DOI:
10.1039/D0QM00992J
(Research Article)
Mater. Chem. Front., 2021,
5, 1863-1871
Facile construction of well-defined radical metallacycles through coordination-driven self-assembly†
Received
27th November 2020
, Accepted 8th January 2021
First published on 12th January 2021
Abstract
Recently, the marriage of organic radicals and supramolecular chemistry has given birth to a booming research field of supramolecular radical chemistry. However, the well-developed supramolecular radical systems are still limited, especially at present the supramolecular coordination radical systems are rarely explored. The construction of discrete supramolecular radical coordination complexes such as radical metallacycles and metallacages still remains a great challenge mainly because of the intrinsically unstable nature of organic radicals and the lack of suitable coordination method. Herein, we design and synthesize a new organic radical ligand CzBTM-Py with well-defined coordination geometry and persistent stability. Subsequently, two discrete radical metallacycles of M1 and M2 are successfully and efficiently constructed via the coordination-driven self-assembly approach. Due to the mild coordination reaction conditions the open-shell nature of CzBTM-Py is preserved as indicated by the electron paramagnetic resonance (EPR) spectroscopy. The chemical structures of CzBTM-Py, M1 and M2 are successfully determined by X-ray crystallography. Interestingly, a chiral self-sorting behavior is observed in this coordination-driven self-assembly process, i.e., M1 exists as MM/PP enantiomers couple while M2 exists as the heterochiral PM form. Moreover, the photophysical properties of the radicals have changed significantly after coordination, making the color and emission of M1 and M2 differ from those of CzBTM-Py. Additionally, metallacycles M1 and M2 show better photostability than radical ligand CzBTM-Py. This work successfully demonstrates a highly efficient coordination-driven self-assembly approach to synthesize radical-based supramolecular coordination complexes, which could modulate the chemical and physical properties of organic radicals, and thus to develop new functional metal–organic radical materials.
Introduction
Free radicals are compounds that feature unpaired electrons or open-shell electronic configurations. Since the landmark discovery of the persistent organic radicals by Moses Gomberg in 1900,1 the design and synthesis of stable organic radicals has been very attractive but also great challenge to chemists.2 Most organic radicals are highly reactive and readily undergo dimerization, hydrogen abstraction, disproportionation and other reactions, therefore usually unstable.3,4 Due to their distinct open-shell nature and special reactivity, organic radicals have played increasingly important roles in the field of organic synthesis, biology, and functional materials.5,6 For example, organic radicals are involved in many synthetic transformations and have been widely used as (co)catalysts for the oxidation, mediators for living radical polymerization, and antioxidants.7–9 Organic free radicals are actually produced continuously in biosystems and play an important role in a number of biological processes, e.g., efficiently transforming air and food into chemical energy in the human body largely depends on the chain reactions of free radicals.10 Organic radicals with intrinsic magnetic nature have also demonstrated the wide application in the field of spin trapping, spin labelling, EPR imaging, and dynamic nuclear polarization (DNP).11–14 Moreover, organic radicals have been intensively employed as functional building blocks to construct a variety of radical-based functional materials such as organic magnetic materials, organic spintronics, organic conductors, organic radical batteries, and so on.15,16 Particularly, organic radicals are recently emerging as peculiar luminescent materials and have demonstrated great success in the application of highly efficient radical-based organic light-emitting diodes (OLEDs).17–22 Notably, organic radicals have also aroused extensive attention in the field of supramolecular chemistry, leading to a booming research field of supramolecular radical chemistry.23–25 The most pioneer work in this field is the study of the various organic radical cation dimerization and their host–guest chemistry and self-assembly.26–30 However, the supramolecular radical systems reported so far are still limited to radical cation-based host–guest systems, while the supramolecular coordination radical systems are still less explored.31–37
The marriage of organic radical chemistry and coordination chemistry, namely radical coordination chemistry, could potentially spark some new design ideas and eventually create new radical-based molecules and materials.38 It has demonstrated that the coordination of organic radicals has a profound effect on the properties of the resultant radical complexes such as the ligand radical reactivity and their magnetic exchange coupling, which are very important to implement their practical applications in organic synthesis and magnetic properties. Specifically, organic radicals can react with transition-metal complexes in many different ways accompanied by various valuable organic reactions and important biological processes.38 Organic radicals, as unique bridging ligands, could efficiently facilitate strong magnetic exchange coupling between high-anisotropy paramagnetic metal centers, which represents a classical rule to design high temperature single-molecule magnets.39–41 Moreover, recent reports have also demonstrated that the coordination of the luminescent radicals to gold(I) could affect the electronic states of radicals and significantly enhance their fluorescence quantum yield and photostability.42,43 To date, a wide variety of metal-radical coordination complexes ranging from simple radical-molecule complexes to infinite one-dimensional coordination chains and two- and three-dimensional metal-radical networks have been successfully constructed.41,44 However, to the best of our knowledge, discrete radical metallacycles are rarely reported and documented, probably due to the challenging design and synthesis of the suitable radical ligands with well-defined coordination geometry and persistent stability.37,45,46 Moreover, the novel mild and efficient coordination method is also highly anticipated to prepare discrete radical metallacycles.
Coordination-driven self-assembly has proven to be a powerful approach to construct a diverse range of supramolecular coordination complexes with well-controlled shapes and sizes, including metallacycles, metallacages, and metal–organic frameworks (MOFs), through the combination of a range of diverse functional ligands and complementary metal salts.47–61 The employment of coordination-driven self-assembly methodology to construct discrete radical metallacycles (Fig. 1) is expected to show several distinct merits including: (1) the coordination-driven supramolecular self-assembly reaction conditions are very mild without adding any acid/base or catalyst; (2) the coordination-driven self-assembly approach is very efficient with no need for the tedious purification steps; (3) the combination of different organometallic acceptors with organic radical ligand could result in a diverse library of metal–organic coordination complexes with different conformations; and (4) the introduction of heavy atoms like Pt may lead to some interesting photophysical properties of organic radicals. In addition to the coordination method, the rational design of the suitable ditopic or polytopic organic radical ligands with well-defined coordination geometry and persistent stability is also crucially important because radical ligands not only dictate the structure and topology of the resulting radical-based supramolecular architectures but also determine their properties and applications.
 |
| | Fig. 1 Cartoon representation of the construction of discrete radical metallacycle through the coordination-driven self-assembly approach in this work and its advantages. | |
In this work, we design and synthesize a new dipyridyl-functionalized (N-carbazolyl)bis(2,4,6-trichlorophenyl) methyl radical (CzBTM-Py) which serves as a stable ligand with conformational rigidity and well-defined coordinate vectors. Subsequently, efficient coordination-driven self-assembly of the radical ligand CzBTM-Py with the complementary metal acceptors resulted in two well-defined radical metallacycles, namely M1 and M2. Notably, continuous wave electron paramagnetic resonance (CW-EPR) spectroscopy revealed that the open-shell nature of radical ligand CzBTM-Py was preserved in the resultant metallacycles, mainly because of the relatively long persistence of radical CzBTM-Py and the mild reaction conditions in such coordination-driven self-assembly process. The crystal structure of CzBTM-Py is determined by single crystal X-ray diffraction and shows that CzBTM-Py exists in two different enantiomeric forms of P and M. Interestingly, metallacycles M1 and M2 displayed distinct well-defined shapes and sizes as well as intriguing chiral self-sorting behavior as indicated in their single crystals. With regard to their photophysical properties, the radical ligand CzBTM-Py and metallacycles M1 and M2 differ in their color and emission. Moreover, the radical stability studies revealed that metallacycles M1 and M2 showed better photostability than those of radical ligand CzBTM-Py. Therefore, this study demonstrates that coordination-driven self-assembly is a facile and versatile strategy for the preparation of discrete radical supramolecular coordination complexes. The preliminary results in this work also successfully prove the importance of supramolecular coordination approach in modulation of the chemical and physical properties of organic radicals, which may open up a new avenue to develop functional metal–organic radical materials.
Results and discussion
Synthesis and characterization of the radical ligand CzBTM-Py
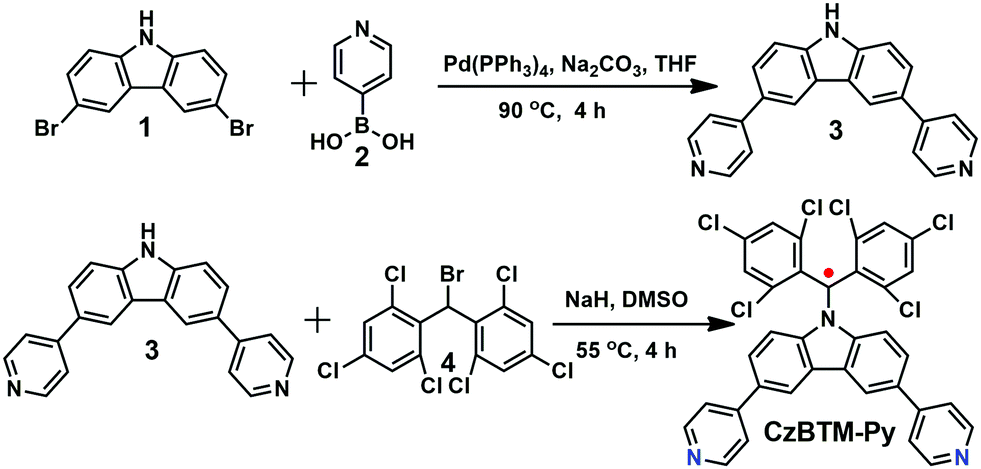
Polychlorotriphenylmethyl radicals such as perchlorotriphenyl methyl (PTM) and tris(2,4,6-trichlorophenyl)methyl (TTM) radicals are reported to be very stable and easily chemically modified. Thus, a new ditopic TTM-based radical ligand, namely CzBTM-Py bearing two 4-pyridyl groups, was designed in this work. CzBTM-Py was successfully synthesized by a two-step procedure as illustrated in Scheme 1. First, a Suzuki coupling reaction of 3,6-dibromocarbazole 1 with 4-pyridinylboronic acid 2 afforded 3,6-di(4-pyridyl) carbazole 3 in 41% yield. Subsequently, a nucleophilic substitution reaction of 3 with (bromomethylene)bis(2,4,6-trichlorobenzene) 4 accompanying a DMSO-mediated oxidation resulted in CzBTM-Py in moderate yield (24%).62 Notably, CzBTM-Py is very stable and could be purified by column chromatography. Due to the paramagnetic nature, the proton nuclear magnetic resonance (1H NMR) spectrum of CzBTM-Py experienced seriously signal broadening (see the ESI,† Fig. S12), leading to very difficult to identify its exact structure. Nevertheless, the structure of CzBTM-Py was unambiguously confirmed by electrospray ionization mass spectrometry (ESI-MS) (Fig. S19, ESI†) and X-ray crystallographic analysis (vide infra). The single crystal of CzBTM-Py further confirmed its good conformational rigidity and well-defined coordinate vectors that is readily able to coordinate with suitable di-Pt(II) acceptors.
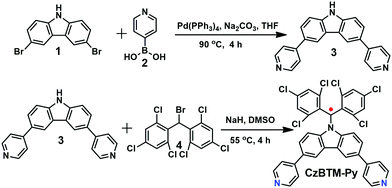 |
| | Scheme 1 Synthetic route to CzBTM-Py. | |
Self-assembly of radical metallacycles M1 and M2 and their characterization
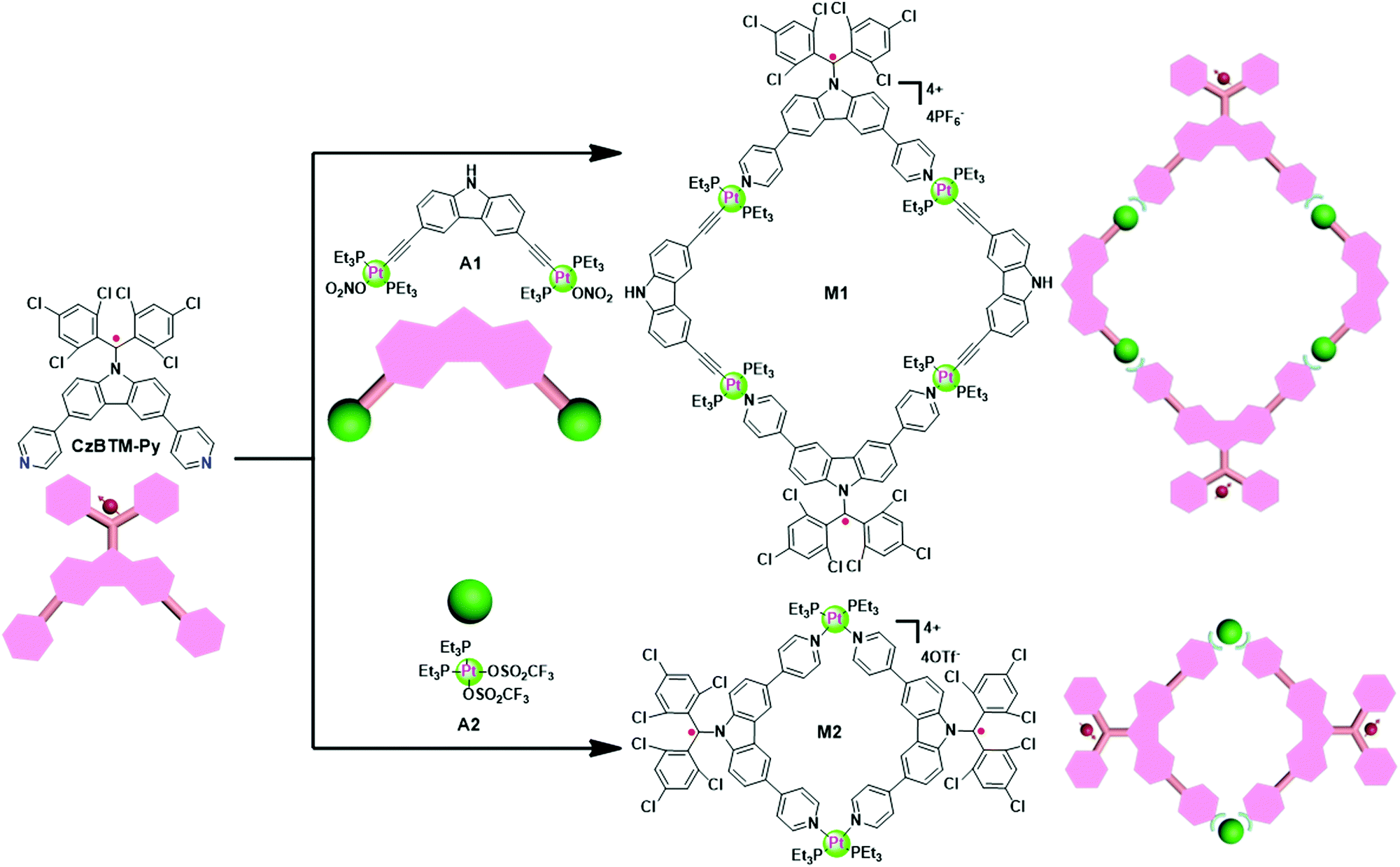
Since CzBTM-Py is a nearly 90° dipyridyl acceptor, two complementary metal acceptors of A1 and A2 are elaborately chosen to construct two metallacycles with different shapes and sizes.63,64 As shown in Scheme 2, metallacycle M1 with PF6− counterion was prepared by simply mixing the radical ligand CzBTM-Py with the 90° diplatinum(II) acceptor A1 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in a mixed solvent of acetone and water at 50 °C, and then adding a saturated aqueous solution of KPF6 to precipitate the dark red products with high yield up to 90%. Similarly, metallacycle M2 with OTf− (OTf− = trifluoromethanesulfonate) counterion was prepared in high yield (∼90%) via coordination-driven self-assembly from CzBTM-Py and corresponding acceptor of A2 in CH2Cl2 at room temperature. It is also worth noting that the open-shell nature of CzBTM-Py is preserved in the resultant metallacycles M1 and M2, mainly because of the relatively long persistence of radical CzBTM-Py and the mild reaction conditions in such coordination-driven self-assembly process. Consequently, the signal peak broadening was also significant in the 1H NMR spectra of M1 and M2, making it difficult to characterize their structure by conventional NMR spectroscopy (Fig. S14–S17, ESI†). Nevertheless, the ESI-MS-TOF mass spectrometry provided further evidence for the formation of the target of M1 and M2 (Fig. S20 and S21, ESI†). Specifically, the observed peaks for M1 and M2 were all isotopically resolved and agreed well with their corresponding simulated isotope pattern, supporting the formation of a discrete structure as the sole assembly product. Finally, the absolute structures of M1 and M2 were successfully determined by X-ray crystallographic analysis (vide infra).
:1 ratio in a mixed solvent of acetone and water at 50 °C, and then adding a saturated aqueous solution of KPF6 to precipitate the dark red products with high yield up to 90%. Similarly, metallacycle M2 with OTf− (OTf− = trifluoromethanesulfonate) counterion was prepared in high yield (∼90%) via coordination-driven self-assembly from CzBTM-Py and corresponding acceptor of A2 in CH2Cl2 at room temperature. It is also worth noting that the open-shell nature of CzBTM-Py is preserved in the resultant metallacycles M1 and M2, mainly because of the relatively long persistence of radical CzBTM-Py and the mild reaction conditions in such coordination-driven self-assembly process. Consequently, the signal peak broadening was also significant in the 1H NMR spectra of M1 and M2, making it difficult to characterize their structure by conventional NMR spectroscopy (Fig. S14–S17, ESI†). Nevertheless, the ESI-MS-TOF mass spectrometry provided further evidence for the formation of the target of M1 and M2 (Fig. S20 and S21, ESI†). Specifically, the observed peaks for M1 and M2 were all isotopically resolved and agreed well with their corresponding simulated isotope pattern, supporting the formation of a discrete structure as the sole assembly product. Finally, the absolute structures of M1 and M2 were successfully determined by X-ray crystallographic analysis (vide infra).
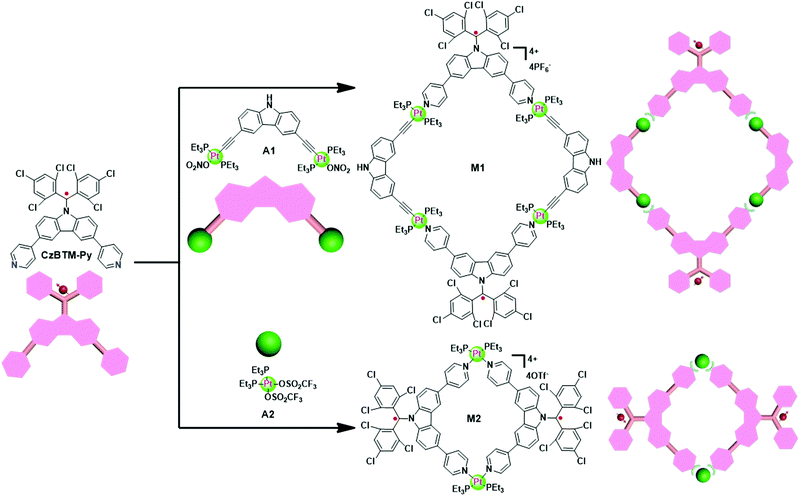 |
| | Scheme 2 Self-assembly of metallacycles M1 and M2. | |
CW-EPR spectroscopy studies
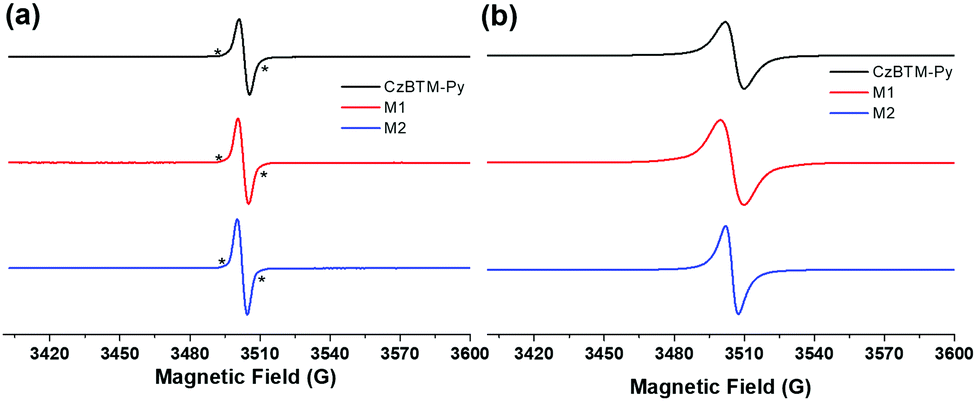
CW-EPR is then employed to characterize the radical signature of CzBTM-Py, M1 and M2. As shown in Fig. 2a, solution-state CW-EPR spectra of CzBTM-Py, M1 and M2 all revealed a dominant sharp peak with g ≈ 2.003, illustrating the existence of the unpaired electron in their structures. The sharp peak was accompanied by very weak satellite lines, which were attributed to the natural abundant 13C hyperfine coupling. The hyperfine coupling with 1H or 14N was hardly observed, implying that the spin density is mainly distributed on the central trivalent C atom. Similarly, solid-state EPR spectra of CzBTM-Py, M1 and M2 also showed a single peak, while the peak became significantly broadened due to their dipole–dipole interactions in the solid state (Fig. 2b). No forbidden half-field transition was observed in the EPR spectra of M1 and M2 despite of their potential biradical nature in the ground state. The absence of the forbidden half-field transition implies the very week spin–spin interactions in M1 and M2, mainly attributed to the very long distance between the two radicals in M1 and M2, which do not favor the spin–spin interaction.65
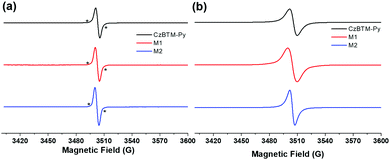 |
| | Fig. 2 Room-temperature CW X-band EPR spectra of CzBTM-Py, M1 and M2 in the solution state (a) and solid state (b). * indicates 13C hyperfine coupling. | |
X-ray crystallographic analysis
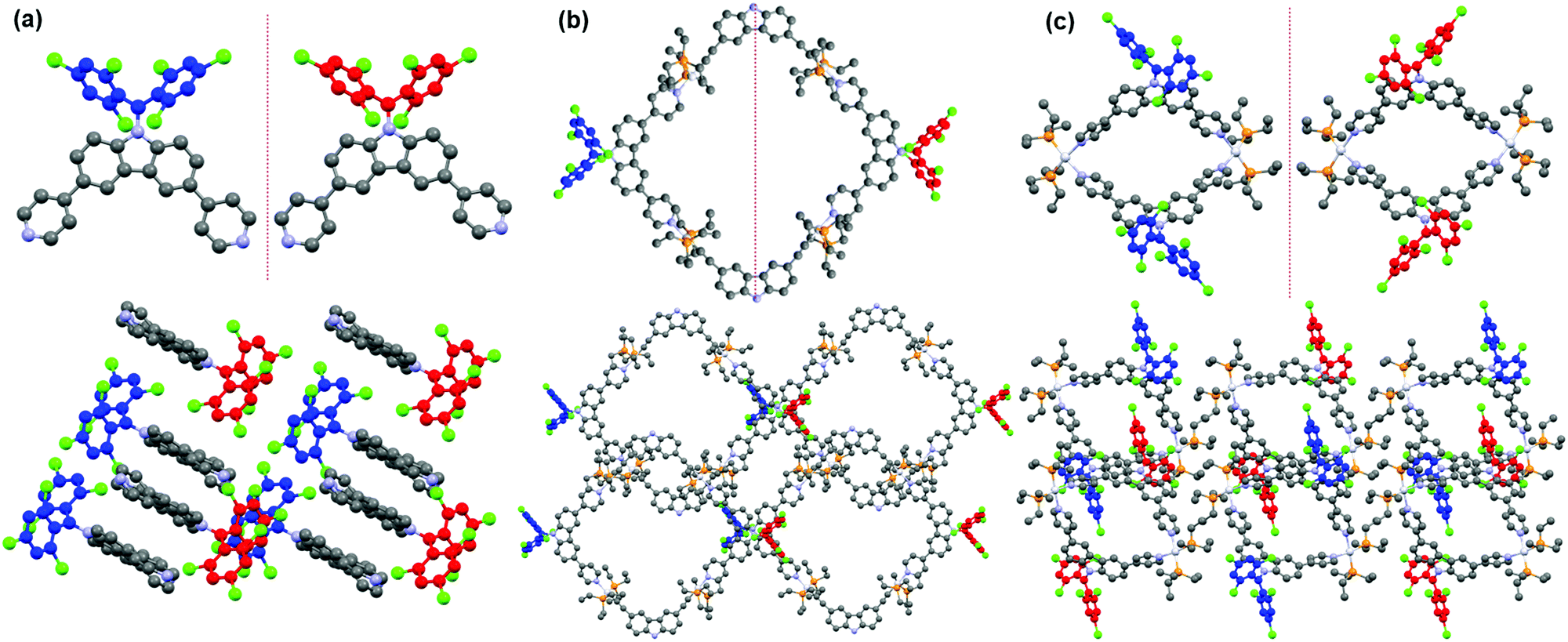
X-ray-quality single crystal of radical ligand CzBTM-Py was easily grown by slow diffusion of methanol into chloroform solution at room temperature. As shown in Fig. 3a, the crystal structure of CzBTM-Py consists of a trivalent, sp2-hybridized carbon atom that is connected to three sterically hindered aromatic rings, implying its open-shell nature. The bulky chlorine atoms caused the two 2,4,6-trichlorophenyl groups to have a highly twist orientation, i.e., the torsion angles between the plane of carbazole group and the plane of trichlorophenyl group were determined to be approximately 48.12° and 44.63° (Fig. 3a). CzBTM-Py crystalized in layers with strong intermolecular π–π stacking interaction (Fig. 3a). Single crystals of metallacycles M1 and M2 were also grown by slow diffusion of isopropyl ether into dichloromethane solution at the room temperature. After coordination, the structural integrity of CzBTM-Py is maintained in M1 and M2, further implied that the radical is persistent under this mild self-assembly reaction condition. In their crystal forms, metallacycle M1 was nearly square and planar, while metallacycle M2 exhibited a bowl shape as indicated from the side view (Fig. 3b and c). The non-planar conformation of M2 is mainly due to the reversible thermodynamic nature of the coordination-driven self-assembly process wherein CzBTM-Py and A2 are prone to adopt the most comfortable coordination geometry and form the thermodynamically stable [2+2] metallacycle M2. The diameters of the internal cavity of M1 and M2 were determined to be approximately 13 Å and 6 Å, respectively. Notably, the spin–spin distances between the two radicals in M1 and M2 were around 30 Å and 14 Å, respectively. Such ultra-long distance could account for the weak spin–spin interactions in M1 and M2 as revealed by their single-line EPR spectra.65 The metallacycles M1 and M2 further packed into columns with well-defined channel size. No obvious intermolecular stacking interaction was existed between the radical centers, implying the weak intermolecular spin–spin interactions in M1 and M2. To the best of our knowledge, this represents one of very few examples of the discrete radical metallacycles being characterized by X-ray crystallography.
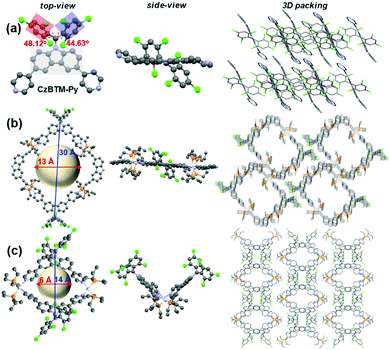 |
| | Fig. 3 ORTEP drawings and 3D packing structures of (a) CzBTM-Py, (b) M1, and (c) M2. Hydrogen atoms, anions and solvent are omitted for clarity. | |
Chiral self-sorting behavior
Polychlorotriphenylmethyl radicals and their derivatives are intrinsically chiral because of their propeller-like conformation with a considerable rotational energy barrier.66 As a result, these radicals have two atropisomeric forms of Plus (P) and Minus (M) according to the Cahn–Ingold–Prelog (CIP) nomenclature.67 Similarly, radical ligand CzBTM-Py also exists in two different enantiomeric forms M (blue) and P (red) as indicated in its single crystal (Fig. 4a). The energy barrier of CzBTM-Py was calculated to be ∼23.23 kcal mol−1 (Fig. S27, ESI†), which also agree well with the literature.66 Upon self-assembly with the complementary metal acceptors, several possible stereoisomers are expected in the resultant [2+2] metallacycles M1 and M2, including the homochiral assemblies of MM and PP, and the heterochiral assembly of PM. Interestingly, metallacycle M1 is heterochiral assembly consisting of a M-enantiomer and a P-enantiomer of CzBTM-Py, while no homochiral assemblies of MM or PP enantiomer is observed in this process (Fig. 4b). In comparison, self-assembly of two CzBTM-Py units with A2 results in only the MM/PP enantiomers couple of metallacycle M2 (Fig. 4c), whereas the heterochiral assembly of PM is not observed in this coordination-driven self-assembly process. Therefore, these results imply the distinct chiral self-sorting behavior of the metallacycles M1 and M2 along their self-assembly process. The underlying mechanism of such chiral self-sorting behavior is not clear and might be related to the reversible thermodynamic nature of the coordination-driven self-assembly process in which the final coordination species tend to form the thermodynamically preferred discrete supramolecular entities in their single crystals.68 Notably, the two enantiomers of CzBTM-Py and M2 are difficult to separate because of their relatively small energy barrier at room temperature, and also the lack of the suitable chiral stationary phase (CSP) columns.69
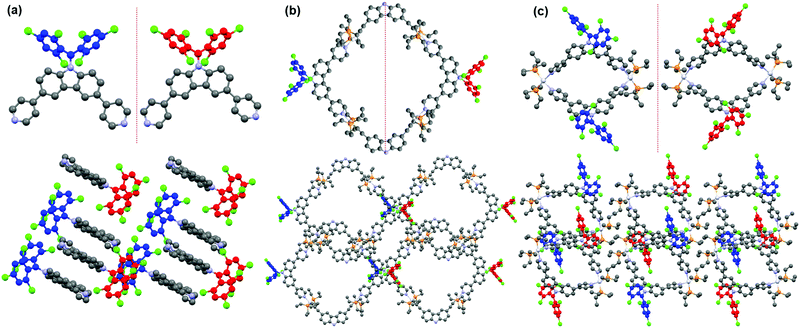 |
| | Fig. 4 (a) M (blue) and P (red) enantiomer of CzBTM-Py. Chiral self-sorting behavior of metallacycles M1 (b) and M2 (c). | |
Photophysical properties
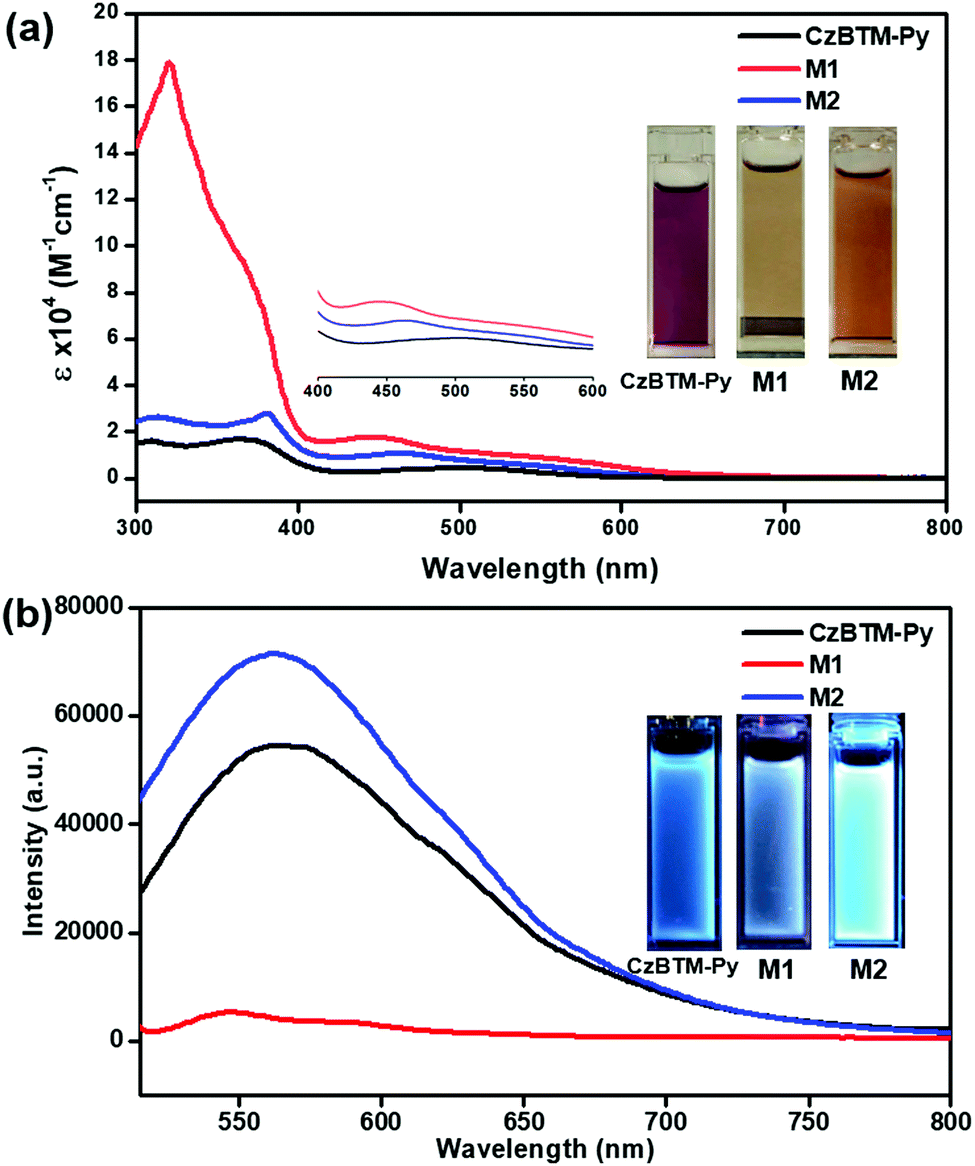
Very recently, polychlorotriphenylmethyl radicals and their derivatives have demonstrated to exhibit peculiar luminescent feature, which is very important to improve the efficiency of conventional OLED.17–22 Thus, the photophysical properties of CzBTM-Py, M1 and M2 have been surveyed in this study. As shown in Fig. 5a, CzBTM-Py featured two relatively strong absorption peaks between 300 nm and 400 nm and a very weak broad absorption band extending from ca. 420 nm into ca. 650 nm. The resultant broad absorption band can be assigned to the characteristic absorption of carbon-centered radicals. After coordination, metallacycle M2 experienced a very similar absorption profile to that of CzBTM-Py, while metallacycle M1 exhibited strong absorption under 400 nm. The strong absorption peak at 320 nm is very likely caused by the acceptor ligand of A1 which contains carbazole moiety since the later one showed absorption peak in this region (Fig. S22, ESI†). Notably, the molar absorption coefficient of metallacycles M1 and M2 is significantly enhanced compared with that of radical ligand CzBTM-Py largely because the metallacycles contain two CzBTM-Py chromophores (Fig. S23, ESI†). As a consequence, the solution of CzBTM-Py is somewhat red while the color of metallacycles M1 and M2 is yellow brown. With regard to their emission properties (Fig. 5b), CzBTM-Py is very weak luminescent in contrast to its precursor analogue CzBTM featuring strong emission.62 The weak emissive property of CzBTM-Py might be attributed to the potential enhanced intersystem crossing induced by the 4-pyridyl substituent, thus quenching the ligand fluorescence.70 The preliminary results also revealed that the supramolecular coordination seemed to have an effect on the luminescent properties of radicals, e.g., coordination-driven self-assembly process decreased the fluorescence intensity of metallacycle M1 while increased the fluorescence intensity of M2. Notably, the fluorescence quantum yield (Φ) of CzBTM-Py, M1 and M2 was extremely low and the exact Φ value was virtually impossible to probe by the relative method (Fig. S24, ESI†). Interestingly, CzBTM-Py, M1 and M2 displayed very weak blue emission in dark upon UV-light irradiation (365 nm), which is mainly derived from the carbazole chromophore (Fig. 5b). The underlying mechanism of the coordination effect on the emissive properties of radicals is unknown and might be related to the metal-to-ligand charge transfer (MLCT) or ligand-to-metal charge transfer (LMCT),71 and the related investigation is in progress in our laboratory.
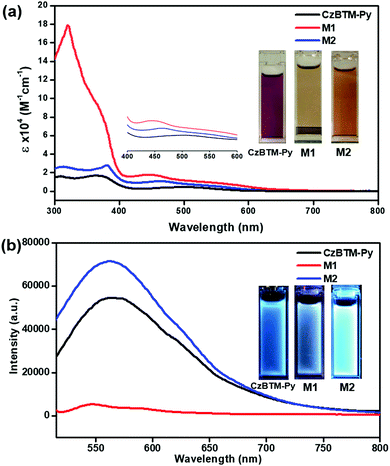 |
| | Fig. 5 (a) UV-Vis absorption spectra of 10 μM CzBTM-Py, M1 and M2 in CH2Cl2 solution; (b) emission spectra of CzBTM-Py (10 μM), M1 (5 μM) and M2 (5 μM) in CH2Cl2 solution (λex = 490 nm); insert are the photos of their solutions under daylight (a) and UV light (365 nm) (b). | |
Stability studies
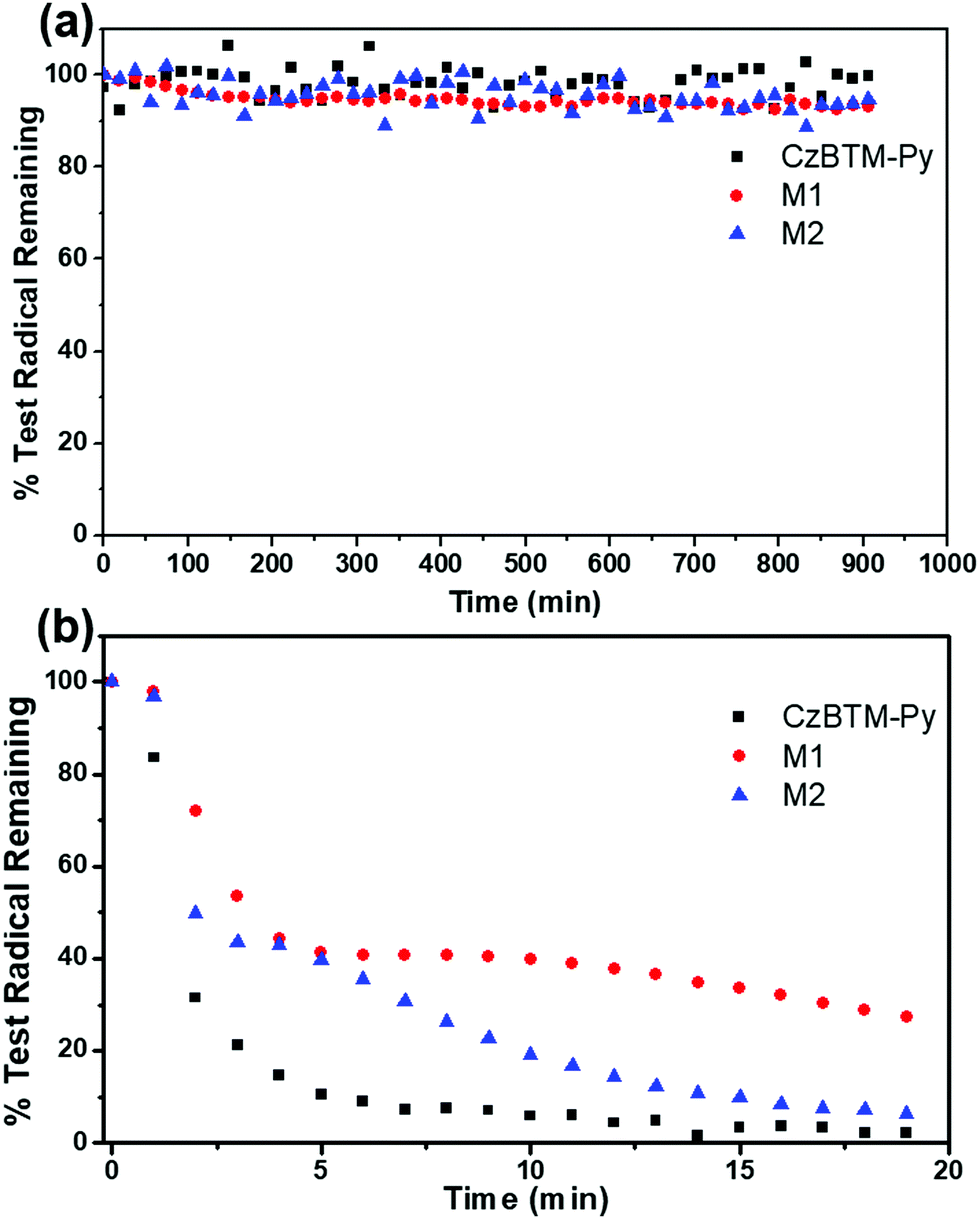
Polychlorotriphenylmethyl radicals are reported to be persistent and stable to air and moisture, and can be kept for a long period in the absence of light.66 As shown in Fig. 6a and Fig. S2–S4 (ESI†), the EPR intensity of the solution of CzBTM-Py, M1 and M2 was measured over 1000 min in the dark condition. The results revealed that CzBTM-Py, M1 and M2 were very stable even after long-term storage under ambient condition, which is consistent with the literature. However, upon irradiation (200 to 2000 nm) CzBTM-Py was thoroughly decomposed within 5 min, while a much longer time (∼20 min) was needed for M2 to be completely degraded (Fig. 6a and Fig. S5–S8, ESI†). Notably, metallacycle M1 had a higher resistance to light compared to CzBTM-Py and M2, e.g., M1 still kept about 30% while the other two were totally decomposed under this irradiation condition. The enhancement of the photostability of metallacycles was probably due to their rigid and tightly packed architectures, which decreases its exposed molecular surface area and protects it from oxygen and moisture under light condition. In addition, CzBTM-Py, M1 and M2 could be heated up to 250 °C as confirmed from their thermogravimetric analysis (TGA), indicating their good thermal stability (Fig. S9, ESI†).
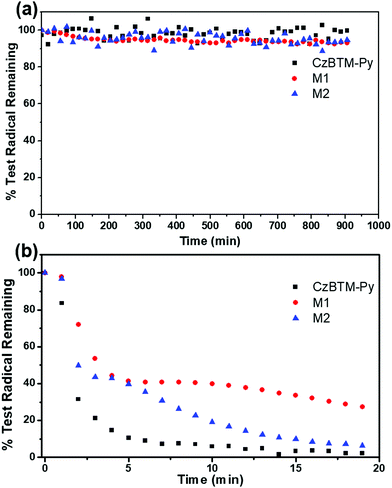 |
| | Fig. 6 EPR signal fading of CzBTM-Py, M1 and M2 in the dark condition (a) and upon irradiation (b). | |
Experimental
Compound 4 and metal acceptors of A1 and A2 were prepared according to the literature.62–64,72
Synthesis of 3
A degassed solution of the 3,6-dibromocarbazole 1 (5.0 g, 15.0 mmol) in THF/H2O (4:1, 200 mL) were added 4-pyridinylboronic acid 2 (11.4 g, 90.0 mmol), Pd(PPh3)4 (1.8 g, 1.5 mmol) and Na2CO3 (6.5 g, 61.0 mmol). The resultant solution was stirred at 80 °C for 48 h under an inert atmosphere. The solvent was then removed by evaporation on a rotary evaporator. The residue was purified by column chromatography on silica gel (dichloromethane/acetone = 1:1, two drops of NEt3) to get the product 3 as a pale-yellow solid (2.4 g, 50% yield). 1H NMR (500 MHz, DMSO-d6, ppm): δ 11.67 (s, 1H), 8.83 (s, 2H), 8.64 (d, J = 5.4 Hz, 4H), 7.91 (d, J = 8.5 Hz, 2H), 7.85 (d, J = 5.5 Hz, 4H), 7.64 (d, J = 8.5 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 150.23, 147.95, 141.01, 127.97, 124.93, 123.46, 121.01, 119.39, 111.99. HR-EI-MS: m/z calculated for C22H15N3 [M]+: 321.1266, found: 321.1270.
Synthesis of CzBTM-Py
Under N2 atmosphere, sodium hydride (60% in oil, 58.7 mg, 1.5 mmol) was dispersed in a solution of anhydrous dimethyl sulfoxide (20 mL), compound 3 (500.0 mg, 1.6 mmol) dissolved in anhydrous dimethyl sulfoxide (5 mL) was added dropwise and stirred until that no gas was generated. Compound 4 (458.3 mg, 1.0 mmol) was added into the mixture and stirred for 3 h under 55 °C. Next, the mixture was cooled to room temperature and saturated ammonium chloride solution (50 mL) was added slowly. The precipitate was collected by suction filtration and purified by flash column chromatography (silica gel, dichloromethane/petroleum ether = 1:8), after that, preparative thin layer chromatography (PTLC) was carried out to get the pure product as a dark red solid (150.0 mg, 24% yield). 1H NMR and 13C NMR of CzBTM-Py are not detected owing to its strong paramagnetic broadening. HR-ESI-MS: m/z calculated for C35H18Cl6N3 [M + H]+: 692.9602, found: 692.9672.
Self-assembly of metallacycle M1
CzBTM-Py (20.0 mg, 28.8 μmol) and diplatinum(II) acceptor A1 (34.6 mg, 28.8 μmol) were weighed accurately into a glass vial, and then added 5.0 mL acetone and 1.0 mL water. The reaction solution was then stirred at 55 °C in dark overnight to yield a homogeneous fuchsia solution. Then the saturated KPF6 solution was added into the bottle with continuous stirring (5 min) to precipitate the product. The reaction mixture was centrifuged, washed several times with water, and dried by freeze-dryer. The product M1 was obtained as a dark red solid (53.4 mg, 90% yield).
Self-assembly of metallacycle M2
CzBTM-Py (10.0 mg, 14.4 μmol) and organoplatinum acceptor A2 (10.5 mg, 14.4 μmol) were weighed accurately into a glass vial, and then added 2.0 mL CH2Cl2. The reaction solution was then stirred at room temperature in the dark for 12 hours. After removing most of the solvent, some drops of ether was added to precipitate the product. The reaction mixture was then centrifuged, washed several times with ether. Dark red powder M2 was gained in 90% yield (18.4 mg) after drying the residue.
Conclusions
In summary, we have designed and synthesized a new ditopic radical ligand CzBTM-Py bearing two 4-pyridyl groups, which has persistent stability and well-defined coordination geometry that is readily able to coordinate with suitable di-Pt(II) acceptors. Two well-defined radical metallacycles M1 and M2 were then successfully constructed via the coordination-driven self-assembly approach. The structures of CzBTM-Py, M1 and M2 were unambiguously determined by mass spectrometry and X-ray crystallography. Metallacycle M1 was nearly square and planar while metallacycle M2 exhibited a bowl shape in the solid state, illustrating the metal ligand effect on the geometry of assemblies. Intriguingly, a distinct chiral self-sorting behavior of the metallacycles M1 and M2 was also observed in their single crystals. Moreover, this study also proves that the supramolecular coordination approach could affect the photophysical properties and stabilities of CzBTM-Py. Therefore, this work has demonstrated the power of coordination-driven self-assembly approach to construct radical-based supramolecular coordination complexes which may serve as functional metal–organic radical materials in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
H.-B. Y. thanks Innovation Program of Shanghai Municipal Education Commission (No. 2019-01-07-00-05-E00012), Program for Changjiang Scholars and Innovative Research Team in University for financial support. X. S. acknowledges the financial supports sponsored by NSFC/China (No. 22071061), Shanghai Sailing Program (19YF1412900), and the Fundamental Research Funds for the Central Universities. We thank the staffs from BL17B beamline of National Facility for Protein Science in Shanghai (NFPS) at Shanghai Synchrotron Radiation Facility, for assistance during data collection.
Notes and references
- M. Gomberg, An instance of trivalent carbon: triphenylmethyl, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- R. G. Hicks, What's new in stable radical chemistry?, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC.
- K. Kato and A. Osuka, Platforms for Stable Carbon-Centered Radicals, Angew. Chem., Int. Ed., 2019, 58, 8978–8986 CrossRef CAS.
- Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Pro-aromatic and anti-aromatic π-conjugated molecules: an irresistible wish to be diradicals, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- I. Ratera and J. Veciana, Playing with organic radicals as building blocks for functional molecular materials, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- M. Abe, Diradicals, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS.
- S. Mukherjee and B. List, Radical catalysis, Nature, 2007, 447, 152–153 CrossRef CAS.
- D. Leifert and A. Studer, The Persistent Radical Effect in Organic Synthesis, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, Radicals: Reactive Intermediates with Translational Potential, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS.
- O. I. Aruoma, Nutrition and health aspects of free radicals and antioxidants, Food Chem. Toxicol., 1994, 32, 671–683 CrossRef CAS.
- E. G. Janzen and Spin Trapping, Acc. Chem. Res., 1971, 4, 31–40 CrossRef CAS.
- M. R. Fleissnera, E. M. Brustad, T. Kalaic, C. Altenbacha, D. Casciod, F. B. Petersb, K. Hidegc, S. Peukere, P. G. Schultzb and W. L. Hubbe, Site-directed spin labeling of a genetically encoded unnatural amino acid, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21637–21642 CrossRef.
- P. Kuppusamy, M. Chzhan, K. Vu, M. Shteynbuk, D. J. Lefer, E. Giannella and J. L. Zweier, Three-dimensional spectral-spatial EPR imaging of free radicals in the heart: A technique for imaging tissue metabolism and oxygenation, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3388–3392 CrossRef CAS.
- A. S. L. Thankamony, J. J. Wittmann, M. Kaushik and B. Corzilius, Dynamic nuclear polarization for sensitivity enhancement in modern solid-state NMR, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 102-103, 120–195 CrossRef.
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Air-Stable Organic Radicals: New-Generation Materials for Flexible Electronics?, Adv. Mater., 2020, 32, 1908015 CrossRef CAS.
- G.-F. Huo, Q. Tu, X.-L. Zhao, X. Shi and H.-B. Yang, Synthesis and characterization of an unexpected mechanochromicbistricyclic aromatic ene, Chin. Chem. Lett., 2020, 31, 1847–1850 CrossRef CAS.
- S. Castellanos, D. Velasco, F. Llpez-Calahorra, E. Brillas and L. Juliá, Taking Advantage of the Radical Character of Tris(2,4,6-trichlorophenyl)methyl To Synthesize New Paramagnetic Glassy Molecular Materials, J. Org. Chem., 2008, 73, 3759–3767 CrossRef CAS.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Organic Light-Emitting Diodes Using a Neutral π Radical as Emitter: The Emission from a Doublet, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Efficient radical-based light-emitting diodes with doublet emission, Nature, 2018, 563, 536–540 CrossRef CAS.
- Z. Cui, A. Abdurahman, X. Ai and F. Li, Stable Luminescent Radicals and Radical-Based LEDs with Doublet
Emission, CCS Chem., 2020, 2, 1129–1145 CrossRef CAS.
- S. Kimura, T. Kusamoto, S. Kimura, K. Kato, Y. Teki and H. Nishihara, Magnetoluminescence in a Photostable, Brightly Luminescent Organic Radical in a Rigid Environment, Angew. Chem., Int. Ed., 2018, 57, 12711–12715 CrossRef CAS.
- K. Kato, S. Kimura, T. Kusamoto, H. Nishihara and Y. Teki, Luminescent Radical-Excimer: Excited-State Dynamics of Luminescent Radicals in Doped Host Crystals, Angew. Chem., Int. Ed., 2019, 58, 2606–2611 CrossRef CAS.
-
M. Lucarini, Supramolecular Radical Chemistry, Basic Concepts and Methodologies, in Encyclopedia of Radicals in Chemistry, Biology and Materials, Wiley, 2012, vol. 2, p. 229 Search PubMed.
- B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Tuning the stability of organic radicals: from covalent approaches to non-covalent approaches, Chem. Sci., 2020, 11, 1192–1204 RSC.
- Y. Wang, M. Frasconi and J. F. Stoddart, Introducing Stable Radicals into Molecular Machines, ACS Cent. Sci., 2017, 3, 927–935 CrossRef CAS.
- W. S. Jeon, H.-J. Kim, C. Lee and K. Kim, Control of the stoichiometry in host–guest complexation by redox chemistry of guests: Inclusion of methylviologen in cucurbit[8]uril, Chem. Commun., 2002, 1828–1829 RSC.
- A. Y. Ziganshina, Y. H. Ko, W. S. Jeon and K. Kim, Stable π-dimer of a tetrathiafulvalene cation radical encapsulated in the cavity of cucurbit[8]uril, Chem. Commun., 2004, 806–807 RSC.
- A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Cotí, D. Benítez, E. Tkatchouk, J.-C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, Radically enhanced molecular recognition, Nat. Chem., 2010, 2, 42–49 CrossRef CAS.
- B. Tang, W.-L. Li, Y. Chang, B. Yuan, Y. Wu, M.-T. Zhang, J.-F. Xu, J. Li and X. Zhang, A Supramolecular Radical Dimer: High-Efficiency NIR-II Photothermal Conversion and Therapy, Angew. Chem., Int. Ed., 2019, 58, 15526–15531 CrossRef CAS.
- D.-W. Zhang, J. Tian, L. Chen, L. Zhang and Z.-T. Li, Dimerization of Conjugated Radical Cations: An Emerging Non-Covalent Interaction for Self-Assembly, Chem. – Asian J., 2015, 10, 56–68 CrossRef CAS.
- K. Nakabayashi, Y. Ozaki, M. Kawano and M. Fujita, A Self-Assembled Spin Cage, Angew. Chem., Int. Ed., 2008, 47, 2046–2048 CrossRef CAS.
- Y. Ozaki, M. Kawano and M. Fujita, Engineering noncovalent spin–spin interactions in an organic-pillared spin cage, Chem. Commun., 2009, 4245–4247 RSC.
- Y. Ueda, H. Ito, D. Fujita and M. Fujita, Permeable Self-Assembled Molecular Containers for Catalyst Isolation Enabling Two-Step Cascade Reactions, J. Am. Chem. Soc., 2017, 139, 6090–6093 CrossRef CAS.
- K. Yazaki, S. Noda, Y. Tanaka, Y. Sei, M. Akita and M. Yoshizawa, An M2L4 Molecular Capsule with a Redox Switchable Polyradical Shell, Angew. Chem., Int. Ed., 2016, 55, 15031–15034 CrossRef CAS.
- L.-X. Cai, S.-C. Li, D.-N. Yan, L.-P. Zhou, F. Guo and Q.-F. Sun, Water-Soluble Redox-Active Cage Hosting Polyoxometalates for Selective Desulfurization Catalysis, J. Am. Chem. Soc., 2018, 140, 4869–4876 CrossRef CAS.
- J. Wang, C. He, P. Wu, J. Wang and C. Duan, An Amide-Containing Metal-Organic Tetrahedron Responding to a Spin-Trapping Reaction in a Fluorescent Enhancement Manner for Biological Imaging of NO in Living Cells, J. Am. Chem. Soc., 2011, 133, 12402–12405 CrossRef CAS.
- G.-F. Huo, X. Shi, Q. Tu, Y.-X. Hu, G.-Y. Wu, G.-Q. Yin, X. Li, L. Xu, H.-M. Ding and H.-B. Yang, Radical-Induced Hierarchical Self-Assembly Involving Supramolecular Coordination Complexes in Both Solution and Solid States, J. Am. Chem. Soc., 2019, 141, 16014–16023 CrossRef CAS.
- R. Poli, Radical Coordination Chemistry and Its Relevance to Metal-Mediated Radical Polymerization, Eur. J. Inorg. Chem., 2011, 1513–1530 CrossRef CAS.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M. Tong, A. Mansikkamaki and R. A. Layfield, Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet, Science, 2018, 362, 1400–1403 CrossRef CAS.
- K. Chakarawet, T. D. Harris and J. R. Long, Semiquinone radical-bridged M2 (M = Fe, Co, Ni) complexes with strong magnetic exchange giving rise to slow magnetic relaxation, Chem. Sci., 2020, 11, 8196–8203 RSC.
- S. Demir, I.-R. Jeon, J. F. Long and T. D. Harris, Radical ligand-containing single-molecule magnet, Coord. Chem. Rev., 2015, 289–290, 149–176 CrossRef CAS.
- Y. Hattori, T. Kusamoto and H. Nishihara, Enhanced Luminescent Properties of an Open-Shell (3,5-Dichloro-4-pyridyl)bis(2,4,6-trichlorophenyl)methyl Radical by Coordination to Gold, Angew. Chem., Int. Ed., 2015, 54, 3731–3734 CrossRef CAS.
- Y. Hattori, T. Kusamoto, T. Sato and H. Nishihara, Synergistic luminescence enhancement of a pyridyl-substituted triarylmethyl radical based on fluorine substitution and coordination to gold, Chem. Commun., 2016, 52, 13393–13396 RSC.
- T. B. Faust and D. M. D’Alessandro, Radicals in metal–organic frameworks, RSC Adv., 2014, 4, 17498–17512 RSC.
- D. I. Alexandropoulos, B. S. Dolinar, K. R. Vignesh and K. R. Dunbar, Putting a New Spin on Supramolecular Metallacycles: Co3 Triangle and Co4 Square Tetrazine-Based Radicals as Bridges, J. Am. Chem. Soc., 2017, 139, 11040–11043 CrossRef CAS.
- W.-L. Jiang, Z. Peng, B. Huang, X.-L. Zhao, D. Sun, X. Shi and H.-B. Yang, TEMPO Radical-Functionalized Supramolecular Coordination Complexes with Controllable Spin–Spin Interactions, J. Am. Chem. Soc., 2021, 143, 433–441 CrossRef CAS.
- Y.-F. Han, W.-G. Jia, W.-B. Yu and G.-X. Jin, Stepwise formation of organometallic macrocycles, prisms and boxes from Ir, Rh and Ru-based half-sandwich units, Chem. Soc. Rev., 2009, 38, 3419–3434 RSC.
- W. Xuan, C. Zhu, Y. Liu and Y. Cui, Mesoporous metal-organic framework materials, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC.
- X. Han, C. Yuan, B. Hou, L. Liu, H. Li, Y. Liu and Y. Cui, Chiral Covalent Organic Frameworks: Design, Synthesis and Property, Chem. Soc. Rev., 2020, 49, 6248–6272 RSC.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Supramolecular Coordination: Self-Assembly of Finite Two- and Three-Dimensional Ensembles, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Metal–Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal–Organic Materials, Chem. Rev., 2013, 113, 734–777 CrossRef CAS.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Stimuli-Responsive Metal-Ligand Assemblies, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS.
- L.-J. Chen and H.-B. Yang, Construction of Stimuli-Responsive Functional Materials via Hierarchical Self-Assembly Involving Coordination Interactions, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS.
- M. Pan, K. Wu, J.-H. Zhang and C.-Y. Su, Chiral metal-organic cages/containers (MOCs): from structural and stereochemical design to applications, Coord. Chem. Rev., 2019, 378, 333–349 CrossRef CAS.
- M. Pan, W.-M. Liao, S.-Y. Yin, S.-S. Sun and C.-Y. Su, Single-Phase White-Light-Emitting and Photoluminescent Color Tuning Coordination Assemblies, Chem. Rev., 2018, 118, 8889–8935 CrossRef CAS.
- H. Cai, Y.-L. Huang and D. Li, Biological metal-organic frameworks: Structures, host–guest chemistry and bio-applications, Coord. Chem. Rev., 2019, 378, 207–221 CrossRef CAS.
- H. Zeng, M. Xie, Y.-L. Huang, Y. Zhao, X.-J. Xie, J.-P. Bai, M.-Y. Wan, R. Krishna, W. Lu and D. Li, Induced Fit of C2H2 in a Flexible MOF Through Cooperative Action of Open Metal Sites, Angew. Chem., Int. Ed., 2019, 58, 8515–8519 CrossRef CAS.
- Z.-Y. Li, Y. Zhang, C.-W. Zhang, L.-J. Chen, C. Wang, H. Tan, Y. Yu, X. Li and H.-B. Yang, Cross-Linked Supramolecular Polymer Gels Constructed from Discrete Multi-pillar[5]arene Metallacycles and Their Multiple Stimuli-Responsive Behavior, J. Am. Chem. Soc., 2014, 136, 8577–8589 CrossRef CAS.
- G.-Y. Wu, X. Shi, H. Phan, H. Qu, Y.-X. Hu, X.-L. Zhao, X. Li, L. Xu, Q. Yu and H.-B. Yang, Efficient self-assembly of heterometallic triangular necklace with strong antibacterial activity, Nat. Commun., 2020, 11, 3178 CrossRef CAS.
- Y. Hu, W. Wang, R. Yao, X.-Q. Wang, Y.-X. Wang, B. Sun, L.-J. Chen, Y. Zhang, X.-L. Zhao, L. Xu, H.-W. Tan, Y. Yu, X. Li and H.-B. Yang, Facile synthesis of diverse rotaxanes via successive supramolecular transformations, Mater. Chem. Front., 2019, 3, 2397–2402 RSC.
- W. Zheng, G. Yang, S.-T. Jiang, N. Shao, G.-Q. Yin, L. Xu, X. Li, G. Chen and H.-B. Yang, A tetraphenylethylene (TPE)-based supra-amphiphilic organoplatinum(II) metallacycle and its self-assembly behavior, Mater. Chem. Front., 2017, 1, 1823–1828 RSC.
- X. Ai, Y. Chen, Y. Feng and F. Li, A Stable Room-Temperature Luminescent Biphenylmethyl Radical, Angew. Chem., Int. Ed., 2018, 57, 2869–2873 CrossRef CAS.
- S. Shanmugaraju, A. K. Bar, K.-W. Chi and P. S. Mukherjee, Coordination-Driven Self-Assembly of Metallamacrocycles via a New PtII2 Organometallic Building Block with 90° Geometry and Optical Sensing of Anions, Organometallics, 2010, 29, 2971–2980 CrossRef CAS.
- S. Shanmugaraju, V. Vajpayee, S. Lee, K.-W. Chi, P. J. Stang and P. S. Mukherjee, Coordination-Driven Self-Assembly of 2D-Metallamacrocycles Using a New Carbazole-Based Dipyridyl Donor: Synthesis, Characterization, and C60 Binding Study, Inorg. Chem., 2012, 51, 4817–4823 CrossRef CAS.
- S. S. Eaton, K. M. More, B. M. Sawant and G. R. Eaton, Use of the ESR half-field transition to determine the interspin distance and the orientation of the interspin vector in systems with two unpaired electrons, J. Am. Chem. Soc., 1983, 105, 6560–6567 CrossRef CAS.
-
J. Veciana and I. Ratera, Polychlorotriphenylmethyl Radicals: Towards Multifunctional Molecular Materials. in Stable Radicals Fundamentals and Applied Aspects of Odd-Electron Compounds, Wiley, 2010, Chapter 2 Search PubMed.
- R. S. Cahn, S. C. Ingold and V. Prelog, Specification of Molecular Chirality, Angew. Chem., Int. Ed., 1966, 5, 385–415 CrossRef CAS.
- M. M. Safont-Sempere, G. Fernandez and F. Wurthner, Self-Sorting Phenomena in Complex Supramolecular Systems, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS.
- P. M. Burrezo, V. G. Jiménez, D. Blasi, I. Ratera, A. G. Campaña and J. Veciana, Organic Free Radicals as Circularly Polarized Luminescence Emitters, Angew. Chem., Int. Ed., 2019, 58, 16282–16288 CrossRef.
- Y. Teki, Excited-State Dynamics of Non-Luminescentand Luminescent π-Radicals, Chem. – Eur. J., 2020, 26, 980–996 CrossRef CAS.
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Highly emissive platinum(II) metallacages, Nat. Chem., 2015, 7, 342–348 CrossRef CAS.
- F. Fochi, P. Jacopozzi, E. Wegelius, K. Rissanen, P. Cozzini, E. Marastoni, E. Fisicaro, P. Manini, R. Fokkens and E. Dalcanale, Self-Assembly and Anion Encapsulation Properties of Cavitand-Based Coordination Cages, J. Am. Chem. Soc., 2001, 123, 7539–7552 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2045293 (CzBTM-Py), 2045295 (M1) and 2045294 (M2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qm00992j |
|
| This journal is © the Partner Organisations 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Haitao
Sun
a,
Haitao
Sun