
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmart active antibiotic nanocarriers with protease surface functionality can overcome biofilms of resistant bacteria†
Paul J.
Weldrick
 a,
Matthew J.
Hardman
b and
Vesselin N.
Paunov
*a
a,
Matthew J.
Hardman
b and
Vesselin N.
Paunov
*a
aDepartment of Chemistry and Biochemistry, University of Hull, Hull, HU6 7RX, UK. E-mail: v.n.paunov@hull.ac.uk; Tel: +44 (0)1482 465660
bCentre for Atherothrombosis and Metabolic Disease, Hull York Medical School, Hull, HU6 7RX, UK
First published on 7th December 2020
Abstract
Treating bacterial infections with species demonstrating antibiotic resistance to the chosen antibiotic is often hindered due to the ability of certain bacteria to grow biofilms where they can effectively hide and resist the antibiotic action. We report an innovative solution for overcoming both antibiotic resistance and biofilm formation by designing active antibiotic nanocarriers with protease surface functionality. We show that this active nanocarrier of common antibiotics can efficiently degrade biofilms of resistant bacteria and bypass their defences. The cationic protease coating, whilst allowing electrostatic adhesion of the nanoparticle to the cell, simultaneously also degrades the biofilm and helps the active nanocarriers to reach the entrapped bacterial cells. We demonstrated this concept by encapsulating Penicillin G and Oxacillin into shellac nanoparticles, subsequently coated with a serine endo-peptidase protease, Alcalase 2.4 L FG. We show for the first time that these active nanocarriers can destroy biofilms of S. aureus resistant to Penicillin G and are significantly more effective in killing the bacterial cells within compared to an equivalent concentration of free antibiotic. The approach of concentrating the antibiotic by encapsulating it into a nanocarrier allows a localised delivery of the antibiotic to the anionic cell wall, facilitated by coating the NPs with a cationic protease. This approach allowed the antibiotic to restore its effectiveness against S. aureus, characterised as resistant to the same antibiotic as well as to cause a rapid degradation of the bacterial biofilm. This approach could be potentially used to revive old antibiotics which have already limited clinical use due to developed resistance.
Introduction
Penicillin is a commonly administered antibiotic used to treat staphylococci and streptococci infections.1 Penicillin is part of the beta-lactam cyclic amine antibiotic family, and all antibiotics in the family used the same bacterial cell wall growth inhibition to kill bacteria.2 There is a range of resistance mechanisms that bacteria have evolved against such antibiotics, some of which are discussed in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3–7 The production of beta-lactamase (which hydrolyses penicillin into penicilloic acid) is common in Staphylococcus aureus, a pathogen that can cause infection in skin, lungs and blood.8 Most strains of S. aureus are now resistant to first-generation penicillin's and so second-generation penicillin's such as oxacillin, nafcillin and methicillin are administered.9 A report by the World Health Organisation in 2014 stated that antibiotic resistance is a growing worldwide threat to public health, driven largely by the lack of new therapeutic agents for treating bacterial infections.10 Overcoming resistance to beta-lactam antibiotics is most commonly achieved by using a beta-lactamase inhibitor such as clavulanic acid, which can bind to the active site of beta-lactamases and provide form a stable acyl enzyme complex, thereby inhibiting the enzyme.11 Another approach is to target the pathways of Gram-positive cell wall production. Tunicamycin and ticlopidine have been shown to inhibit the enzymatic pathway for teichoic acid, a key component of the Gram-positive cell wall.12
3–7 The production of beta-lactamase (which hydrolyses penicillin into penicilloic acid) is common in Staphylococcus aureus, a pathogen that can cause infection in skin, lungs and blood.8 Most strains of S. aureus are now resistant to first-generation penicillin's and so second-generation penicillin's such as oxacillin, nafcillin and methicillin are administered.9 A report by the World Health Organisation in 2014 stated that antibiotic resistance is a growing worldwide threat to public health, driven largely by the lack of new therapeutic agents for treating bacterial infections.10 Overcoming resistance to beta-lactam antibiotics is most commonly achieved by using a beta-lactamase inhibitor such as clavulanic acid, which can bind to the active site of beta-lactamases and provide form a stable acyl enzyme complex, thereby inhibiting the enzyme.11 Another approach is to target the pathways of Gram-positive cell wall production. Tunicamycin and ticlopidine have been shown to inhibit the enzymatic pathway for teichoic acid, a key component of the Gram-positive cell wall.12
The ability of bacteria to form biofilms further complicates their treatment.13 A biofilm is an extracellular polymeric substance (EPS) which is secreted by bacterial communities and surrounds them in a protective layer.14 EPS is a structured matrix of exopolysaccharides, lipids, nucleic acids and protein.15 Biofilms protect the bacterial cells within and allow them to tolerate treatments due to the quenching of antimicrobial agents at concentrations that would otherwise kill planktonic cells.16 Impaired diffusion of antimicrobial agents through the EPS increases the tolerance to treatments.17 Opportunistic pathogens, such as S. aureus, can cause chronic infection in dermal wounds, potentially caused by a combination of biofilm production and antimicrobial resistance.18,19
Nanoparticles (NPs) have been investigated for their anti-biofilm action and ability to overcome antimicrobial resistance. A variety of different NPs such as Au NPs, silica NPs, Mg(OH)2 NPs and CuONPs have all been shown to demonstrate increased toxicity towards bacteria.20–24,45 AgNPs has been researched in anti-biofilm treatments due to the silver ions intrinsic antimicrobial property.25,43 Nanocarriers for antimicrobial agents have also been shown to increase their efficacy against a wide range of microorganisms, including resistant species26–29 and deliver drugs specifically to a bacterial target.35,42–44 This can potentially reduce unwanted drug interaction in healthy tissue and lower general toxicity.36 They can also protect the antimicrobial agent from premature degradation and inactivation.37,38 Biofilm degradation has been investigated using a range of biologically inspired hydrolases, amylases and proteases.30,46 The disruption of the biofilm formation by NPs can improve the ability of antimicrobial agents to kill the cells within.28 Proteases can be particularly useful for degrading biofilms due to their ability to hydrolyse ESP matrix proteins and bacterial adhesins.31–33 Disruption of the synthesis of biofilm formation using small molecules to inhibit key signalling pathways such as c-dis-GMP and indole has also been investigated.34
In this study, we present a novel concept for an active nanocarrier delivery of Penicillin G and Oxacillin to biofilms by their encapsulation into a protease-functionalised shellac NPs. The NPs surface coating of a cationic protease, Alcalase 2.4 L FG, performs a dual purpose to both degrade the biofilms and increase the electrostatic interaction between the cationic NPs and the anionic cell walls of the entrapped bacteria. Fig. 1A illustrates the scheme for fabrication of the composite shellac NPs, and Fig. 1B shows their mechanism of action. Fig. S1 (see ESI†) provides a detailed information about the individual components of the NP and the chemical structure of the antibiotics. S. aureus was chosen as the model pathogen for its ability to form a biofilm. This S. aureus strain (ATCC® 29213™) was selected due to its resistance to Penicillin G (PenG) and its sensitivity to oxacillin (Oxa). The gene blaZ which produces the protein product beta-lactamase (EC number: 3.5.2.6) was shown to be in the genome of this strain, as confirmed by the ATCC One Codex depository.39 Additionally, this stain does not carry the mecA gene which encodes an altered penicillin-binding protein (PBP2a) which confers resistance to oxacillin due to reduced beta-lactamase affinity.40
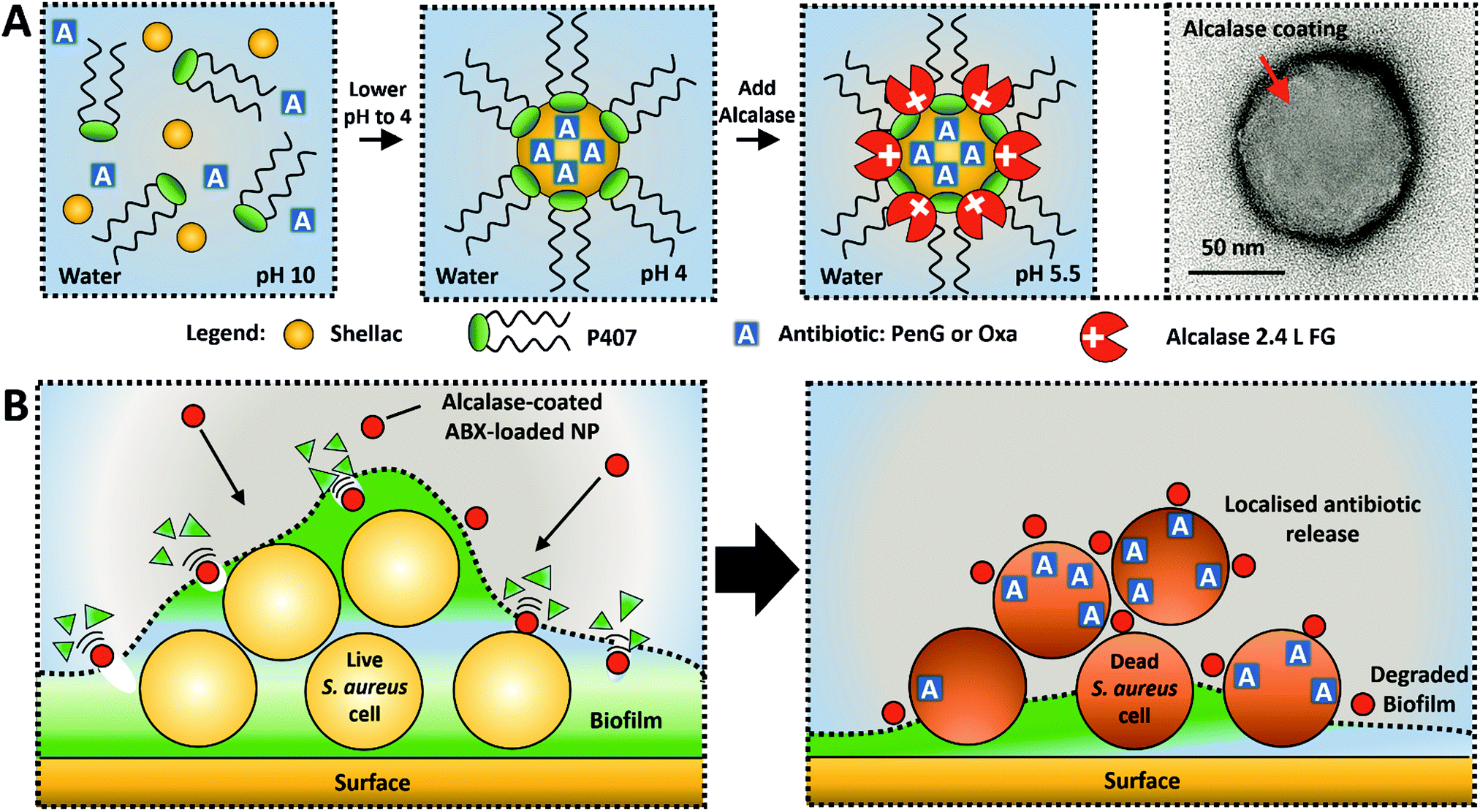 | ||
| Fig. 1 (A) Schematic for the synthesis of functionalized shellac NPs as nanocarriers for Benzylpenicillin or Oxacillin (PenG or Oxa). The PenG/Oxa-loaded shellac NPs are sterically stabilized with Poloxamer 407 (P407) and have cationic surface functionality achieved by coating with the cationic protease Alcalase 2.4. L FG. (B) Diagram of the mechanism of action for PenG/Oxa-loaded Alcalase functionalized shellac NPs. Alcalase coating degrades the biofilm exposing the cells and allowing the cationic NPs to adhere to the cell surface. Gradual release of antibiotic is highly localised and results in rapid cell death. | ||
Here we aim to prove that its antimicrobial resistance can be overcome by using highly concentrated antibiotics loaded inside the shellac NPs, both in planktonic cells and biofilm treatment experiments. NP characterisation, including preparation, encapsulation efficiency, release kinetics and Alcalase functionalisation of PenG/Oxa-loaded shellac NPs can be found in ESI.†
Results and discussion
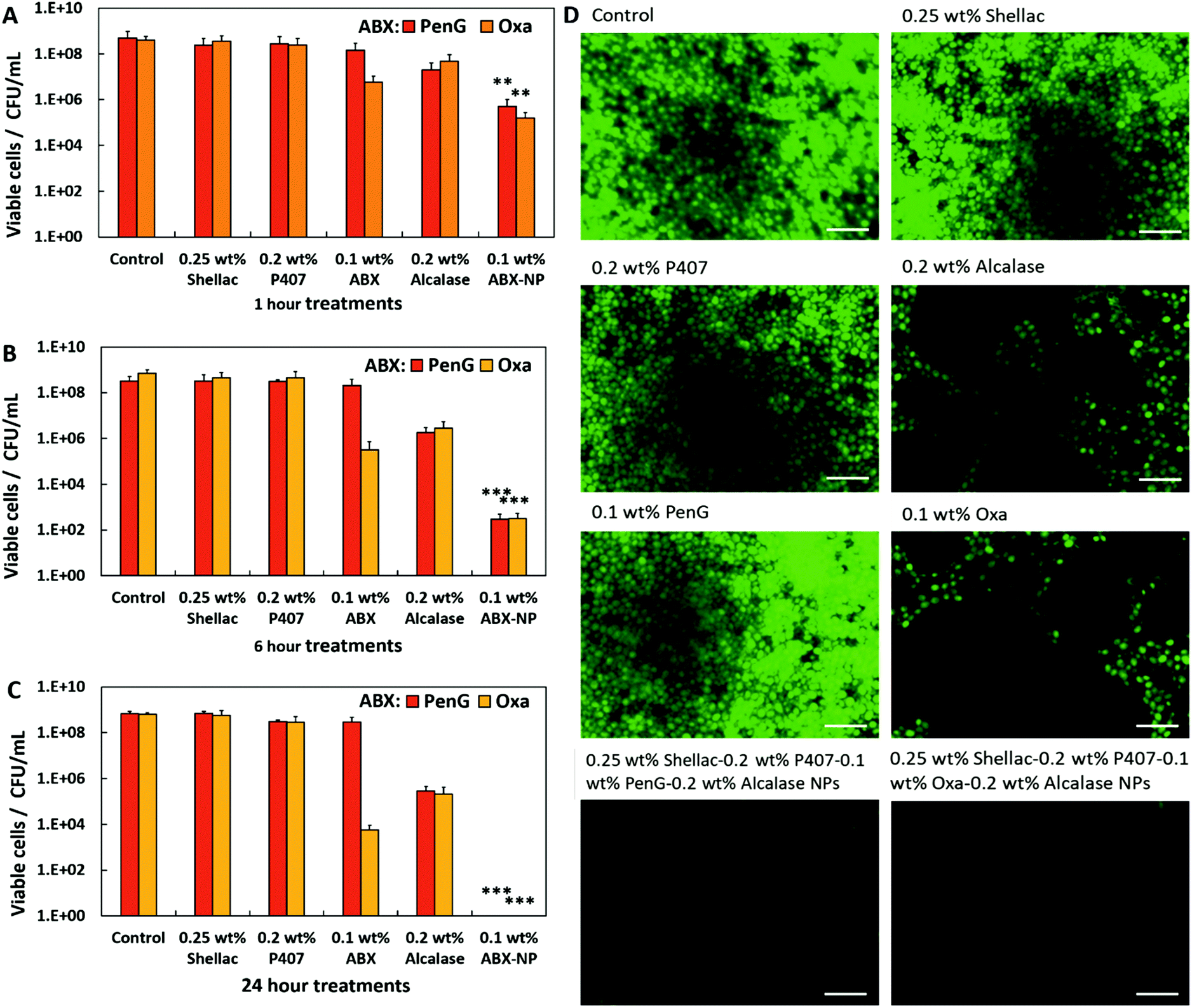
A formulation of 0.25 wt% shellac NPs loaded with 0.1 wt% PenG/Oxa and coated with 0.2 wt% P407 and subsequently with 0.2 wt% Alcalase was chosen as the stock treatment for testing against S. aureus. Here, we show that PenG and Oxacillin encapsulated in the shellac NPs, functionalised with a surface coating of a cationic protease, are significantly more effective than the free antibiotics administered at equivalent concentration. We aim to demonstrate that antibiotics encapsulated into such active NPs have an increased efficacy that can overwhelm the enzymatic inhibition of the antibiotic (specifically PenG). This is investigated on S. aureus in planktonic and biofilm forms.Firstly, we confirmed S. aureus as a suitable candidate by performing an antimicrobial susceptibility screen using VITEK®2 ID and AST microwell cards (Fig. S16, ESI†). Table S2 (ESI†) shows the range of antibiotics tested for susceptibility and their interpretation as resistant or susceptible. This S. aureus strain was confirmed to be susceptible to Oxa and resistant to PenG. This was verified using antimicrobial disk diffusion for both Oxa and PenG (Fig. S17, ESI†). The zone diameter breakpoint for PenG is <26 mm, as stated by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) testing breakpoint tables.41
There is a clear zone of inhibition diameter around the oxacillin disk (27 mm) confirming its susceptibility to that antibiotic. Most staphylococci are now beta-lactamase producers and this is the mechanism of resistance to penicillin antibiotics, requiring treatment with isoxazolylpenicillins (e.g. oxacillin). To confirm this a simple beta-lactamase detection strip was used on a S. aureus suspension (Fig. S18, ESI†). This test is based on beta-lactamase hydrolysing the beta lactam ring (present in PenG and all penicillin antibiotics) into penicillioc acid. A yellow solution was generated after 10 min indicating the presence of an acid in the solution (acid–base indicator), and therefore beta-lactamase produced by S. aureus.
The effectiveness of the base formulation of 0.25 wt% shellac–0.2 wt% P407–0.1 wt% PenG/Oxa–0.2 wt% Alcalase NPs (0.1 wt% PenG-NPs or 0.1 wt% Oxa-NPs) was tested against the equivalent concentration of free PenG or Oxa (0.1 wt%) using a time-kill assay.
Fig. 2A and B show that S. aureus starting at a concentration of 1 × 106 cells per mL increased to approximately 1 × 109 cells per mL when untreated. Treatment with 0.1 wt% free PenG had a minimal effect on bacterial growth, with 1 × 107 cells after a 24 h incubation time, confirming the resistance of the cells to this treatment (Fig. 2A). However, when treated with 0.1 wt% PenG-NPs there were no viable cells after 4 h, a 4 log+ reduction from the free PenG sample. This confirms the PenG-loaded NPs are overcoming the resistance and restoring the effectiveness of the antibiotic. This is due to the high concentration of the agent within the NPs cores, which is localised onto the bacterial cell wall due to the electrostatic attraction of the cationic NPs to the anionic surface of the bacterial cell. Fig. 2B shows the same time kill assay performed against S. aureus, this time with 0.1 wt% free Oxa and 0.1 wt% Oxa-NPs. As described earlier, this strain of S. aureus is susceptible to Oxa and so after 6 h treatment with 0.1 wt% Oxa (free) there were no viable cells detected. However, the 0.1 wt% Oxa-NPs were faster-acting, killing the cells within 4 h, providing further evidence of their ability to increase the effectiveness of the encapsulated antibiotic. Fig. S19 (ESI†) shows the time-kill results for all the individual components of the NP. The 0.25 wt% shellac and 0.2 wt% Alcalase NP components alone had only a mild effect on reducing the viable cell count after 24 h (∼1 × 108 and 1 × 107 CFU per mL, respectively). The free 0.2 wt% P407 alone reduced the cell viability concentration to ∼1 × 105 CFU per mL after 24 h, however, when combined with 0.25 wt% shellac (0.25 wt% shellac–0.2 wt% P407) empty NPs, the toxicity was reduced, with ∼1 × 108 viable cells per mL detected after 24 h of treatment. This indicates that the complexing of P407 onto shellac cores reduces the toxicity of this Pluronic surfactant and that the shellac-NPs are a safer antibiotic delivery vehicle. Surfactants are intrinsically toxic to life due to their amphipathic nature which can dissolve lipids in water. The free P407 demonstrates a bacteriostatic effect against S. aureus by disrupting the cell membrane, however, the lack of cell specificity makes them unsuitable for use in a therapeutic context.
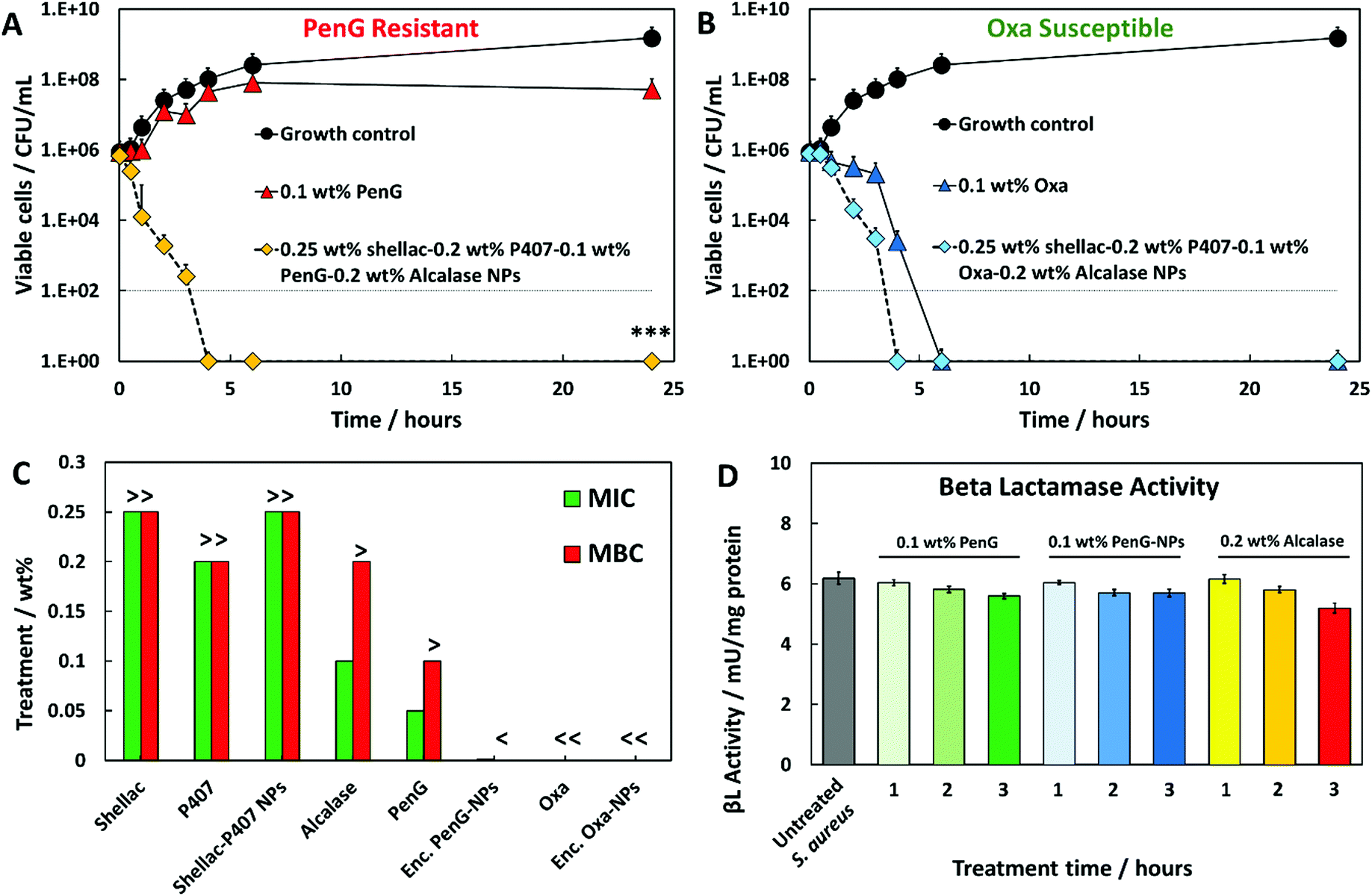 | ||
| Fig. 2 Time kill assays against S. aureus. Growth control (untreated). (A) 0.1 wt% free PenG (Penicillin G) (B) and 0.1 wt% Oxa (Oxacillin) were compared to the same antibiotic concentration encapsulated into Alcalase-coated shellac NPs. <0.05, **P < 0.01, ***P < 0.001 compared to the free antibiotic. Table S4 (ESI†) shows the statistical analysis P-values. (C) S. aureus MIC and MBC of free PenG/Oxa, encapsulated PenG/Oxa NPs and individual constituents of the NP. 0.25 wt% shellac–0.2 wt% P407–0.1 wt% PenG/Oxa-0.2 wt% Alcalase was the stock treatment solution. Treatment concentrations lower than this were 2-fold dilutions from the stock. “>” indicates that a greater and “<” a lower concentration is needed to either inhibit or kill S. aureus. The MIC was determined to be the lowest concentration of active antimicrobial agent which inhibited the growth of each strain. The MBC was the lowest concentration in which no viable cells were detected after streaking the contents onto fresh MHA plates. (D) Beta lactamase activity of S. aureus after 1 to 3 h PenG, PenG-NP and Alcalase treatments expressed per mg of protein. Experiment was capped at 3 hours due to lack of remaining cells to analyse. Unit definition: 1 Unit Beta lactamase activity = amount of enzyme that generates 1 μmol of nitrocefin per minute at pH 7 at 25 °C. | ||
A further investigation into the wt% PenG/Oxa-NPs (2-fold serial dilutions of the stock treatment) was performed to elucidate the MIC/MBC of S. aureus after 24 h of treatment.
Fig. 2C shows that PenG-NPs had a minimal inhibitory concentration (MIC) of 0.015 wt%, a 5-fold improvement of 0.05 wt% for free PenG. The minimal bactericidal concentration (MBC) for PenG-NPs was less than 0.0003 wt%, an 8-fold decreased from free PenG showing >0.1 wt% (due to its resistance). Oxa-NP and free oxacillin had both MIC and MBC values of <0.0003 wt% after 24 h (oxacillin sensitive), however as described above the Oxa-NP showed increased efficacy by the faster killing of the bacteria. Shellac, P407, shellac–P407 NPs all had MIC and MBC of >0.25 wt%, 0.2 wt% and 0.25 wt% respectively (stock NP concentrations). Alcalase showed a MIC of 0.1 wt% and an MBC of > 0.1 wt%, this agrees with the time-kill results which show that the free protease did not have a meaningful impact on cell viability. Table S3 (ESI†) shows tabulated data for the MIC/MBC results.
Fig. 3A and B shows SEM images of untreated planktonic S. aureus which appear to have good morphology with no visible NPs on their surface. When compared to Fig. 3C and D in which planktonic S. aureus was treated for 1 hour with 0.1 wt% PenG-NPs, there is a clear coating of NPs on the surface of the bacteria. This provides further evidence of the cationic NPs adhering to the anionic bacterial cell via electrostatic interactions. This highly localised concentration of antibiotic encapsulated NPs is apparently able to overcome the enzymatic resistance mechanism of the S. aureus. Additionally, we investigated S. aureus using TEM and EDS (Energy-dispersive X-ray spectroscopy) elemental mapping. Fig. 4 shows the elemental distribution of carbon, oxygen, nitrogen and sulphur in S. aureus. When treated with 0.1 wt% free PenG there is an increased concentration of sulphur compared to the natural sulphur concentration contained in the untreated sample. This indicates the presence of PenG, which contains the element sulphur in its thiazolidine ring. The sample treated with 0.1 wt% PenG-NPs contains a further increase in sulphur, with small pockets located where the NP has adhered to the bacterial cell wall. This further supports the increased concentration of PenG, highly localised on the cell wall.
 | ||
| Fig. 3 (A) and (B) SEM images of planktonic S. aureus after 24 hours growth (no treatment) and (C), (D) SEM images of S. aureus after 24 hours growth (0.1 wt% PenG-NPs, 1 hour treatment). 10 nm gold coating. Samples were imaged using a Carl Zeiss Evo-60 (Germany) with a variable pressure 100-micron aperture at 40 Pa. EHT was 20 kV with probe current of 100 pA. Images were captured with Zeiss smartSEM software. | ||
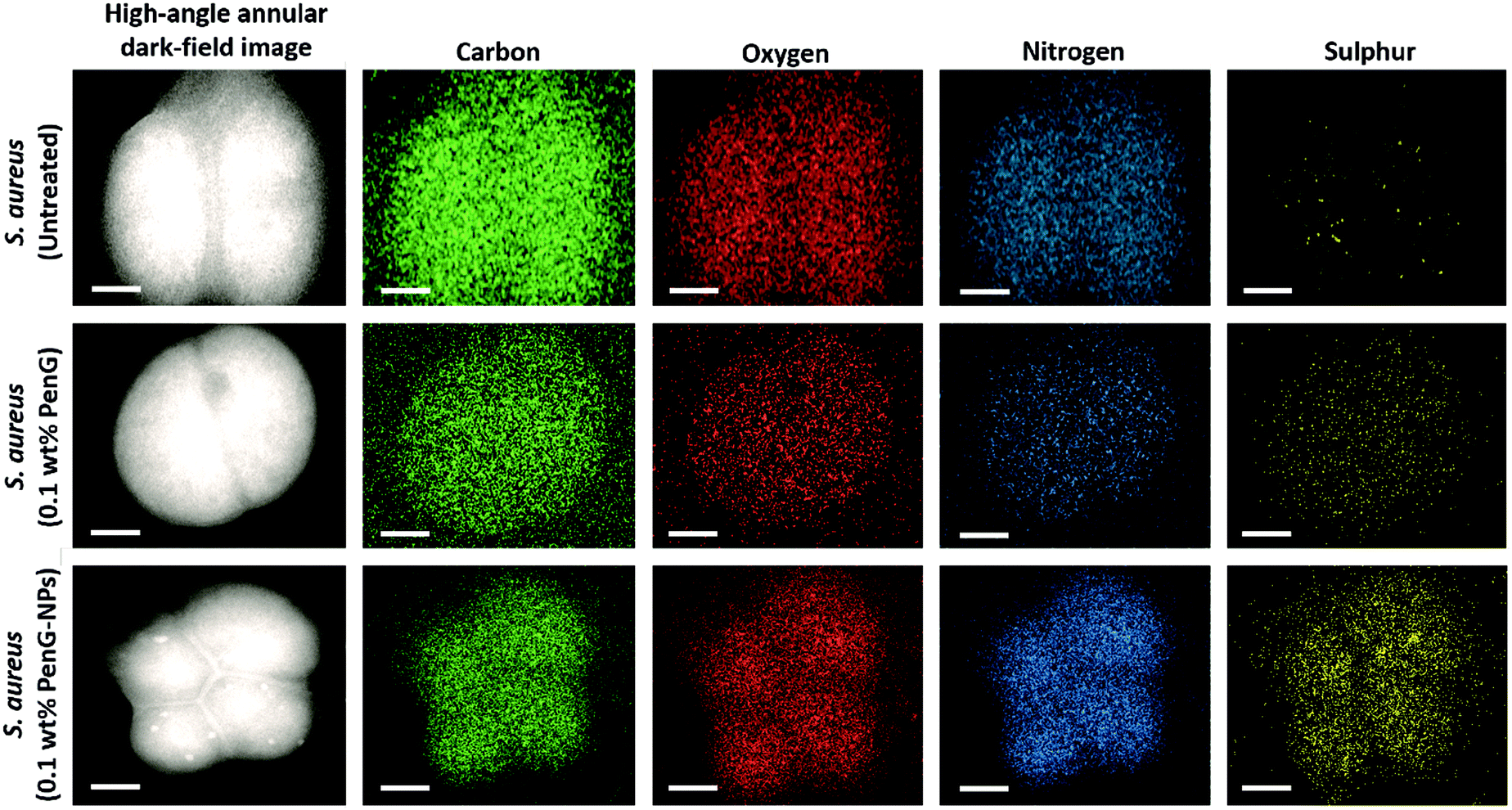 | ||
| Fig. 4 TEM/EDS images/scans of S. aureus after 24 hour treatment with either 0.1 wt% PenG or 0.1 wt% PenG-NPs. TEM images were obtained using a JEM 2010 (JOEL, Japan) at 120 kV and a Gatan Ultrascan 4000 digital camera. A nanoanalysis X-Max 65-T detector and the INCA software were used to produce the elemental analysis maps. White scale bar = 250 nm. | ||
We also investigated the beta-lactamase activity of S. aureus after treatment with either 0.1 wt% PenG, 0.1 wt% PenG-NPs and 0.2 wt% Alcalase. Fig. 2D shows the beta-lactamase activity measured in S. aureus cells after 1, 2 and 3 h treatments (compared to a 3 h S. aureus sample which was untreated). The activity was obtained from the hydrolysis of nitrocefin (a beta-lactamase substrate). Fig. S20 (ESI†) shows the nitrocefin calibration curve. The untreated S. aureus had beta-lactamase of an enzyme activity of 6 mU per mg protein per min. Interestingly, there was only a marginal decrease in activity in the free PenG and PenG-NPs over 3 h, and no discernible difference between free PenG and PenG-NPs. This indicates that S. aureus is not actively increasing the production and secretion of the beta-lactamase during treatment.
This lack of PenG defence activity may explain a mechanism for why PenG-loaded NPs can be effective against S. aureus, when an equivalent concentration of free PenG is not. The bacterial cell is not able to produce enough beta-lactamase enzyme activity to inhibit the PenG, which is locally delivered and highly concentrated on the cell surface, leached by the adhered PenG-NPs. Fig. S21 (ESI†) shows the beta-lactamase activity S. aureus cell and the culture media (containing any secreted enzyme). There is no notable difference in beta-lactamase activity of S. aureus when treated with either free PenG or PenG-NPs (both 0.1 wt%) between 1 h and 3 h. The effect of 0.2 wt% Alcalase was also investigated. There is a marginal decrease in beta-lactamase activity after 3 h, compared to the control, 0.1 wt% PenG and 0.1 wt% PenG-NPs, which are almost identical. This is likely due to the Alcalase hydrolysing a small amount of beta-lactamase produced by the bacteria, and therefore lowering the detected enzymatic activity. This provides evidence that the Alcalase itself can confer benefits in the context of reducing a beta-lactamase producing bacteria's resistance mechanism.
Next, we investigated the effectiveness of the 0.1 wt% PenG/Oxa NPs on S. aureus biofilms. Biofilms were grown in 96-well plates for 24 h, and then treated with free antibiotic and antibiotic-loaded shellac NPs for further 24 h. Fig. 5A and B show the residual biofilm after staining the wells with Crystal Violet (CV). Compared to the control of an untreated biofilm, there was a ∼90% biofilm reduction for both 0.1 wt% PenG-NPs and 0.1 wt% Oxa-NPs treatments. The treatment with 0.1 wt% free PenG showed no significant reduction in residual biofilm, due doubly to the resistance of S. aureus to this antibiotic and the protection a biofilm confers to exogenous treatments. 0.1 wt% free Oxa reduced the biofilm by ∼50% compared to the growth control, significantly less the 90% achieved by the 0.1 wt% Oxa-NPs. The 0.25 wt% shellac and 0.2 wt% P407 showed only a negligible reduction in residual biofilm in the wells. The 0.2 wt% Alcalase reduced the biofilm to ∼50% compared to the growth control, likely due to the protease activity degrading the EPS matrix protein structure. Fig. S22 (ESI†) shows images of the biofilm wells with the CV staining, there is a clear reduction in staining in the 0.1 wt% PenG/Oxa-NPs compared to the 0.1 wt% free antibiotics. Fig. 5C and D show the biofilm mass after treatment, where the results corroborate the ones obtained with CV staining. Again, the 0.1 wt% PenG-NPs reduced the biofilm mass significantly (∼1 mg compared to the ∼3.4 mg untreated control). The free PenG showed only a marginal reduction of ∼0.5 mg due to the resistance of S. aureus.
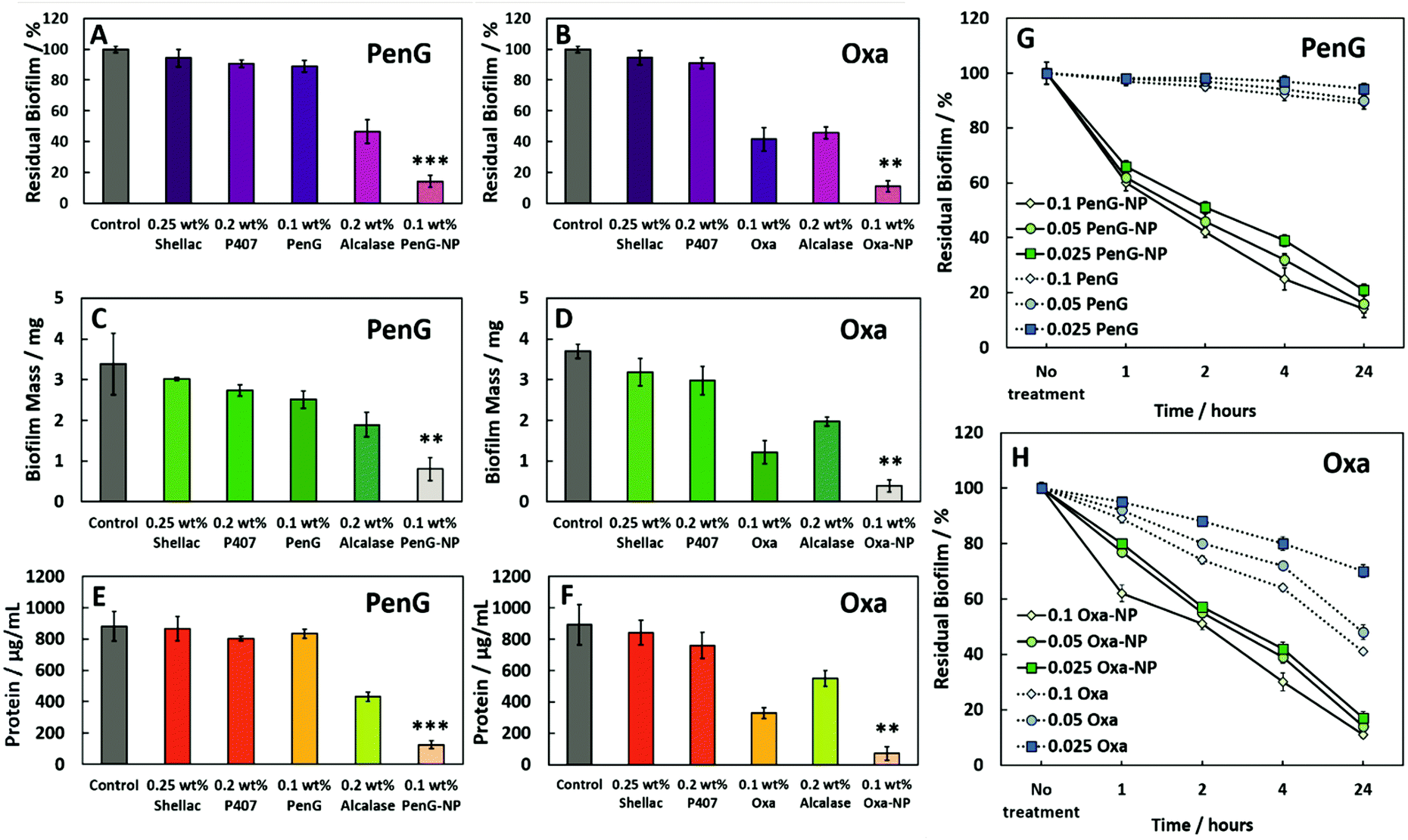 | ||
| Fig. 5 The effect of (A), (C) and (E) 0.25 wt% shellac–0.2 wt% P407–0.1 wt% PenG–0.2 wt% Alcalase and (B), (D), (F) 0.25 wt% shellac–0.2 wt% P407–0.1 wt% Oxa–0.2 wt% Alcalase treatments on S. aureus biofilms after 24 h. Residual biofilm staining, biofilm mass and protein concentration respectively. 0.1 wt% PenG/Oxa-NP; 0.25 wt% shellac–0.2 wt% P407–0.1 wt% PenG/Oxa–0.2 wt% Alcalase. <0.05, **P < 0.01, ***P < 0.001 compared to the free antibiotic. Table S5 (ESI†) shows the statistical analysis P-values. (G) Residual biofilm (CV staining) after treatment with shellac–P407–ABX formulations after 1, 2, 4 and 24 h treatment times. Treatments were made from a stock of 0.25 wt% shellac–0.2 wt% P407–0.1 wt% PenG–0.02 wt% Alcalase (referred as PenG-NP) or (H) 0.25 wt% shellac–0.2 wt% P407–0.1 wt% Oxa–0.02 wt% Alcalase (referred as Oxa-NP) and diluted 2-fold. All tests were performed in triplicates (N = 3 with ± S.D.). | ||
The 0.1 wt% Oxa-NPs reduced the biofilm mass to 0.3 mg, a significant improvement against 0.1 wt% free Oxa which reduced the mass to ∼1.1 mg. The shellac, P407 and Alcalase showed a similar trend when measuring the biofilm mass to the biofilm stained with CV. Fig. 5E and F measured the protein concentration of the biofilms after treatment. Fig. S23 (ESI†) shows the BCA calibration curve used to interpolate protein concentration in the samples. The 0.1 wt% PenG/Oxa-NP treatments reduced the protein concentration to ∼150–100 μg mL−1, respectively, significantly more than the ∼800 μg mL−1 for 0.1 wt% free PenG and 0.1 wt% ∼400 μg mL−1 for free Oxa. The 0.2 wt% Alcalase treatment alone was effective in reducing protein concentration (∼500 μg mL−1). A time and concentration-dependent examination of the free 0.1 wt% PenG/Oxa treatments vs. the 0.1 wt% PenG/Oxa-NPs was performed. Fig. 5G shows that free PenG (at 0.1, 0.05 and 0.025 wt% concentrations) was no discernable effect at reducing the residual biofilm. In comparison, the PenG-NPs after 24 h had reduced the residual biofilm to ∼20%. After 1 h the reduction was ∼40% lower compared to no treatment indicating the antibiotic-loaded NPs are fast-acting on the biofilms. There was a very little difference between the 0.025 wt% and the 0.1 wt% stock NPs treatment. This reveals that the PenG-NPs remain potent at lower concentrations. Fig. 5H shows the time and concentration-dependent free Oxa vs. Oxa-NPs. There is a similar trend in the effectiveness of the Oxa-NPs (across 0.1, 0.05 and 0.025 wt% concentrations), with the residual biofilm being reduced by ∼90% compared to no treatment after 24 h. The free Oxa showed a gradual reduction in residual biofilm over 24 h, culminating in a reduction of 20%, 50% and 60% in residual biofilm for the 0.1, 0.05 and 0.025 wt% free Oxa treatments. We also investigated the viability of the cells within the biofilm after 1, 6 and 24 h treatments. Fig. 6A shows the results after 1 h. The untreated control, 0.25 wt% shellac and 0.2 wt% P407 showed no reduction in cell viability, with all samples retaining ∼1 × 109 CFU per mL. The results were similar in the enumeration of the biofilm cells after 6 and 24 h, respectively. 0.1 wt% free PenG showed no reduction in viable cells, however, the 0.1 wt% free Oxa had reduced the cells to ∼1 × 107 CFU per mL.
 | ||
| Fig. 6 24 h grown S. aureus biofilm cellular viability after anti-biofilm treatments. CFU per mL of colony biofilms after timed exposure after (A) 1 hour, (B) 6 hours and (C) 24 hours. Post-treatment, the colony biofilms were glass bead beaten in 1 mL of MHB with serial dilutions. 0.1 wt% ABX = NP; 0.25 wt% shellac–0.2 wt% P407–0.1 wt% PenG/Oxa-0.2 wt% Alcalase. The agar drop plate method was used to elucidate the CFU per mL. N = 3 with ± S.D. <0.05, **P < 0.01, ***P < 0.001 compared to the free antibiotic. Table S6 (ESI†) shows the statistical analysis P-values. (D) Fluorescent images of S. aureus biofilm stained with Acridine Orange after 24 h of treatment. The white inset bar represents 5 μm. | ||
The 0.1 wt% PenG/Oxa-NPs showed a significant improvement compared to the free antibiotic, reducing the viable cells to ∼1 × 106 CFU per mL.
Fig. 6B shows the viable cells after 6 h of treatment. Both the 0.1 wt% PenG and Oxa-NPs reduced the viable cells to ∼1 × 102 CFU per mL. 0.1 wt% PenG remained unchanged compared to the untreated control, the 0.1 wt% free Oxa treatment has reduced the viable cells to ∼1 × 105 CFU per mL. The 0.2 wt% Alcalase treatment had reduced the viable cells to ∼1 × 106 CFU per mL, likely due to the degradation of the biofilm by the protease releasing cells from biofilm. Fig. 6C shows the viable cells after 24 h. There were no detectable cells in the 0.1 wt% PenG/Oxa-NP treatments, a significant reduction compared to ∼1 × 109 CFU per mL for the 0.1 wt% free PenG and ∼1 × 104 CFU per mL for the free Oxa. After 24 h treatment with 0.2 wt% Alcalase the viable cells were reduced to ∼1 × 105 CFU per mL. These results indicate that both the 0.1 wt% PenG-NPs and the 0.1 wt% Oxa-NPs are significantly more effective then and equivalent concentration of the free antibiotic. Crucially, the 0.1 wt% PenG-NP being effective against a strain of S. aureus classified as resistant to PenG. Fig. 6D shows fluorescence microscopy images of the biofilms stained with Acridine Orange (AO) after 24 h treatment. It is clear to see that there is a much-decreased concentration in cells in the PenG/Oxa-NP treatment compared to the control, 0.25 wt% shellac and 0.2 wt% P407 treatments. The 0.1 wt% free PenG remains relatively unchanged compared to the untreated sample, with 0.1 wt% free Oxa and 0.2 wt% Alcalase reducing the AO staining by ∼50%. Fig. S24 (ESI†) shows the percentage of AO orange staining per sample.
The treatment/biofilm growth media was also examined. It is known that cells within biofilms are able to disperse during a biofilms natural formation.19 Fig. S25 (ESI†) shows the number of viable cells after 24 h treatments as described in Fig. 6A. The untreated control and 0.25 wt% shellac contained ∼1 × 104 CFU per mL, much lower than the ∼1 × 109 CFU per mL than found in the biofilms. Biofilms treated with 0.2 wt% and 0.2 wt% Alcalase had fewer cells, ∼1 × 103 CFU per mL, likely due to the moderate cytotoxic nature of these compounds. 0.1 wt% PenG treatment produced ∼1 × 104 CFU per mL due to the resistance of S. aureus to this antibiotic. Treatment with 0.1 wt% free Oxa treatment showed no viable cells in the media. No viable cells were also found in the treatment media of biofilms treated with 0.1 wt% PenG/Oxa-NPs. These results mirror the CFU counts of planktonic cells shown in Fig. 2. This indicated that the ABX-NP treatments are able to effectively kill cells within a biofilm and planktonic cells outside. It is worth noting that metabolically inactive persister cells are known to be present in biofilms.47 These cells are very difficult to culture and highly tolerant to antibiotics which target a metabolic mechanism, such as beta-lactam antibiotics. Therefore it is possible that some cells survived treatment but were not detected during enumeration.
To understand the structure of the biofilm after treatment we used confocal laser scanning microscopy (CLSM) to image S. aureus biofilms stained with AO. Fig. 7 shows images of S. aureus after 1 h and 24 h treatments as administered in Fig. 6. The images show that 0.25 wt% and 0.1 wt% PenG had very little effect on the structure or thickness of the biofilms compared to the control, which was untreated. 0.2 wt% P407 and 0.2 wt% Alcalase treatments showed some reduction in biofilm thickness, more notably after 24 h of treatment. The structure appears less dense particularly on the surface of the biofilm. 0.1 wt% PenG had very little effect on the S. aureus PenG resistant biofilm, which agrees with the CV staining and the CFU viability counts. 0.1 wt% Oxa showed a slightly decreased thickness and less dense structure after 24 h of treatment. The 0.1 wt% PenG/Oxa-NPs showed a much-improved reduction in biofilm mass in comparison the antibiotics alone at equivalent concentrations. There appears to the patches of completely cleared biofilm indicating the NPs are able to degrade the biofilm and reveal the cells within. The NPs showed most reduction after 24 h, confirming the CFU viability counts in Fig. 6. Crucially, the PenG-NPs were effective against a species with PenG resistance. To further collaborate the CSLM 3D images we cryosectioned 10 μm slices of the membrane biofilm and examined the thickness and structure of the EPS matrix with concanavalin A (ConA) dye under brightfield and fluorescent microscopy. Samples were treated for 24 h before examination. Fig. 8A shows the biofilm lateral structure and Fig. 8B shows the thickness of the sectioned biofilms measured across 3 standardised sections of the image. The sections show a densely packed biofilm, in the control (untreated) and 0.25 wt% shellac sections (57 μm and 59 μm, respectively). The thickness is reduced when treated with 0.2 wt% P407 and 0.2 wt% Alcalase (40 μm and 38 μm) which is consistent with the CSLM results. 0.1 wt% PenG showed very little change from the untreated control and 0.25 wt% shellac treatment, with a thickness of 55 μm. Treatment with 0.1 wt% Oxa reduced the mass to 40 μm. The samples treated with 0.1 wt% PenG/Oxa-NPs showed a completely cleared biofilm with no ConA staining detected. This agrees with CFU viability count results in Fig. 6. The reduction is also consistent with the much-reduced biofilm mass seen in Fig. 7 using CLSM. The biofilm remnants still visible in the 3D image is likely due to the differences in visualisation techniques, 3D visualisation vs. 2D cryo-sectioned lateral fluorescent imaging. However, both Fig. 7 and 8A both provide visual support to the CFU viability counts and provide evidence the PenG/Oxa-NPs are much more effective biofilm clearing treatments than equivalent concentration of the free antibiotic. This provides evidence that formulations based on NP-encapsulated antibiotics with targeted delivery could be a useful tool in repurposing old antibiotics. This option is becoming increasingly attractive in the antibiotic resistance era.48,49
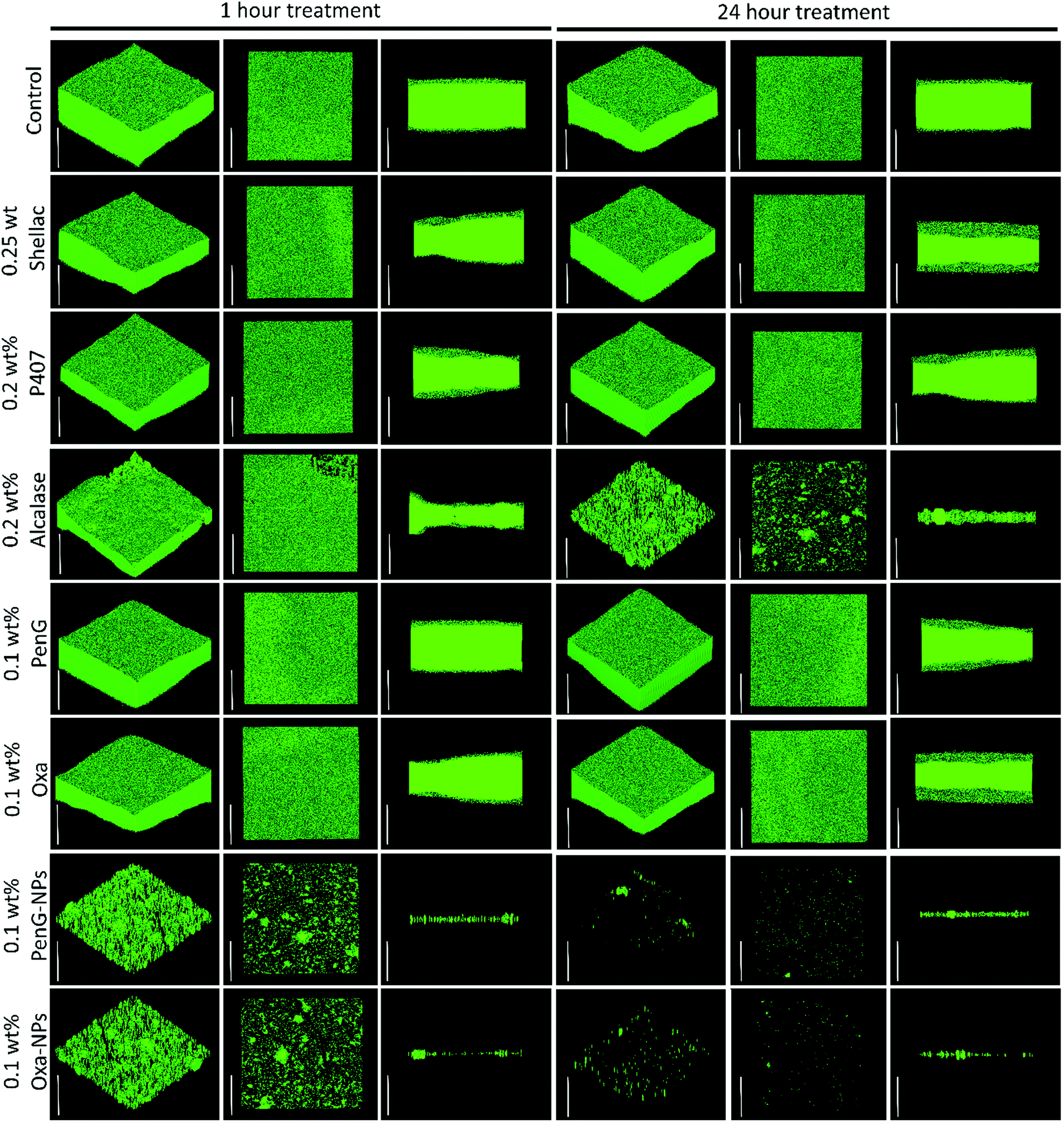 | ||
| Fig. 7 Confocal laser scanning microscopy. S. aureus 24 h grown biofilms treated for 1 or 24 h with various treatments. Biofilms were stained with 0.2 wt% Acridine Orange for 5 mins in darkness before gently washing away. The white inset bar represents 50 μm. 0.1 wt% PenG-NP and 0.1 wt% Oxa-NP refers to 0.25 wt% shellac–0.2 wt% P407–0.1 wt% PenG–0.2 wt% Alcalase NPs and 0.25 wt% shellac–0.2 wt% P407–0.1 wt% Oxa–0.2 wt% Alcalase NPs. Confocal laser scanning microscopy images (CLSM) were obtained with a Carl Zeiss LSM 710 confocal microscope with Z-series images taken in 1 μm slices. | ||
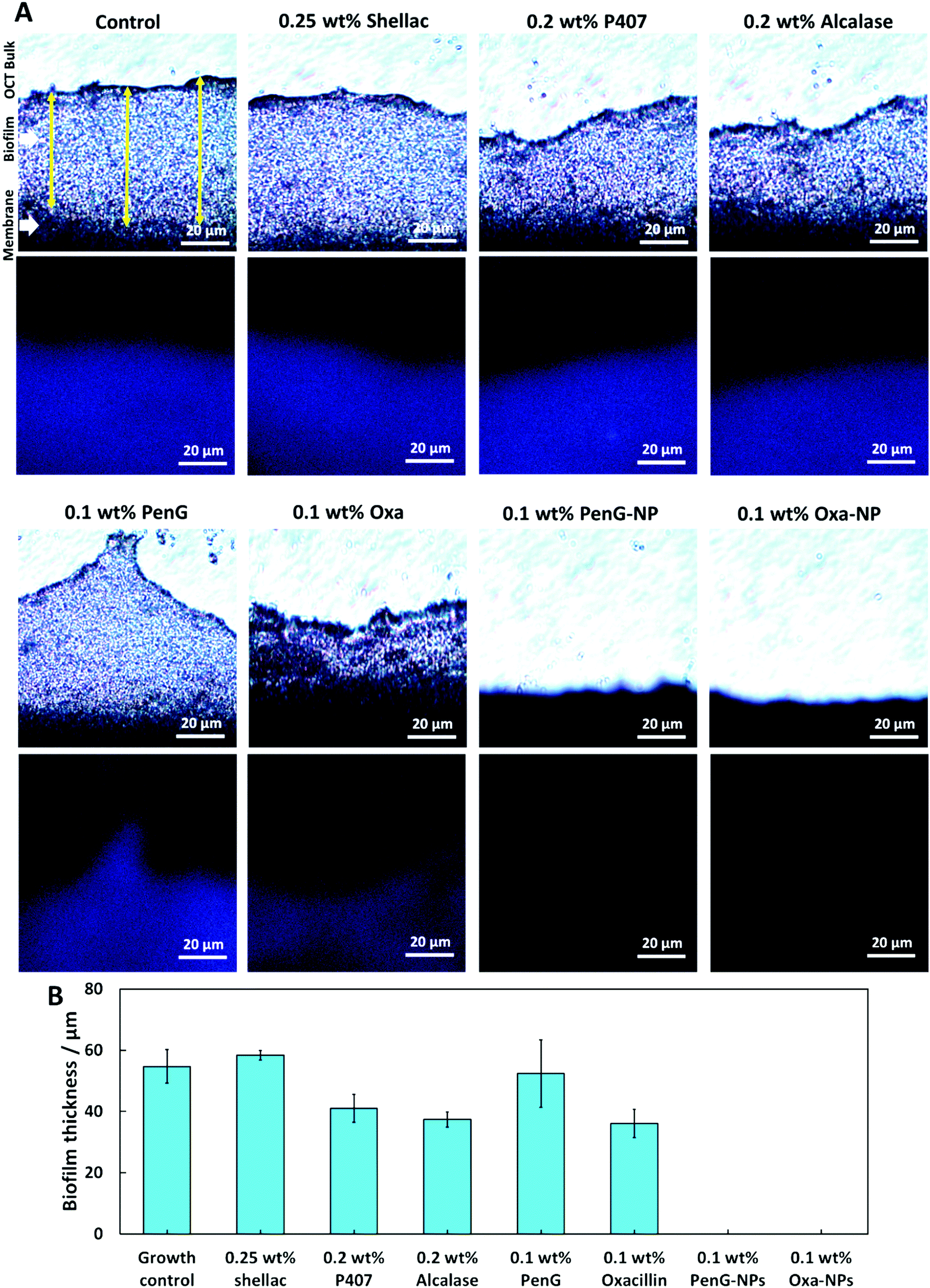 | ||
| Fig. 8 (A) Cryostat lateral 10 μm sliced images of S. aureus biofilms after 24 h treatments. Images were taken a 200× magnification. (B) Biofilm thickness measured over three locations (illustrated with yellow arrows) in Image J v1.52a. 0.1 wt% PenG-NPs and 0.1 wt% Oxa-NPs refers to 0.25 wt% shellac–0.2 wt% P407–0.1 wt% PenG–0.2 wt% Alcalase NPs and 0.25 wt% shellac–0.2 wt% P407–0.1 wt% Oxa–0.2 wt% Alcalase NPs. | ||
A preliminary human cell cytotoxicity experiment was performed on HaCaT and HEP G2 cells to investigate if the shellac, P407, free Alcalase, free PenG/Oxa and PenG/Oxa-NPs were toxic to human cells. HaCaT cells were chosen as they a good proxy for studying human keratinocytes, a cell type which would be exposed to the treatment in topical applications. HEP G2 were chosen as they are good proxy for studying the effects of xenobiotics. A full analysis can be found in the ESI.†
Conclusions
In summary, we have developed an innovative nanocarrier system with a protease coating which can overcome an antibiotic-resistant pathogen in both planktonic and biofilm form. We demonstrate the effectiveness of Penicillin G and oxacillin encapsulated in shellac NPs against a strain of S. aureus verified as Penicillin G resistant. The functionalisation of the NPs with a coating of the cationic protease Alcalase creates a dual application NPs which allows the degradation of the biofilms and electrostatic adhesion to the anionic bacterial cell wall. The attachment of the antibiotic-loaded NPs to the bacterial cells allows a highly localised continuous release of concentrated antibiotic on the bacteria cell wall which is much higher than an equivalent concentration of the free antibiotic. Note that the Alcalase coating allows the antibiotic-packed nanocarriers to reach the bacterial cells which are “deeply buried” in the biofilm matrix. This novel method of bacteria-targeting active NPs with an additional anti-biofilm function offers an important opportunity to revive the usefulness of antibiotics considered obsolete (i.e. Penicillin G) and target biofilm embedded bacteria which are particularly prevalent in chronic wounds. We conclude that Alcalase-coated and PenG/Oxa-loaded shellac NPs are an effective and safe method of clearing antibiotic resistant biofilms, and propose this could be an alternative approach to topical biofilm treatments, particularly in chronic wounds. We believe that further experimental investigation into the application of these NPs on biofilm-infected chronic wounds is required, and could provide a new therapeutic approach for treating antibiotic resistant biofilms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. J. W. acknowledges the University of Hull for funding his PhD studentship as part of the Advanced Wound Care PhD cluster. The authors thank SEM technicians Tim Dunstan and Ann Lowry (University of Hull) with their assistance during this project.References
- C. B. Creech, D. N. Al-Zubeidi and S. A. Fritz, Prevention of Recurrent Staphylococcal Skin Infections, Infect. Dis. Clin. North Am., 2015, 29, 429–464 CrossRef.
- G. Kapoor, S. Saigal and A. Elongavan, Action and resistance mechanisms of antibiotics: A guide for clinicians, J. Anaesthesiol., Clin. Pharmacol., 2017, 33, 300 CrossRef CAS.
- O. Irazoki, S. B. Hernandez and F. Cava, Peptidoglycan Muropeptides: Release, Perception, and Functions as Signaling Molecules, Front. Microbiol., 2019, 10, 1–10 CrossRef.
- M. Mihelič, K. Vlahoviček-Kahlina, M. Renko, S. Mesnage, A. Doberšek, A. Taler-Verčič, A. Jakas and D. Turk, The mechanism behind the selection of two different cleavage sites in NAG-NAM polymers, IUCrJ, 2017, 4, 185–198 CrossRef.
- A. K. Yadav, A. Espaillat and F. Cava, Bacterial Strategies to Preserve Cell Wall Integrity Against Environmental Threats, Front. Microbiol., 2018, 9, 2064 CrossRef.
- Ç. Yılmaz and G. Özcengiz, Antibiotics: Pharmacokinetics, toxicity, resistance and multidrug efflux pumps, Biochem. Pharmacol., 2017, 133, 43–62 CrossRef.
- A. M. Smith, C. Feldman, O. Massidda, K. McCarthy, D. Ndiweni and K. P. Klugman, Altered PBP 2A and its role in the development of penicillin, cefotaxime, and ceftriaxone resistance in a clinical isolate of Streptococcus pneumoniae, Antimicrob. Agents Chemother., 2005, 49, 2002–2007 CrossRef CAS.
- C. C. S. Fuda, J. F. Fisher and S. Mobashery, β-Lactam resistance in Staphylococcus aureus: the adaptive resistance of a plastic genome, Cell. Mol. Life Sci., 2005, 62, 2617–2633 CrossRef CAS.
- K. T. Bæk, A. Gründling, R. G. Mogensen, L. Thøgersen, A. Petersen, W. Paulander and D. Frees, β-Lactam resistance in methicillin-resistant Staphylococcus aureus USA300 is increased by inactivation of the ClpXP protease, Antimicrob. Agents Chemother., 2014, 58, 4593–4603 CrossRef.
- W. H. O. Antimicrobial Resistance and Surveillance Report. 2014, https://apps.who.int/iris/bitstream/handle/10665/112642/9789241564748_eng.pdf;jsessionid=67D9BD148E9C9EFCA4EB1D2F3B4C32C3?sequence=1.
- R. Worthington and C. Melander, Overcoming Resistance to β-Lactam Antibiotics, J. Org. Chem., 2013, 78, 4207–4213 CrossRef CAS.
- R. Worthington and C. Melander, Combination approaches to combat multidrug-resistant bacteria, Trends Biotechnol., 2013, 31, 177–184 CrossRef CAS.
- G. Gebreyohannes, A. Nyerere, C. Bii and D. B. Sbhatu, Challenges of intervention, treatment, and antibiotic resistance of biofilm-forming microorganisms, Heliyon, 2019, 5, e02192 CrossRef.
- A. D. Verderosa, M. Totsika and K. E. Fairfull-Smith, Bacterial Biofilm Eradication Agents: A Current Review, Front. Chem., 2019, 7, 824 CrossRef CAS.
- D. H. Limoli, C. J. Jones and D. J. Wozniak, Bacterial Extracellular Polysaccharides in Biofilm Formation and Function, Microbiol. Spectrum, 2015, 3, MB-0011-2014 Search PubMed.
- D. Sharma, L. Misba and A. U. Khan, Antibiotics versus biofilm: an emerging battleground in microbial communities, Antimicrob. Resist. Infect. Control, 2019, 8, 76 CrossRef.
- S. Singh, S. K. Singh, I. Chowdhury and R. Singh, Understanding the Mechanism of Bacterial Biofilms Resistance to Antimicrobial Agents, Open Microbiol. J., 2017, 11, 53–62 CrossRef CAS.
- J. L. Lister and A. R. Horswill, Staphylococcus aureus biofilms: recent developments in biofilm dispersal, Front. Cell. Infect. Microbiol., 2014, 4, 178 Search PubMed.
- J. Richards and C. Melander, Controlling bacterial biofilms, ChemBioChem, 2009, 10, 2287–2294 CrossRef CAS.
- S. Newase and A. Banker, Synthesis Of Bio-Inspired Ag–Au Nanocomposite And Its Anti-Biofilm Efficacy, Bull. Mater. Sci., 2017, 40, 157–162 CrossRef CAS.
- K. Markowska, A. Grudniak and K. Wolska, Silver Nanoparticles As An Alternative Strategy Against Bacterial Biofilms, Acta Biochim. Pol., 2013, 60, 523–530 Search PubMed.
- A. F. Halbus, T. S. Horozov and V. N. Paunov, Strongly Enhanced Antibacterial Action of Copper Oxide Nanoparticles with Boronic Acid Surface Functionality, ACS Appl. Mater. Interfaces, 2019, 11, 38519–38530 CrossRef CAS.
- A. F. Halbus, T. S. Horozov and V. N. Paunov, Controlling the Antimicrobial Action of Surface Modified Magnesium Hydroxide Nanoparticles, Biomimetics, 2019, 4, 1–20 CrossRef.
- A. F. Halbus, T. S. Horozov and V. N. Paunov, Self-grafting copper oxide nanoparticles show a strong enhancement of their anti-algal and anti-yeast action, Nanoscale Adv., 2019, 1, 2323–2336 RSC.
- H. N. Wilkinson, S. Iveson, P. Catherall and M. J. Hardman, A Novel Silver Bioactive Glass Elicits Antimicrobial Efficacy Against Pseudomonas aeruginosa and Staphylococcus aureus in an ex Vivo Skin Wound Biofilm Model, Front. Microbiol., 2018, 9, 1450 CrossRef.
- M. Al-Awady, P. J. Weldrick, M. J. Hardman, G. M. Greenway and V. N. Paunov, Amplified antimicrobial action of chlorhexidine encapsulated in PDAC-functionalized acrylate copolymer nanogel carriers, Mater. Chem. Front., 2018, 2, 2032–2044 RSC.
- P. J. Weldrick, S. Iveson, M. J. Hardman and V. N. Paunov, Breathing new life into old antibiotics: overcoming antibacterial resistance by antibiotic-loaded nanogel carriers with cationic surface functionality, Nanoscale, 2019, 11, 10472–10485 RSC.
- P. J. Weldrick, M. J. Hardman and V. N. Paunov, Enhanced Clearing of Wound-Related Pathogenic Bacterial Biofilms Using Protease-Functionalized Antibiotic Nanocarriers, ACS Appl. Mater. Interfaces, 2019, 11, 43902–43919 CrossRef CAS.
- S. S. M. Al-Obaidy, A. F. Halbus, G. M. Greenway and V. N. Paunov, Boosting the antimicrobial action of vancomycin formulated in shellac nanoparticles of dual-surface functionality, J. Mater. Chem. B, 2019, 7, 3119–3133 RSC.
- R. Singh, M. Kumar, A. Mittal and P. Mehta, Microbial enzymes: industrial progress in 21st century, 3 Biotech, 2016, 6, 174 CrossRef.
- D. R. Baidamshina, E. Y. Trizna, M. G. Holyavka, M. I. Bogachev, V. G. Artyukhov, F. S. Akhatova, E. V. Rozhina, R. F. Fakhrullin and A. R. Kayumov, Targeting Microbial Biofilms Using Ficin, A Nonspecific Plant Protease, Sci. Rep., 2017, 7, 46068 CrossRef CAS.
- O. Mitrofanova, A. Mardanova, V. Evtugyn, L. Bogomolnaya and M. Sharipova, Effects Of Bacillus Serine Proteases On The Bacterial Biofilms, BioMed Res. Int., 2017, 1–10 Search PubMed.
- C. Leroy, C. Delbarre, F. Ghillebaert, C. Compere and D. Combes, Effects Of Commercial Enzymes On The Adhesion Of A Marine Biofilm-Forming Bacterium, Biofouling, 2008, 24, 11–22 CrossRef CAS.
- R. Melander and C. Melander, Innovative Strategies for Combating Biofilm-Based Infections, Adv. Exp. Med. Biol., 2014, 69–91 Search PubMed.
- F. U. Din, W. Arman, L. Ullah, O. S. Qureshi, O. Mustapha, S. Shafique and A. Zeb, Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors, Int. J. Nanomed., 2017, 12, 7291–7309 CrossRef.
- X. Yu, I. Trase, M. Ren, K. Duval, X. Guo and Z. J. Chen, Design of Nanoparticle-Based Carriers for Targeted Drug Delivery, J. Nanomater., 2016, e31718141 Search PubMed.
- A. Kumari, R. Singla, A. Guliani and S. K. Yadav, Nanoencapsulation for drug delivery, EXCLI J., 2014, 13, 265–286 Search PubMed.
- H. Jahangirian, E. G. Lemraski, T. J. Webster, R. RafieeMoghaddam and Y. Abdollahi, A review of drug delivery systems based on nanotechnology and green chemistry: green nanomedicine, Int. J. Nanomed., 2017, 12, 2957–2978 CrossRef CAS.
- ATCC Genome Portal, 2020, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC154455/.
- F. Lowy, Antimicrobial resistance: the example of Staphylococcus aureus, J. Clin. Invest., 2003, 111, 1265–1273 CrossRef CAS.
- EUCAST Breakpoint Tables v 10.0, 2020, http://www.eucast.org/clinicalbreakpoints/.
- S. S. M. Al-Obaidy, G. M. Greenway and V. N. Paunov, Dual-functionalised shellac nanocarriers give a super-boost of the antimicrobial action of berberine, Nanoscale Adv., 2019, 1, 858–872 RSC.
- A. P. Richter, J. S. Brown, B. Bharti, A. Wang, S. Gangwal, K. Houck, E. A. Cohen Hubal, V. N. Paunov, S. D. Stoyanov and O. D. Velev, Synthesis and Characterization of Biodegradable Lignin Nanoparticles with Tunable Surface Properties, Nat. Nanotechnol., 2015, 10, 817–823 CrossRef CAS.
- M. J. Al-Awady, A. Fauchet, G. M. Greenway and V. N. Paunov, Enhanced antimicrobial effect of berberine in nanogel carriers with cationic surface functionality, J. Mater. Chem. B, 2017, 5, 7885–7897 RSC.
- M. J. Al-Awady, G. M. Greenway and V. N. Paunov, Nanotoxicity of polyelectrolyte-functionalized titania nanoparticles towards microalgae and yeast: role of the particle concentration, size and surface charge, RSC Adv., 2015, 5, 37044–37059 RSC.
- M. Blackledge, R. Worthington and C. Melander, Biologically inspired strategies for combating bacterial biofilms, Curr. Opin. Pharmacol., 2013, 13, 699–706 CrossRef CAS.
- K. Lewis, Persister cells, dormancy and infectious disease, Nat. Rev. Microbiol., 2006, 5, 48–56 CrossRef.
- M. Farha and E. Brown, Drug repurposing for antimicrobial discovery, Nat. Microbiol., 2019, 4, 565–577 CrossRef CAS.
- E. Brown and G. Wright, Antibacterial drug discovery in the resistance era, Nature, 2016, 529, 336–343 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qm00874e |
| This journal is © the Partner Organisations 2021 |