
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular aggregation properties of 4-(1-morpholino)-1,8-naphthalimide based fluorescent materials†
June I.
Lovitt
 *ab,
Tumpa
Gorai
a,
Emanuele
Cappello
a,
Jason M.
Delente
a,
Sebastian T.
Barwich
c,
Matthias E.
Möbius
c,
Thorfinnur
Gunnlaugsson
*abd and
Chris S.
Hawes
*e
*ab,
Tumpa
Gorai
a,
Emanuele
Cappello
a,
Jason M.
Delente
a,
Sebastian T.
Barwich
c,
Matthias E.
Möbius
c,
Thorfinnur
Gunnlaugsson
*abd and
Chris S.
Hawes
*e
aSchool of Chemistry and Trinity Biomedical Sciences Institute, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland. E-mail: jlovitt@tcd.ie; gunnlaut@tcd.ie
bAMBER (Advanced Materials and Bioengineering Research) Centre, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
cSchool of Physics, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
dSynthesis and Solid State Pharmaceutical Centre (SSPC), Ireland
eSchool of Chemical and Physical Sciences, Keele University, Keele ST5 5BG, UK. E-mail: c.s.hawes@keele.ac.uk
First published on 17th March 2021
Abstract
Here we report the synthesis of two morpholino-substituted naphthalimide ligands, N-(3-picolyl)-4-(1-morpholino)-1,8-naphthalimide L1 and N-benzyl-4-(1-morpholino)-1,8-naphthalimide L2, and study their supramolecular properties in the crystalline, solution and gel phases. These ligands were designed through incorporation of the morpholino group to enhance their photophysical and pH-responsive properties following recently reported N-(3-picolyl) naphthalimide metallogels. L1 was found to form metallogels on reaction with either Mn2+ or Co2+. The gels were found to be thermally and chemically responsive to various stimuli including pH. Conversely, L2 showed no reaction or coordination with transition metals, and did not gel under analogous conditions to L1. In the solution state, the fluorescence of both L1 and L2 exhibited pH responsiveness and counterion-influenced aggregation. The microparticle formation over the pH range was further investigated through Dynamic Light Scattering and Scanning Electron Microscopy. These two ligands illustrate how a modular ligand family can derive structure–function relationships and allow for systematic tuning, thus allowing for the future development of luminescent pH responsive soft materials.
Introduction
The development of stimuli-responsive materials using functional supramolecular constituents is an intriguing area within the materials science field.1a–g Weak noncovalent interactions can be fractured and reformed in response to several external chemical stimuli such as light, sonication, temperature, mechanical force and addition of other chemical components.2a–c Chemical changes to local environments yielding fluorometric and colorimetric responses have been widely incorporated into sensors for signalling in biological media, pH and ions.3a–e The ligand design can incorporate motifs which behave as fluorescent sensors for Lewis acids, including protons and/or metal ions.4a,bThe 1,8-naphthalimide fluorophore has been consistently utilised in supramolecular chemistry as a versatile building block to develop diverse libraries of organic ligands and metal–organic motifs.5a–d This framework is an attractive building block due to ease of functionalisation and excellent photophysical properties which result in a wide range of applications.6a,b In particular, 4-amino-1,8-naphthalimide derivatives are popular as fluorescent probes due to the delocalisation that can occur from the amino group into the naphthalimide causing a ‘push–pull’ based internal charge transfer (ICT) to occur in a fluorophore-spacer-receptor design.7a,b The related 4-morpholino-1,8-naphthalimides are mainly used as fluorescent probes for biological systems, including in diverse applications in cell imaging, trace analyte detection and signalling in biological media but the coordination chemistry of these ligands remains relatively underexplored.8a–d
The preparation of supramolecular gels and their subset, metallogels, using low-molecular-weight-gelators (LMWGs) has been successfully demonstrated in various applications such as drug delivery, catalysis and photonics, with commercial importance in a range of industries.9a–e Stimuli responsive behaviour can be introduced into LMWGs containing metal binding groups (such as pyridine, bipyridine, triazole or carboxylate) or metal ions from tuning the underlaying coordination chemistry.10a–e A wealth of metallogels containing d-block metal ions have been reported including a nickel(II) metallogel formed from a bis(3-pyridylimine) ligand reported by Gloe et al.,11 and a metallogel formed from a trimesic amide ligand induced by iron(III)/iron(II) was prepared by Huang et al.12 Another nickel(II) based metallogel formed from a triazole ligand by Mitra et al. has illustrated the dual function of reversible adsorption of toxic gases and sequestration of heavy metal ions, via gel to gel transformations due to the dynamic nature of Ni–Ntriazole interactions.9b An alternative design method is utilising the donor–acceptor properties of the ligands to form a thermally responsive silver(I) gel based on charge-transfer complex formation which has been reported by Bhattacharya et al.13a,b We have previously reported responsive gels based on various scaffolds, including the 1,3,5-benzenetricarboxamide (BTA) motif, which itself is often functionalised to form supramolecular gelator ligands and can form luminescent metallogels through subsequent metalation with lanthanide ions.14a–c,15a–c In another study, we generated a series of luminescent lanthanide metallogels from both 2,6-bis(1,2,3-triazol-4-yl)pyridine (BTP) and pyridine-2,6-dicarboxamide (DPA) chelators.16a,b More recently, we have reported a reversibly formed silver(I) metallogel with a 3-pyridyl substituted 1,2,3-triazol-4-yl-picolinamide (tzpa) ligand.17 However, despite a multitude of stimuli responsive materials there are relatively few examples in the literature of pH responsive metallogels incorporating a fluorescence sensor motif into the metallogels design.
We have recently reported a series of N-picolyl-1,8-naphthalimide derivatives and explored their coordination chemistry with d-block metal ions in the solid and solution states as well as exploited the structural and chemical properties of these ligands to generate a family of robust metallogels.18 The gels were found to be thermally and chemically reversible under various stimuli and similar materials could be generated by installing a piperidinyl substituent at the naphthalimide 4-position, giving intensely coloured gels. However, the effect of supramolecular aggregation on the photophysical properties of this system could not be measured.
Herein, we present a new study into the behaviour of these 3-picolylimide-substituted naphthalimide derivatives with the synthesis of two new fluorophores N-(3-picolyl)-4-(1-morpholino)-1,8-naphthalimide and N-(benzyl)-4-(1-morpholino)-1,8-naphthalimide, as shown in Scheme 1, and study their supramolecular properties in the crystalline, solution and gel phases. The tunability of this system is realised in both the solution and gelation phases, showing that this system has future potential in the development and application of luminescent pH responsive materials.
 | ||
| Scheme 1 Synthesis and structure of the ligands L1 and L2. Reagents and conditions: (i) morpholine, DMF, 140 °C. | ||
Results and discussion
Synthesis and solid-state characterisation of L1–L2
The ligands N-(3-picolyl)-4-(1-morpholino)-1,8-naphthalimide L1 and N-(benzyl)-4-(1-morpholino)-1,8-naphthalimide L2 were prepared based on modified literature procedures.18 By using morpholino substituted naphthalimides we envisaged analogous coordination chemistry to that previously observed for the N-(3-picolyl)-1,8-naphthalimide analogues, involving either monodentate coordination through the pyridine nitrogen atom as well as potential coordination through the morpholino nitrogen or oxygen atoms to softer Lewis acids such as silver(I).19 However, unlike the previous systems, the morpholino substituent allows new observations of the supramolecular aggregation effect on the photophysical properties of the system to be investigated by improving solubility while maintaining electron donor capabilities. The tunability of this system was examined through substituting the N-position with benzylamine. This illustrated a structure–function relationship in the ligand design which can be used as a handle for developing the photophysical properties of this system.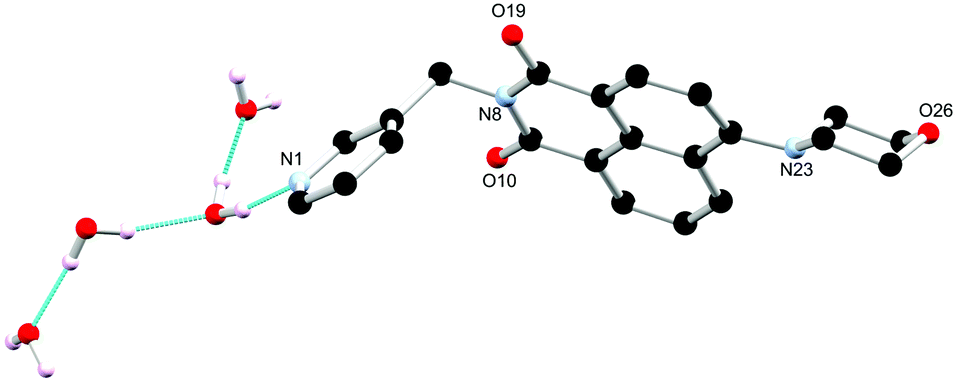 | ||
| Fig. 1 Structure of L1 with heteroatom labelling scheme. Selected hydrogen atoms omitted for clarity. | ||
Ligands L1 and L2 were prepared in two steps from the commercially available 4-nitro-1,8-naphthalic anhydride as shown in Scheme 1. Reaction of 3-picolyl amine with 4-nitro-1,8-naphthalic anhydride in acetic acid at reflux for 6 hours yielded L1a and L2a in 85% and 90% yield respectively. These could then be treated with the relevant amine giving the products L1 and L2 in yields 65% and 53%. All precursors were characterised using conventional techniques, and the data were consistent with that previously reported.18 The characteristic morpholino resonances were present at 3.91 and 3.23 ppm for L1 and 4.04 and 3.28 ppm for L2 indicating conversion. The high-resolution mass spectrometry (HRMS) analysis of L1 and L2 showed peaks at m/z = 374.1494 and m/z = 373.1547 respectively which corresponded to the protonated molecular ions [M + H]+.
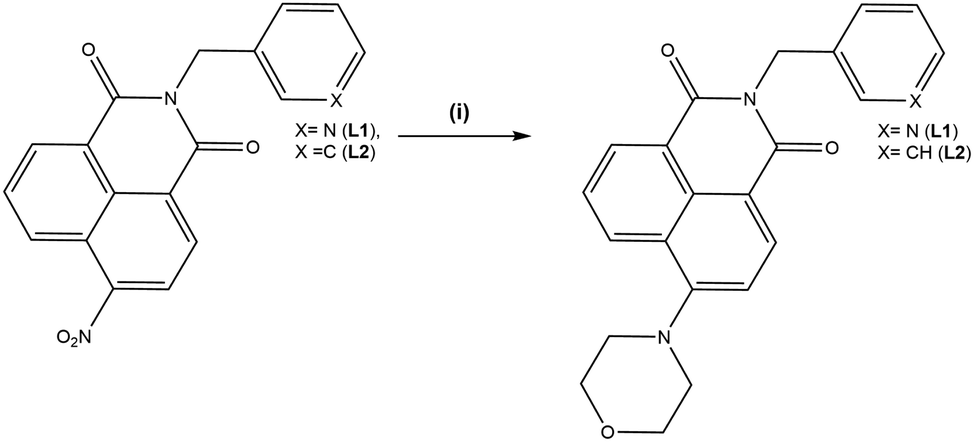
Single crystals of L1 suitable for X-ray diffraction analysis were prepared through dissolution in dilute aqueous AcOH (5% v/v) and standing at room temperature for 7 days. Crystallographic data and refinement parameters are summarised in Table S1 (ESI†). The X-ray diffraction analysis of colourless needle crystals provided a structural model in the chiral monoclinic space group P21. The asymmetric unit contained one complete ligand molecule with four fully occupied water molecules per ligand molecule, as shown in Fig. 1. The morpholino group adopts a chair conformation with the nitrogen atom resisting full planarization with the naphthalimide, being located 0.40 Å from the mean plane of its substituent carbon atoms. Nonetheless, in solution it is expected that this group may partially conjugate with the naphthalimide ring, lowering its basicity compared to free morpholine. The 3-pyridyl substituent attached to the imide nitrogen atom faces outwards, with an angle around the methylene group (N8–C7–C5) of 112.2(3)°. The orientation of the morpholino group results in a complementary hydrogen bonding array between four fully occupied water molecules in the asymmetric unit, as illustrated in Fig. 2.
 | ||
| Fig. 2 Extended structure of L1 featuring hydrogen bonding and π⋯π stacking interactions between adjacent ligand moieties. Hydrogen atoms not involved in hydrogen bonding are omitted for clarity. | ||
The extended structure of L1 features offset symmetric face-to-face π⋯π stacking interactions between adjacent naphthalimide rings, the distance between the rings (C13 to C17) is 3.712 Å. There is also an extensive hydrogen bonding network with both ring motifs such as R66(12) as well as chain motifs, C33(19) and C22(4), between the ligand units and the water molecules in each unit cell present.20 These intermolecular interactions link the naphthalimide oxygen to the water molecules (O19 to O32 is 2.851(4) Å), and pyridyl nitrogen atom to the water molecules (N1 to O30 is 2.759(4) Å) between the adjacent units. These interactions support the face-to-face π⋯π stacking interactions within sheets and head to tail π⋯π stacking interactions between adjacent sheets of the ligand. This is illustrated in Fig. 2 and Fig. S2 (ESI†).
The acentric packing in this structure relates to the steric bulk of the morpholino group combined with the stabilising interactions of the lattice solvent molecules. Previously, with other substituents (such as nitro or halogen groups) we see alternating positions between two semi-equivalent naphthalimide 4-positions, which often also relates to crystallographic disorder.18,21 In the extended structure of L1, however, all the morpholino substituents stack on the same side of the naphthalimide. To limit steric clash between adjacent columns, the morpholine substituents are unidirectional along b, prohibiting the expected glide symmetry and implying some level of long-range ordering in the columnar arrangement of this species. The acentric packing also influences the hydrogen bonding network which orients parallel to the naphthalimide stacks. While only a single crystalline phase could be observed visually, PXRD indicated the presence of several additional microcrystalline phases within the bulk sample isolated from the mother liquor. We were unable to fully ascertain the nature of the additional phase(s), though several of the Bragg reflections matched those expected from a small quantity of morpholinium acetate (ESI,† Fig. S7 and S8).
In order to probe the coordination behaviour of L1, first row transition metals were screened such as cobalt(II) chloride in acetonitrile, which subsequently formed a metallogel, Co-L1. The chemical properties of the gel were investigated (vide infra), including the pH responsiveness. A small quantity of single crystals of L1-gel were generated from acid/base additions to the Co-L1 gel. Following the addition of HCl (0.1 M, 100 μL) and subsequently of NaOH (0.1 M, 100 μL) and brief sonication, the gel broke into a green solution which upon standing for 7 days formed yellow needle crystals. The X-ray diffraction analysis provided a structural model of L1-gel in the monoclinic P21/c space group. The asymmetric unit contained one complete ligand molecule and no solvent molecules. The substituents adopt a similar conformation to those described above; the morpholino group adopts a chair conformation with an equivalent deviation from planarity for the morpholine nitrogen atom of 0.40 Å. The N-substituted 3-pyridyl group faces outwards and exhibits an equivalent angle about the methylene group of 112.8(5)° as illustrated in Fig. 3a.
 | ||
| Fig. 3 (a) Structure of L1-gel from dissolution of the gel sample. Hydrogen atoms are omitted for clarity. (b) Extended structure of L1 featuring offset head-to-tail π⋯π stacking interactions between adjacent naphthalimide rings. Hydrogen atoms are omitted for clarity. | ||
The extended structure of L1-gel features offset head-to-tail π⋯π stacking interactions between adjacent naphthalimide rings with centroid–centroid distance of 3.58 Å and a perpendicular slip distance of 2.17 Å. Additionally, there are edge-to-face C–H⋯O interactions in the b direction between the naphthalimide oxygen atoms and the adjacent naphthalimide C–H groups (O10⋯C14 is 3.320(6) Å, C14–H14⋯O10 angle is 137.5(3)°) which forms corrugated layers as illustrated in Fig. 3b. The structure of L1-gel is an anhydrate, with no water or other guest molecules present in the lattice. The role of solvation is apparent here as in the absence of the water molecules a very different arrangement is adopted. A centrosymmetric packing mode is observed in this structure; although each column maintains a directional orientation of the morpholine group, adjacent columns along b are inverted with respect to one another. This arrangement contrasts greatly from the L1 hydrate structure described above and indicates that the acentric inter-column ordering may be mediated by the lattice hydrogen bonding in the hydrate. These structural changes illustrate the influence solvation can have on the packing mode adopted in these species and may be consequential for the behaviour of the subsequent materials.
Single crystals suitable for X-ray diffraction analysis of L2a, the nitro precursor to L2, were prepared through dissolution in hot ethyl acetate and obtained after slow evaporation over 24 hours. The X-ray diffraction analysis of orange block crystals provided a structural model in the orthorhombic Pbca space group. There is one complete molecule of L2a in the asymmetric unit and no solvent or guest molecules present, as illustrated in Fig. 4. The extended structure of L2a features offset face-to-face π⋯π stacking interactions between adjacent naphthalimide rings in which adjacent naphthalimide ring are twisted antiparallel to one another. The mean interplanar distance is 3.38 Å with a slip distance of 3.13 Å. Additional C–H⋯O/N interactions provide further stabilisation to the overall architecture in the structure. The phase purity was determined through X-ray powder diffraction. The measured diffraction pattern of L2a matches well with the simulated powder diffraction pattern at 100 K of the crystal indicating that the crystallinity of the ligand was retained in the bulk structure and that this species crystallises as a single phase, as illustrated in Fig. S9 (ESI†).
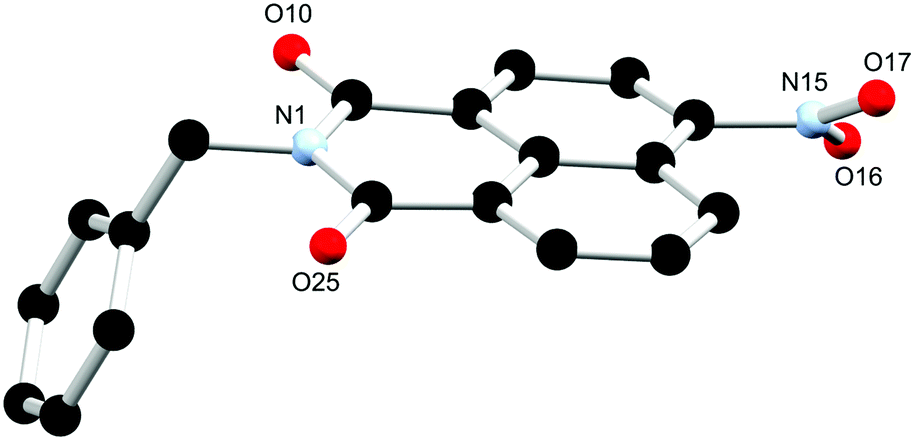 | ||
| Fig. 4 Structure of L2a with heteroatom labelling scheme. All hydrogen atoms are omitted for clarity. | ||
Single crystals suitable for X-ray diffraction analysis of L2 were prepared through dissolution in hot ethyl acetate and obtained after slow evaporation over 24 hours. The X-ray diffraction analysis of yellow block crystals provided a structural model in the monoclinic P21/c space group. There is one complete molecule of L2 in the asymmetric unit and no solvent or guest molecules present within the lattice as illustrated in Fig. 5. Similarly to the two phases of L1 the morpholino group again adopts a chair conformation, though the nitrogen atom is slightly more planarized, residing 0.35 Å from the mean plane of its carbon substituents. The benzyl substituent at the N-position of the naphthalimide exhibits an angle about the methylene group (C4–C7–N8) of 113.86(15)° which is comparable to that observed in the 3-pyridyl morpholino derivative L1.
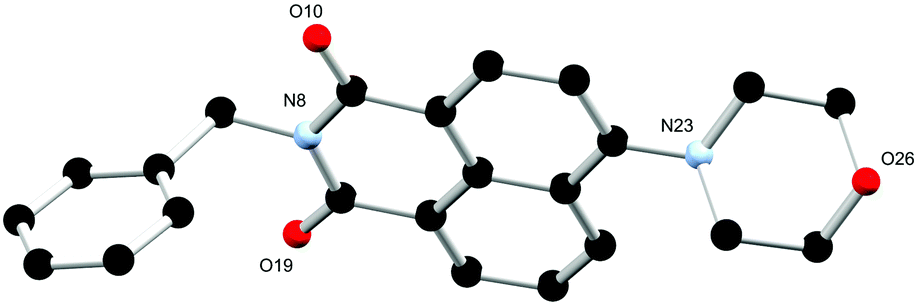 | ||
| Fig. 5 Structure of L2 with heteroatom labelling scheme. All hydrogen atoms are omitted for clarity. | ||
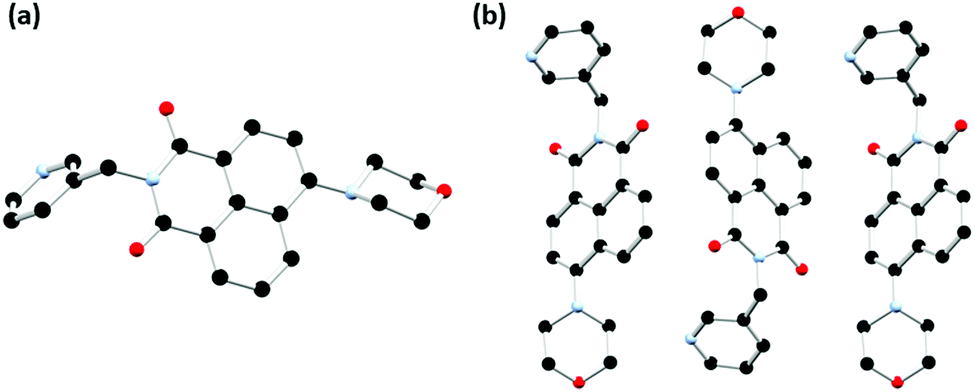
The chosen N-substituent influences the structure directionality. In the L1 structures, the pyridyl nitrogen atom can be further implicated in C–H⋯N interactions as well as interactions with the lattice water molecules in the case of the hydrated ligand structure. Whereas the benzyl substituted ligands, L2a and L2, favour more C–H⋯O interactions as well as π⋯π interactions. This is particularly evident when comparing the calculated Hirshfeld surfaces and resultant fingerprint plots of each molecule (ESI,† Fig. S5 and S6). The extended structure of L2 features offset head-to-tail π⋯π stacking interactions between adjacent naphthalimide rings with a mean interplanar distance of 3.23 Å and a slip distance of 4.66 Å between the two naphthyl centroids. The large slip distance between the two naphthyl centroids indicates that direct interaction is only achieved at the periphery of the molecule. There are few significant C–H⋯π interactions that occur between adjacent ligand moieties. In addition to the π⋯π stacking interactions another C–H⋯O interaction occurs between the naphthalimide oxygen and the adjacent benzyl hydrogen atom (O19⋯C29 is 3.294(3) Å, C28–H28B⋯O19 angle is 149.9(19)°) which supports this offset conformation adopted by the ligand. This is illustrated in Fig. 6 and Fig. S4 (ESI†). The X-ray powder diffraction pattern of L2 matches well with the simulated powder diffraction pattern at 100 K of the crystal indicating the presence of a single phase and that the crystallinity of the ligand was retained on drying, as illustrated in Fig. S10 (ESI†).
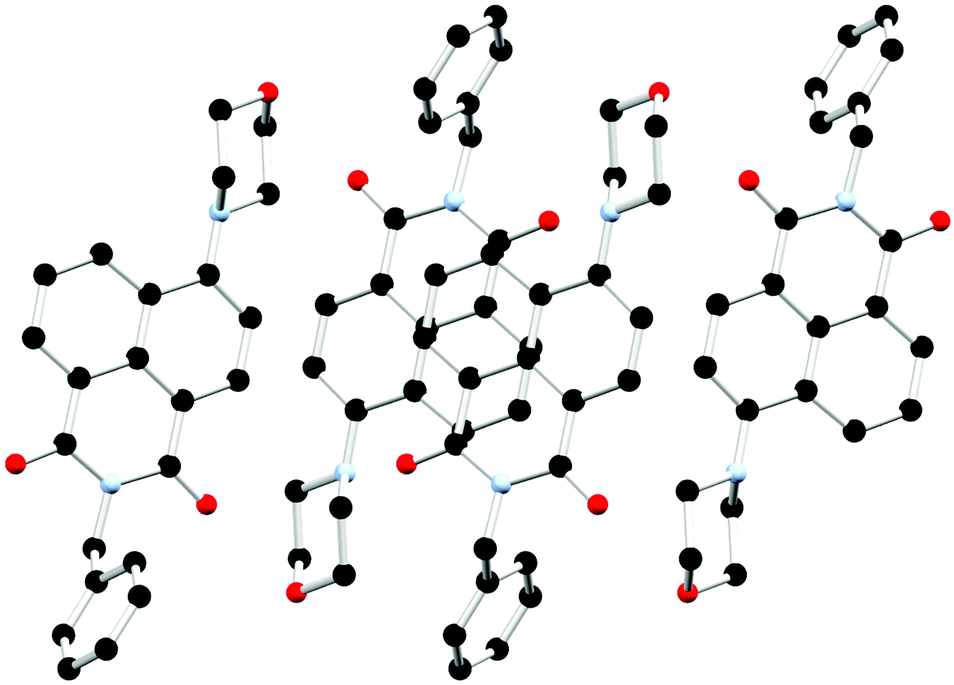 | ||
| Fig. 6 Extended structure of L2 featuring offset head-to-tail π⋯π stacking interactions between adjacent naphthalimide rings. Hydrogen atoms are omitted for clarity. | ||
The thermal stabilities of both L1 and L2 were probed through thermogravimetric analysis. Both ligands had moderate melting points (ca. 170 °C) and maintained stable mass in TGA until above this temperature. A rapid single step decomposition of L1 above 280 °C was then observed, while a two-step decomposition was observed for L2 above 200 °C. This further corroborates that both ligands have good thermal stabilities (Fig. S12 and S13, ESI†).
Solution studies of L1–L2
A UV-visible absorption study of L1 and L2 was initially carried out in acetonitrile following solid state characterisation of these ligands. As illustrated in (Fig. S14, ESI†), L1 and L2 shows several absorbance bands in the UV region with λmax/nm (εmax × 103/L mol−1 cm−1): 255 nm (16.6 ± 0.2), 400 nm (11.1 ± 0.1) for L1, and 400 nm (4.8 ± 0.08) for L2. We initially attempted to determine the coordination chemistry of L1 and L2 in solution but addition of manganese(II) and cobalt(II) chloride in acetonitrile to these solutions led only to minor variations in the absolute absorbance intensity corresponding to build up of unreacted metal salt, with no significant changes to indicate that coordination was occurring at these concentrations.As previously mentioned, 4-amino-1,8-naphthalimide derivatives are known to be strongly coloured and fluorescent due to the ICT. There is a large change in dipole between the ground and excited states with an associated reorganization of the solvent sphere, and therefore the Stokes shift is solvent polarity dependent. Hence, the absorption and emission spectra of L1 and L2 were recorded in different solvents with varying polarity including CH3CN, DMSO, THF, CH3OH, CH2Cl2, toluene and THF. The UV-vis absorption spectra of L1 and L2 in both protic and aprotic solvents showed a high-energy transition at 250 nm assigned to the π–π* transition and a low energy band at 400 nm assigned to the ICT transition. The fluorescence emission spectra of L1 exhibited modest solvatochromism; a red shift in the emission maxima is observed upon increasing the solvent polarizability. In addition to this, there is a large difference in fluorescence intensity as the solvent polarity increases, in solvents such as toluene and DCM there is a high fluorescence intensity, but the intensity decreases significantly when the study was repeated with CH3OH and DMSO. For example, in toluene (ε = 2.4), L1 exhibited blue emission at 508 nm, while in CH3CN (ε = 37.5), a yellow-green emission at 532 nm was observed. A similar solvatochromatic effect is observed for L2; in dichloromethane (ε = 8.9), L2 exhibited blue emission at 511 nm, while in CH3CN (ε = 37.5), a yellow-green emission at 530 nm was observed. The Lippert–Mataga equation was used to examine the influence of solvent on the emission spectra of L1 and L2.22 This is visualised in the Lippert–Mataga plots, see Fig. S15 and S16 (ESI†). We also observed that the Stokes shift increased significantly from 5900 cm−1 to 6700 cm−1 and 5700 cm−1 to 6300 cm−1 across the solvent polarity range for L1 and L2 respectively, in addition to the expected spectral broadening, consistent with the higher energy cost of solvent sphere reorganisation on increasing solvent polarity as illustrated in Tables S2 and S3 (ESI†) and (Fig. 7 and Fig. S17, ESI†).
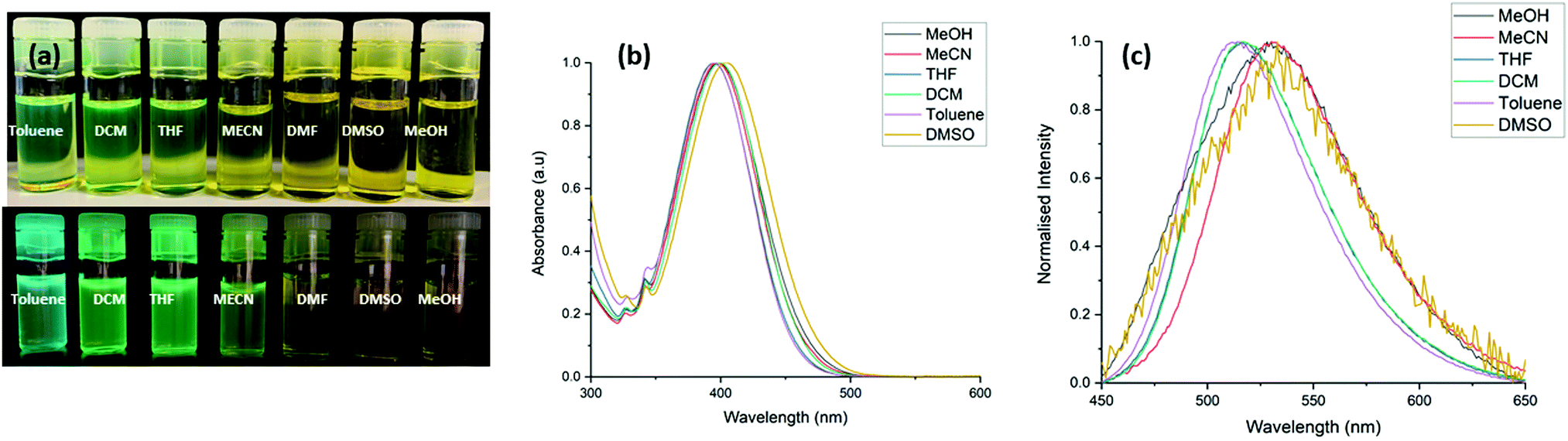 | ||
| Fig. 7 (a) Corresponding photograph taken in room light and under UV light, (b) UV absorption spectra of L1 in solvents of varying polarity (c) normalised fluorescence spectra of L1 in solvents of varying polarity. | ||
To investigate potential aggregation-induced emission (AIE) behaviour of L1 and L2, the UV-vis absorption spectra and fluorescence emission spectra were measured of CH3CN/H2O mixtures, with the expectation that increased water fraction would induce aggregation. As the percentage of H2O increased, however, no enhancement of fluorescence was observed. Instead, this led to a reduction in the observed emission intensity (ESI,† Fig. S18–S20), implying that aggregation is likely not a mechanism for emission enhancement in this system, which instead is most emissive as the free molecule in solution. A concentration dependent fluorescence study of L1 and L2 over the range 1 × 10−4 M to 1 × 10−6 M was also carried out in both H2O and CH3CN; the normalised spectra remained essentially invariant over this range and no significant change to band shape was observed, further supporting that this system is not prone to AIE behaviour (ESI,† Fig. S21 and S22).
pH solution studies of L1–L2
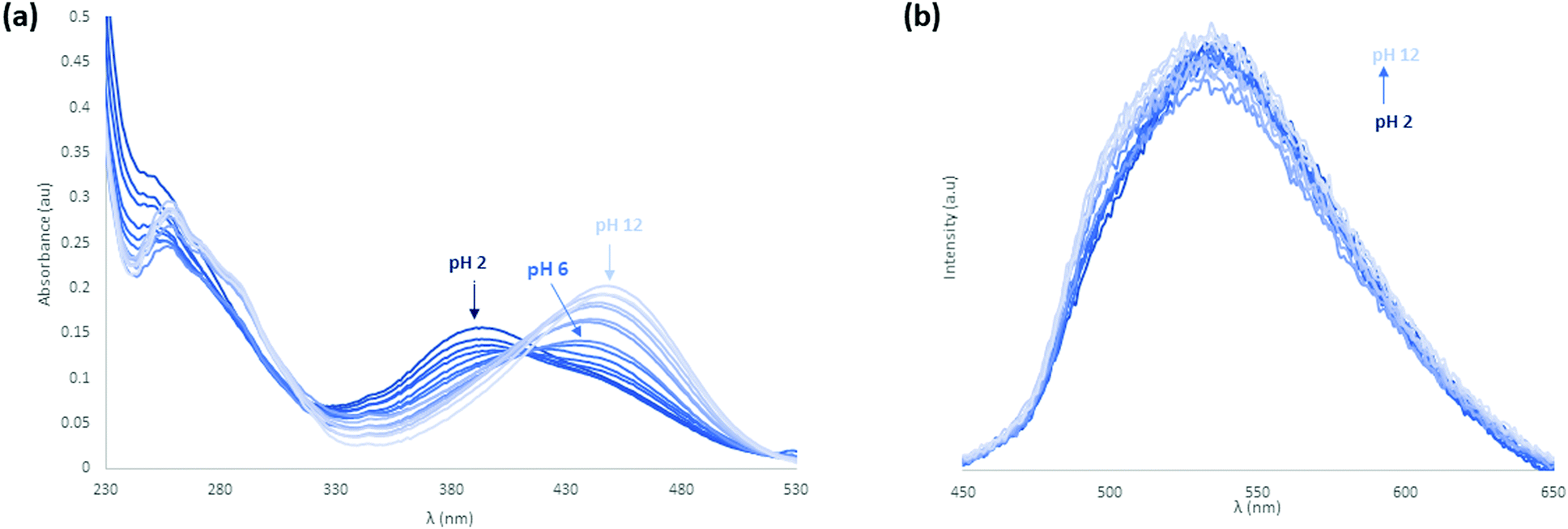
Following the study of the fundamental photophysical properties of L1 and L2 we next investigated the changes in the photophysical properties of the molecules in aqueous solution as a function of pH, by carrying out pH titrations from either acidic media to alkaline, or from alkaline to acidic media. The photophysical properties of L1 and L2 were first investigated as a function of pH in water, in the presence of 100 mM NaCl(aq) to maintain constant ionic strength. Initially at pH 2, L1 is protonated and upon sequential additions of NaOH(aq) deprotonation occurs. As can be seen in Fig. 8 there are significant absorbance changes observed both in the high energy transitions as well as in the ICT band from 380 nm to 450 nm. As the pH increases from pH 2 to pH 12, particularly at pH 4.5–5, there is a sharp decrease observed in UV absorbance with no corresponding new absorbance bands, suggesting the removal of chromophores from solution. A sharp increase in fluorescence emission accompanies the change in protonation state. As the pyridyl group is electronically isolated from the naphthalimide fluorophore, this change in photophysical behaviour could relate to aggregation once the cation–cation repulsion is removed and the solubility decreases. The increase in fluorescence emission could also relate to an aggregation process, either through the accessibility of new energy transfer modes, or the loss of a non-radiative decay pathway relating to the change in protonation state. Repeating this titration in reverse, starting in alkaline conditions at pH 12, L1 is deprotonated and following sequential additions of HCl(aq) protonation occurs, as shown in Fig. S24 and S25 (ESI†). A hysteresis is also evident in the absorbance response of L1 to pH, as illustrated in Fig. S26 (ESI†), as the full intensity of the absorbance band is not recovered until pH 2.5 in the reverse titration. This observation is also consistent with a phase change for L1 between the protonated and deprotonated forms, rather than a fully solution phase acid–base equilibrium. The effect of counterions on the system was also investigated, and the titrations were repeated with a HNO3(aq) spike (Fig. S27–S30, ESI†) as well as an initial CH3COOH(aq) spike (Fig. S31–S34, ESI†) and hysteresis in the forward and reverse titrations was consistently observed. We also noted a difference in the onset pHs as a function of counterion, further consistent with an aggregation/precipitation process rather than simple protonation equilibria. The counterion titrations were repeated in pure H2O media and the same effect was observed as previously described (Fig. S35–S38 for HCl(aq), Fig. S39–S42 for HNO3(aq) and Fig. S43–S46 for CH3COOH(aq), ESI†). The changes occurring in the ICT band could also indicate that some aggregation or slight dilution effects are being observed. Although photoinduced electron transfer (PET)-type behaviour might be expected from this class of fluorophore, both the direction of emission enhancement (i.e., higher intensity at higher pH) and the electronic isolation of the pyridyl nitrogen atom from the fluorophore would seem to disfavour such a process.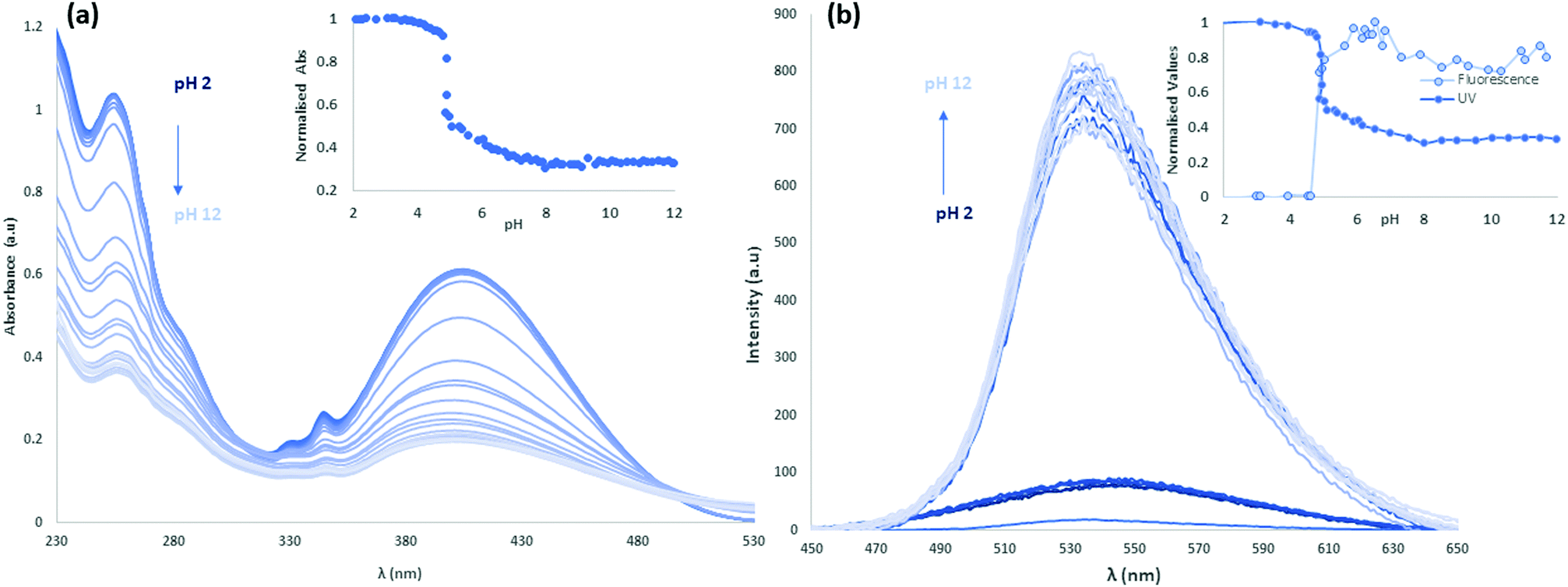 | ||
| Fig. 8 (a) UV-vis pH titration of L1 with NaOH(aq) at 0.1 pH unit intervals and an initial HCl(aq) acid spike in 100 mM NaCl(aq) medium. (inset) Normalised absorbance (λmax = 400 nm) of the titration. (b) Fluorescence pH titration of L1 with NaOH(aq) at 0.1 pH unit intervals and an initial HCl(aq) acid spike in 100 mM NaCl(aq) medium. (inset) Normalised absorbance (λmax = 400 nm) and fluorescence of the titration (λex = 400 nm, λmax = 550 nm). | ||
To further investigate the effect of the pyridyl group on the pH responsiveness of this motif, this study was repeated with the analogous benzyl derivative, L2, where one of the two possible protonation sites is removed. Initially in acidic conditions with HCl(aq) spike at pH 2, L2 has an absorbance λmax at 400 nm and upon sequential additions of NaOH(aq), there is a gradual decrease in absorbance until pH 5.5. In contrast to L1, above this pH into the alkaline region there is the formation of a new ICT band with λmax at 455 nm which blue-shifts and gradually increases in intensity with sequential additions until pH 12 (Fig. S47, ESI†). This effect is reversible; repeating the titration initially in alkaline conditions at pH 12, L2 has a λmax at 455 nm which gradually decreases in intensity until pH 6 and below this pH into the acidic region there is growth of the original ICT band with λmax at 400 nm. The fluorescence intensity increases with excitation at 450 nm during the formation of this new band as the pH is increased (Fig. S48, ESI†).
To investigate the effect of counterions, these titrations were repeated with a HNO3(aq) spike (Fig. S49 and S50, ESI†) as well as an initial CH3COOH(aq) spike (Fig. S51 and S52, ESI†) and the same phenomenon was observed. However, with acetic acid the fluorescence intensity was invariant in the same pH range. Finally, the L2 counterion titrations were repeated in H2O medium and the same effect was observed as described above (Fig. S53, S54 for HCl(aq), Fig. S55, S56 for HNO3(aq), Fig. 9 and Fig. S57 for CH3COOH(aq), ESI†). The counterion effect is summarised in Fig. S58 (ESI†). There is variation in the isosbestic point in the titrations that is also indicative of aggregation. From the observations above it does not appear that L1 and L2 are undergoing a simple protonation and deprotonation process while remaining fully dissolved as there is both a change in the total absorbance with pH and a hysteresis with pH, both of which are anion-dependent. The degree at which this occurs changes between L1 and L2, which suggests it is related to the pyridine functionality and not isolated to the morpholine group which is strongly coupled to the emission properties of the naphthalimide fluorophore. However, as no solids were observed to form in the cuvette and the total absorbance at higher wavelengths was not significantly affected by background scattering from the sample this suggests that the aggregates remain well dispersed, but also potentially larger than 1 μm. Therefore, this hypothesis was pursued by using Dynamic Light Scattering (DLS) and Scanning Electron Microscopy (SEM).
 | ||
| Fig. 9 (a) Representative UV-vis pH titration of L2 with additions of NaOH(aq) at 0.1 pH unit intervals and an initial CH3COOH(aq) acid spike. (b) Representative fluorescence pH titration (λex = 400 nm) with additions of NaOH(aq) and an initial CH3COOH(aq) acid spike in H2O medium. | ||
To examine the formation of aggregates over the pH range, samples of L1 and L2 were prepared for DLS measurements and SEM at pH 4, 6 and 8 with HCl(aq), HNO3(aq) and HOAc(aq) initial acid spikes. The formation of micrometer-scale aggregates in solution was indicated as scattering occurred during the DLS measurements for both L1 and L2. However, in the DLS measurements the apparent polydispersity index for all samples is high (>0.4) and therefore it is not possible to ascertain accurate size information or draw meaningful conclusions on trends of the particle size between samples. Following this, SEM was carried out, for the solutions at pH 4, pH 6 and pH 8 with HCl(aq), HNO3(aq) and HOAc(aq) spikes. The samples were deposited onto silicon wafers, evaporated under vacuum and subsequent imaging of the dried samples showed the presence of aggregates. In the dried L1 and L2 samples initially spiked with HCl(aq) and HNO3(aq) at pH 4 there is the formation of branched aggregates with some ordered morphology and as the pH increases to pH 6 there is the formation of a very high density of aggregates which lose their structured morphology. Upon a further increase to pH 8 there is the formation of micrometer-scale aggregates which is consistent with the DLS measurements as illustrated in Fig. 10 and Fig. S59 (ESI†). In contrast to this, the dried CH3COOH(aq) samples for L1 and L2 have a very low number of aggregates at pH 4 but more aggregation occurs at pH 6 and then the presence of micrometre-sized aggregates is observed at pH 8. The morphology of the dried CH3COOH(aq) samples also differs from HNO3 and HCl samples as the CH3COOH(aq) samples have a crystalline morphology at pH 4 and pH 6 as illustrated in Fig. 10 and Fig. S59, S60 (ESI†). This further illustrates the role that the counterions play in the deprotonation and protonation events, correlating to the spectroscopic findings.
 | ||
| Fig. 10 (a) SEM image of L2 sample spiked with HNO3 at pH 4 showing a branching morphology of aggregates, (b) SEM image of L2 sample spiked with HNO3 at pH 6 showing a higher density of aggregates (c) SEM image of L2 sample spiked with HNO3 at pH 8 showing micrometer aggregates. | ||
Metallogel formation
From the observations above, it is evident that L1 and L2 are likely exhibiting various solution-phase aggregation processes which are sensitive to the protonation state. In order to further probe the supramolecular assembly characteristics of these systems we examined the interaction of L1 and L2 with other Lewis acids, using first row transition metal ions as our test case based on successful gelation experiments with similar compounds. By incorporating a fluorescent sensor motif into the metallogel design a pH responsive material could be generated, with potential applications in luminescent sensing.In order to probe the coordination behaviour of L1 and L2, a range of solvent systems (toluene, hexane, chloroform, dichloromethane, ethyl acetate, acetonitrile, acetone, THF, methanol and mixtures of these solvents), various first-row transition metal ions with different counterions as well as a variety of standard crystallisation techniques were screened but only amorphous or microcrystalline powders were generated.
The reaction of L1 with manganese(II) (1), cobalt(II) (2) and nickel(II) (3) as their chloride salts in acetonitrile at 10 mM concentration (ca. 1.0 wt%) gave viscous, homogeneous solutions which set into clear and homogeneous metallogels in sealed sample tubes over a 48 hour period. The green cobalt and pale-yellow manganese gels resist inversion in wide-bore sample tubes indefinitely as illustrated in Fig. 11. In comparison, the nickel equivalent forms a considerably less robust gel and is unstable to inversion over a matter of seconds. This is consistent with what has been previously observed for this system. In contrast, reaction of L2 under analogous conditions with the same metal salts yielded amorphous or microcrystalline precipitate, indicating the requirement for the strongly coordinating 3-picolyl group in the metallogel formation. The reaction of L2 with manganese(II) chloride was analysed by powder X-ray diffraction and the pattern matched the simulated of the ligand as illustrated in Fig. S10 (ESI†). This indicated that only unreacted ligand was recovered, and no other (crystalline) material was formed.
 | ||
| Fig. 11 (left) Co-L1 (gel 2) and Mn-L1 (gel 1) undergoing an inversion test, (right) SEM image indicating the fibrous morphology of the xerogel 2. | ||
The gels were examined by thermogravimetric analysis and showed a residual non-volatile mass of between 1.0% and 0.9% after desolvation, completed by 100 °C as illustrated in Fig. S61 and S62 (see ESI†). These residual masses were consistent with the combined masses of the gelator mixture from the original synthesis showing the entire solvent content is encapsulated in the continuous gel phase. SEM analysis was carried out on the materials after desolvation under dynamic vacuum. The resulting xerogels undergo compression into nearly homogeneous films during the drying process. On closer inspection, a rough texture and densely packed fibrous morphology can be observed, conceivably as a remnant of the original gel network as illustrated in Fig. 11 and Fig. S63 (ESI†).
Chemical properties
A series of experiments were carried out to measure the response of the gel to external stimuli such as mechanical force, temperature or chemical stimuli. Thermoreversibility properties were exhibited by each of the gels 1 and 2. At 25 °C, the gels are stable and resistant to inversion indefinitely. On heating, the gels revert to the sol phase, and re-form on standing at room temperature overnight. The gel–sol transition temperature Ts–g for each gel was measured by holding the set gels in a temperature-controlled bath in glass tubes at 45° inclination and slowly heating until the transition to a homogeneous free flowing liquid was observed. Gel 2 displayed the lower thermal stability, reverting to the liquid phase at 52–54 °C. Gel 1 however retained its form until 74–76 °C, these values are tabulated with measured wt% in Table S4 (ESI†).We carried out investigations into the responsiveness of the gels to a variety of chemical stimuli; in particular pH responsiveness. The pH responsiveness of the cobalt gel 2 was examined by adding an HCl spike (100 μL, 0.1 M) in which the gel broke up and turned pale green, as illustrated in Fig. S64 (ESI†). Upon addition of a NaOH spike (100 μL, 0.1 M) and brief sonication to disperse the addition evenly, the solution turned yellow. Upon heating to 50 °C the solution changed colour to green, when sonicated again back to yellow indicating a thermochromic response of the dissolved species. This study was repeated with H2O, after the first 100 μL addition, a yellow layer diffused through the gel indicating the generation of free ligand and [Co(H2O)6] species, similarly on the second 100 μL addition of H2O, the same observation was made. Heating and cooling lead to a thermochromic response through the cycled colour change from green to yellow solutions.
These experiments were repeated with gel 1 and similarly upon an HCl spike (100 μL, 0.1 M) the gel broke up into a yellow solution generating the free ligand and [Mn(H2O)6]2+ species. Subsequent addition of an NaOH spike (100 μL, 0.1 M) was added and the gel broke up into a yellow solution. This was repeated with H2O as described above and the gel is stable for 4 hours but slowly breaks up into a yellow solution upon standing overnight. In contrast to gel 2, heating and cooling incited no thermochromic response. A similar process occurs in solution, but the colour change is not observed due to the weak absorbance from the manganese, which is saturated by the yellow absorbance from the ligand, unlike in the strongly absorbing tetrahedral polychloro-cobaltate ions likely present in solution 2 above.
Following the pH responsiveness of the gels, a metal sequestration experiment was carried out. Upon addition of a (100 μL, 0.1 mmol) disodium ethylenediaminetetraacetate dihydrate (EDTA-Na2) acetonitrile solution broke up gel 1 into a yellow solution. Following this triethylamine (100 μL, 0.1 mmol) acetonitrile solution was added dropwise to the gel forming a green suspension, as illustrated in Fig. S65 (ESI†). Upon standing for 7 days single yellow needle crystals were formed. The crystals are a second phase of the L1-gel ligand crystals described above.
The fluorescence emission of gels 1 and 2 were measured as well as their responses to pH through the addition of a HCl spike (100 μL, 0.1 M), NaOH spike (100 μL, 0.1 M) and H2O (2 × 100 μL) spikes to each sample. The normalised fluorescence spectra of gels 1 and 2 indicated a small shift in λmax from 536 nm to 544 nm upon addition of HCl and NaOH. Similarly, the fluorescence was invariant with the first addition of H2O (100 μL) but a similar shift was observed upon the second addition of H2O as illustrated in Fig. 12 and Fig. S66 (ESI†). The free ligand emission at pH 2 is very similar to the measured spectra for the 200 μL of water with a λmax of 545 nm. However, at pH 12 the ligand emission is blue shifted to 536 nm, which is the opposite trend to that observed in the gels.
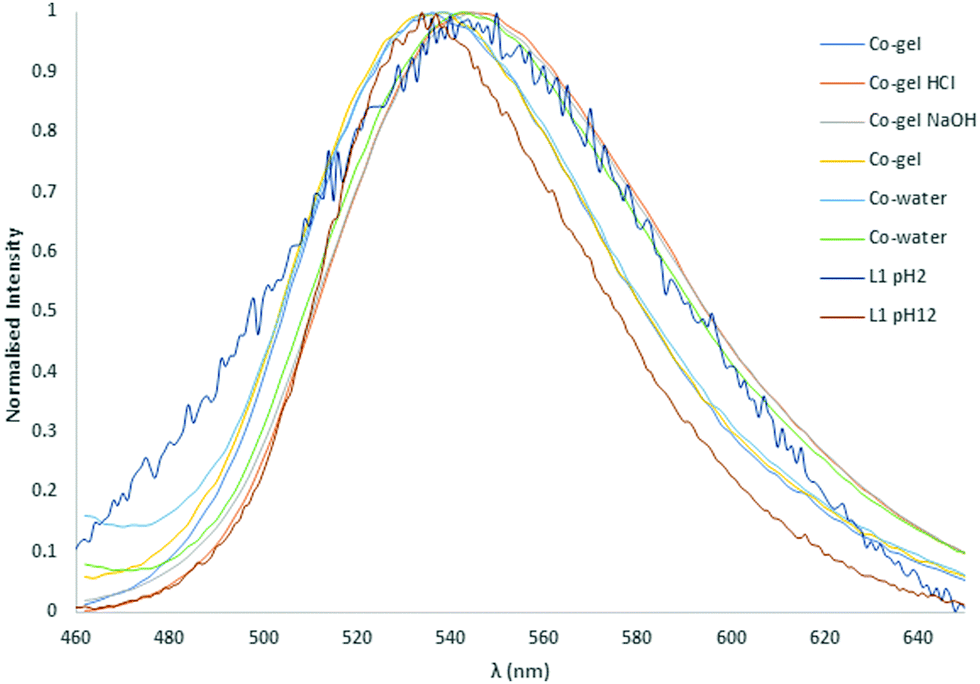 | ||
| Fig. 12 Normalised solid-state fluorescence of the L1 metallogels, compared with the parent ligand, and the response to pH changes. | ||
To investigate the aggregation behaviour of the metallogels, the solution state UV-vis and fluorescence spectra were measured of gels 1 and 2. The normalised spectra were compared to the free ligand spectra measured over the range 1 × 10−4 M to 1 × 10−6 M (ESI,† Fig. S67 and S68) and indicated a small shift in λem from the free ligand in H2O (520 nm) to the gel (527 nm) to the free ligand in CH3CN (533 nm). No change was observed in the band shape compared to the free ligand. Additionally, Fourier Transform infrared (FTIR) spectroscopy was also used to probe the dried xerogels of 1 and 2 and compared to the free ligand L1. In both cases there was a small shift of approx. 8 cm−1 in the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band from 1694 cm−1 to 1686 cm−1. In gel 1, there was also the formation of a weak band at 1739 cm−1 observed as illustrated in Fig. S69 and S70 (ESI†), though no other notable changes to the ligand absorbances were observed.
O stretching band from 1694 cm−1 to 1686 cm−1. In gel 1, there was also the formation of a weak band at 1739 cm−1 observed as illustrated in Fig. S69 and S70 (ESI†), though no other notable changes to the ligand absorbances were observed.
To measure the effect of the metal ion on the aggregation of L1, the UV-vis and fluorescence emission spectra was measured of sequential 0.2 eq. additions of cobalt(II) or manganese(II) to L1 until 1 eq. was reached (as illustrated in Fig. S71 and S72, ESI†). On comparison of this normalised spectra to the free ligand L1 as well as spectra of gels 1 and 2 no significant changes were observed (Fig. S69 and S70, ESI†). FTIR spectra were also measured of dried samples of the M:L mixtures, as illustrated in the overlaid spectra (Fig. S73 and S74, ESI†) no significant changes were observed and the spectra remained largely similar to that of the free ligand.
Rheological studies
Rheological studies (strain and frequency sweeps) and recovery tests were performed on the most robust gels 1 and 2 (Mn-L1 and Co-L1) to quantitatively study the mechanical properties of the metallogels. Each material studied was prepared using identical procedures to maintain equivalent ligand fractions for each sample. The gels were illustrated to possess viscoelastic properties often associated with supramolecular gel-phase materials.The frequency sweep for gel 2 showed the G′ value (1520 Pa) which is 1300 Pa higher than the G′′ value (220 Pa), which is characteristic of gel behaviour. Similarly, the frequency sweep for gel 1 showed the G′ value (1400 Pa) which is 1200 Pa higher than the G′′ value (200 Pa), which is also characteristic of gel behaviour. Following this, to test the strength of the gel, strain tests were carried out. At low strain amplitudes, the storage modulus, G′ and the loss modulus G′′, of the gel remains constant until the onset of liquid-like behaviour. For gel 2, the intersection of the G′ and G′′ curves occurred at 172 Pa with 25% strain amplitude. For gel 1, the intersection of the G′ and G′′ curves occurred at 130 Pa with 40% strain amplitude. The values of G′ and G′′ for gels 1 and 2 did not vary significantly with frequency under a frequency sweep in the range 0.1–100 rad s−1, Fig. 13 and Fig. S75 (ESI†).
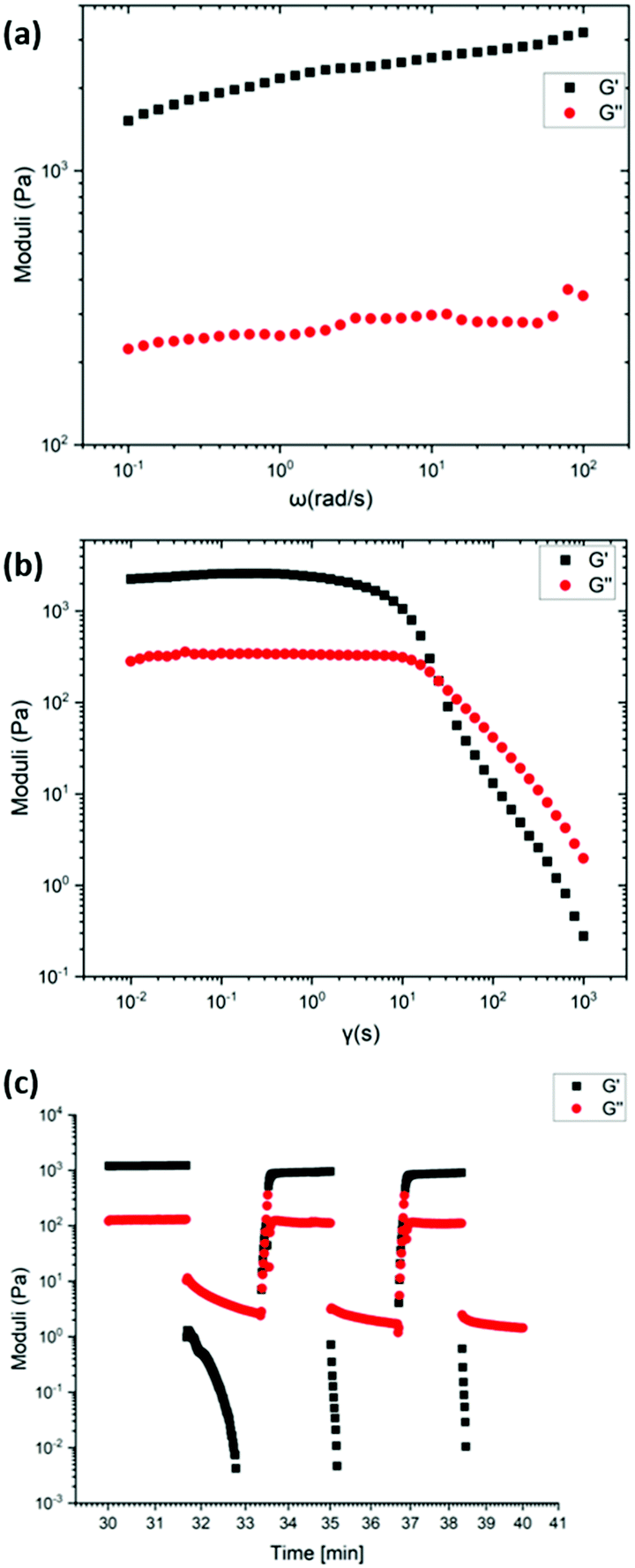 | ||
| Fig. 13 Rheological studies for gel 2 prepared at 1.7 wt% in acetonitrile, showing (a) frequency sweep over consecutive cycles (γ = 0.1%), (b) amplitude sweep (ω = 1 rad s−1) and (c) recovery test at alternating 0.1% and 500% strain amplitudes. | ||
The recovery properties of the gels were tested in an oscillatory recovery test, by imposing alternating strain amplitudes of 500% and 0.1% at constant 1 Hz oscillation frequency. Each of the gels displayed transitions from liquid-like (G′′ > G′) to solid-like (G′ > G′′) behaviour within ca. 100 s. While the gels do not fully recover, the gels recover 80% of the original low-strain values of the storage and the loss moduli.
Conclusions
We have prepared and structurally characterised two new 4-(N-morpholino)-1,8-naphthalimide ligands and reported on their solution and gelation chemistry in response to pH and the presence of d-block metal ions. The solid-state characterisation of L1 and L2 illustrated both the influence of solvent and substituent on the structure directionality. In the solution state L1 was observed to undergo a pH and anion-dependent aggregation process which contributes to a sharp increase in fluorescence emission, and a hysteresis effect. This indicates that the change in the absorption and fluorescence is not only associated with a simple protonation and deprotonation process but also the additional effect of supramolecular organization, which led to the formation of microparticles. The tunability of this system was examined through substituting the N-position with benzyl amine and in the solution state L2 also undergoes an aggregation process. The formation of aggregates over the pH range was investigated further by using DLS and SEM measurements which demonstrated the presence of micrometre-scale aggregates in solution. Following this, the supramolecular assembly characteristics of these systems were investigated by examining the interaction of L1 and L2 with other Lewis acids. By using first row divalent transition metals as our test basis, two metallogels were generated from L1 and the gels response to various physical and chemical stimuli, including pH, metal sequestration and thermal responsiveness were examined. The ligands presented here illustrate a structure–function relationship in the ligand design which can be used as a handle for developing the unique variation in photophysical properties of this system, which provides a future role in the design of pH responsive supramolecular materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Science Foundation Ireland (PI award 13/IA/1865 to T. G.), CDA award 17/CDA/4704 to M. E. M., the SFI Centre AMBER (for Research Centres Phase 2 12/RC/2278_P2) and the SFI Centre SSPC (for Research Centres Phase 2 12/RC/2275_P2) to T. G., Trinity College Dublin and the School of Chemical and Physical Sciences, Keele University.Notes and references
- (a) B. O. Okesola and D. K. Smith, Applying low-molecular weight supramolecular gelators in an environmental setting – self-assembled gels as smart materials for pollutant removal, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC; (b) J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Supramolecular materials based on AIE luminogens (AIEgens): construction and applications, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC; (c) G. Tseberlidis, D. Intrieri and A. Caselli, Catalytic Applications of Pyridine-Containing Macrocyclic Complexes, Eur. J. Inorg. Chem., 2017, 3589–3603 CrossRef CAS; (d) D. Écija, J. I. Urgel, A. P. Seitsonen, W. Auwärter and J. V. Barth, Lanthanide-Directed Assembly of Interfacial Coordination Architectures – From Complex Networks to Functional Nanosystems, Acc. Chem. Res., 2018, 51, 365–375 CrossRef PubMed; (e) I. V. Kolesnichenko and E. V. Anslyn, Practical applications of supramolecular chemistry, Chem. Soc. Rev., 2017, 46, 2385–2390 RSC; (f) T. Gorai, W. Schmitt and T. Gunnlaugsson, Highlights of the development and application of luminescent lanthanide based coordination polymers, MOFs and functional nanomaterials, Dalton Trans., 2021, 50, 770–784 RSC; (g) A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. Ó’Máille and T. Gunnlaugsson, Supramolecular Chemistry: A Toolkit for Soft Functional Materials and Organic Particles, Chem, 2017, 3, 764–811 CrossRef CAS.
- (a) C. D. Jones and J. W. Steed, Gels with sense: supramolecular materials that respond to heat, light and sound, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC; (b) T. Feldner, M. Häring, S. Saha, J. Esquena, R. Banerjee and D. D. Díaz, Supramolecular Metallogel That Imparts Self-Healing Properties to Other Gel Networks, Chem. Mater., 2016, 28, 3210–3217 CrossRef CAS; (c) M. Externbrink, S. Riebe, C. Schmuck and J. Voskuhl, A dual pH-responsive supramolecular gelator with aggregation-induced emission properties, Soft Matter, 2018, 14, 6166–6170 RSC.
- (a) Z. Li, J. R. Askim and K. S. Suslick, The Optoelectronic Nose: Colorimetric and Fluorometric Sensor Arrays, Chem. Rev., 2019, 119, 231–292 CrossRef CAS PubMed; (b) P. A. Gale and C. Caltagirone, Fluorescent and colorimetric sensors for anionic species, Coord. Chem. Rev., 2018, 354, 2–27 CrossRef CAS; (c) L. M. Mesquita, V. André, C. V. Esteves, T. Palmeira, M. N. Berberan-Santos, P. Mateus and R. Delgado, Dinuclear Zinc(II) Macrocyclic Complex as Receptor for Selective Fluorescence Sensing of Pyrophosphate, Inorg. Chem., 2016, 55, 2212–2219 CrossRef CAS PubMed; (d) J. J. Lee, Y. S. Kim, E. Nam, S. Y. Lee, M. H. Lim and C. Kim, A PET-based fluorometric chemosensor for the determination of mercury(ii) and pH, and hydrolysis reaction-based colorimetric detection of hydrogen sulfide, Dalton Trans., 2016, 45, 5700–5712 RSC; (e) D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Fluorescent chemosensors: the past, present and future, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- (a) R. Parkesh, T. Clive Lee and T. Gunnlaugsson, Highly selective 4-amino-1,8-naphthalimide based fluorescent photoinduced electron transfer (PET) chemosensors for Zn(ii) under physiological pH conditions, Org. Biomol. Chem., 2007, 5, 310–317 RSC; (b) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Signaling Recognition Events with Fluorescent Sensors and Switches, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS.
- (a) E. E. Langdon-Jones, D. Lloyd, A. J. Hayes, S. D. Wainwright, H. J. Mottram, S. J. Coles, P. N. Horton and S. J. A. Pope, Alkynyl-naphthalimide Fluorophores: Gold Coordination Chemistry and Cellular Imaging Applications, Inorg. Chem., 2015, 54, 6606–6615 CrossRef CAS; (b) D. L. Reger, A. P. Leitner and M. D. Smith, Supramolecular Metal–Organic Frameworks of s- and f-Block Metals: Impact of 1,8-Naphthalimide Functional Group, Cryst. Growth Des., 2016, 16, 527–536 CrossRef CAS; (c) A. B. Carter, N. Zhang, I. A. Kühne, T. D. Keene, A. K. Powell and J. A. Kitchen, Layered Ln(III) Complexes from a Sulfonate-Based 1,8-Naphthalimide: Structures, Magnetism and Photophysics, ChemistrySelect, 2019, 4, 1850–1856 CrossRef CAS; (d) D. L. Reger, J. J. Horger and M. D. Smith, Copper(ii) carboxylate tetramers formed from an enantiopure ligand containing a π-stacking supramolecular synthon: single-crystal to single-crystal enantioselective ligand exchange, Chem. Commun., 2011, 47, 2805–2807 RSC.
- (a) M. Martínez-Calvo, S. A. Bright, E. B. Veale, A. F. Henwood, D. C. Williams and T. Gunnlaugsson, 4-Amino-1,8-naphthalimide based fluorescent photoinduced electron transfer (PET) pH sensors as liposomal cellular imaging agents: The effect of substituent patterns on PET directional quenching, Front. Chem. Sci. Eng., 2020, 14, 61–75 CrossRef; (b) Y. Ni, Z. Sun, Y. Wang, H. F. Nour, A. C. H. Sue, N. S. Finney, K. K. Baldridge and M. A. Olson, Versatile hydrochromic fluorescent materials based on a 1,8-naphthalimide integrated fluorophore-receptor system, J. Mater. Chem., 2019, 7, 7399–7410 CAS.
- (a) R. M. Duke and T. Gunnlaugsson, 3-Urea-1,8-naphthalimides are good chemosensors: a highly selective dual colorimetric and fluorescent ICT based anion sensor for fluoride, Tetrahedron Lett., 2011, 52, 1503–1505 CrossRef CAS; (b) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Colorimetric and fluorescent anion sensors: an overview of recent developments in the use of 1,8-naphthalimide-based chemosensors, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- (a) Y. Tang, Y. Ma, J. Yin and W. Lin, Strategies for designing organic fluorescent probes for biological imaging of reactive carbonyl species, Chem. Soc. Rev., 2019, 48, 4036–4048 RSC; (b) H.-J. Lee, C.-W. Cho, H. Seo, S. Singha, Y. W. Jun, K.-H. Lee, Y. Jung, K.-T. Kim, S. Park, S. C. Bae and K. H. Ahn, A two-photon fluorescent probe for lysosomal zinc ions, Chem. Commun., 2016, 52, 124–127 RSC; (c) S.-H. Park, N. Kwon, J.-H. Lee, J. Yoon and I. Shin, Synthetic ratiometric fluorescent probes for detection of ions, Chem. Soc. Rev., 2020, 49, 143–179 RSC; (d) C. L. Fleming, T. D. Ashton, C. Nowell, M. Devlin, A. Natoli, J. Schreuders and F. M. Pfeffer, A fluorescent histone deacetylase (HDAC) inhibitor for cellular imaging, Chem. Commun., 2015, 51, 7827–7830 RSC.
- (a) J. Mayr, C. Saldías and D. Díaz Díaz, Release of small bioactive molecules from physical gels, Chem. Soc. Rev., 2018, 47, 1484–1515 RSC; (b) E. Saha and J. Mitra, Multistimuli-Responsive Self-Healable and Moldable Nickel(II)-Based Gels for Reversible Gas Adsorption and Palladium Sequestration via Gel-to-Gel Transformation, ACS Appl. Mater. Interfaces, 2019, 11, 10718–10728 CrossRef CAS PubMed; (c) J. Li, L. Geng, G. Wang, H. Chu and H. Wei, Self-Healable Gels for Use in Wearable Devices, Chem. Mater., 2017, 29, 8932–8952 CrossRef CAS; (d) S. Ganta and D. K. Chand, Multi-Stimuli-Responsive Metallogel Molded from a Pd2L4-Type Coordination Cage: Selective Removal of Anionic Dyes, Inorg. Chem., 2018, 57, 3634–3645 CrossRef CAS; (e) E. R. Draper and D. J. Adams, Low-Molecular-Weight Gels: The State of the Art, Chem, 2017, 3, 390–410 CrossRef CAS.
- (a) S. Panja, S. Bhattacharya and K. Ghosh, Pyridine coupled mono and bisbenzimidazoles as supramolecular gelators: selective metal ion sensing and ionic conductivity, Mater. Chem. Front., 2018, 2, 385–395 RSC; (b) A. Pal, S. Chand and M. C. Das, A Water-Stable Twofold Interpenetrating Microporous MOF for Selective CO2 Adsorption and Separation, Inorg. Chem., 2017, 56, 13991–13997 CrossRef CAS; (c) S. Chand, A. Pal, S. C. Pal and M. C. Das, A Trifunctional Luminescent 3D Microporous MOF with Potential for CO2 Separation, Selective Sensing of a Metal Ion, and Recognition of a Small Organic Molecule, Eur. J. Inorg. Chem., 2018, 2785–2792 CrossRef CAS; (d) D. Dietrich, C. Licht, A. Nuhnen, S.-P. Höfert, L. De Laporte and C. Janiak, Metal–Organic Gels Based on a Bisamide Tetracarboxyl Ligand for Carbon Dioxide, Sulfur Dioxide, and Selective Dye Uptake, ACS Appl. Mater. Interfaces, 2019, 11, 19654–19667 CrossRef CAS; (e) C. K. Karan, M. C. Sau and M. Bhattacharjee, A copper(ii) metal–organic hydrogel as a multifunctional precatalyst for CuAAC reactions and chemical fixation of CO2 under solvent free conditions, Chem. Commun., 2017, 53, 1526–1529 RSC.
- N. Kelly, K. Gloe, T. Doert, F. Hennersdorf, A. Heine, J. März, U. Schwarzenbolz, J. J. Weigand and K. Gloe, Self-assembly of [2+2] Co(II) metallomacrocycles and Ni(II) metallogels with novel bis(pyridylimine) ligands, J. Organomet. Chem., 2016, 821, 182–191 CrossRef CAS.
- J.-L. Zhong, X.-J. Jia, H.-J. Liu, X.-Z. Luo, S.-G. Hong, N. Zhang and J.-B. Huang, Self-assembled metallogels formed from N,N′,N′′-tris(4-pyridyl)trimesic amide in aqueous solution induced by Fe(iii)/Fe(ii) ions, Soft Matter, 2016, 12, 191–199 RSC.
- (a) C. K. Karan and M. Bhattacharjee, Self-Healing and Moldable Metallogels as the Recyclable Materials for Selective Dye Adsorption and Separation, ACS Appl. Mater. Interfaces, 2016, 8, 5526–5535 CrossRef CAS PubMed; (b) S. Bhattacharjee, B. Maiti and S. Bhattacharya, First report of charge-transfer induced heat-set hydrogel. Structural insights and remarkable properties, Nanoscale, 2016, 8, 11224–11233 RSC.
- (a) A. D. Lynes, C. S. Hawes, E. N. Ward, B. Haffner, M. E. Möbius, K. Byrne, W. Schmitt, R. Pal and T. Gunnlaugsson, Benzene-1,3,5-tricarboxamide n-alkyl ester and carboxylic acid derivatives: tuneable structural, morphological and thermal properties, CrystEngComm, 2017, 19, 1427–1438 RSC; (b) O. Kotova, R. Daly, C. M. G. dos Santos, M. Boese, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Europium-Directed Self-Assembly of a Luminescent Supramolecular Gel from a Tripodal Terpyridine-Based Ligand, Angew. Chem., Int. Ed., 2012, 51, 7208–7212 CrossRef CAS PubMed; (c) R. Daly, O. Kotova, M. Boese, T. Gunnlaugsson and J. J. Boland, Chemical Nano-Gardens: Growth of Salt Nanowires from Supramolecular Self-Assembly Gels, ACS Nano, 2013, 7, 4838–4845 CrossRef CAS PubMed.
- (a) O. Kotova, S. J. Bradberry, A. J. Savyasachi and T. Gunnlaugsson, Recent advances in the development of luminescent lanthanide-based supramolecular polymers and soft materials, Dalton Trans., 2018, 47, 16377–16387 RSC; (b) O. Kotova, R. Daly, C. M. G. dos Santos, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Cross-Linking the Fibers of Supramolecular Gels Formed from a Tripodal Terpyridine Derived Ligand with d-Block Metal Ions, Inorg. Chem., 2015, 54, 7735–7741 CrossRef CAS; (c) M. Klein-Hitpaß, A. D. Lynes, C. S. Hawes, K. Byrne, W. Schmitt and T. Gunnlaugsson, A Schiff-base cross-linked supramolecular polymer containing diiminophenol compartments and its interaction with copper(II) ions, Supramol. Chem., 2018, 30, 93–102 CrossRef.
- (a) S. J. Bradberry, G. Dee, O. Kotova, C. P. McCoy and T. Gunnlaugsson, Luminescent lanthanide (Eu(iii)) cross-linked supramolecular metallo co-polymeric hydrogels: the effect of ligand symmetry, Chem. Commun., 2019, 55, 1754–1757 RSC; (b) E. P. McCarney, J. P. Byrne, B. Twamley, M. Martínez-Calvo, G. Ryan, M. E. Möbius and T. Gunnlaugsson, Self-assembly formation of a healable lanthanide luminescent supramolecular metallogel from 2,6-bis(1,2,3-triazol-4-yl)pyridine (btp) ligands, Chem. Commun., 2015, 51, 14123–14126 RSC.
- I. N. Hegarty, H. L. Dalton, A. D. Lynes, B. Haffner, M. E. Möbius, C. S. Hawes and T. Gunnlaugsson, Balancing connectivity with function in silver(i) networks of pyridyltriazole (tzpa) ligands results in the formation of a metallogel, Dalton Trans., 2020, 49, 7364–7372 RSC.
- J. I. Lovitt, C. S. Hawes, A. D. Lynes, B. Haffner, M. E. Möbius and T. Gunnlaugsson, Coordination chemistry of N-picolyl-1,8-naphthalimides: colourful low molecular weight metallo-gelators and unique chelation behaviours, Inorg. Chem. Front., 2017, 4, 296–308 RSC.
- L. J. Beeching, C. S. Hawes, D. R. Turner and S. R. Batten, The influence of anion, ligand geometry and stoichiometry on the structure and dimensionality of a series of AgI-bis(cyanobenzyl)piperazine coordination polymers, CrystEngComm, 2014, 16, 6459–6468 RSC.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- E. Calatrava-Pérez, S. Acherman, L. Stricker, G. McManus, J. Delente, A. D. Lynes, A. F. Henwood, J. I. Lovitt, C. S. Hawes, K. Byrne, W. Schmitt, O. Kotova, T. Gunnlaugsson and E. M. Scanlan, Fluorescent supramolecular hierarchical self-assemblies from glycosylated 4-amino- and 4-bromo-1,8-naphthalimides, Org. Biomol. Chem., 2020, 18, 3475–3480 RSC.
- N. Mataga, Y. Kaifu and M. Koizumi, Solvent Effects upon Fluorescence Spectra and the Dipolemoments of Excited Molecules, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Thermogravimetric analysis data, X-ray powder diffraction plots, UV-vis and NMR spectroscopy. CCDC 2048284–2048287. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qm00091h |
| This journal is © the Partner Organisations 2021 |