
An aqueous rechargeable lithium ion battery with long cycle life and overcharge self-protection†

Abstract
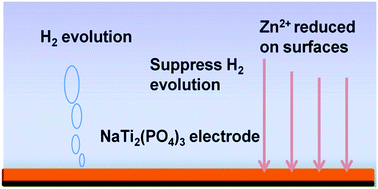
Aqueous lithium ion batteries are receiving increasing attention for large-scale energy storage applications because of their intrinsic safety and environmental friendliness. However, they suffer from severe irreversibility issues, such as sustained water consumption, and especially low resistance to overcharging, which has rarely been studied. We find that the kinetics of H2 evolution on the anode is much more facile than O2 evolution on the cathode, resulting in limited resistance to overcharging. When the battery is overcharged, there is severe H2 evolution on the anode, triggering O2 evolution. The structure collapse of the anode, resulting from oxidation by O2, leads to rapid capacity fading. Here, Zn2+ is added into the electrolyte to solve these problems without any loss of battery energy density. When charged in the end, the Zn2+ will be reduced to Zn metal exhibiting high over potential for H2 evolution, which can suppress H2 evolution. A LiMn2O4/NaTi2(PO4)3 full cell exhibits enhanced overcharging performance and excellent cycling stability up to 10 000 cycles at 10C.
- This article is part of the themed collection: 2021 Materials Chemistry Frontiers HOT articles
 Please wait while we load your content...
Please wait while we load your content...

