
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoscale chemical speciation of β-amyloid/iron aggregates using soft X-ray spectromicroscopy†
James
Everett
 *ab,
Jake
Brooks
b,
Joanna F.
Collingwood
b and
Neil D.
Telling
a
*ab,
Jake
Brooks
b,
Joanna F.
Collingwood
b and
Neil D.
Telling
a
aSchool of Pharmacy and Bioengineering, Guy Hilton Research Centre, Thornburrow Drive, Keele University, Staffordshire, ST4 7QB, UK. E-mail: j.everett@keele.ac.uk
bSchool of Engineering, Library Road, University of Warwick, Coventry, CV4 7AL, UK
First published on 15th January 2021
Abstract
Iron (Fe) is an essential trace element required for healthy brain function. Yet, disrupted iron neurochemistry, and the associated formation of aberrantly aggregated protein lesions has been implicated in the development of multiple degenerative brain disorders including Alzheimer's disease (AD). Here, nanoscale resolution soft X-ray spectromicroscopy is used to examine the interaction of β-amyloid (Aβ), a peptide fundamentally implicated in the development of Alzheimer's, and ferric (Fe3+) iron. Crucially, by probing the carbon K (280–320 eV) and iron L2,3 (700–740 eV) edges, both the organic and inorganic (iron) sample chemistry was established. The co-aggregation of Aβ and iron is known to influence iron chemistry, resulting in the chemical reduction of Fe3+ into reactive and potentially toxic ferrous (Fe2+) and zero-oxidation (Fe0) states. Here, nanoscale (i.e. sub-micron) variations in both iron oxidation state and the organic composition of Aβ were observed, replicating in vitro the diverse iron chemistry documented in amyloid plaques from human brain, with the chemical state of iron linked to the conformation state of Aβ. Furthermore, aggregates were formed that were morphologically and chemically distinct dependent on the treatment of Aβ prior to the addition of ferric iron. These findings support the hypothesis that Aβ is responsible for altering iron neurochemistry, and that this altered chemistry is a factor in neurodegenerative processes documented in AD. The methods applied here, combining nanoscale-resolution imaging and high chemical sensitivity, enabled discovery of the nanoscale heterogeneity in the iron and carbon chemistry of in vitro aggregates, and these approaches have scope for wider application in metallomics.
Introduction
The transition metal iron is vital for healthy brain function, acting as a co-factor in numerous brain processes.1–4 The maintenance of brain iron homeostasis is fundamental to its well-being, and multiple complex mechanisms exist to maintain this balance.1,4 Although essential, iron can convey neurotoxic effects when mishandled in the brain.4–6 Reactive labile iron can partake in Fenton redox chemistry, producing reactive oxygen species (ROS) capable of inducing oxidative stress, and ultimately cell death.5–7 Chemically-reduced, low-oxidation-state iron (<3+) can catalyse these reactions, driving ROS production.6Perturbed iron homeostasis has been observed in numerous degenerative brain disorders including Alzheimer's disease (AD),3,8–10 indicating a link between altered brain iron handling and the onset of neurodegeneration. Disrupted iron homeostasis has also been implicated in the formation of the two hallmark protein lesions of AD: neurofibrillary tangles composed of hyperphosphorylated tau,11–13 and amyloid plaques composed of the peptide β-amyloid (Aβ).13–15 More specifically, chemically-reduced, low-oxidation-state iron has been observed co-localized to amyloid plaques in Alzheimer's affected tissues.16–18 Such metal forms are not commonplace in disease-free brains,18 suggesting their formation to be a result of Aβ interaction with endogenous brain iron. As increased oxidative burdens and related oxidative stress are recognised as key early stage events in the development of AD,19 the aberrant formation of low-oxidation-state iron may be fundamental to Alzheimer's disease pathogenesis. Moreover, iron has been shown to influence the aggregation dynamics of Aβ,20–23 potentially enhancing the toxicity of Aβ, further implicating Aβ/iron interaction in Alzheimer's disease aetiology.
Despite these observations, the chemistry of Aβ and iron interaction is not fully understood, whilst the chemical state and origin of iron associated with amyloid pathologies in AD requires further investigation. To date, metal-targeting therapies in AD and other neurodegenerative diseases have been limited in their success.24 One reason may be a lack of drug specificity, resulting in depletion of metal stores required to sustain neuronal health. Thus it is of paramount importance to characterise metal phases solely associated with disease states, and distinguish them from normally metabolized metals.
Synchrotron-based X-ray techniques offer a unique means to characterize metal (bio)chemistry in AD to an exceptional level of chemical and spatial detail.16,17 In our previous publications we demonstrated Aβ to chemically-reduce ferric (Fe3+) iron into a ferrous (Fe2+) state in vitro using X-ray absorption spectroscopy,25,26 providing a potential origin for the low-oxidation-state iron observed in AD tissues in vivo. Using this approach, we provided an averaged iron oxidation state arising from Aβ/iron aggregate structures over a spatial scale of ca. 100 μm2.
However, our more recent chemical speciation of amyloid pathology from AD tissues17 has demonstrated metal and organic sample chemistry to vary dramatically over small (<100 nm) spatial scales. Using nanoscale-resolution soft X-ray spectromicroscopy in the form of scanning transmission X-ray microscopy (STXM), we demonstrated complex sample heterogeneity, resulting in the identification of sub-micron deposits of zero-oxidation-state iron (Fe0) within these amyloid structures,17 that would have been otherwise overlooked using microfocus techniques.
Thus, to adequately understand the chemistry of Aβ/iron interaction, nanoscale resolution imaging coupled with precise chemical sensitivity is of vital importance. In this microscopy study, STXM and transmission electron microscopy (TEM) was used to examine the nanoscale chemical speciation of aggregate structures formed through the interaction of Aβ(1–42) [the full length form of the Aβ peptide] and iron(III). Carbon K-edge (280–320 eV) and iron L2,3-edge (700–740 eV) spectroscopic analysis was combined with high resolution (ca. 20 nm) microscopy, to monitor the organic composition and iron oxidation state of the Aβ/iron aggregates. We report dramatic nanoscale variations in both the carbon (peptide) and iron chemistry of Aβ/iron aggregates, and the discrimination of individual sub-micron iron deposits.
Experimental
Preparation of samples
Frozen synthetic Aβ(1–42) (Bachem) was thawed and dissolved in 100 mM NaOH for 30 minutes at room temperature to create a 1 mg mL−1 (220 μM) Aβ stock solution. NaOH was used to dissolve any peptide aggregates that may have formed during peptide storage, ensuring peptide dissolution to a monomeric state. The Aβ stock was then diluted in a modified, phosphate free, Krebs-Hensleit buffer (pH 7.4; 100 mM PIPES (piperazine-N,N′-bis(2-ethanesulfonic acid), 118.5 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 1.4 mM CaCl2, 11 mM glucose; all Sigma Aldrich) modelled on the cerebrospinal fluid of the central nervous system,23 and consistently used in our prior examination of Aβ/metal interactions.25–27 This buffer contains physiologically relevant concentrations of both calcium and magnesium, metals that are known to influence the aggregation dynamics of β-amyloid.28,29 The buffer PIPES was chosen as it does not strongly interact with metal ions.30Two Aβ/iron treatments were then prepared:
(i) To establish how iron(III) integrates into pre-formed Aβ aggregates, Aβ solutions in KH buffer were allowed to incubate at 37 °C for a minimum of 48 hours before the addition of iron(III) nitrate (PerkinElmer). The resulting Aβ/iron suspensions were then incubated for a further 30 minutes at 37 °C before sampling.
(ii) To assess the effect of Aβ and iron(III) co-incubation on a monomeric Aβ solution, iron(III) nitrate was added to the KH buffer immediately after Aβ, and the resulting Aβ/iron suspensions were allowed to incubate at 37 °C until the time of sampling (incubation times are stated in the respective figure legends).
These two treatment types were designed based on previous studies where differences in Aβ/iron chemistry were observed dependent upon the aggregation state of amyloid at the point of iron loading.22,23
For both treatments, final peptide and iron concentrations were 35 μM and 370 μM respectively. These concentrations were chosen to allow efficient detection of Aβ/iron structures using soft X-ray spectromicroscopy.26 35 μM Aβ solutions were prepared in KH buffer as an amyloid reference, and 370 μM iron(III) nitrate suspensions were prepared in KH buffer as an iron(III) reference. All solutions/suspensions of Aβ/iron were incubated within sealed microcentrifuge tubes. A flow chart summarizing the preparation methods used for all sample types is provided in the ESI (Fig. S1†).
Sampling was performed by depositing 15 μL of Aβ/iron solutions/suspensions onto silicon oxide membranes (75 nm window thickness; DuneSciences), and removing excess liquid using filter paper. Membranes containing Aβ/iron structures were then mounted onto aluminium plates for soft X-ray spectromicroscopy (STXM) examination. As STXM experiments were being performed to assess iron oxidation state following co-incubation with Aβ, precautions were taken to maintain anaerobic conditions during sampling and examination of Aβ/iron material. All sampling was performed in a nitrogen filled glove bag maintained under a positive pressure with respect to the ambient conditions. To further prevent changes in iron oxidation states, membranes containing Aβ/iron structures were stored within nitrogen filled, O-ring sealed jars. The mounting of membranes onto STXM plates was performed within a nitrogen filled glove bag, and anoxic conditions were maintained during sample transportation to the beamline by using a nitrogen filled vessel for sample transfer, and purging of the STXM endstation with nitrogen prior to sample loading.
Scanning transmission X-ray microscopy (STXM)
To assess the nanoscale morphology and chemical composition of Aβ/iron structures, X-ray spectromicroscopy was performed using STXM.STXM experiments were conducted on the Swiss Light Source (Villigen, Switzerland) PolLux beamline using the STXM endstation, with a focussed X-ray beam spot size of ca. 20 nm. Energy-specific images of the Aβ/iron structures were obtained by raster scanning the sample across the focussed X-ray beam, and recording the intensity of the transmitted X-rays. To best preserve sample chemistry, scanning (exposure) times were kept to a minimum (<4 ms per pixel), to minimise potential photoreduction effects.
To determine the chemical composition of Aβ/iron structures at a nanoscale spatial resolution, speciation maps were created by collecting paired images: (i) a “peak” image at the energy of the feature of interest, and (ii) an “off peak” image a few eV below this feature. The off peak image was then subtracted from the peak image to create an artefact-free contrast map (see ESI Fig. S2†), providing the chemical speciation for the examined sample region. Speciation mapping was performed at the carbon K-edge (288.3 eV) to determine Aβ morphology, and the iron L3-edge (710 eV) to show iron distribution.
X-ray absorption spectra, providing detailed information regarding the chemical state of the sample material (e.g. iron oxidation state) were generated by collecting a series of images, called a stack, of a given sample area at multiple energies across a desired energy range (e.g. the iron L2,3-absorption edge [700–740 eV], or the carbon K-absorption edge [280–320 eV]). For each stack image, transmitted X-ray intensities were converted to optical density using regions that did not contain any sample material. This conversion removes any background absorption features attributable to the beamline. By using this approach, X-ray absorption spectra can be generated from every pixel of a stack image. This allows the chemical composition of highly localized regions (20 nm) of interest to be determined to an exceptionally high level of detail. To minimize X-ray induced damage to carbon-based structures, carbon K-edge spectromicroscopy was performed prior to higher energy iron L2,3-edge spectroscopy.
STXM data processing was performed using the aXis 2000 software package (http://unicorn.mcmaster.ca/aXis2000.html). X-ray microscopy images were adjusted for brightness and contrast using ImageJ software. Grey scale X-ray microscopy images were converted to false colour and recombined as overlays to create coloured composite images.
Analysis of X-ray absorption spectra
To provide a quantitative estimate of the relative proportion of iron phases contributing to measured iron L2,3-edge X-ray absorption spectra in these experiments, spectra were fitted to reference iron X-ray absorption spectra using a non-linear least squares fitting procedure.Four reference iron L2,3-edge X-ray absorption spectra obtained from ferritin, FeCl2, magnetite and an Fe film (see Fig. S3†) were used to estimate the relative contribution of Fe3+, Fe2+, Fe3O4 and Fe0 to the displayed experimental iron L2,3-edge spectra. The iron L2,3-edge absorption features from ferritin are consistent with a pure ferric phase and it was therefore chosen as a suitable ferric (Fe3+) standard for fitting. Furthermore, this ferritin spectrum is representative of a biologically-sourced ferric phase. The iron L2,3-edge absorption features of FeCl2 are consistent with a pure ferrous (Fe2+) phase and it was used as a ferrous standard. Magnetite (Fe3O4) was chosen as this phase has previously been observed in amyloid structures from Alzheimer's disease tissue.17,31,32 Inclusion of a magnetite reference spectrum for fitting was further required as this mixed-valence (Fe2+/3+) phase provides a unique iron L2,3-edge absorption spectrum, that is not simply a sum of its principal Fe2+ and Fe3+components. The Fe0 reference spectrum used for fitting was obtained from Fe0 film standards prepared and measured under vacuum to prevent oxidation.33 Like magnetite, Fe0 was included in the fitting process owing to our previous observation of Fe0 nanoparticles in amyloid structures from Alzheimer's disease tissue.17
Reference spectra were appropriately scaled by normalizing the X-ray absorption intensity for each iron phase to the integrated intensity over the iron L2,3 absorption edges, as described in our previous work (see also Fig. S4†).17 A visualisation of the energy ranges used for fitting, and an example overlay of an experimental spectrum and its corresponding fit are provided in ESI Fig. S5 and S6† respectively.
A table listing the strength of fit for all displayed experimental iron L2,3-edge absorption spectra is provided in the ESI (Table S1†). An error value of ±2% for each iron phase is used in spectral fitting. This error value is due to ambiguity in the uniqueness of fit owing to noise in the experimental iron L2,3-edge X-ray absorption spectra.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed using a JEOL 1230 microscope operating at 100 kV. No positive or negative stains were used during TEM microscopy. TEM was performed last, to avoid any scope for electron beam induced changes to sample chemistry prior to STXM analysis.Results & discussion
TEM images and STXM analysis of four Aβ/iron aggregates formed under the two described incubation conditions are provided in Fig. 1–4. A further four Aβ/iron aggregates are displayed in the ESI Fig. S8–11.† TEM images from Aβ aggregates formed in the absence of iron are shown in Fig. 5.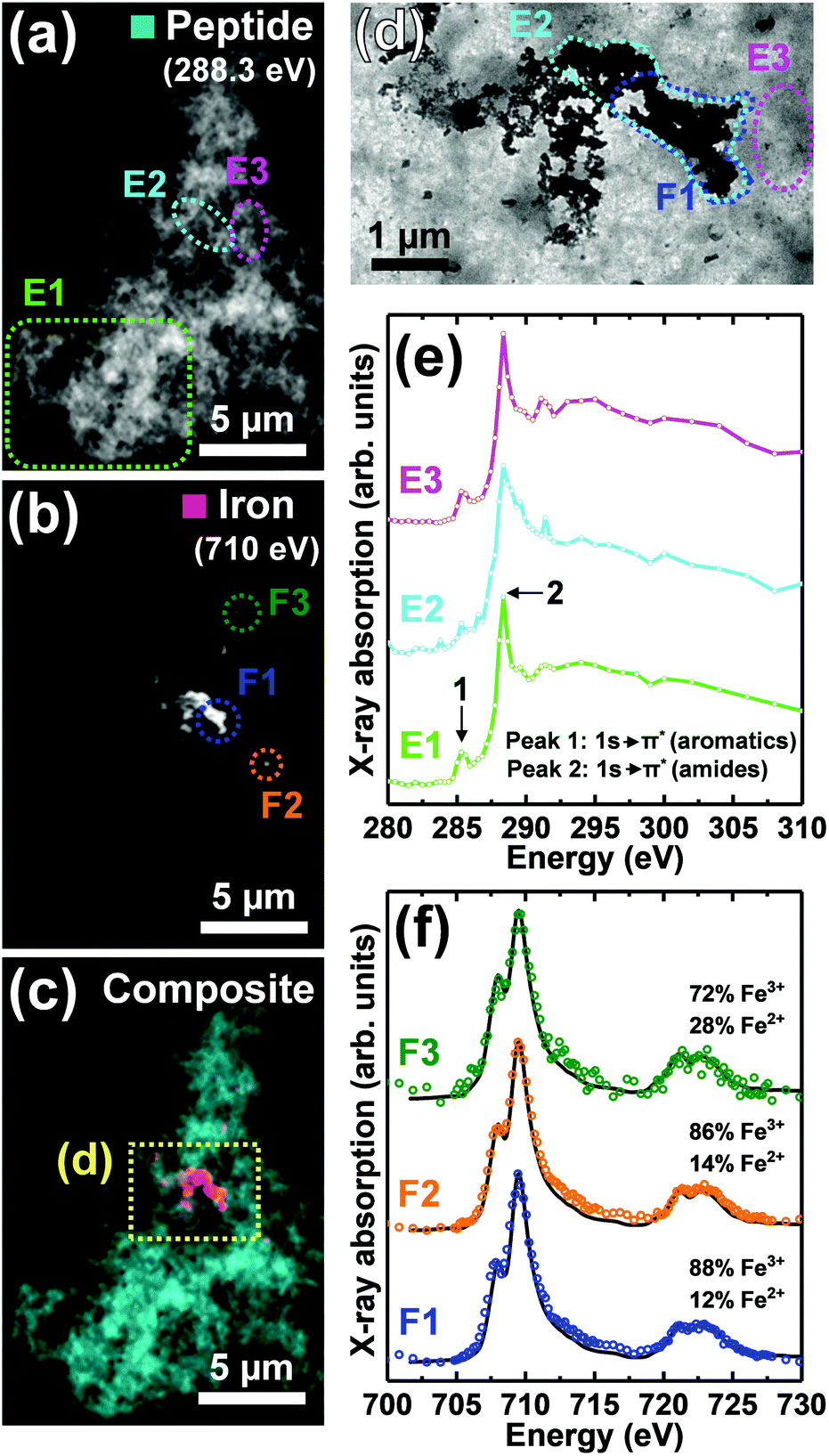 | ||
| Fig. 1 STXM speciation maps, TEM image, carbon K-edge X-ray absorption spectra and iron L2,3-edge X-ray absorption spectra from a Aβ/iron aggregate, formed following 72 hours of Aβ incubation prior to the addition of iron(III). (a) Carbon K-edge peptide map. (b) Iron L3-edge map. (c) Composite image showing peptide (cyan) and iron (red) content. (d) TEM image of the aggregate region highlighted in (c). A TEM image of the whole aggregate is also shown in ESI Fig. S2.† (e) Carbon K-edge X-ray absorption spectra from the aggregate regions highlighted in panels (a) and (d). (f) Iron L2,3-edge X-ray absorption spectra (coloured circles) from the regions highlighted in panel (b). The solid lines for the spectra correspond to best fit curves created by superposition of suitably scaled iron reference X-ray absorption spectra (see Fig. S4† and Everett et al., Nanoscale17). | ||
 | ||
| Fig. 2 TEM image, STXM speciation maps, carbon K-edge and iron L2,3-edge X-ray absorption spectra from a Aβ/iron aggregate, formed following 48 hours of Aβ incubation prior to the addition of iron(III). (a) TEM image. (b) Carbon K-edge peptide map. (c) Iron L3-edge map. (d) Composite image showing peptide (cyan) and iron (red) content. (e) Carbon K-edge X-ray absorption spectra from the regions highlighted in (b). The Aβ(1–42) reference spectrum displayed in ESI Fig. S7† is also provided [black trace]. (f) Iron L2,3-edge X-ray absorption spectra from the regions highlighted in (c). The solid lines for the spectra correspond to best fit curves created by superposition of suitably scaled iron reference X-ray absorption spectra. | ||
 | ||
| Fig. 3 TEM image, STXM speciation maps, carbon K-edge and iron L2,3-edge X-ray absorption spectra from a Aβ/iron aggregate, formed following 48 hours of Aβ/iron co-incubation. (a) TEM image. (b) Carbon K-edge peptide map. (c) Iron L3-edge map. (d) Composite image showing peptide (cyan) and iron (red) content. (e) Carbon K-edge X-ray absorption spectrum from the region highlighted in (b). (f) Iron L2,3-edge X-ray absorption spectra from the regions highlighted in (c). The solid lines for the spectra correspond to best fit curves created by superposition of suitably scaled iron reference X-ray absorption spectra. | ||
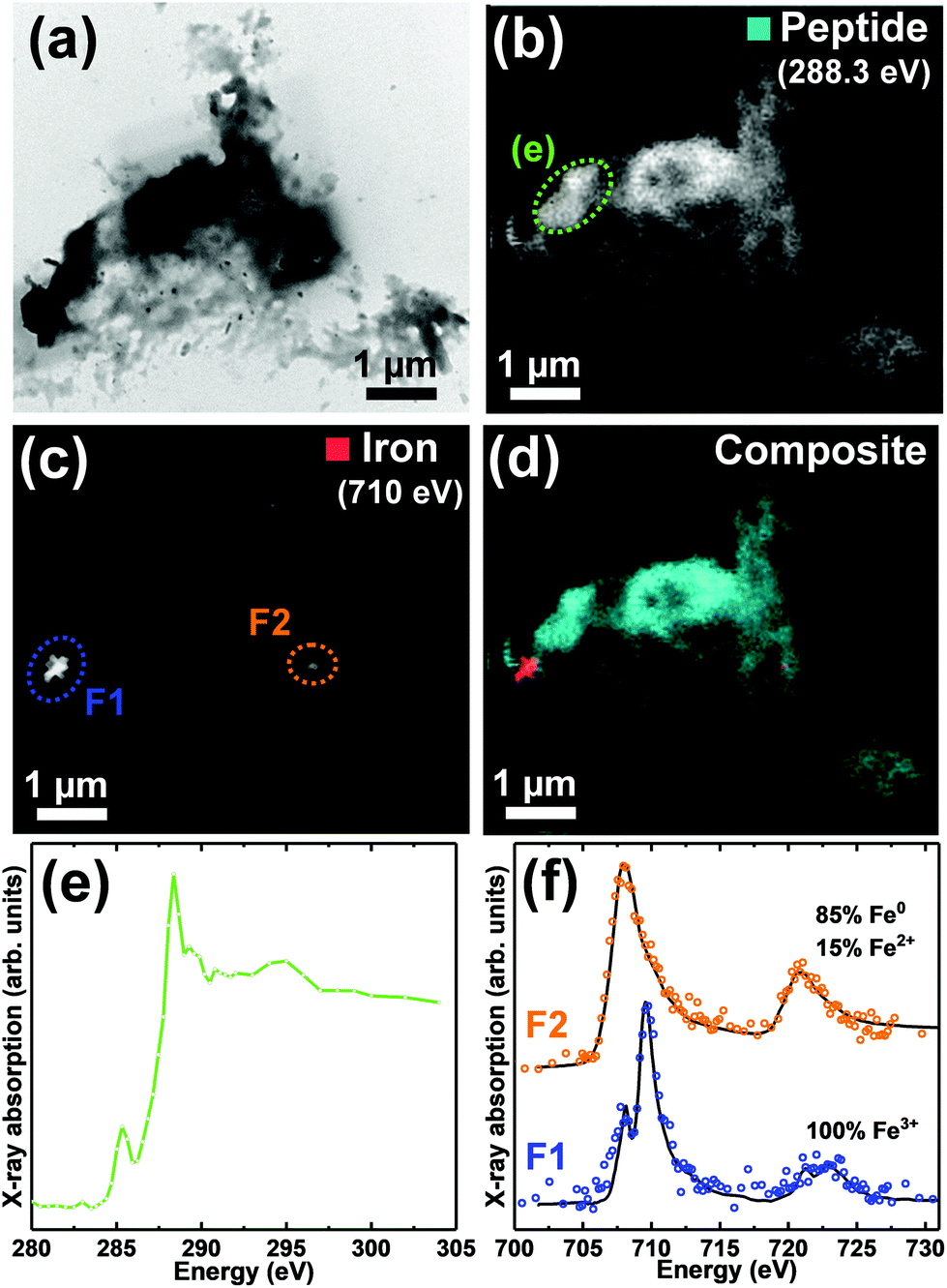 | ||
| Fig. 4 TEM image, STXM speciation maps, carbon K-edge and iron L2,3-edge X-ray absorption spectra from a further Aβ/iron aggregate formed following 48 hours of Aβ/iron co-incubation. (a) TEM image. (b) Carbon K-edge peptide map. (c) Iron L3-edge map. (d) Composite image showing peptide (cyan) and iron (red) content. (e) Carbon K-edge X-ray absorption spectrum from the region highlighted in (b). (f) Iron L2,3-edge X-ray absorption spectra from the regions highlighted in (c). The solid lines for the spectra correspond to best fit curves created by superposition of suitably scaled iron reference X-ray absorption spectra. | ||
 | ||
| Fig. 5 TEM images of Aβ aggregates formed without addition of iron. | ||
Pre-incubated Aβ
As previously described,26,34 TEM examination of Aβ samples allowed to incubate in KH buffer for a minimum of 48 h prior to the addition of iron(III) led to the observation of extensive firbillar aggregates containing electron dense regions (Fig. 1– and 2, S1†; see also Fig. 3 in ref. 34). STXM speciation mapping performed at the carbon K-edge to observe peptide distribution, showed peptide structure to closely resemble the aggregates observed under TEM (Fig. 1a and 2b), confirming these aggregates to be composed of Aβ. Iron L3-edge speciation mapping of the aggregates (Fig. 1b and 2c) showed regions of iron accumulation spanning several microns, with iron distribution resembling the peptide morphology in these regions, indicating that iron had incorporated into the aggregated Aβ fibrils. For the aggregate shown in Fig. 1, iron was present as discrete regions of iron deposition, whereas in Fig. 2 iron was found to be distributed throughout the entire aggregate structure. These results demonstrate a heterogeneity in iron loading between Aβ aggregates formed under the same starting experimental conditions. By comparing the iron L3-edge speciation maps with the TEM images, it was apparent that the most electron dense regions in these fibrillar aggregates corresponded to highest levels of iron loading. In addition, Aβ fibrils associated with iron were poorly defined compared to regions within the same aggregate devoid of iron (see also Fig. S12†), and Aβ-only control aggregates formed without the addition of iron(III) (Fig. 5).To probe the organic composition of the Aβ/iron structures in more detail, X-ray spectromicroscopy was performed over the entire carbon K-edge (280–320 eV). The resulting X-ray absorption spectra obtained from the aggregates displayed in Fig. 1 and 2 are shown in panels 1e and 2e respectively. By examining these spectra, clear differences can be seen, dependent on the level of iron loading in the aggregate region being examined.
In Fig. 1, spectrum E1 was collected from an aggregate region containing no detectable iron. This spectrum was composed of two sharp peak features at ca. 285.2 eV and 288.3 eV, corresponding to the 1s to π* transitions for peptide aromatic and amide groups respectively, and closely resembled the Aβ reference spectrum (Fig. 2e [black trace] and ESI Fig. S7†). Conversely, spectrum E2 was collected from a region of high iron loading, and displayed a suppressed aromatic feature at 285.2 eV, and a broadening of the amide feature at 288.3 eV, when compared to the iron-free aggregate region. These results suggest alterations in Aβ chemical composition upon iron loading. Spectrum E3, which was collected from an iron-free region immediately adjacent to the iron-rich region E2 (see Fig. 1d), was consistent with spectrum E1 and the Aβ reference, demonstrating variations in Aβ composition to occur over spatial scales 100s of nm in length.
Consistent with the spectrum from the iron-rich aggregate region of Fig. 1 (spectrum E2), carbon K-edge spectra from multiple regions of the aggregate in Fig. 2 (where iron was spread throughout the entire structure) showed a broadened amide peak and suppressed aromatic peak compared to the Aβ standard. This altered Aβ carbon K-edge spectra associated with iron loading was also observed in the Aβ/aggregate shown in Fig. 3 of ref. 34, which was formed under identical experimental conditions to the aggregates shown in Fig. 1 and 2 here.
The “building block principle” states that X-ray absorption spectra acquired from complex structures can be divided into individual contributions from specific functional groups/bonds.35 Important caveats to this approach are the effects of delocalization of electronic charge across functional groups giving rise to new molecular orbitals,35 the effects of changing bond length on overlap between orbitals36 and practical restrictions on available energy resolution. We hypothesise that observed reproducible broadening of the π* amide peak in carbon K-edge spectra acquired from iron-rich amyloid regions may be attributable to the formation of an iron–amyloid composite within these regions giving rise to new molecular orbitals.
To establish the oxidation state of iron within the amyloid aggregates, STXM measurements were performed over the iron L2,3-edge (700–740 eV). In order to provide an estimate of the relative proportion of iron phases contributing to the measured iron L2,3-edge X-ray absorption spectra, spectra were fitted to suitably scaled reference X-ray absorption spectra using a non-linear least squares fitting procedure (see ref. 17).
Iron L2,3-edge X-ray absorption spectra from different iron-rich regions of the Aβ/iron aggregates displayed in Fig. 1 and 2, are shown in panels 1f and 2f respectively. For all of these regions, iron was found to be in a predominantly ferric (Fe3+) state, with minor contributions from ferrous (Fe2+) iron. Ferric materials display a low intensity peak at 708 eV followed by a dominant high-intensity 709.5 eV peak at the L3-edge, and two further low intensity L2-absorption edge peaks at 721 and 723 eV (see Fig. S3† for reference iron L2,3-edge X-ray absorption spectra).37 These findings show that the oxidation state of the starting iron(III) was predominantly preserved where iron was added to the pre-formed Aβ aggregates.
Aβ/iron co-incubation
Fig. 3 and 4 show TEM images and STXM data from aggregate structures formed where Aβ and iron(III) were added simultaneously to the KH buffer, and allowed to co-incubate for 48 hours.TEM imaging (Fig. 3a and 4a) showed these aggregates to be largely amorphous, displaying no clear fibrillar structure. This was in stark contrast to the aggregate structures shown in Fig. 1 and 2, which displayed fibril morphology. Carbon K-edge speciation mapping (Fig. 3b and 4b) and spectromicroscopy showed both aggregates to be carbon-dense, providing carbon K-edge X-ray absorption spectra (Fig. 3e and 4e) largely consistent with the Aβ reference, confirming these amorphous structures to be composed of the peptide.
Iron L3-edge mapping (Fig. 3c and 4c) showed iron distribution in these amorphous aggregates to be confined to discrete sub-micron deposits, 100s of nm in diameter. This was again in contrast to the aggregates shown in Fig. 1 and 2, where regions of iron loading spanned several microns, within which iron distribution mirrored peptide morphology. Iron L2,3-edge X-ray absorption spectra from the aggregate regions highlighted in Fig. 3c and 4c, are shown in Fig. 3f and 4f respectively, and reveal a dramatic variation in iron oxidation state within the aggregates; with both oxidized and chemically-reduced phases being present.
For the aggregate shown in Fig. 3, fitting of the iron L2,3-edge X-ray absorption spectrum F1 showed this region to be composed of pure ferric iron. Fitting of the spectra from regions F2 and F4 showed evidence of ferrous and/or zero-oxidation-state iron (Fe0), made apparent by an enhanced intensity of the principal Fe2+/Fe0 absorption feature at 708 eV,37 with respect to the Fe3+ absorption feature at 709.5 eV. Although Fe2+ and Fe0 share a principal L3 absorption energy at 708 eV, the two phases are distinguishable by the broader line-shape for the Fe0 spectrum which lacks the splitting seen for oxide spectra, and the more prominent L2 peak and post-L2 edge absorption intensity for Fe0 (see ESI Fig. S3†).33,37
Fitting of spectrum F3 showed this region to be primarily composed of the mixed-valence phase magnetite. The presence of magnetite was determined by a decreased clarity of separation between the L3-edge absorption features at 708 eV and 709.5 eV when compared to a ferric standard. In magnetite the low energy Fe3+ peak feature at 708 eV is not discernible, instead appearing as a shoulder on the 709.5 eV peak feature.37
Iron L2,3-edge X-ray absorption spectra from the two strongest absorbing regions of the aggregate shown in Fig. 4 are displayed in Fig. 4f. Fitting of spectrum F1 showed this region to be composed of pure ferric iron, whereas region F2 was composed primarily of Fe0, with a minor contribution from Fe3+.
Interestingly, fibrillar aggregates containing extensive regions of iron accumulation (similar to those shown in Fig. 1 and 2) were also observed where amyloid and iron(III) were added simultaneously and allowed to co-incubate in the KH buffer. These aggregates are shown in ESI Fig. S8–S11,† and further demonstrate a heterogeneity in Aβ/iron aggregate structures formed under the same initial incubation conditions. In these fibrillar aggregates, iron was observed to remain in a predominantly ferric state, suggesting a possible link between Aβ/iron aggregate morphology and iron oxidation state (vide infra). Additional higher magnification TEM images are provided in ESI Fig. S12,† with Aβ/iron aggregates being categorised according to fibril containing or amorphous morphology.
Addition of iron(III) nitrate to the KH buffer medium in the absence of Aβ resulted in the precipitation of an insoluble Fe(OH)3 species as shown in ESI Fig. S13.† Examination of this iron across the iron L2,3-edge X-ray absorption edge provided spectra consistent with ferric iron (ESI Fig. S14†).
The data presented shows the chemical reduction of iron(III) following co-aggregation with Aβ. A clear heterogeneity in the morphology and chemistry of Aβ/iron aggregates was apparent, even where aggregates were formed under the same initial experimental conditions, with two predominant Aβ/iron aggregate subtypes being observed:
(i) Fibrillar aggregates containing extensive regions of iron loading, in which iron distribution closely resembled peptide morphology, indicating that iron was incorporated into the aggregating Aβ(1–42). This aggregate type was observed both where Aβ was incubated prior to the addition of iron(III), and where Aβ and iron(III) were added simultaneously and allowed to co-incubate. Within these aggregates, iron loading was found to be heterogeneous and to affect the organic composition of Aβ. In aggregate regions free of detectable iron, Aβ provided a carbon K-edge X-ray absorption spectrum consistent with experimental Aβ references. However, in aggregate regions loaded with iron, the Aβ spectrum was altered, with a suppression of the aromatic absorption peak, and a broadened amide peak being recorded.34 This altered Aβ spectrum was highly localized to regions of iron accumulation, with nanoscale variations in carbon X-ray absorption spectra being observed between adjacent iron loaded and iron-free aggregate regions. Iron L2,3-edge measurements showed iron within these fibrillar aggregates to remain in a predominantly ferric state, albeit with a minor ferrous component, which varied subtly in intensity across the aggregate regions, again demonstrating a heterogeneity in sample chemistry even within individual aggregate structures.
(ii) Amorphous aggregates, containing discrete, sub-micron deposits of iron. This aggregate type was observed only where Aβ and iron(III) had been added simultaneously and allowed to co-incubate. The oxidation state of these iron deposits was found to vary dramatically, with pure ferric and predominantly low-oxidation-state (<3+) iron regions being found within the same aggregate structures. This included the presence of zero-oxidation-state iron (Fe0), a phase not observed in the fibrillar aggregate type.
This variation in the organic and inorganic composition of Aβ/iron structures, evident over spatial scales 100s of nm in length, highlights the necessity of combined nanoscale resolution imaging and chemical speciation (as provided by STXM) to examine the role played by metal/protein interactions in neurodegenerative disorders. Whilst chemically sensitive, microfocus X-ray techniques typically applied to characterize metal chemistry in pathological structures (e.g. ref. 38 and 39) do not offer nanoscale spatial resolution, resulting in a signal averaging on the micrometre (μm) scale. The use of such techniques would have precluded the identification of the nanoscale low-oxidation-state iron deposits, including the Fe0 that we report here, as the signal from Fe0 would have been dominated by the (more) oxidized signal from the surrounding iron. The highly localized nature of the Fe0 deposits also offers an explanation as to why such minor phases were not observed in our previous examination of equivalent Aβ/iron aggregates using X-ray absorption spectroscopy,26 where the iron signal was averaged over ca. 100 μm2.
Comparatively, although spatially sensitive, electron-based measurements induce a significantly higher degree of beam damage when compared to STXM,40 preventing the quantification of iron oxidation state described here. Thus, the combined chemical sensitivity and nanoscale resolution of STXM provides a powerful tool to establish new insights into the biochemistry of neurodegeneration, and more broadly the role of metals in biology. Furthermore, anticipated technical developments to synchrotron facilities (including the completion of 4th generation synchrotron light sources) promise reduced measurement times and greater levels of sensitivity, allowing for high-throughput studies of biological systems with STXM, and the generation of sufficiently large datasets for robust statistical analysis.
The observation of low-oxidation-state iron phases following the co-incubation of Aβ and iron(III) is in keeping with previous publications from our group,25–27 from others who have described Aβ-mediated reduction of iron(III) in bulk measurements via spectrophotometry,23,41 and cyclic voltammetry evidence reporting the high reduction potential of Aβ–metal complexes.42,43 Furthermore, these results provide direct evidence that Aβ/iron interaction may serve as an origin for the nanoscale, chemically-reduced iron phases observed within amyloid pathology from human AD tissues including Fe0, which we recently discovered to exist within amyloid plaque cores for the first time.17 What remains unclear is whether iron(III) binding to Aβ was required for the chemical reduction of iron to occur. Determining Aβ–iron binding was not possible with STXM, and was therefore beyond the scope of this study. However numerous other works, including those conducted by our group, have examined Aβ–metal iron binding using more viable techniques for this purpose (e.g. ref. 34, 44 and 45).
It has been proposed that Aβ–metal interactions occurring in the AD brain could lead to the localized overproduction of ROS via Fenton chemistry,5,6 resulting in oxidative stress and disease progression through neuronal failure, and may therefore be relevant to disease pathogenesis. Indeed, previous in vitro evidence shows the chemical reduction of iron(III) by Aβ to result in the production ROS,41,46 at sufficiently high levels as to induce cytotoxic effects.22,47 Interestingly this reported cytotoxic effect was found to be influenced by the aggregation state of Aβ at the point of iron loading, with toxicity only being observed where iron was present throughout the aggregation process.22 This is consistent with the results we present here, where Fe0 was observed only in aggregates formed through the co-incubation of Aβ and iron(III).
The identification of differing aggregate subtypes dependent upon the aggregation state of Aβ at the point of iron loading in vitro, is relevant to the amyloid structures observed in our examination of brain tissues from a transgenic mouse model of AD.16 In these tissues, large fibrillar amyloid structures were found to be uniformly loaded with iron, with iron existing predominantly in a ferric state. These iron loaded structures also displayed an altered carbon signature compared to the surrounding iron-free neuropil. Conversely, amorphous amyloid structures in these tissues contained punctate, nanoscale iron deposits of varying oxidation state, with ferric, ferrous and magnetite phases being observed. The results presented herein, suggest that the differing amyloid plaque types formed in vivo may reflect the conformational state of amyloid at the point at which the peptide comes into contact with iron. Specifically, soluble amyloid fragments interacting with iron may result in the formation of amorphous amyloid plaques containing nanoscale chemically-reduced iron deposits, whereas pre-formed insoluble aggregates may sequester iron into their fibrillar structure upon encountering the metal.
The observation of differing iron oxidation states, dependent upon the aggregation state of Aβ at the point of iron loading, also raises a series of important questions regarding our understanding of Aβ/iron interaction in AD tissues. Are soluble Aβ fragments stronger iron reducing agents than insoluble Aβ plaques? By extension, are these reactions occurring intracellularly, extracellularly or both? Is the secretion of Aβ as insoluble plaques a protective mechanism,48 preventing the redox cycling of iron and production of ROS by soluble Aβ fragments?
Conclusions
In conclusion, the results presented demonstrate the co-incubation of Aβ with iron(III) to result in the formation of aggregate structures displaying a heterogeneity in morphology and nanoscale chemical composition. The chemical reduction of iron(III) upon co-aggregation with the Aβ(1–42) fragment was observed, with the extent of this reduction appearing to be influenced by the conditions of incubation. These findings implicate Aβ in the formation of the chemically-reduced iron phases observed in amyloid pathology from human AD tissues, and suggest Aβ/iron interactions to be a potential source of toxicity in AD-affected brains, through the generation of iron phases capable of ROS production.Low-oxidation-state iron phases associated with the formation of Aβ aggregates may represent a promising target for viable therapies intended to combat oxidative stress in AD tissues, thereby hindering disease progression. This could be achieved by either preventing the co-aggregation of Aβ and iron, or by targeting the low-oxidation-sate iron directly (e.g. through iron chelators). By specifically targeting phases such as Fe0, which have no known biological function, it may be possible to lower oxidative burdens in AD tissues, without negatively impacting upon iron stores required to maintain healthy brain function. Furthermore, phases such as Fe0 are strongly magnetic and could act as an endogenous marker for disease diagnosis and staging using magnetically sensitive imaging techniques such as MRI. Contrast from amyloid plaques is observed in high resolution pre-clinical and clinical MRI, so detailed characterization of iron associated with amyloid deposits may support progress in developing ionizing–radiation-free sequences sensitive to amyloid burden via this endogenous contrast. The X-ray spectromicroscopy methodologies used here also demonstrate the value of combining both chemical sensitivity and nanoscale spatial resolution when examining pathological inclusions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Diamond Light Source, EPSRC doctoral training grant EP/P503981, and EPSRC project grants EP/N033191/1 and EP/N033140/1. Measurements were performed on the PolLux beamline at the Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland, and we thank Dr Joerg Raabe and Dr Benjamn Watts for their support. We thank Karen Walker (Keele University) for TEM support.Notes and references
- J. R. Connor, S. L. Menzies, J. R. Burdo and P. J. Boyer, Iron and iron management proteins in neurobiology, Pediatr. Neurol., 2001, 25, 118–129 CrossRef CAS.
- D. J. Piñero and J. R. Connor, Iron in the Brain: An Important Contributor in Normal and Diseased States, Neuroscientist, 2000, 6, 435–453 CrossRef.
- L. Zecca, M. B. H. Youdim, P. Riederer, J. R. Connor and R. R. Crichton, Iron, brain ageing and neurodegenerative disorders, Nat. Rev. Neurosci., 2004, 5, 863–873 CrossRef CAS.
- R. J. Ward, F. A. Zucca, J. H. Duyn, R. R. Crichton and L. Zecca, The role of iron in brain ageing and neurodegenerative disorders, Lancet Neurol., 2014, 13, 1045–1060 CrossRef CAS.
- D. B. Kell, Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases, BMC Med. Genomics, 2009, 2, 2 CrossRef.
- J. Prousek, Fenton chemistry in biology and medicine, Pure Appl. Chem., 2007, 79, 2325–2338 CAS.
- A. I. Bush, The metallobiology of Alzheimer's disease, Trends Neurosci., 2003, 26, 207–214 CrossRef CAS.
- H. Kozlowski, M. Luczkowski, M. Remelli and D. Valensin, Copper, zinc and iron in neurodegenerative diseases (Alzheimer's, Parkinson's and prion diseases), Coord. Chem. Rev., 2012, 256, 2129–2141 CrossRef CAS.
- Y. Ke and Z. M. Qian, Iron misregulation in the brain: a primary cause of neurodegenerative disorders, Lancet Neurol., 2003, 2, 246–253 CrossRef CAS.
- R. J. Castellani, P. I. Moreira, G. Liu, J. Dobson, G. Perry, M. A. Smith and X. Zhu, Iron: The redox-active center of oxidative stress in Alzheimer disease, Neurochem. Res., 2007, 32, 1640–1645 CrossRef CAS.
- R. J. Castellani, R. K. Rolston and M. A. Smith, Alzheimer Disease, Dis.-Mon., 2010, 56, 484–546 CrossRef.
- M. Goedert, S. S. Sisodia and D. L. Price, Neurofibrillary tangles and β-amyloid deposits in Alzheimer's disease, Curr. Opin. Neurobiol., 1991, 1, 441–447 CrossRef CAS.
- C. Ballard, S. Gauthier, A. Corbett, C. Brayne, D. Aarsland and E. Jones, Alzheimer's disease, Lancet, 2011, 377, 1019–1031 CrossRef.
- J. C. Fiala, Mechanisms of amyloid plaque pathogenesis, Acta Neuropathol., 2007, 114, 551–571 CrossRef.
- J. Rogers and J. H. Morrison, Quantitative morphology and regional and laminar distributions of senile plaques in Alzheimer's-disease, J. Neurosci., 1985, 5, 2801–2808 CrossRef CAS.
- N. D. Telling, J. Everett, J. F. Collingwood, J. Dobson, G. van der Laan, J. J. Gallagher, J. Wang and A. P. Hitchcock, Iron Biochemistry is Correlated with Amyloid Plaque Morphology in an Established Mouse Model of Alzheimer's Disease, Cell Chem. Biol., 2017, 24, 1205–1215 CrossRef CAS.
- J. Everett, J. F. Collingwood, V. Tjendana-Tjhin, J. Brooks, F. Lermyte, G. Plascencia-Villa, I. Hands-Portman, J. Dobson, G. Perry and N. D. Telling, Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer's disease subjects, Nanoscale, 2018, 10, 11782–11796 RSC.
- M. A. Smith, P. L. R. Harris, L. M. Sayre and G. Perry, Iron accumulation in Alzheimer disease is a source of redox-generated free radicals, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9866–9868 CrossRef CAS.
- A. Nunomura, G. Perry, G. Aliev, K. Hirai, A. Takeda, E. K. Balraj, P. K. Jones, H. Ghanbari, T. Wataya, S. Shimohama, S. Chiba, C. S. Atwood, R. B. Petersen and M. A. Smith, Oxidative damage is the earliest event in Alzheimer disease, J. Neuropathol. Exp. Neurol., 2001, 60, 759–767 CrossRef CAS.
- X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer's Aβ peptides, JBIC, J. Biol. Inorg. Chem., 2004, 9, 954–960 CrossRef CAS.
- E. House, J. Collingwood, A. Khan, O. Korchazkina, G. Berthon and C. Exley, Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Aβ 42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease, J. Alzheimer's Dis., 2004, 6, 291–301 CAS.
- B. Liu, A. Moloney, S. Meehan, K. Morris, S. E. Thomas, L. C. Serpell, R. Hider, S. J. Marciniak, D. A. Lomas and D. C. Crowther, Iron Promotes the Toxicity of Amyloid β Peptide by Impeding Its Ordered Aggregation, J. Biol. Chem., 2011, 286, 4248–4256 CrossRef CAS.
- A. Khan, J. P. Dobson and C. Exley, Redox cycling of iron by Aβ42, Free Radicals Biol. Med., 2006, 40, 557–569 CrossRef CAS.
- M. T. Nuñez and P. Chana-Cuevas, New Perspectives in Iron Chelation Therapy for the Treatment of Neurodegenerative Diseases, Pharmaceuticals, 2018, 11, 109 CrossRef.
- J. Everett, E. Cespedes, L. R. Shelford, C. Exley, J. F. Collingwood, J. Dobson, G. van der Laan, C. A. Jenkins, E. Arenholz and N. D. Telling, Evidence of Redox-Active Iron Formation Following Aggregation of Ferrihydrite and the Alzheimer's Disease Peptide beta-Amyloid, Inorg. Chem., 2014, 53, 2803–2809 CrossRef CAS.
- J. Everett, E. Cespedes, L. R. Shelford, C. Exley, J. F. Collingwood, J. Dobson, G. van der Laan, C. A. Jenkins, E. Arenholz and N. D. Telling, Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide beta-amyloid (1–42), J. R. Soc., Interface, 2014, 11, 20140165 CrossRef CAS.
- J. Everett, J. Brooks, F. Lermyte, P. B. O'Connor, P. J. Sadler, J. Dobson, J. F. Collingwood and N. D. Telling, Iron stored in ferritin is chemically reduced in the presence of aggregating Aβ(1-42), Sci. Rep., 2020, 10, 10332 CrossRef CAS.
- A. M. Isaacs, D. B. Senn, M. Yuan, J. P. Shine and B. A. Yankner, Acceleration of Amyloid β-Peptide Aggregation by Physiological Concentrations of Calcium, J. Biol. Chem., 2006, 281, 27916–27923 CrossRef CAS.
- K. Garai, P. Sengupta, B. Sahoo and S. Maiti, Selective destabilization of soluble amyloid β oligomers by divalent metal ions, Biochem. Biophys. Res. Commun., 2006, 345, 210–215 CrossRef CAS.
- M. H. Benn, A. Rauk and T. W. Swaddle, Measurement of the interaction of aqueous copper(II) with a model amyloid-β protein fragment—Interference from buffers, Can. J. Chem., 2011, 89, 1429–1444 CrossRef CAS.
- J. F. Collingwood, R. K. K. Chong, T. Kasama, L. Cervera-Gontard, R. E. Dunin-Borkowski, G. Perry, M. Pósfai, S. L. Siedlak, E. T. Simpson, M. A. Smith and J. Dobson, Three-Dimensional Tomographic Imaging and Characterization of Iron Compounds within Alzheimer's Plaque Core Material, J. Alzheimer's Dis., 2008, 14, 235–245 CAS.
- G. Plascencia-Villa, A. Ponce, J. F. Collingwood, M. J. Arellano-Jiménez, X. Zhu, J. T. Rogers, I. Betancourt, M. José-Yacamán and G. Perry, High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer's disease, Sci. Rep., 2016, 6, 24873 CrossRef CAS.
- N. D. Telling, G. v. d. Laan, M. T. Georgieva and N. R. S. Farley, Facility for combined in situ magnetron sputtering and soft X-ray magnetic circular dichroism, Rev. Sci. Instrum., 2006, 77, 073903 CrossRef.
- F. Lermyte, J. Everett, J. Brooks, F. Bellingeri, K. Billimoria, P. J. Sadler, P. B. O'Connor, N. D. Telling and J. F. Collingwood, Emerging Approaches to Investigate the Influence of Transition Metals in the Proteinopathies, Cells, 2019, 8, 1231 CrossRef CAS.
- K. Kaznacheyev, A. Osanna, C. Jacobsen, O. Plashkevych, O. Vahtras, H. Ågren, V. Carravetta and A. P. Hitchcock, Innershell Absorption Spectroscopy of Amino Acids, J. Phys. Chem. A, 2002, 106, 3153–3168 CrossRef CAS.
- J. Stöhr, F. Sette and A. L. Johnson, Near-Edge X-Ray-Absorption Fine-Structure Studies of Chemisorbed Hydrocarbons: Bond Lengths with a Ruler, Phys. Rev. Lett., 1984, 53, 1684–1687 CrossRef.
- T. J. Regan, H. Ohldag, C. Stamm, F. Nolting, J. Lüning, J. Stöhr and R. L. White, Chemical effects at metal/oxide interfaces studied by X-ray-absorption spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 214422 CrossRef.
- L. M. Miller, Q. Wang, T. P. Telivala, R. J. Smith, A. Lanzirotti and J. Miklossy, Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer's disease, J. Struct. Biol., 2006, 155, 30–37 CrossRef CAS.
- J. F. Collingwood, A. Mikhaylova, M. Davidson, C. Batich, W. J. Streit, J. Terry and J. Dobson, In situ characterization and mapping of iron compounds in Alzheimer's disease tissue, J. Alzheimers Dis., 2005, 7, 267–272 CAS.
- J. F. Collingwood and F. Adams, Chemical imaging analysis of the brain with X-ray methods, Spectrochim. Acta, Part B, 2017, 130, 101–118 CrossRef CAS.
- X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, The Aβ Peptide of Alzheimer's Disease Directly Produces Hydrogen Peroxide through Metal Ion Reduction, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS.
- D. Jiang, X. Li, R. Williams, S. Patel, L. Men, Y. Wang and F. Zhou, Ternary Complexes of Iron, Amyloid-β, and Nitrilotriacetic Acid: Binding Affinities, Redox Properties, and Relevance to Iron-Induced Oxidative Stress in Alzheimer's Disease, Biochemistry, 2009, 48, 7939–7947 CrossRef CAS.
- L. Guilloreau, S. Combalbert, A. Sournia-Saquet, H. Mazarguil and P. Faller, Redox Chemistry of Copper–Amyloid-β: The Generation of Hydroxyl Radical in the Presence of Ascorbate is Linked to Redox-Potentials and Aggregation State, ChemBioChem, 2007, 8, 1317–1325 CrossRef CAS.
- F. Lermyte, J. Everett, Y. P. Y. Lam, C. A. Wootton, J. Brooks, M. P. Barrow, N. D. Telling, P. J. Sadler, P. B. O'Connor and J. F. Collingwood, Metal Ion Binding to the Amyloid β Monomer Studied by Native Top-Down FTICR Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2019, 30, 2123–2134 CrossRef CAS.
- F. Bousejra-ElGarah, C. Bijani, Y. Coppel, P. Faller and C. Hureau, Iron(II) Binding to Amyloid-β, the Alzheimer's Peptide, Inorg. Chem., 2011, 50, 9024–9030 CrossRef CAS.
- S. C. Bondy, S. X. Guo-Ross and A. T. Truong, Promotion of transition metal-induced reactive oxygen species formation by β-amyloid, Brain Res., 1998, 799, 91–96 CrossRef CAS.
- D. Schubert and M. Chevion, The Role of Iron in Beta Amyloid Toxicity, Biochem. Biophys. Res. Commun., 1995, 216, 702–707 CrossRef CAS.
- P. Carrillo-Mora, R. Luna and L. Colin-Barenque, Amyloid Beta: Multiple Mechanisms of Toxicity and Only Some Protective Effects?, Oxid. Med. Cell. Longevity, 2014, 2014, 15 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qi01304h. The datasets for this paper are available through the Keele Research Repository at https://doi.org/10.21252/vvn7-yf50. |
| This journal is © the Partner Organisations 2021 |