
Polymer light-emitting electrochemical cells with ultralow salt content: performance enhancement through synergetic chemical and electrochemical doping actions†
Jun
Gao  *a
*a
*a
Abstract
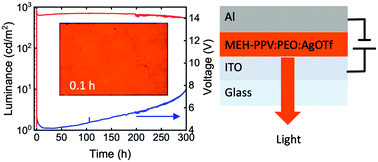
Polymer light-emitting electrochemical cells (PLECs) employing a silver trifluoromethanesulfonate (Ag triflate) salt are demonstrated. The red-emitting PLECs contained 0.2–1 wt% salt, but exhibited a peak luminance of 6000 cd m−2, with high efficiency and minimal efficiency roll-off. Notably, the Ag triflate cells activate more than 100 times faster than an optimized reference cell containing a potassium triflate salt. The high performance of the Ag triflate PLECs can be attributed to the partial chemical doping of the light-emitting polymer by the silver cations. The synergetic action of chemical and electrochemical doping provides a new avenue to design high-performance mixed conductor devices.
- This article is part of the themed collection: 2021 Materials Chemistry Frontiers HOT articles
 Please wait while we load your content...
Please wait while we load your content...

