
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insight into the structure–property relationship of UO2 nanoparticles†
Evgeny
Gerber
 abc,
Anna Yu.
Romanchuk
c,
Stephan
Weiss
b,
Stephen
Bauters
ab,
Bianca
Schacherl
d,
Tonya
Vitova
d,
René
Hübner
b,
Salim
Shams Aldin Azzam
b,
Dirk
Detollenaere
ef,
Dipanjan
Banerjee
fg,
Sergei M.
Butorin
h,
Stepan N.
Kalmykov
c and
Kristina O.
Kvashnina
*abc
abc,
Anna Yu.
Romanchuk
c,
Stephan
Weiss
b,
Stephen
Bauters
ab,
Bianca
Schacherl
d,
Tonya
Vitova
d,
René
Hübner
b,
Salim
Shams Aldin Azzam
b,
Dirk
Detollenaere
ef,
Dipanjan
Banerjee
fg,
Sergei M.
Butorin
h,
Stepan N.
Kalmykov
c and
Kristina O.
Kvashnina
*abc
aThe Rossendorf Beamline at ESRF – The European Synchrotron, CS40220, 38043 Grenoble Cedex 9, France. E-mail: kristina.kvashnina@esrf.fr
bHelmholtz-Zentrum Dresden-Rossendorf (HZDR), Institute of Resource Ecology, PO Box 510119, 01314, Dresden, Germany
cLomonosov Moscow State University, Department of Chemistry, 119991 Moscow, Russia
dInstitute for Nuclear Waste Disposal (INE), Karlsruhe Institute of Technology, P.O. 3640, D-76021 Karlsruhe, Germany
eDepartment of Chemistry, X-ray Imaging and Microspectroscopy Research Group, Ghent University, Ghent, Belgium
fDutch-Belgian Beamline (DUBBLE), European Synchrotron Radiation Facility, 71 Avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France
gDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, Box 2404, B-3001 Leuven, Belgium
hMolecular and Condensed Matter Physics, Department of Physics and Astronomy, Uppsala University, P.O. Box 516, Uppsala, Sweden
First published on 15th December 2020
Abstract
Highly crystalline UO2 nanoparticles (NPs) with sizes of 2–3 nm were produced by fast chemical deposition of uranium(IV) under reducing conditions at pH 8–11. The particles were then characterized by microscopy and spectroscopy techniques including high-resolution transmission electron microscopy (HRTEM), X-ray diffraction (XRD), high-energy resolution fluorescence detection (HERFD) X-ray absorption spectroscopy at the U M4 edge and extended X-ray absorption fine structure (EXAFS) spectroscopy at the U L3 edge. The results of this investigation show that despite U(IV) being the dominant oxidation state of the freshly prepared UO2 NPs, they oxidize to U4O9 with time and under the X-ray beam, indicating the high reactivity of U(IV) under these conditions. Moreover, it was found that the oxidation process of NPs is accompanied by their growth in size to 6 nm. We highlight here the major differences and similarities of the UO2 NP properties to PuO2, ThO2 and CeO2 NPs.
Introduction
Uranium dioxide remains one of the most essential uranium compounds due to its application as a nuclear fuel in most of the commercial nuclear reactors worldwide.1 The structural chemistry and physics of the U/O system are very complicated but highly important for reactor performance, spent nuclear fuel storage and its further geological disposal. While bulk UO2 has been intensively studied, it is still not clear if the investigated properties remain the same at the nanoscale.2,3 It is known that actinide (An) nanoparticles (NPs) form aggregates of various sizes.4,5 In particular, UO2 NPs may be formed by redox reactions from either the reduction of U(VI) by γ-irradiation,6 minerals,7–10 microorganisms,11–17 and redox-active chemicals18 or due to corrosion of metallic U in contact with water.4,19,20 They can also be formed via hydrolysis of U(IV) solutions21–23 or by decomposition of U(IV) compounds.24Under environmental conditions, uranium mineral NPs are found to be ubiquitous and have been identified in a number of studies.7,25–28 As a highly hydrolysable cation, U(IV) migrates predominantly in the form of pseudo-colloids and intrinsic colloids rather than in the soluble complexed form. UO2 NPs formed as a result of bacteria mediated redox reactions have an influence on U migration in the far-field conditions of repositories. Accidental (like Chernobyl and Fukushima) and routine releases of radionuclides into the environment result in the formation of U oxide NPs.29–32 It has also been shown that the dissolution of spent nuclear fuel may result in the formation of UO2 NPs that should be taken into account in the performance assessment of repositories,33,34 considering that conditions in deep geological repositories are expected to be reducing.
The peculiarities of nanoscale objects affect their properties.2 Nanoscale UO2 is readily oxidized with the formation of UO2+x, while the crystal structure does not significantly alter.35–37 Similar AnO2+x NPs with a structure close to bulk AnO2 were also observed for plutonium,38 which is not surprising as both UO2 and PuO2 are isostructural to the fluorite-type fcc structure with a very similar lattice parameter. However, recent publications show that PuO2 NPs do not contain other oxidation states except for Pu(IV)39,40 and their structural properties are close to the bulk. Similar predictions were made for CeO2 NPs, with the suggestion that CeO2−x NPs were expected to be predominantly composed of Ce(IV), which can be reduced to Ce(III).41 Later, the absence of the Ce(III) oxidation state was confirmed for NPs even for 2 nm particles.42,43 This could lead to the assumption that there is a similar trend for all highly-hydrolyzed tetravalent Ln or An cations. However, to the best of our knowledge, the pure tetravalent oxidation state for UO2 NPs has never been proven.
The main difference between U and Pu lies in multivalent behaviour. Under oxidizing conditions, PuO2 is the sole stable oxide, but more than ten stable U binary oxides – UO2+x – are known. Similar to plutonium, CeO2 is the only stable oxide under oxidizing conditions, however both Ce(IV) and Ce(III) ions may be present in solution. Later we briefly compare the differences and similarities of various An and Ln oxide NP properties, based on the results reported here.
Experimental
Nanoparticle synthesis
UO2 NPs were synthesized from U(IV) aqueous solution by adding ammonia under reducing conditions. Due to the highly sensitive nature of U(IV) towards oxidation, all synthetic processes, including the preparation of samples for the following characterization methods, were done in a glovebox under a nitrogen atmosphere (<10 ppm O2).Special care was taken to avoid any contact with oxygen before and during the measurements. U(IV) stock was prepared by galvanostatic reduction of 0.1 M U(VI) in 0.5 M HClO4 (5 hours, 20 mA). The presence of only U(IV) and the stability of the solution were verified by UV-vis spectrometry (AvaSpec-2048x14, Avantes, Fig. S1†). Each U(IV) solution (0.1 M and 0.01 M) was divided into two parts. The first set of aliquots was added to 3 M NH3 in the volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 under continuous stirring. The pH of the 3 M ammonia solution was 12.5, but the pH slightly decreased due to the interaction with the U(IV) solution, most likely due to hydrolysis reactions. This set of samples was named “0.1 M/0.01 M U(IV) pH > 11”. The second set of aliquots of stock U(IV) was added to water in the volume ratio of 1:10, after which several drops of 3 M NH3 were added under continuous stirring to reach pH 8. This set of samples was named “0.1 M/0.01 M U(IV) pH 8”. In all syntheses, the mixing rate and vessel geometry were kept constant. The precipitation process for all samples started shortly (within ten minutes) after addition of all reagents. A black precipitate was formed, and the reaction was continued for about 2 hours to reach equilibrium. Then, the pH and redox potential of the formed suspensions were measured (Table S1†). The UO2 reference was made by pressing industrially obtained uranium dioxide powder into a pellet followed by sintering at 1700 °C under a H2/Ar stream. The industrial uranium dioxide, in its turn, was obtained from UF6 by the gas-flame method, followed by annealing under reducing conditions at 600–650 °C.44 The reference was characterized by X-ray diffraction (XRD) and polarography; the oxygen coefficient of UO2+x was found to be on the order of x = 0.001.
:10 under continuous stirring. The pH of the 3 M ammonia solution was 12.5, but the pH slightly decreased due to the interaction with the U(IV) solution, most likely due to hydrolysis reactions. This set of samples was named “0.1 M/0.01 M U(IV) pH > 11”. The second set of aliquots of stock U(IV) was added to water in the volume ratio of 1:10, after which several drops of 3 M NH3 were added under continuous stirring to reach pH 8. This set of samples was named “0.1 M/0.01 M U(IV) pH 8”. In all syntheses, the mixing rate and vessel geometry were kept constant. The precipitation process for all samples started shortly (within ten minutes) after addition of all reagents. A black precipitate was formed, and the reaction was continued for about 2 hours to reach equilibrium. Then, the pH and redox potential of the formed suspensions were measured (Table S1†). The UO2 reference was made by pressing industrially obtained uranium dioxide powder into a pellet followed by sintering at 1700 °C under a H2/Ar stream. The industrial uranium dioxide, in its turn, was obtained from UF6 by the gas-flame method, followed by annealing under reducing conditions at 600–650 °C.44 The reference was characterized by X-ray diffraction (XRD) and polarography; the oxygen coefficient of UO2+x was found to be on the order of x = 0.001.
Characterization
The U L3 edge (17166 eV) EXAFS spectra were collected at BM26A, the Dutch–Belgium beamline (DUBBLE) at the ESRF (the European Synchrotron) in Grenoble, France.48 The energy of the X-ray beam was tuned by using a double-crystal monochromator operating in a fixed-exit mode using a Si(111) crystal pair. Measurements were performed in transmission mode with N2/He and Ar/He filled ionization chambers. Energy calibration was performed by recording the EXAFS spectrum of the K-edge of metallic Y (∼17038 eV) which was collected simultaneously with the sample scans for each sample. The samples were measured at room temperature using a double-confined, heat-sealed polyethylene holder. Energy calibration, averaging of the individual scans, EXAFS data extraction and fitting were performed with the software package Demeter.49
Results and discussion
Due to the chemical reactivity, the ideal structure of UO2 can be easily perturbed (even in the bulk crystal).35,50,51 The particle size distribution and crystallinity could also differ depending on the synthesis route.6,11,52–54 The HRTEM data reported in Fig. 1a and Fig. S3† confirm that regardless of U(IV) concentration and pH conditions, similar NPs are formed (with respect to their size distribution and crystallinity). A comparison of the SAED patterns (Fig. 1a and Fig. S3† (insets)) and the diffractograms from XRD measurements (Fig. 1b) with bulk UO2 shows that the crystalline structure of the NPs is similar to that of bulk UO2 (ICDD 03-065-0285). However, the diffraction peaks are broad, indicating the nanosize dimensions of the samples. The crystallite size was estimated from XRD with Scherrer's equation and found to be similar for all samples, varying in the range of 1.7–2.5 nm (Table S2†) with respect to the fact that Scherrer's equation is supposed to give information about coherent domains rather than crystallites. Nevertheless, the diffraction peaks of NPs obtained at pH 8 are slightly narrower than those for pH > 11, indicating that pH has a small but notable effect on the NP size. It was unexpected as previous research showed that synthesis conditions highly impact the UO2 shape and size. For example, Hu and coauthors55 made UO2 in the form of NPs, nanoribbons and nanowires, changing the precursor/organic solvent ratio and temperature. There are many other examples, where the size of the obtained UO2 NPs varied from several nm up to several microns depending on the synthesis route (radiolytic reduction, organic precursor-assisted syntheses, U(IV) hydrolysis, biogenic reduction etc.) as well as starting precursors and reaction conditions.8,13,52–54,56–63 Our HRTEM results (Table S2†) also confirm the nanosize of the crystallites.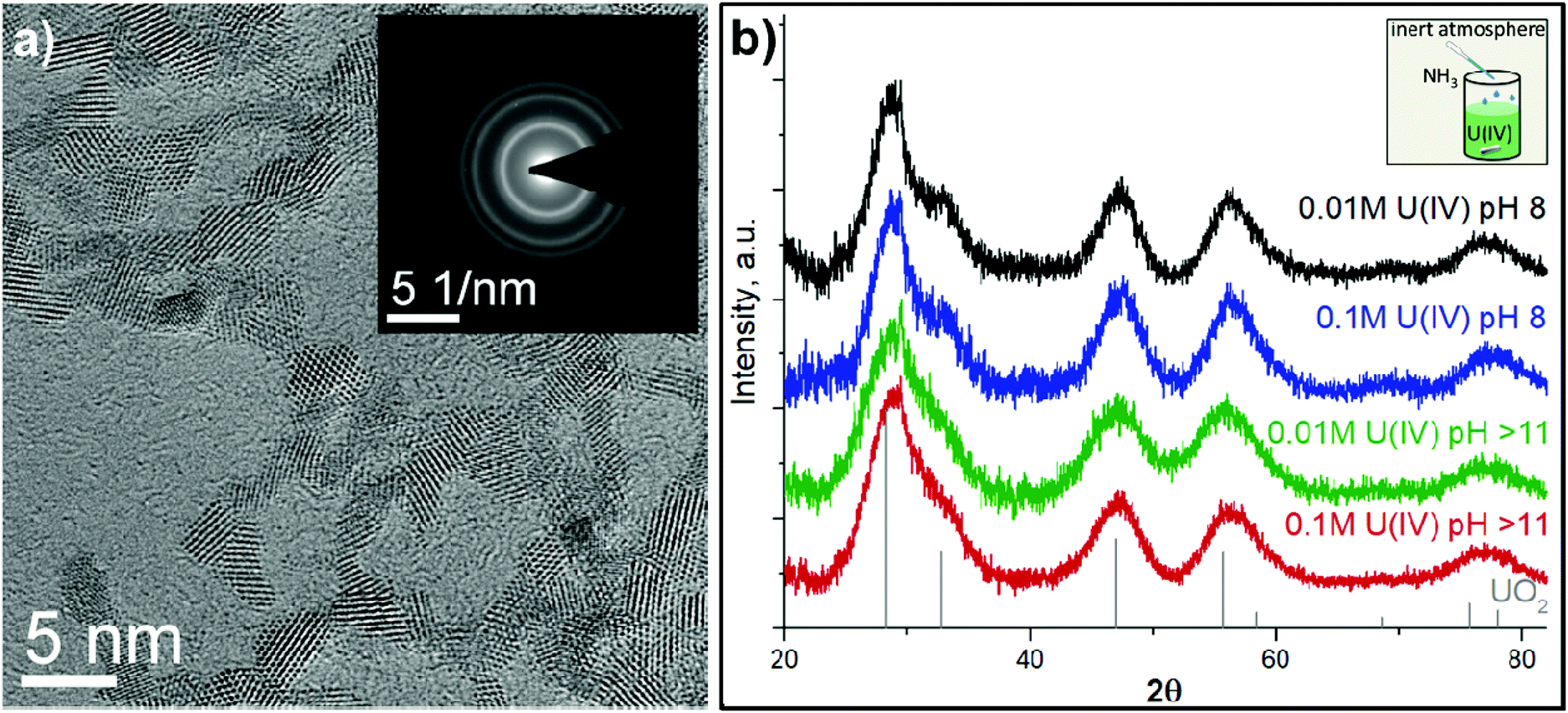 | ||
| Fig. 1 (a) HRTEM image of 0.01 M U(IV) pH 8 NPs and the corresponding SAED pattern (inset), (b) XRD patterns of UO2 reference and the precipitates from U(IV) with different pH and concentrations. The inset shows the schematic drawing of UO2 NP synthesis. | ||
For U–O systems, the number of stoichiometric binary oxides and solid solutions with various compositions are known. Uranium upon oxidation may form various oxides with mixed oxidation states of U (like U2O5, U3O7, U4O9, U3O8).64,65 The fluorite structure of UO2 can accommodate a large amount of excess oxygen up to UO2.25, therefore XRD, giving information about coherent scattering domains, is generally less sensitive to this kind of alteration. In other words, the XRD patterns of U4O9 and UO2 are very similar and the presence of those species in UO2 NPs cannot be detected by XRD (Fig. S4†). Further oxidation of UO2.25 (U4O9) may lead to the formation of UO2+x oxides with 0.25 < x ≤ 0.33 accompanied by a change of the crystal structure. Subsequent oxidation may proceed through the formation of UO2.5 (U2O5) and UO2.67 (U3O8) until UO3 is formed.
To further complicate the matter, there is a peak broadening effect for NPs in XRD, making reliable analysis nearly impossible, especially in the case of extremely small NPs. This is where synchrotron-based high-energy resolution fluorescence detection (HERFD) X-ray absorption spectroscopy at the U M4 edge really comes into use. Thanks to its high sensitivity, it can easily detect even the tiniest oxidation state impurities, which are present in many uranium oxides (U4O9, U3O8, U3O7).9,10,46,66–69 The HERFD method at the An M4 edge probes An 3d–5f electronic transitions and is thus highly effective for the detection of the 5f electron configuration and for oxidation state identification.9,10,39,40,46,66,77 Moreover, the shapes of the recorded data on various mixed uranium oxides are so distinct66,67 that recorded HERFD spectra on uranium systems can be straightforwardly analyzed by a fingerprint approach to detect the presence of U4O9/U3O8 impurities in UO2 NPs.
Fig. 2 shows the U M4 edge measurements on four UO2 NP samples compared to the spectrum of the UO2 reference. All spectral features of UO2 NPs are very similar, corresponding to those of the UO2 reference, thus confirming the results from XRD and HRTEM. The shape of the main absorption peak in the HERFD spectrum of the UO2 reference shows an asymmetric profile, which was observed before,66 however for NP spectra the asymmetry of the peak increases, leading to a high-energy shoulder (Fig. S5†). It is expected that the asymmetry of the peak originated from partial oxidation and the presence of oxidized uranium species. However, the theoretical calculations of the U(IV) M4 HERFD spectra (cf. ESI, Fig. S6†) show that nanoscale distortion or even different coordination environments have a strong correlation with the high-energy shoulder in U M4 HERFD. It is not easy to distinguish between the influence of the presence of a higher oxidation state in NPs and the distortion contributions to the asymmetry of the peak. However, theoretical results clearly indicate that the distortion at the surface and random changes of the coordination number for surface atoms will affect the intensity (increase and decrease) of the higher energy U M4 HERFD shoulder. Regardless of the asymmetry origin, one can conclude that U(IV) is the dominant oxidation state for UO2 NPs. To the best of our knowledge, this has never been shown and reported for UO2 NPs at a high sensitivity that HERFD gives for redox speciation.
 | ||
| Fig. 2 U M4 HERFD experimental data recorded for the four UO2 NP samples and compared with a UO2 reference. | ||
The crystallinity of the NPs was investigated for structural disorder by uranium L3-edge EXAFS studies (cf. Fig. S7 and Table S3†). EXAFS is actively used for uranium8,10,63,68,70 to investigate the local chemical environment. It has also been used previously to determine the oxidation state of uranium due to the different U(X)–O bond lengths (where X is the oxidation state of uranium) and static disorder contributions. Our U L3-EXAFS data and shell fit results indicate a UO2-like structure, with characteristic distances of 2.33 Å and 3.85 Å for U–O and U–U, respectively (Fig. S7†). The absence of other shorter or longer U–O distances suggests that there is no need to invoke different U oxidation states or a substantially different structure (e.g. U4O9 or U(V)–O). However, the reduced CN for the U–O shell and high Debye–Waller factors are suggestive of the particle-size effect and static and thermal disorder, which was previously observed on similar particles.8,9,13,63,71
Taking into account: (1) the significant disorder revealed by EXAFS and (2) the theoretical prediction of the distortion effects on the high-energy shoulder of the U M4 HERFD spectra, surface distortion might be the predominant reason for the experimental observation of the intensity variation of the high-energy shoulder in U M4 HERFD data between various UO2 NPs.
Reactivity of the UO2 NPs
The reactivity of UO2 NPs under different conditions was studied previously. It was found that sintering leads to NP growth.6,13 Rath et al. found that UO2 NPs obtained with γ-irradiation oxidize in several hours under air conditions, while Singer et al. and Wang et al. did not find any changes after 2 months of ageing or several days of air exposure.11,53,60 However, visual observations of the change in residue colour by UV-vis spectroscopy or the U L3 edge XANES (used in previous studies) may not be sufficient to detect other oxidation states of uranium.Here we investigate in detail the reactivity and phase stability of the synthesized UO2 NPs. First, we noticed the impact of the synchrotron X-ray beam on the freshly synthesized materials. In order to verify the damage of samples by X-ray irradiation, all samples were scanned several times at the exact same position to examine how the beam exposure affects the samples. Fig. 3a shows the average of the first U M4 spectral scans on UO2 NPs synthesized from 0.01 M U(IV) pH > 11 and its comparison with the last scans after 45–60 minutes of X-ray exposure. With each subsequent scan (which takes 5 min) the shoulder of the HERFD high-energy side increases and can even be resolved as an individual component, indicating the partial oxidation of the sample to U(V) and U(VI). Other samples were oxidized under exposure as well, leading to the conclusion that beam exposure is responsible for uranium oxidation in NPs. In order to obtain data on freshly made materials, HERFD measurements have been made on the newly synthesized UO2 NPs (reported in Fig. 2), with a short X-ray exposure time (15 min in total over several scans) on samples sealed in a special inert gas-filled container (more info is given in the Experimental section and ESI†).
 | ||
| Fig. 3 HERFD M4 edge spectra: (a) for 0.01 M U(IV) pH > 11 before and after beam damage with references. (b) For fresh and 1-year-aged 0.1 M samples with references. The HERFD spectra of the UO3 and U4O9 references have been reproduced from data, reported by Leinders et al. and have shown here for clarity.67 | ||
The recorded U M4 HERFD data on oxidized UO2 NPs are reported in Fig. 3 and compared with the spectra of UO2, U4O9 and UO3 (reproduced from Leinders et al.67). It should be noted that despite the longer duration of L3-edge EXAFS measurements beam damage does not take place in this case due to the lower unfocused beam intensity, compared to M4-edge HERFD measurements. Several scans made on the same sample position were reproducible and oxidation (or significant differences from the UO2 structure) was not detected.
Moreover, the stability of NPs over time has also been studied. Two samples synthesized from 0.1 M U(IV) concentrations were kept as wet pastes under inert conditions and ambient temperature for a year in closed 2 mL plastic tubes with a tiny amount of water left after washing the NPs. Afterwards these samples were analysed with HRTEM, XRD and M4 edge HERFD techniques. It was found that the size of NPs increases (Table S2 and Fig. S8, S9†) after aging (likely due to the dissolution–precipitation processes57), while partial oxidation was observed by HERFD (Fig. 3b). An increase of U(V) contribution in the damaged NPs is shown (Fig. 3a), though it is clear by the difference in peak intensity ratios that the NPs have not fully converted to pure U4O9. Bulk U4O9 has equal amounts of U(IV) and U(V), yet the peak intensities in HERFD are not the same due to the different probability of the absorption process, i.e. different absolute absorption cross-sections of U(IV) and U(V).67 Therefore, a significant amount of U(IV) is still retained in our damaged samples. The HERFD spectra on the aged NPs show higher contribution of U(V) compared to U(IV). Overall, it leads to the conclusion that small NPs oxidize to U4O9 and grow up to 6 nm over time. The size of 6 nm was determined from the XRD data and is in agreement with the HRTEM size estimations (Table S2†).
The strong influence of the X-ray beam and the aging behaviour of the UO2 NPs are clear evidence of the low stability of the samples; therefore, special care must be taken to avoid sample oxidation and destructive effects of the X-ray beam. The reasonable solution is to keep samples under reducing conditions as long as possible before performing any experiments, to record relatively quick scans and to choose new sample positions for every scan to limit sample exposure. Measurements under cryogenic conditions might also overcome the issue of beam damage, but this has not been tested yet on the UO2 NPs.
Comparison between various An and Ln oxide NPs
Tetravalent cations (Cat) undergo extensive hydrolysis accompanied by the formation of mono- and oligomeric species. Eventually CatO2 NPs originate from aqueous solutions. Besides uranium, the formation of small (2–4 nm) crystalline NPs was observed for cerium,43 thorium,72 neptunium73 and plutonium.74 Dioxides of these elements demonstrate similar crystallographic properties: a fluorite-type structure with a similar lattice parameter. However, the redox properties of these elements are different. Thorium is redox inactive, while Ce, U, Np and Pu may be present in different oxidation states. The correlation between UO2 and PuO2 NPs is of high interest as both U(IV) and Pu(IV) are mobile in their colloid form.5,75,76 The redox conditions in deep geological repositories are expected to be reducing. Therefore, U(IV), being stable under these conditions, could be a reference for Pu(IV) as well, due to the similarities in An(IV) ionic radii and crystallographic properties of their dioxides. Our investigation shows that there are many resemblances between these An(IV) NPs. It was shown39 that neither super stoichiometric AnO2+x nor other higher oxide phases are present in PuO2 NPs though it could be expected due to the stability of Pu(IV) under these conditions. Similar behaviour can be predicted for NpO2 NPs; however, to the best of our knowledge the presence of other oxidation states in NpO2 NPs has not been studied yet by the HERFD method.In contrast to Pu and especially Np, Ce(III) is stable in aqueous solutions; therefore, one can expect that Ce(III) is present in the hydrolysis products. Nevertheless, it was found42,43 that CeO2 NPs do not contain even slight amounts of Ce(III), leading to the conclusion that CatO2 NPs formed by fast chemical deposition retain Cat(IV) as the dominating oxidation state regardless of their redox affinity. In this study we confirmed that it is also valid for UO2 NPs, synthesized by the fast chemical deposition method at pH 8–11. These results do not include the possibility that the formation of other phases or other oxidation states takes place under different synthesis conditions.
Conclusions
It is reasonable to believe that the properties of UO2 in bulk and at the nanoscale are different. Due to a larger surface-to-volume ratio, UO2 NPs are expected to be more reactive and, therefore, to exist as UO2+x, with some of the U oxidized at the surface.35–37 However, it was found that U(IV) is the dominant oxidation state of the UO2 NPs, synthesized by the fast chemical deposition method at pH 8–11, but their stability is significantly lower than bulk UO2 in terms of time and oxidation sensitivity. They are easily oxidized not only in air, but also slowly under inert conditions or during X-ray exposure. Therefore, special care has to be taken while investigating reactions with UO2 NPs and their properties.The electronic and local structures of the freshly synthesized UO2 NPs with a size of 2–3 nm were revealed by combination of the U L3-edge EXAFS, U M4-edge HERFD, XRD and HRTEM methods. We show here that the structural and electronic properties of fresh ultra-small UO2 NPs (2–3 nm) are similar to those of bulk UO2 when inert or reducing conditions are maintained. It was found that high reactivity of UO2 NPs in time and under X-ray beam exposure leads to the formation of the U4O9 species complemented by the growth of the NP size to 6 nm. We believe that these findings are beneficial for the fundamental understanding of nuclear fuels and for tailoring the functionality of UO2 since most previous studies focused on large-bulk UO2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the European Commission Council under ERC [grant no. 759696]. E. G. acknowledges the support from RFBR (project number no. 19-33-90127). S. N. K. acknowledges the support by the Russian Ministry of Science and Education under grant no. 075-15-2019-1891. S. M. B. acknowledges the support from the Swedish Research Council (grant 2017-06465). The authors thank HZDR for the beamtime at the CAT-ACT beamline of KARA. Moreover, we acknowledge the help of J. Rothe, A. Beck, J. Galanzew and T. Prüßmann at the CAT-ACT beamline of KARA during the HERFD experiment at the U M4 edge and we would like to thank the Institute for Beam Physics and Technology (IBPT) for the operation of the storage ring, the Karlsruhe Research Accelerator (KARA). Furthermore, the use of the HZDR Ion Beam Centre TEM facilities is acknowledged.References
- R. C. Ewing, Long-term storage of spent nuclear fuel, Nat. Mater., 2015, 14, 252–257 CrossRef CAS.
- M. F. Hochella, D. W. Mogk, J. Ranville, I. C. Allen, G. W. Luther, L. C. Marr, B. P. McGrail, M. Murayama, N. P. Qafoku, K. M. Rosso, N. Sahai, P. A. Schroeder, P. Vikesland, P. Westerhoff and Y. Yang, Natural, incidental, and engineered nanomaterials and their impacts on the Earth system, Science, 2019, 363, eaau8299 CrossRef.
- H. Wu, Y. Yang and Y. C. Cao, Synthesis of Colloidal Uranium–Dioxide Nanocrystals, J. Am. Chem. Soc., 2006, 128, 16522–16523 CrossRef CAS.
- T. S. Neill, K. Morris, C. I. Pearce, L. Abrahamsen-Mills, L. Kovarik, S. Kellet, B. Rigby, T. Vitova, B. Schacherl and S. Shaw, Silicate stabilisation of colloidal UO2 produced by uranium metal corrosion, J. Nucl. Mater., 2019, 526, 151751 CrossRef CAS.
- Actinide Nanoparticle Research, ed. S. N. Kalmykov and M. A. Denecke, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011 Search PubMed.
- T. M. Nenoff, B. W. Jacobs, D. B. Robinson, P. P. Provencio, J. Huang, S. Ferreira and D. J. Hanson, Synthesis and Low Temperature In Situ Sintering of Uranium Oxide Nanoparticles, Chem. Mater., 2011, 23, 5185–5190 CrossRef CAS.
- J. R. Bargar, K. H. Williams, K. M. Campbell, P. E. Long, J. E. Stubbs, E. I. Suvorova, J. S. Lezama-Pacheco, D. S. Alessi, M. Stylo, S. M. Webb, J. A. Davis, D. E. Giammar, L. Y. Blue and R. Bernier-Latmani, Uranium redox transition pathways in acetate-amended sediments, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4506–4511 CrossRef CAS.
- E. J. O'Loughlin, S. D. Kelly, R. E. Cook, R. Csencsits and K. M. Kemner, Reduction of Uranium(VI) by Mixed Iron(II)/Iron(III) Hydroxide (Green Rust): Formation of UO2 Nanoparticles, Environ. Sci. Technol., 2003, 37, 721–727 CrossRef.
- I. Pidchenko, K. O. Kvashnina, T. Yokosawa, N. Finck, S. Bahl, D. Schild, R. Polly, E. Bohnert, A. Rossberg, J. Göttlicher, K. Dardenne, J. Rothe, T. Schäfer, H. Geckeis and T. Vitova, Uranium Redox Transformations after U(VI) Coprecipitation with Magnetite Nanoparticles, Environ. Sci. Technol., 2017, 51, 2217–2225 CrossRef CAS.
- Z. Pan, B. Bártová, T. LaGrange, S. M. Butorin, N. C. Hyatt, M. C. Stennett, K. O. Kvashnina and R. Bernier-Latmani, Nanoscale mechanism of UO2 formation through uranium reduction by magnetite, Nat. Commun., 2020, 11, 4001 CrossRef CAS.
- D. M. Singer, F. Farges and G. E. Brown, in AIP Conference Proceedings, AIP, 2007, vol. 882, pp. 277–279 Search PubMed.
- Y. Suzuki, S. D. Kelly, K. M. Kemner and J. F. Banfield, Nanometre-size products of uranium bioreduction, Nature, 2002, 419, 134–134 CrossRef CAS.
- D. M. Singer, F. Farges and G. E. Brown, Biogenic nanoparticulate UO2: Synthesis, characterization, and factors affecting surface reactivity, Geochim. Cosmochim. Acta, 2009, 73, 3593–3611 CrossRef CAS.
- M. I. Boyanov, K. E. Fletcher, M. J. Kwon, X. Rui, E. J. O'Loughlin, F. E. Löffler and K. M. Kemner, Solution and Microbial Controls on the Formation of Reduced U(IV) Species, Environ. Sci. Technol., 2011, 45, 8336–8344 CrossRef CAS.
- L. Newsome, K. Morris and J. R. Lloyd, The biogeochemistry and bioremediation of uranium and other priority radionuclides, Chem. Geol., 2014, 363, 164–184 CrossRef CAS.
- J. R. Bargar, R. Bernier-Latmani, D. E. Giammar and B. M. Tebo, Biogenic Uraninite Nanoparticles and Their Importance for Uranium Remediation, Elements, 2008, 4, 407–412 CrossRef.
- L. Newsome, K. Morris, D. Trivedi, N. Atherton and J. R. Lloyd, Microbial reduction of uranium(VI) in sediments of different lithologies collected from Sellafield, Appl. Geochem., 2014, 51, 55–64 CrossRef CAS.
- K.-U. Ulrich, A. Singh, E. J. Schofield, J. R. Bargar, H. Veeramani, J. O. Sharp, R. Bernier-Latmani and D. E. Giammar, Dissolution of Biogenic and Synthetic UO 2 under Varied Reducing Conditions, Environ. Sci. Technol., 2008, 42, 5600–5606 CrossRef CAS.
- E. L. Fuller, N. R. Smyrl, J. B. Condon and M. H. Eager, Uranium oxidation: Characterization of oxides formed by reaction with water by infrared and sorption analyses, J. Nucl. Mater., 1984, 120, 174–194 CrossRef CAS.
- M. D. Kaminski, N. M. Dimitrijevic, C. J. Mertz and M. M. Goldberg, Colloids from the aqueous corrosion of uranium nuclear fuel, J. Nucl. Mater., 2005, 347, 77–87 CrossRef CAS.
- G. Rousseau, M. Fattahi, B. Grambow, F. Boucher and G. Ouvrard, Coprecipitation of thorium with UO2, Radiochim. Acta, 2002, 90, 523–527 CAS.
- T. Fanghänel and V. Neck, Aquatic chemistry and solubility phenomena of actinide oxides/hydroxides, Pure Appl. Chem., 2002, 74, 1895–1907 Search PubMed.
- V. Neck and J. I. Kim, Solubility and hydrolysis of tetravalent actinides, Radiochim. Acta, 2001, 89, 1–16 CAS.
- O. Walter, K. Popa and O. D. Blanco, Hydrothermal decomposition of actinide(IV) oxalates: a new aqueous route towards reactive actinide oxide nanocrystals, Open Chem., 2016, 14, 170–174 CAS.
- P. Liu, X. Luo, M. Wen, J. Zhang, C. Zheng, W. Gao and F. Ouyang, Geoelectrochemical anomaly prospecting for uranium deposits in southeastern China, Appl. Geochem., 2018, 97, 226–237 CrossRef CAS.
- R. M. Hazen, R. C. Ewing and D. A. Sverjensky, Evolution of uranium and thorium minerals, Am. Mineral., 2009, 94, 1293–1311 CrossRef CAS.
- M. Schindler, A. J. Lussier, J. Bellrose, S. Rouvimov, P. C. Burns and T. K. Kyser, Mobilization and agglomeration of uraninite nanoparticles: A nano-mineralogical study of samples from the Matoush Uranium ore deposit, Am. Mineral., 2017, 102, 1776–1787 CrossRef.
- S. Fuchs, D. Schumann, A. E. Williams-Jones and H. Vali, The growth and concentration of uranium and titanium minerals in hydrocarbons of the Carbon Leader Reef, Witwatersrand Supergroup, South Africa, Chem. Geol., 2015, 393–394, 55–66 CrossRef CAS.
- B. Salbu, K. Janssens, O. C. Lind, K. Proost, L. Gijsels and P. R. Danesi, Oxidation states of uranium in depleted uranium particles from Kuwait, J. Environ. Radioact., 2005, 78, 125–135 CrossRef CAS.
- P. Pöml and B. Burakov, Study of the redistribution of U, Zr, Nb, Tc, Mo, Ru, Fe, Cr, and Ni between oxide and metallic phases in the matrix of a multiphase Chernobyl hot-particle extracted from a soil sample of the Western Plume, Radiochim. Acta, 2018, 106, 985–990 Search PubMed.
- A. Ochiai, J. Imoto, M. Suetake, T. Komiya, G. Furuki, R. Ikehara, S. Yamasaki, G. T. W. Law, T. Ohnuki, B. Grambow, R. C. Ewing and S. Utsunomiya, Uranium Dioxides and Debris Fragments Released to the Environment with Cesium-Rich Microparticles from the Fukushima Daiichi Nuclear Power Plant, Environ. Sci. Technol., 2018, 52, 2586–2594 CrossRef CAS.
- V. Kashparov, B. Salbu, S. Levchuk, V. Protsak, I. Maloshtan, C. Simonucci, C. Courbet, H. L. Nguyen, N. Sanzharova and V. Zabrotsky, Environmental behaviour of radioactive particles from chernobyl, J. Environ. Radioact., 2019, 208–209, 106025 CrossRef CAS.
- K. Spahiu, J. Devoy, D. Cui and M. Lundström, The reduction of U(VI) by near field hydrogen in the presence of UO2(s), Radiochim. Acta, 2004, 92, 597–601 CAS.
- M. Rovira, S. El Aamrani, L. Duro, J. Giménez, J. de Pablo and J. Bruno, Interaction of uranium with in situ anoxically generated magnetite on steel, J. Hazard. Mater., 2007, 147, 726–731 CrossRef CAS.
- G. Leinders, J. Pakarinen, R. Delville, T. Cardinaels, K. Binnemans and M. Verwerft, Low-Temperature Oxidation of Fine UO2 Powders: A Process of Nanosized Domain Development, Inorg. Chem., 2016, 55, 3915–3927 CrossRef CAS.
- S. Szenknect, D. Alby, M. López García, C. Wang, R. Podor, F. Miserque, A. Mesbah, L. Duro, L. Zetterström Evins, N. Dacheux, J. Bruno and R. C. Ewing, Coffinite formation from UO2+x, Sci. Rep., 2020, 10, 12168 CrossRef CAS.
- S. R. Spurgeon, M. Sassi, C. Ophus, J. E. Stubbs, E. S. Ilton and E. C. Buck, Nanoscale oxygen defect gradients in UO2+x surfaces, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17181–17186 CrossRef CAS.
- J. M. Haschke, T. H. Allen and L. A. Morales, Reaction of Plutonium Dioxide with Water: Formation and Properties of PuO2+x, Science, 2000, 287, 285–287 CrossRef CAS.
- E. Gerber, A. Y. Romanchuk, I. Pidchenko, L. Amidani, A. Rossberg, C. Hennig, G. B. M. Vaughan, A. Trigub, T. Egorova, S. Bauters, T. Plakhova, M. O. J. Y. Hunault, S. Weiss, S. M. Butorin, A. C. Scheinost, S. N. Kalmykov and K. O. Kvashnina, The missing pieces of the PuO2 nanoparticle puzzle, Nanoscale, 2020, 12, 18039–18048 RSC.
- K. O. Kvashnina, A. Y. Romanchuk, I. Pidchenko, L. Amidani, E. Gerber, A. Trigub, A. Rossberg, S. Weiss, K. Popa, O. Walter, R. Caciuffo, A. C. Scheinost, S. M. Butorin and S. N. Kalmykov, A Novel Metastable Pentavalent Plutonium Solid Phase on the Pathway from Aqueous Plutonium(VI) to PuO2 Nanoparticles, Angew. Chem., Int. Ed., 2019, 58, 17558–17562 CrossRef CAS.
- O. N. Batuk, D. V. Szabo, M. A. Denecke, T. Vitova and S. N. Kalmykov, Synthesis and characterization of thorium, uranium and cerium oxide nanoparticles, Radiochim. Acta, 2013, 101, 233–239 CrossRef CAS.
- J.-D. Cafun, K. O. Kvashnina, E. Casals, V. F. Puntes and P. Glatzel, Absence of Ce3+ Sites in Chemically Active Colloidal Ceria Nanoparticles, ACS Nano, 2013, 7, 10726–10732 CrossRef CAS.
- T. V. Plakhova, A. Y. Romanchuk, S. M. Butorin, A. D. Konyukhova, A. V. Egorov, A. A. Shiryaev, A. E. Baranchikov, P. V. Dorovatovskii, T. Huthwelker, E. Gerber, S. Bauters, M. M. Sozarukova, A. C. Scheinost, V. K. Ivanov, S. N. Kalmykov and K. O. Kvashnina, Towards the surface hydroxyl species in CeO2 nanoparticles, Nanoscale, 2019, 11, 18142–18149 RSC.
- O. N. Batuk, S. N. Kalmykov, V. G. Petrov, E. V. Zakharova, Y. A. Teterin, A. Y. Teterin, V. I. Shapovalov and M. J. Haire, Neptunium interaction with uranium dioxide in aqueous solution, J. Nucl. Mater., 2007, 362, 426–430 CrossRef CAS.
- M. Wojdyr, Fityk : a general-purpose peak fitting program, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- J. Rothe, M. Altmaier, R. Dagan, K. Dardenne, D. Fellhauer, X. Gaona, E. G.-R. Corrales, M. Herm, K. O. Kvashnina, V. Metz, I. Pidchenko, D. Schild, T. Vitova and H. Geckeis, Fifteen Years of Radionuclide Research at the KIT Synchrotron Source in the Context of the Nuclear Waste Disposal Safety Case, Geosciences, 2019, 9, 91 CrossRef CAS.
- A. Zimina, K. Dardenne, M. A. Denecke, D. E. Doronkin, E. Huttel, H. Lichtenberg, S. Mangold, T. Pruessmann, J. Rothe, T. Spangenberg, R. Steininger, T. Vitova, H. Geckeis and J.-D. Grunwaldt, CAT-ACT—A new highly versatile X-ray spectroscopy beamline for catalysis and radionuclide science at the KIT synchrotron light facility ANKA, Rev. Sci. Instrum., 2017, 88, 113113 CrossRef CAS.
- M. Borsboom, W. Bras, I. Cerjak, D. Detollenaere, D. Glastra van Loon, P. Goedtkindt, M. Konijnenburg, P. Lassing, Y. K. Levine, B. Munneke, M. Oversluizen, R. van Tol and E. Vlieg, The Dutch–Belgian beamline at the ESRF, J. Synchrotron Radiat., 1998, 5, 518–520 CrossRef CAS.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- L. Desgranges, G. Baldinozzi, G. Rousseau, J.-C. Nièpce and G. Calvarin, Neutron Diffraction Study of the in Situ Oxidation of UO2, Inorg. Chem., 2009, 48, 7585–7592 CrossRef CAS.
- D. A. Andersson, G. Baldinozzi, L. Desgranges, D. R. Conradson and S. D. Conradson, Density Functional Theory Calculations of UO2 Oxidation: Evolution of UO2+x, U4O9−y, U3O7, and U3O8, Inorg. Chem., 2013, 52, 2769–2778 CrossRef CAS.
- S. Hasan and T. K. Ghosh, Synthesis of Uranium Oxide Nanoparticles in Aqueous Solutions, Nucl. Technol., 2011, 173, 310–317 CrossRef CAS.
- Y. Wang, Q. Chen and X. Shen, Preparation of low-temperature sintered UO2 nanomaterials by radiolytic reduction of ammonium uranyl tricarbonate, J. Nucl. Mater., 2016, 479, 162–166 CrossRef CAS.
- M. C. Rath, S. Keny and D. B. Naik, Direct Synthesis of UO2 Nanoparticles in Aqueous Solutions Through Photochemical Method, J. Nanosci. Nanotechnol., 2016, 16, 9575–9582 CrossRef CAS.
- S. Hu, H. Li, H. Liu, P. He and X. Wang, Nanocrystals of Uranium Oxide: Controlled Synthesis and Enhanced Electrochemical Performance of Hydrogen Evolution by Ce Doping, Small, 2015, 11, 2624–2630 CrossRef CAS.
- A. Leticia Soldati, D. Carolina Lago and M. Oscar Prado, in Nuclear Materials, IntechOpen, 2020 Search PubMed.
- A. J. Popel, B. T. Tan, T. Gouder, G. I. Lampronti, J. Day, R. Eloirdi, A. Seibert and I. Farnan, Surface alteration evidence for a mechanism of anoxic dissolution of UO2, Appl. Surf. Sci., 2019, 464, 376–379 CrossRef CAS.
- L. Balice, D. Bouëxière, M. Cologna, A. Cambriani, J. Vigier, E. De Bona, G. Domenico, C. Kübel, O. Walter and K. Popa, Nano and micro U1−xThxO2 solid solutions : From powders to pellets, J. Nucl. Mater., 2018, 498, 307–313 CrossRef CAS.
- N. P. Martin, C. Volkringer, N. Henry, X. Trivelli, G. Stoclet, A. Ikeda-Ohno and T. Loiseau, Formation of a new type of uranium poly-oxo cluster {U38} based on a controlled release of water via esterification reaction, Chem. Sci., 2018, 9, 5021–5032 RSC.
- M. C. Rath, D. B. Naik and S. K. Sarkar, Reversible growth of UO2 nanoparticles in aqueous solutions through 7 MeV electron beam irradiation, J. Nucl. Mater., 2013, 438, 26–31 CrossRef CAS.
- D. Hudry, C. Apostolidis, O. Walter, T. Gouder, E. Courtois, C. Kübel and D. Meyer, Non-aqueous Synthesis of Isotropic and Anisotropic Actinide Oxide Nanocrystals, Chem. – Eur. J., 2012, 18, 8283–8287 CrossRef CAS.
- D. Hudry, C. Apostolidis, O. Walter, T. Gouder, E. Courtois, C. Kübel and D. Meyer, Controlled Synthesis of Thorium and Uranium Oxide Nanocrystals, Chem. – Eur. J., 2013, 19, 5297–5305 CrossRef CAS.
- L. M. Moreau, A. Herve, M. D. Straub, D. R. Russo, R. J. Abergel, S. Alayoglu, J. Arnold, A. Braun, G. J. P. Deblonde, Y. Liu, T. D. Lohrey, D. T. Olive, Y. Qiao, J. A. Rees, D. K. Shuh, S. J. Teat, C. H. Booth and S. G. Minasian, Structural properties of ultra-small thorium and uranium dioxide nanoparticles embedded in a covalent organic framework, Chem. Sci., 2020, 11, 4648–4668 RSC.
- D. L. Clark, M. P. Neu, W. Runde and D. W. Keogh, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2006 Search PubMed.
- T. Gouder, R. Eloirdi and R. Caciuffo, Direct observation of pure pentavalent uranium in U2O5 thin films by high resolution photoemission spectroscopy, Sci. Rep., 2018, 8, 8306 CrossRef CAS.
- K. O. Kvashnina, S. M. Butorin, P. Martin and P. Glatzel, Chemical State of Complex Uranium Oxides, Phys. Rev. Lett., 2013, 111, 253002 CrossRef CAS.
- G. Leinders, R. Bes, J. Pakarinen, K. Kvashnina and M. Verwerft, Evolution of the Uranium Chemical State in Mixed-Valence Oxides, Inorg. Chem., 2017, 56, 6784–6787 CrossRef CAS.
- G. Leinders, R. Bes, K. O. Kvashnina and M. Verwerft, Local Structure in U(IV) and U(V) Environments: The Case of U3O7, Inorg. Chem., 2020, 59, 4576–4587 CrossRef CAS.
- L. Desfougeres, É. Welcomme, M. Ollivier, P. M. Martin, J. Hennuyer, M. O. J. Y. Hunault, R. Podor, N. Clavier and L. Favergeon, Oxidation as an Early Stage in the Multistep Thermal Decomposition of Uranium(IV) Oxalate into U3O8, Inorg. Chem., 2020, 59, 8589–8602 CrossRef CAS.
- S. D. Conradson, D. Manara, F. Wastin, D. L. Clark, G. H. Lander, L. A. Morales, J. Rebizant and V. V. Rondinella, Local Structure and Charge Distribution in the UO2 –U4O9 System, Inorg. Chem., 2004, 43, 6922–6935 CrossRef CAS.
- J. C. Renshaw, L. J. C. Butchins, F. R. Livens, I. May, J. M. Charnock and J. R. Lloyd, Bioreduction of Uranium: Environmental Implications of a Pentavalent Intermediate, Environ. Sci. Technol., 2005, 39, 5657–5660 CrossRef CAS.
- L. Amidani, T. V. Plakhova, A. Y. Romanchuk, E. Gerber, S. Weiss, A. Efimenko, C. J. Sahle, S. M. Butorin, S. N. Kalmykov and K. O. Kvashnina, Understanding the size effects on the electronic structure of ThO2 nanoparticles, Phys. Chem. Chem. Phys., 2019, 21, 10635–10643 RSC.
- R. Husar, R. Hübner, C. Hennig, P. M. Martin, M. Chollet, S. Weiss, T. Stumpf, H. Zänker and A. Ikeda-Ohno, Intrinsic formation of nanocrystalline neptunium dioxide under neutral aqueous conditions relevant to deep geological repositories, Chem. Commun., 2015, 51, 1301–1304 RSC.
- A. Y. Romanchuk, T. V. Plakhova, A. V. Egorov, T. B. Egorova, P. V. Dorovatovskii and Y. V. Zubavichus, Redox-mediated formation of plutonium oxide nanoparticles, Dalton Trans., 2018, 47, 11239–11244 RSC.
- H. Geckeis, B. Grambow, A. Loida, B. Luckscheiter, E. Smailos and J. Quinones, Formation and Stability of Colloids under Simulated Near Field Conditions, Radiochim. Acta, 1998, 82, 123–128 CAS.
- H. Zänker and C. Hennig, Colloid-borne forms of tetravalent actinides: A brief review, J. Contam. Hydrol., 2014, 157, 87–105 CrossRef.
- G. Kauric, O. Walter, A. Beck, B. Schacherl, O. Dieste Bianco, J.-F. Vigier, E. Zuleger, T. Vitova and K. Popa, Synthesis and characterization of nanocrystalline U1−xPuxO2(+y) mixed oxides, Mater. Today Adv., 2020, 8, 1000105 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qi01140a |
| This journal is © the Partner Organisations 2021 |