initial reordering to amorphous zirconia starting around 200 °C. At 460 °C, the tetragonal phase starts to form until the evolution of m-ZrO2 is observable at approximately 700 °C. The two separate approaches employing either isothermal calcination (characterised by ex situ XRD in the ESI† in Fig. S5) or a heating ramp in the in situ XRD experiments (ESI,† Fig. S6) lead to apparently different temperature stability regions of the polymorphs, which is a consequence of the different time the samples are exposed to a certain temperature. This is visible in the appearance of m-ZrO2 already at 600 °C following isothermal calcination for 2 h, while it is formed at 700 °C in the in situ XRD characterisation.
The Cu/ZrO2 samples display similar trends, but the effect of copper on the stability of the zirconia polymorph is clearly visible. After isothermal calcination at 400 °C for 2 h in air, small amounts of t-ZrO2 are already present in the sample (see ex situ XRD characterisation in the ESI† in Fig. S7), next to a mostly amorphous state. At 600 °C, pure tetragonal zirconia is obtained, whereas calcination at 800 °C yields pure m-ZrO2. Comparison to the isothermal calcination treatments of pure ZrO2 shows that the stability region of t-ZrO2 is expanded by the addition of copper, but the transformation kinetics are also accelerated. This means that t-ZrO2 is stabilised in the presence of Cu at an extended temperature range, but the transformation to m-ZrO2 occurs faster at high temperatures as well, as compared to pure ZrO2.
The corresponding in situ XRD calcination treatment of the amorphous Cu/ZrO2 post-combustion powder in 20 vol% O2 in Ar from 25–800 °C (Fig. 2) reveals that the onset temperature of t-ZrO2 formation is higher than in pure ZrO2 (≈510 °C vs. ≈460 °C). The same is true for the evolution of m-ZrO2 (≈770 °C in Cu/ZrO2vs. ≈700 °C in ZrO2).
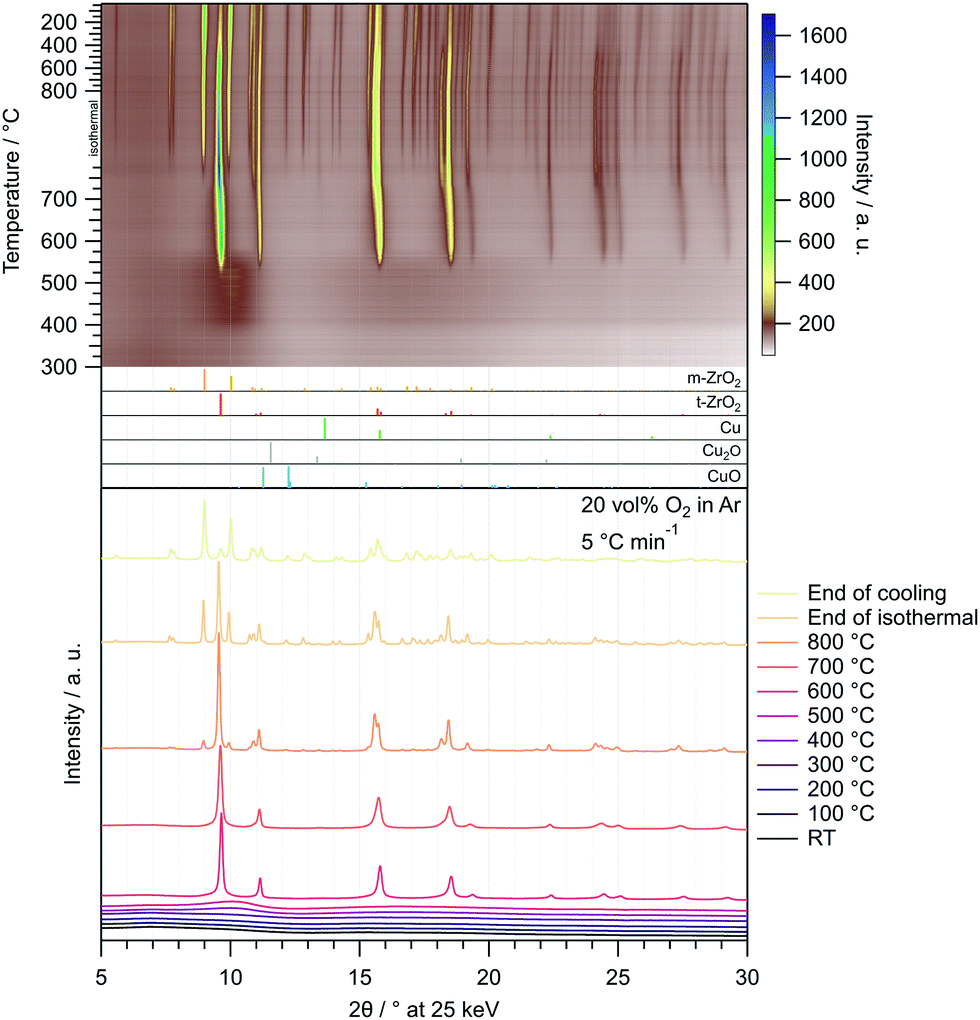 | ||
| Fig. 2 Temperature-resolved in situ XRD investigation of the Cu/ZrO2 precursor after combustion by calcination in 20 vol% O2 in Ar with a heating rate of 5 °C min−1 between 25 °C and 800 °C. In the uppermost panel, the entire treatment consisting of heating to 800 °C (depicted from 300 °C), an isothermal period of 30 min and cooling to 25 °C with 20 °C min−1 is illustrated as a heat map. Selected diffractograms are depicted in the bottom panel. The patterns were recorded utilizing synchrotron radiation at 25 keV (λ = 0.4959 Å). References were taken from the ICDD database.65–70 | ||
No copper phase is detected in the XRD pattern for the samples CZ400 and CtZ600 after isothermal calcination (ESI,† Fig. S7 at 400 °C and 600 °C, respectively), whereas CuO is clearly observable for CmZ800. This further supports the formation of an amorphous CuxZryOz phase as proposed by Tada et al.63,64 After MSR, metallic copper is found in all samples, but only in trace amounts in CZ400 (ESI,† Fig. S7).
The specific surface area of the Cu/ZrO2 catalysts was characterised by N2 adsorption according to the BET method as well as dissociative N2O adsorption followed by H2 titration (Table 2). CZ400 exhibits the highest BET surface area as it was treated at the lowest temperature, whereas CmZ800 has the lowest value. This trend is not directly reflected in the specific copper surface area. Two cycles of dissociative N2O chemisorption were conducted on each sample, where each cycle consists of pre-reduction at 300 °C in pure H2 to convert Cu quantitatively into the metallic state, selective surface oxidation with N2O at 70 °C and reduction of the formed surface Cu2O layer with H2 at 300 °C. The highest Cu surface area is observed for CtZ600, followed by CZ400 and finally CmZ800 with the lowest value. An analogous trend was reported by Wang et al.30 with Cu/ZrO2 catalysts prepared by oxalate gel-coprecipitation, where the precursor was calcined at different temperatures from 350–750 °C. Based on dissociative N2O adsorption studies, they observed the highest specific copper surface area for the sample treated at the intermediate temperature of 550 °C and interpreted this in terms of agglomeration of copper or metal–support interaction in Cu–ZrO2.30 An alternative tentative explanation could be a very high stability of the CuxZryOz in CZ400 in reducing atmosphere, indicating that most of the Cu remains inaccessible in the bulk of the oxide. To circumvent this deficiency, XPS in combination with the BET surface area is utilised to calculate the specific copper surface area (ESI† in Section S3). The comparison of the obtained SACu(XPS) and SACu(N2O) confirms the magnitude of the specific copper surface area, but the trend of SACu(XPS) follows the BET surface area, where CZ400 exhibits the largest and CmZ800 the lowest value.
In the second cycle, the copper surface area is decreased in all samples, which can be attributed to sintering of metallic Cu under reductive atmosphere. The extent of sintering matches the trend of the calcination temperature. While the decrease of the Cu surface area amounts to approximately 30% in CZ400, it increases to 40% for CtZ600 and 50% for CmZ800. This implies that CZ400 is the catalysts that is most resistant to deactivation by sintering under the applied conditions.
As a measure of the MSR performance of pure ZrO2 prepared via the sol–gel autocombustion, the mostly amorphous ZrO2 obtained by calcination in air at 400 °C for 2 h (termed Z400) was selected as a representative sample. The MSR profiles of Z400 measured between 100 °C and 350 °C in the recirculating batch reactor are depicted in the ESI† in Fig. S8. Two cycles were conducted, one without pre-treatments and the other one including pre-oxidation and pre-reduction. The onset temperatures of all major products – H2, CO, CO2 and CH4 – are located at approximately 300 °C. Additionally, we observe formation of formic acid at around 310 °C. We explain the formation of methane and formic acid on Z400 by different reaction mechanisms occurring with and without Cu. The formation of formic acid on Z400 starts around 310 °C, indicating that these species are tightly bound to the catalyst and persist conversion to CO2 on pure ZrO2. The presence of Cu facilitates the conversion of formate species (as the precursor of formic acid) to CO2, which is evident by the low CO2 onset temperature of ≈150 °C in the Cu-containing catalysts (Fig. 3). We observe the formation of methane at ≈300 °C on Z400, whereby the formation of methane is suppressed for the Cu-containing systems. This is due to the fast conversion of the precursors for methane formation to CO2 and H2 before the mechanism of methane formation plays a significant role. Additionally, we cannot exclude that the reactivity of centres active for methane formation in pure ZrO2 are altered or suppressed by the addition of Cu.19
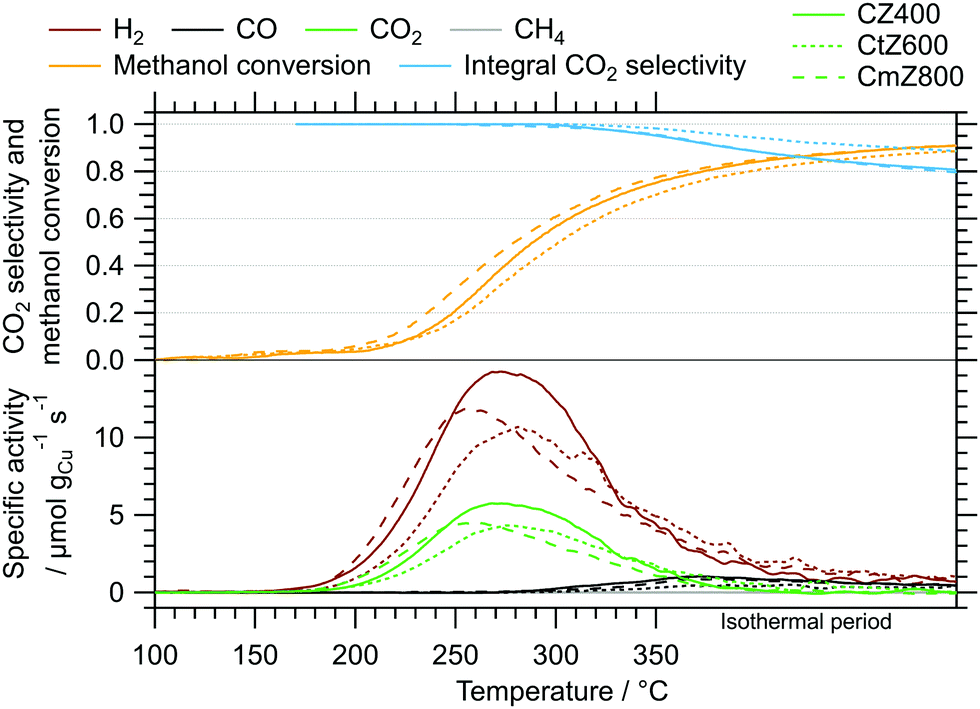 | ||
| Fig. 3 MSR profiles of CZ400, CtZ600 and CmZ800 between 100 °C and 350 °C including an isothermal period of 30 min. Colour code: orange – methanol conversion, blue – integral CO2 selectivity, specific activity of brown: H2, black: CO, green: CO2, grey: CH4. Heating rate: 5 °C min−1; sample mass: CZ400 – 18.6 mg, CtZ600 – 21.6 mg, CmZ800 – 20.2 mg. | ||
The MSR profiles obtained on CZ400, CtZ600 and CmZ800 in the batch reactor are summarized in Fig. 3 and Table 3. In comparison to Z400, these three catalysts exhibit a drastically different behaviour in MSR. The total formation rates of both cycles of Z400 are two orders of magnitude lower than for these copper-containing catalysts. The onset temperatures of H2 (≈150 °C), CO (≈270 °C) and CO2 (≈150 °C) are almost identical for all three samples, as is the general progression of the specific activity, CO2 selectivity and methanol conversion. This implies that the crystal structure of ZrO2, which varies from amorphous over tetragonal to monoclinic in these catalysts, does not significantly impact the selectivity patterns of these systems in MSR. Additionally, the formation of methane and formic acid observed on Z400 (ESI,† Fig. S8 and S9) is absent for CZ400, CtZ600 and CmZ800. In case Cu is present, the onset temperature of CO formation is shifted to lower temperatures (270 °C vs. 300 °C), as well as the higher specific activity towards CO. We interpret this as Cu-enhanced kinetics of the support, yielding more CO already at lower temperatures. Note that the decrease of the CO2 selectivity at higher temperatures in Fig. 3 is characteristic for MSR operation in a recirculating batch reactor. As we provide no constant feed of methanol and water in batch reactor measurements, methanol is successively depleted and the corresponding formation rates of H2 and CO2 decrease, while the reverse water–gas shift reaction, converting H2 and CO2 to CO and water, increasingly occurs. Therefore, the decrease in the CO2 selectivity is mechanistically connected to the transition from MSR to the reverse water–gas shift reaction in a batch reactor and should not be interpreted as a purely MSR-specific value above methanol conversions of ≈40%.
| CZ400 | CtZ600 | CmZ800 | |
|---|---|---|---|
| Onset T(H2)/°C | 150 | 150 | 150 |
| Onset T(CO)/°C | 270 | 270 | 270 |
| Onset T(CO2)/°C | 150 | 150 | 150 |
| T(<95%)/°C | 150–350 (start of isothermal) | 150–350 (8 min isothermal) | 150–350 (1 min isothermal) |
| Max. activity CO2/μmol gCu−1 s−1 | 5.8 | 4.3 | 4.5 |
| 270 °C | 280 °C | 260 °C | |
| Max. activity H2/μmol gCu−1 s−1 | 14.3 | 10.7 | 11.9 |
| 270 °C | 280 °C | 255 °C | |
| E A(CO2)/kJ mol−1 | 78 | 77 | 78 |
The results of the isothermal long-term characterisation of CZ400 in MSR at 300 °C is depicted in Fig. 4. After a slight initial decrease of the specific activity by approximately 25% in the first 15 h on stream, the deactivation trend wanes and the performance remains stable up to 110 h time on stream. The CO2 selectivity remains high at ≈99% throughout the experiment. The comparably low methanol conversion is a result of the small accessible specific copper surface area (Table 2) in combination with the low sample amount.
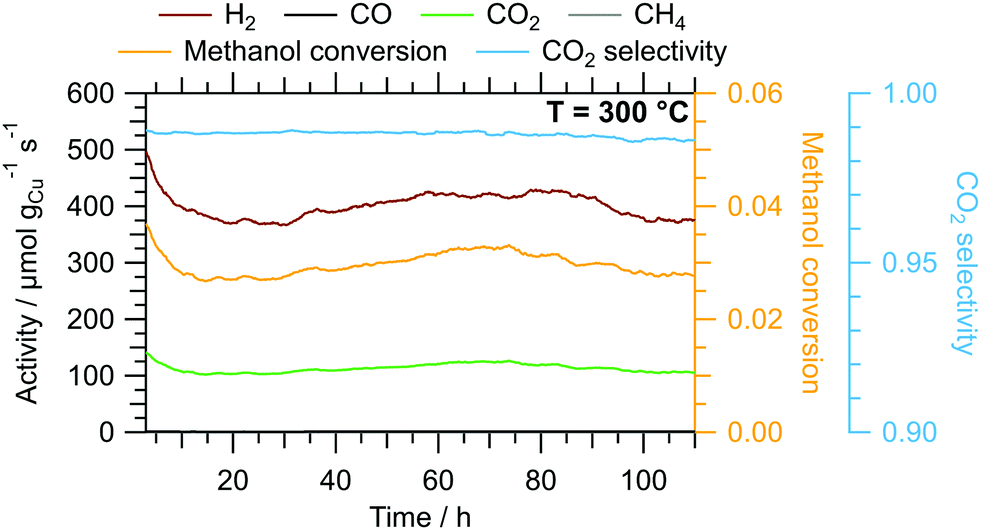 | ||
| Fig. 4 Isothermal long-term MSR test of CZ400 at 300 °C in a continuous flow reactor with a GHSV of 1925 h−1. Colour code: orange – methanol conversion, blue – CO2 selectivity, specific activity of brown: H2, black: CO, green: CO2, grey: CH4. Sample mass: 40.0 mg. We corrected spikes caused by fluctuations in the supply of the initial MSR mixture during the experiment assuming a linear trend between the adjacent data points as well as a similar noise level. | ||
The isothermal MSR flow characterisation of CtZ600 and CmZ800 are visualised in the ESI† in Fig. S10 and S11. Both exhibit a much stronger deactivation trend that continues throughout the entire experiment. The specific activity of CtZ600 decreases by about 90% and of CmZ800 by approximately 80% during 100 h time on stream. In combination with the initially already lower activity of these catalysts, the CO2 selectivity could not be determined reliably after 20 h time on stream, because the CO formation dropped below the detection limit of the GC (20 ppm). These results clearly show that despite the apparent indifference of the ZrO2 polymorph on the CO2 selectivity, the activity as well as the long-term stability are significantly improved by the presence of the amorphous ZrO2 support. The general trends of the deactivation, which can most likely be ascribed to sintering of metallic copper, have already been observed in the dissociative N2O adsorption experiments, where the decrease in specific copper surface area was smaller in CZ400 than in CtZ600 and CmZ800 (see Table 2), although the conditions were different (MSR mixture vs. reduction in H2).
The beneficial properties of CZ400 in terms of specific activity are especially pronounced in a comparison with analogous systems from literature. In Fig. 5, the MSR performance of CZ400 is related to the best performing impregnated Cu/ZrO2 catalysts from our workgroup,19 since they were characterised under identical catalytic conditions in the same batch reactor setup. This guarantees optimal comparability and illustrates the remarkable specific activity of CZ400, which exhibits a maximum specific activity towards H2 that is approximately 8 times higher than the Cu/m-ZrO2 system prepared by aqueous impregnation with a copper loading of 6.9 wt% and 66 times higher than the analogously synthesised Cu/m-ZrO2 catalyst with 80 wt% Cu. Furthermore, a comparison of all five catalysts (CZ400, CtZ600, CmZ800 and the two abovementioned catalysts) in terms of their turnover frequency (TOF) as well as the specific activity in μmol gCat−1 s−1 can be found in the ESI† in Fig. S12 and S13, respectively. The TOF values of CZ400 are comparable with the Cu/m-ZrO2 catalyst prepared by aqueous impregnation with a loading of 6.9 wt% Cu described in ref. 19, where its performance is compared to other systems from literature. The TOF values were not used in the manuscript, because the catalysts are altered significantly upon MSR (cf.Fig. 6). This change caused by exposure to the MSR mixture, however, is not represented in the measurements for the determination of the specific copper surface area, as they were conducted prior to MSR.
 | ||
| Fig. 5 Comparison of CZ400 with other impregnated Cu/ZrO2 catalysts, one from literature19 and another analogous system with a higher Cu loading, measured in the same recirculating batch reactor setup under identical MSR conditions. | ||
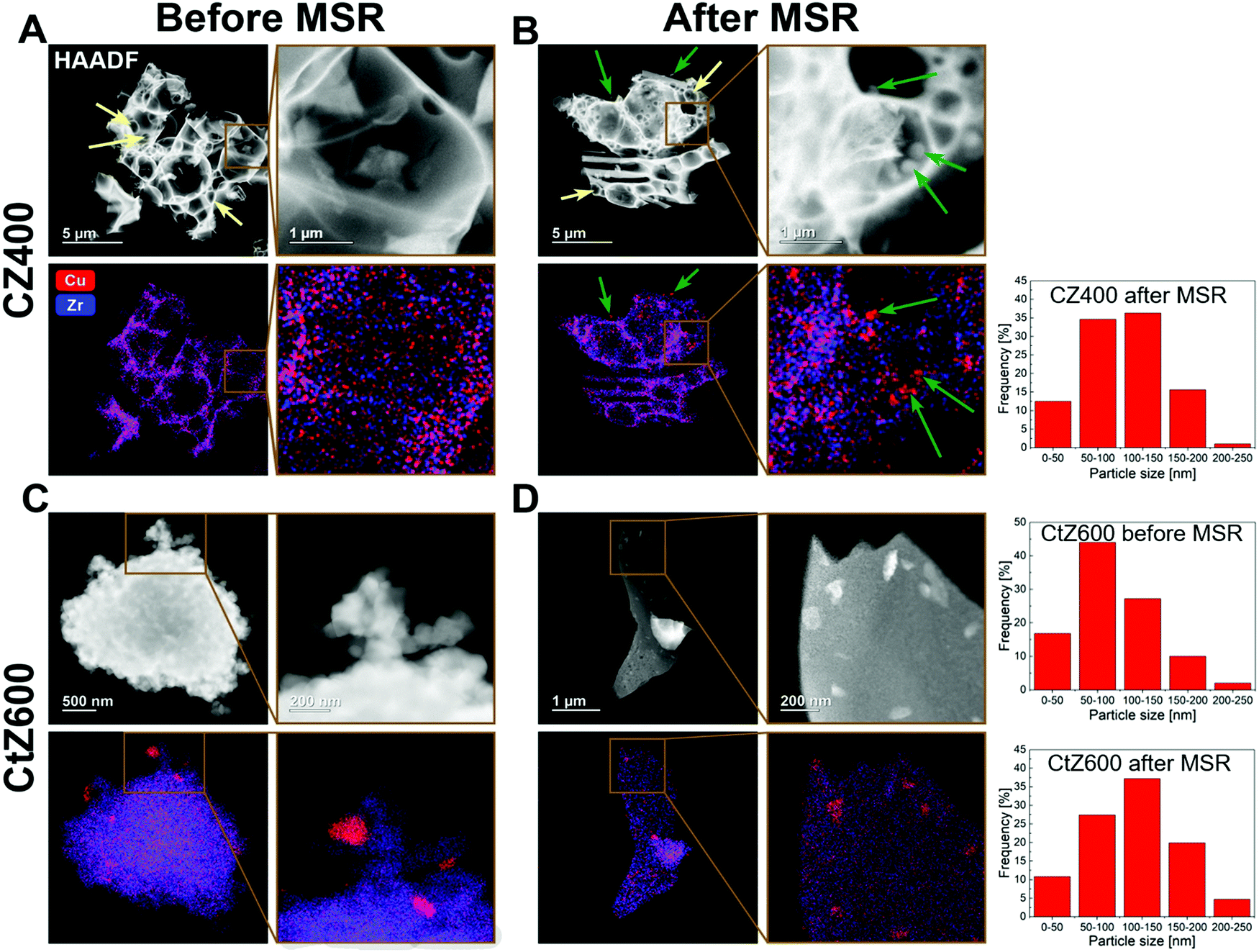 | ||
| Fig. 6 HAADF-STEM images and EDX maps of the samples CZ400 and CtZ600 after calcination and after one MSR cycle in the recirculating batch reactor. For each CZ400 (panels A and B) and CtZ600 (panels C and D) state, one image including an enlarged region is depicted (small brown frames indicate the selected area), each with one HAADF image and the corresponding EDX map of Cu in red and Zr in blue. In panels A and B, combustion pores are indicated with yellow arrows. In panel B, Cu particles observed after MSR are marked with green arrows, where the same particles are highlighted in the respective HAADF-STEM images and EDX maps. Panel A: CZ400 after calcination; panel B: CZ400 after MSR350; panel C: CtZ600 after calcination; panel D: CtZ600 after MSR350. In the right panels, we show the exemplary particle size histograms based on the TEM/EDX analysis for two representative catalysts CZ400 and Ct600. | ||
To identify the reason for the different deactivation behaviour, combined HAADF-STEM and EDX investigations were performed on CZ400 as the most sinter-resistant sample and CtZ600 as the catalyst exhibiting the strongest deactivation. The images are depicted in Fig. 6 and provide a direct comparison of the Cu and ZrO2 morphology and elemental distribution after calcination and after one MSR cycle in the recirculating batch reactor. In Panel A, CZ400 after calcination exhibits a homogeneous distribution of Cu and Zr, with merely sporadic large Cu particles (around 120 nm) being visible. Additionally, this sample exhibits the typical combustion pores.42,44 After MSR (Panel B), an increased number of particles with sizes of up to 200 nm can be found, which are primarily located in the pores. The porous morphology is retained and regions with less agglomeration of Cu can also be observed.
In Fig. 6 Panel C, CtZ600 exhibits a completely different morphology without clearly visible pores. In contrast to CZ400, larger Cu particles of up to 200 nm are visible after calcination in CtZ600. After MSR, these agglomerates become more frequent and the general morphology of the catalyst changes from an apparently loose network of particles to continuous large platelets of zirconia with copper particles on top. This drastic change of the morphology of CtZ600 in combination with the increased frequency of Cu agglomerates could serve as an explanation for the much more severe deactivation of this catalyst, as compared to CZ400. The latter mostly retains its initial morphology, but also shows more Cu particles accounting for the slight initial decrease of the activity in MSR.
The sintering stability argument is further strengthened by the particle size histograms provided for the most active (CZ400) and one deactivating catalyst (CtZ600) before and after MSR operation (note that for CZ400, no such particle size histogram could be reliably provided before MSR, as we assume that for CZ400 a ternary mixed Cu–Zr–O oxide is prevalent before catalysis). However, after MSR operation the amount of smaller particles is clearly higher for CZ400 and for CtZ600, we find an increased amount of significantly larger particles.
4. Conclusions
The adaption of an established synthesis approach of the sol–gel autocombustion to an alternative class of Cu–ZrO2 catalysts enables the tuning of the ZrO2 polymorph and the interface between Cu and ZrO2via the calcination temperature. This creates the possibility to prepare samples with very similar chemical properties arising from identical precursors, which represents one of the key parameters governing the CO2 selectivity in MSR.19 Altering the morphology of the catalysts allows to optimise the activity and long-term stability. Employing this approach, a highly CO2-selective, active and long-term stable Cu/ZrO2 catalyst for MSR was synthesised, which outperforms various impregnated and co-precipitated samples from literature in terms of both specific and long-term stable activity.The advantages of the sol–gel autocombustion were clearly demonstrated by exploiting the typical formation of combustion pores,44 creating a unique catalyst morphology. Upon calcination of the post-combustion precursor at 400 °C (yielding CZ400), this special porous structure stemming from the combustion is still retained and serves as an explanation for its increased resistance towards deactivation by hindering the diffusion of Cu at the surface and sintering. This morphology is lost upon calcination at higher temperatures and, hence, CtZ600 and CmZ800 exhibit increased deactivation through sintering.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Austrian Science Foundation (FWF) and the Deutsche Forschungsgemeinschaft (DFG) under the DACH project initiative (project numbers I2877-N34 and AR 617/12-1). Further support was provided by the framework of the platform Materials- and Nanoscience and the special PhD program “Reactivity and Catalysis” at the University of Innsbruck. We particularly thank the group of H. Huppertz from the Department of General, Inorganic and Theoretical Chemistry of the University of Innsbruck for providing the possibility of using their X-ray diffractometer for ex situ powder XRD measurements. In situ powder XRD measurements in this research were performed using resources of the Advanced Light Source, a U.S. DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. A. Gili appreciates the support of Unifying Systems in Catalysis (UniSysCat), funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 – 390540038.References
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, Weinheim, 3rd edn, 2018 Search PubMed.
- S. Sá, H. Silva, L. Brandão, J. M. Sousa and A. Mendes, Catalysts for methanol steam reforming—A review, Appl. Catal., B, 2010, 99, 43–57, DOI:10.1016/j.apcatb.2010.06.015.
- S. Tosti, A. Basile, F. Borgognoni, V. Capaldo, S. Cordiner, S. Di Cave, F. Gallucci, C. Rizzello, A. Santucci and E. Traversa, Low temperature ethanol steam reforming in a Pd-Ag membrane reactor: Part 1: Ru-based catalyst, J. Membr. Sci., 2008, 308, 250–257, DOI:10.1016/j.memsci.2007.10.001.
- P. Preuster, C. Papp and P. Wasserscheid, Liquid Organic Hydrogen Carriers (LOHCs): Toward a Hydrogen-free Hydrogen Economy, Acc. Chem. Res., 2017, 50, 74–85, DOI:10.1021/acs.accounts.6b00474.
- N. Wagner and M. Schulze, Change of electrochemical impedance spectra during CO poisoning of the Pt and Pt–Ru anodes in a membrane fuel cell (PEFC), Electrochim. Acta, 2003, 48, 3899–3907, DOI:10.1016/S0013-4686(03)00528-0.
- B. Frank, F. Jentoft, H. Soerijanto, J. Kröhnert, R. Schlögl and R. Schomäcker, Steam reforming of methanol over copper-containing catalysts: Influence of support material on microkinetics, J. Catal., 2007, 246, 177–192, DOI:10.1016/j.jcat.2006.11.031.
- M. Behrens and M. Armbrüster, in Catalysis for Alternative Energy Generation, ed. L. Guczi and A. Erdôhelyi, Springer New York, New York, NY, 2012, pp. 175–235 Search PubMed.
- Z. Baysal and S. Kureti, CO2 methanation on Mg-promoted Fe catalysts, Appl. Catal., B, 2020, 262, 118300, DOI:10.1016/j.apcatb.2019.118300.
- S. S. Araya, V. Liso, X. Cui, N. Li, J. Zhu, S. L. Sahlin, S. H. Jensen, M. P. Nielsen and S. K. Kær, A Review of The Methanol Economy: The Fuel Cell Route, Energies, 2020, 13, 596, DOI:10.3390/en13030596.
- J. Zhu, S. S. Araya, X. Cui, S. L. Sahlin and S. K. Kær, Modeling and Design of a Multi-Tubular Packed-Bed Reactor for Methanol Steam Reforming over a Cu/ZnO/Al2O3 Catalyst, Energies, 2020, 13, 610, DOI:10.3390/en13030610.
- L. Mayr, B. Klötzer, D. Schmidmair, N. Köpfle, J. Bernardi, S. Schwarz, M. Armbrüster and S. Penner, Boosting Hydrogen Production from Methanol and Water by in situ Activation of Bimetallic Cu–Zr Species, ChemCatChem, 2016, 8, 1778–1781, DOI:10.1002/cctc.201600361.
- H. Purnama, F. Girgsdies, T. Ressler, J. H. Schattka, R. A. Caruso, R. Schomäcker and R. Schlögl, Activity and Selectivity of a Nanostructured CuO/ZrO2 Catalyst in the Steam Reforming of Methanol, Catal. Lett., 2004, 94, 61–68, DOI:10.1023/B:CATL.0000019332.80287.6b.
- H. F. McMurdie, M. C. Morris, E. H. Evans, B. Paretzkin, W. Wong-NG and C. R. Hubbard, Methods of Producing Standard X-Ray Diffraction Powder Patterns, Powder Diffr., 1986, 1, 40–43, DOI:10.1017/S0885715600011271.
- J. Málek, L. Beneš and T. Mitsuhashi, Powder diffraction data and Rietveld refinement of metastable t-ZrO2 at low temperature, Powder Diffr., 1997, 12, 96–98, DOI:10.1017/S0885715600009519.
- U. Martin, H. Boysen and F. Frey, Neutron powder investigation of tetragonal and cubic stabilized zirconia, TZP and CSZ, at temperatures up to 1400 K, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 403–413, DOI:10.1107/S0108768192011297.
- V. P. Oleshko, J. M. Howe, S. Shukla and S. Seal, High-Resolution and Analytical TEM Investigation of Metastable-Tetragonal Phase Stabilization in Undoped Nanocrystalline Zirconia, J. Nanosci. Nanotechnol., 2004, 4, 867–875, DOI:10.1166/jnn.2004.110.
- M. Kogler, E.-M. Köck, S. Vanicek, D. Schmidmair, T. Götsch, M. Stöger-Pollach, C. Hejny, B. Klötzer and S. Penner, Enhanced kinetic stability of pure and Y-doped tetragonal ZrO2, Inorg. Chem., 2014, 53, 13247–13257, DOI:10.1021/ic502623t.
- T. Chraska, A. H. King and C. C. Berndt, On the size-dependent phase transformation in nanoparticulate zirconia, Mater. Sci. Eng., A, 2000, 286, 169–178, DOI:10.1016/S0921-5093(00)00625-0.
- K. Ploner, M. Watschinger, P. Delir Kheyrollahi Nezhad, T. Götsch, L. Schlicker, E.-M. Köck, A. Gurlo, A. Gili, A. Doran, L. Zhang, N. Köwitsch, M. Armbrüster, S. Vanicek, W. Wallisch, C. Thurner, B. Klötzer and S. Penner, Mechanistic insights into the catalytic methanol steam reforming performance of Cu/ZrO2 catalysts by in situ and operando studies, J. Catal., 2020, 391, 497–512, DOI:10.1016/j.jcat.2020.09.018.
- H. Oguchi, H. Kanai, K. Utani, Y. Matsumura and S. Imamura, Cu2O as active species in the steam reforming of methanol by CuO/ZrO2 catalysts, Appl. Catal., A, 2005, 293, 64–70, DOI:10.1016/j.apcata.2005.07.010.
- B. Lindström, L. J. Pettersson and P. Govind Menon, Activity and characterization of Cu/Zn, Cu/Cr and Cu/Zr on γ-alumina for methanol reforming for fuel cell vehicles, Appl. Catal., A, 2002, 234, 111–125, DOI:10.1016/S0926-860X(02)00202-8.
- S. Patel and K. K. Pant, Activity and stability enhancement of copper–alumina catalysts using cerium and zinc promoters for the selective production of hydrogen via steam reforming of methanol, J. Power Sources, 2006, 159, 139–143, DOI:10.1016/j.jpowsour.2006.04.008.
- S. D. Jones and H. E. Hagelin-Weaver, Steam reforming of methanol over CeO2- and ZrO2-promoted Cu-ZnO catalysts supported on nanoparticle Al2O3, Appl. Catal., B, 2009, 90, 195–204, DOI:10.1016/j.apcatb.2009.03.013.
- P. Udani, P. Gunawardana, H. C. Lee and D. H. Kim, Steam reforming and oxidative steam reforming of methanol over CuO–CeO2 catalysts, Int. J. Hydrogen Energy, 2009, 34, 7648–7655, DOI:10.1016/j.ijhydene.2009.07.035.
- J. Agrell, H. Birgersson, M. Boutonnet, I. Melián-Cabrera, R. M. Navarro and J. L. G. Fierro, Production of hydrogen from methanol over Cu/ZnO catalysts promoted by ZrO2 and Al2O3, J. Catal., 2003, 219, 389–403, DOI:10.1016/S0021-9517(03)00221-5.
- Y. Liu, T. Hayakawa, K. Suzuki, S. Hamakawa, T. Tsunoda, T. Ishii and M. Kumagai, Highly active copper/ceria catalysts for steam reforming of methanol, Appl. Catal., A, 2002, 223, 137–145, DOI:10.1016/S0926-860X(01)00733-5.
- J.-P. Shen and C. Song, Influence of preparation method on performance of Cu/Zn-based catalysts for low-temperature steam reforming and oxidative steam reforming of methanol for H2 production for fuel cells, Catal. Today, 2002, 77, 89–98, DOI:10.1016/S0920-5861(02)00235-3.
- Q. Liu, L.-C. Wang, M. Chen, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Waste-free Soft Reactive Grinding Synthesis of High-Surface-Area Copper–Manganese Spinel Oxide Catalysts Highly Effective for Methanol Steam Reforming, Catal. Lett., 2008, 121, 144–150, DOI:10.1007/s10562-007-9311-6.
- C.-Z. Yao, L.-C. Wang, Y.-M. Liu, G.-S. Wu, Y. Cao, W.-L. Dai, H.-Y. He and K.-N. Fan, Effect of preparation method on the hydrogen production from methanol steam reforming over binary Cu/ZrO2 catalysts, Appl. Catal., A, 2006, 297, 151–158, DOI:10.1016/j.apcata.2005.09.002.
- L.-C. Wang, Q. Liu, M. Chen, Y.-M. Liu, Y. Cao, He and K.-N. Fan, Structural Evolution and Catalytic Properties of Nanostructured Cu/ZrO2 Catalysts Prepared by Oxalate Gel-Coprecipitation Technique, J. Phys. Chem. C, 2007, 111, 16549–16557, DOI:10.1021/jp075930k.
- J. Papavasiliou, G. Avgouropoulos and T. Ioannides, Production of hydrogen via combined steam reforming of methanol over CuO–CeO2 catalysts, Catal. Commun., 2004, 5, 231–235, DOI:10.1016/j.catcom.2004.02.009.
- J. Papavasiliou, G. Avgouropoulos and T. Ioannides, Effect of dopants on the performance of CuO–CeO2 catalysts in methanol steam reforming, Appl. Catal., B, 2007, 69, 226–234, DOI:10.1016/j.apcatb.2006.07.007.
- Z. Shao, W. Zhou and Z. Zhu, Advanced synthesis of materials for intermediate-temperature solid oxide fuel cells, Prog. Mater. Sci., 2012, 57, 804–874, DOI:10.1016/j.pmatsci.2011.08.002.
- T. W. Mammo, N. Murali, C. V. Kumari, S. J. Margarette, A. Ramakrishna, R. Vemuri, Y. B. Shankar Rao, K. L. Vijaya Prasad, Y. Ramakrishna and K. Samatha, Synthesis, structural, dielectric and magnetic properties of cobalt ferrite nanomaterial prepared by sol-gel autocombustion technique, Phys. B, 2020, 581, 411769, DOI:10.1016/j.physb.2019.411769.
- P. Naderi, S. M. Masoudpanah and S. Alamolhoda, Magnetic properties of Li0.5Fe2.5O4 nanoparticles synthesized by solution combustion method, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 702, DOI:10.1007/s00339-017-1304-8.
- Y. Ji, J. Liu, T. He, L. Cong, J. Wang and W. Su, Single intermedium-temperature SOFC prepared by glycine–nitrate process, J. Alloys Compd., 2003, 353, 257–262, DOI:10.1016/S0925-8388(02)01198-2.
- G. Kéranguéven, C. Ulhaq-Bouillet, V. Papaefthimiou, S. Royer and E. Savinova, Perovskite-carbon composites synthesized through in situ autocombustion for the oxygen reduction reaction: the carbon effect, Electrochim. Acta, 2017, 245, 156–164, DOI:10.1016/j.electacta.2017.05.113.
- A. Tarjomannejad, P. R. Zonouz, M. E. Masoumi, A. Niaei and A. Farzi, LaFeO3 Perovskites Obtained from Different Methods for NO + CO Reaction, Modeling and Optimization of Synthesis Process by Response Surface Methodology, J. Inorg. Organomet. Polym., 2018, 28, 2012–2022, DOI:10.1007/s10904-018-0860-5.
- M. Grünbacher, A. Tarjomannejad, P. D. K. Nezhad, C. Praty, K. Ploner, A. Mohammadi, A. Niaei, B. Klötzer, S. Schwarz, J. Bernardi, A. Farzi, M. J. I. Gómez, V. T. Rivero and S. Penner, Promotion of La(Cu0.7Mn0.3)0.98M0.02O3−δ (M = Pd, Pt, Ru and Rh) perovskite catalysts by noble metals for the reduction of NO by CO, J. Catal., 2019, 379, 18–32, DOI:10.1016/j.jcat.2019.09.005.
- Y. Liu, Y. Long, Y. Tang, Z. Gu and K. Li, Effect of Preparation Method on the Structural Characteristics of NiO-ZrO2 Oxygen Carriers for Chemical-looping Combustion, Chem. Res. Chin. Univ., 2019, 35, 1024–1031, DOI:10.1007/s40242-019-9189-z.
- S. Minaei, M. Haghighi, N. Jodeiri, H. Ajamein and M. Abdollahifar, Urea-nitrates combustion preparation of CeO2-promoted CuO/ZnO/Al2O3 nanocatalyst for fuel cell grade hydrogen production via methanol steam reforming, Adv. Powder Technol., 2017, 28, 842–853, DOI:10.1016/j.apt.2016.12.010.
- H. Ajamein and M. Haghighi, Influence of ambient gas on microwave-assisted combustion synthesis of CuO–ZnO–Al2O3 nanocatalyst used in fuel cell grade hydrogen production via methanol steam reforming, Ceram. Int., 2016, 42, 17978–17989, DOI:10.1016/j.ceramint.2016.07.092.
- J. Baneshi, M. Haghighi, H. Ajamein and M. Abdollahifar, Homogeneous precipitation and urea-nitrate combustion preparation of nanostructured CuO/CeO2/ZrO2/Al2O3 oxides used in hydrogen production from methanol for fuel cells, Part. Sci. Technol., 2020, 38, 464–474, DOI:10.1080/02726351.2018.1455778.
- H. Ajamein, M. Haghighi and S. Alaei, The role of various fuels on microwave-enhanced combustion synthesis of CuO/ZnO/Al2O3 nanocatalyst used in hydrogen production via methanol steam reforming, Energy Convers. Manage., 2017, 137, 61–73, DOI:10.1016/j.enconman.2017.01.044.
- H. Ajamein, M. Haghighi, R. Shokrani and M. Abdollahifar, On the solution combustion synthesis of copper based nanocatalysts for steam methanol reforming: Effect of precursor, ultrasound irradiation and urea/nitrate ratio, J. Mol. Catal. A: Chem., 2016, 421, 222–234, DOI:10.1016/j.molcata.2016.05.028.
- C.-L. Yu, S. Sakthinathan, B.-Y. Hwang, S.-Y. Lin, T.-W. Chiu, B.-S. Yu, Y.-J. Fan and C. Chuang, CuFeO2–CeO2 nanopowder catalyst prepared by self-combustion glycine nitrate process and applied for hydrogen production from methanol steam reforming, Int. J. Hydrogen Energy, 2020, 45, 15752–15762, DOI:10.1016/j.ijhydene.2020.04.077.
- S. Esposito, M. Turco, G. Bagnasco, C. Cammarano, P. Pernice and A. Aronne, Highly dispersed sol–gel synthesized Cu–ZrO2 materials as catalysts for oxidative steam reforming of methanol, Appl. Catal., A, 2010, 372, 48–57, DOI:10.1016/j.apcata.2009.10.006.
- A. Doran, L. Schlicker, C. M. Beavers, S. Bhat, M. F. Bekheet and A. Gurlo, Compact low power infrared tube furnace for in situ X-ray powder diffraction, Rev. Sci. Instrum., 2017, 88, 13903, DOI:10.1063/1.4973561.
- L. Schlicker, A. Doran, P. Schneppmüller, A. Gili, M. Czasny, S. Penner and A. Gurlo, Transmission in situ and operando high temperature X-ray powder diffraction in variable gaseous environments, Rev. Sci. Instrum., 2018, 89, 33904, DOI:10.1063/1.5001695.
- C. Prescher and V. B. Prakapenka, DIOPTAS: a program for reduction of two-dimensional X-ray diffraction data and data exploration, High Pressure Res., 2015, 35, 223–230, DOI:10.1080/08957959.2015.1059835.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60, 309–319, DOI:10.1021/ja01269a023.
- J. W. Evans, M. S. Wainwright, A. J. Bridgewater and D. J. Young, On the determination of copper surface area by reaction with nitrous oxide, Appl. Catal., 1983, 7, 75–83, DOI:10.1016/0166-9834(83)80239-5.
- E. S. Vasiliadou, T. M. Eggenhuisen, P. Munnik, P. E. de Jongh, K. P. de Jong and A. A. Lemonidou, Synthesis and performance of highly dispersed Cu/SiO2 catalysts for the hydrogenolysis of glycerol, Appl. Catal., B, 2014, 145, 108–119, DOI:10.1016/j.apcatb.2012.12.044.
- S. Natesakhawat, J. W. Lekse, J. P. Baltrus, P. R. Ohodnicki, B. H. Howard, X. Deng and C. Matranga, Active Sites and Structure–Activity Relationships of Copper-Based Catalysts for Carbon Dioxide Hydrogenation to Methanol, ACS Catal., 2012, 2, 1667–1676, DOI:10.1021/cs300008g.
- J. J. F. Scholten and J. A. Konvalinka, Reaction of nitrous oxide with copper surfaces. Application to the determination of free-copper surface areas, Trans. Faraday Soc., 1969, 65, 2465–2473, 10.1039/TF9696502465.
- M. E. Straumanis and L. S. Yu, Lattice parameters, densities, expansion coefficients and perfection of structure of Cu and of Cu–In α phase, Acta Crystallogr., Sect. A: Found. Crystallogr., 1969, 25, 676–682, DOI:10.1107/S0567739469001549.
- U. Anselmi-Tamburini, J. N. Woolman and Z. A. Munir, Transparent Nanometric Cubic and Tetragonal Zirconia Obtained by High-Pressure Pulsed Electric Current Sintering, Adv. Funct. Mater., 2007, 17, 3267–3273, DOI:10.1002/adfm.200600959.
- H. Zhang, Z. Li, B.-N. Kim, K. Morita, H. Yoshida, K. Hiraga and Y. Sakka, Highly Infrared Transparent Nanometric Tetragonal Zirconia Prepared by High-Pressure Spark Plasma Sintering, J. Am. Ceram. Soc., 2011, 94, 2739–2741, DOI:10.1111/j.1551-2916.2011.04680.x.
- S. Shukla and S. Seal, Mechanisms of room temperature metastable tetragonal phase stabilisation in zirconia, Int. Mater. Rev., 2005, 50, 45–64, DOI:10.1179/174328005X14267.
- R. Ianoş, R. Lazău, I. Lazău and C. Păcurariu, Chemical oxidation of residual carbon from ZnAl2O4 powders prepared by combustion synthesis, J. Eur. Ceram. Soc., 2012, 32, 1605–1611, DOI:10.1016/j.jeurceramsoc.2011.12.028.
- M. Bhagwat, A. V. Ramaswamy, A. K. Tyagi and V. Ramaswamy, Rietveld refinement study of nanocrystalline copper doped zirconia, Mater. Res. Bull., 2003, 38, 1713–1724, DOI:10.1016/S0025-5408(03)00201-0.
- K. Samson, M. Śliwa, R. P. Socha, K. Góra-Marek, D. Mucha, D. Rutkowska-Zbik, J.-F. Paul, M. Ruggiero-Mikołajczyk, R. Grabowski and J. Słoczyński, Influence of ZrO2 Structure and Copper Electronic State on Activity of Cu/ZrO2 Catalysts in Methanol Synthesis from CO2, ACS Catal., 2014, 4, 3730–3741, DOI:10.1021/cs500979c.
- S. Tada, A. Katagiri, K. Kiyota, T. Honma, H. Kamei, A. Nariyuki, S. Uchida and S. Satokawa, Cu Species Incorporated into Amorphous ZrO2 with High Activity and Selectivity in CO2-to-Methanol Hydrogenation, J. Phys. Chem. C, 2018, 122, 5430–5442, DOI:10.1021/acs.jpcc.7b11284.
- S. Tada, S. Kayamori, T. Honma, H. Kamei, A. Nariyuki, K. Kon, T. Toyao, K. Shimizu and S. Satokawa, Design of Interfacial Sites between Cu and Amorphous ZrO2 Dedicated to CO2-to-Methanol Hydrogenation, ACS Catal., 2018, 8, 7809–7819, DOI:10.1021/acscatal.8b01396.
- ICDD Database PDF-4+, International Centre for Diffraction Data, Newtown Square, PA 19073, USA, 2010.
- H. Boysen, F. Frey and T. Vogt, Neutron powder investigation of the tetragonal to monoclinic phase transformation in undoped zirconia, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 881–886, DOI:10.1107/S010876819100856X.
- C. J. Howard, R. J. Hill and B. E. Reichert, Structures of ZrO2 polymorphs at room temperature by high-resolution neutron powder diffraction, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 44, 116–120, DOI:10.1107/S0108768187010279.
- H. E. Swanson and E. Tatge, Standard X-ray diffraction patterns, Natl. Bur. Stand. Circ. (U. S.), 1953, 1, 15 Search PubMed.
- A. Kirfel and K. Eichhorn, Accurate structure analysis with synchrotron radiation. The electron density in Al2O3 and Cu2O, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 271–284, DOI:10.1107/S0108767389012596.
- S. Asbrink and A. Waskowska, CuO: X-ray single-crystal structure determination at 196 K and room temperature, J. Phys.: Condens. Matter, 1991, 3, 8173–8180, DOI:10.1088/0953-8984/3/42/012.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qm00641j |
| This journal is © the Partner Organisations 2021 |