
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox activity of a dissymmetric ligand bridging divalent ytter-bium and reactive nickel fragments†
Ding
Wang
,
Maxime
Tricoire
,
Valeriu
Cemortan
,
Jules
Moutet
and
Grégory
Nocton
 *
*
LCM, CNRS, École Polytechnique, Institut Polytechnique de Paris, Route de Saclay, 91128 Palaiseau, France. E-mail: gregory.nocton@polytechnique.edu; Tel: +33-1-6933-4402
First published on 20th November 2020
Abstract
The reaction of a reactive nickel dimethyl 1 bearing a redox-active, dissymmetric ligand, which is obtained by deprotonation of 2-pyrimidin-2-yl-1H-benzimidazole (Hbimpm) with a divalent lanthanide complex, Cp*2Yb(OEt2), affords an unprecedented, trimeric 2 with C(sp3)–C(sp3) bond formation between two ligands in an exo position. Meanwhile, the transient, dimeric species 3 can be isolated with the same ligand coupling fashion, however, with a drastic distorsion angle of the bimpm ligand and reactive NiMe2 fragment, revealing the possible mechanism of this rearrangement. A much more stable dimeric congener, 5, with an exo ligand coupling, is synthesized in the presence of 18-crown-6, which captures the potassium counter ion. The C–C coupling formation between two bimpm ligands results from the effective electron transfer from divalent lanthanide fragments. Without the divalent lanthanide, the reductive coupling occurs on a different carbon of the ligand, nicely showing the modulation of the spin density induced by the presence of the lanthanide ion. The electronic structures of these complexes are investigated by magnetic study (SQUID), indicating a 2F7/2 ground state for each ytterbium in all the heterometallics. This work firstly reports ligand coupling reactivity in a redox-active, yet dissymmetric system with divalent organolanthanides, and the reactive nickel moiety can impact the intriguing transition towards a stable homoleptic, trinulear lanthanide species.
Introduction
Electron transfer behaviour in organometallic complexes has been well documented over decades. In these complexes, when the electron is accessibly transferred to an empty ligand orbital while the metal centre remains stable, the ligand is regarded as redox-active.1 Generally, N-heterocyclic ligands are excellent candidates as redox-active ligands because of their large delocalization extent added to a low-lying π*-system.2 In redox-active ligand–metal systems, the electron(s) (or hole(s)) can be stored on the ligand, therefore affecting the ground spin state of the complex and ultimately leading to unusual chemical transformation(s) at the metal site,3–5 but also at the ligand site. For the latter situation, a cooperative effect of the ligand is invoked and can develop with or without a redox event.6,7In the extensive studies of complexes with redox active ligands, divalent organolanthanides are of specific interest because of the formation of strongly correlated systems.8–21 In the latter, an important question is the role of the symmetry orbitals and the geometry of the ligand in the electronic structure and the subsequent reactivity of the overall complex. For instance, the electronic configurations of decamethylytterbocenes with 2,2′-bipyridine (bpy) and phenanthroline (phen) are drastically different: Cp*2Yb(bpy) has multiconfigurational singlet ground states, in which Yb is intermediate valent, composed of an open-shell (f13, bpy˙−) and a closed-shell (f14, bpy0) state9 and is stable in solution; while Cp*2Yb(phen) is a ground state triplet (f13, phen˙−), which dimerizes reversibly to form a C–C σ bond at the 4,4′-positions on the phen ligands.13 This reversible coupling has been also been observed with Sm and Tm analogues14,22 and other low-valent metal ions.22–26 The chemical correlation between the ligand design and the reactivity of the complex provides a strategic framework for the usage of this type of complex in further reactivity. Our group has recently taken advantage of this work to develop the synthesis of a series of complexes combining a divalent lanthanide (Ln) fragment with a transition metal (TM) fragment bridged by N-heterocyclic aromatics.27–29
However, these examples remained based on neutral symmetrical N-aromatics. Thus, furthering the study on these heterometallic frameworks by using dissymmetric redox active ligands, such as LX-type ones (Chart 1), is worthy of interest. Particularly, LX-type ligands are widely used in Nickel-catalysed cross-coupling reactions to enhance catalytic efficiency.30–33 They are considered to allow significantly facile activation of unreactive carbon-halogen bonds.34 Inspired by recent work by Diao and co-workers which highlighted the complexity of high-valent nickel-mediated sp3–sp3 and sp2–sp2 C–C bond formation using spectroscopic and computational methods,35 we thought that imidazole-based linkers, such as 2-pyrimidin-2-yl-1H-benzimidazole, namely Hbimpm,36 would be good candidates to construct original bimetallic Ln–TM complexes and study the electronic modulation within these systems. In addition, the bite angle and the dissymmetry of the ligand37 are proved to be key parameters for varying the electronic structures and reactivity of metal complexes.38
 | ||
| Chart 1 Ligands used with divalent lanthanides in previous and this study, containing 4,5,9,10-tetraazaphenanthrene (taphen), 2,2′-bipyrimidine (bipym), and 2-pyrimidin-2-yl-1H-benzimidazole (Hbimpm). | ||
In this article, we present the synthesis and characterization of original organometallic frameworks with the dissymmetric bimpm ligand, which combines a divalent organolanthanide and a dimethyl nickel fragment. Dimerization between two bimpm ligands occurs in an exo position. The lack of stability of the nickel fragment in the dimeric species is demonstrated by the evolution of dimer to a trimer with the identical exo coupling formation, in which the nickel fragment has been replaced by a Cp*Yb fragment.
Results and discussion
Synthesis and structural characterization
The first attempt at combining the nickel precursor, (tmeda)NiMe2, and Hbimpm in chelating solvents such as pyridine or acetonitrile at room temperature led to insoluble solids that were difficult to characterize. Heating up the mixture to higher temperature only yielded decomposition products, which were hardly identifiable, probably due to the temperature sensitivity of the precursor, (tmeda)NiMe2. However, when the Hbimpm pro-ligand was deprotonated beforehand with a strong base (potassium hexamethyldisilane) prior to an addition of (tmeda)NiMe2 in THF, dark red microcrystalline solid K(bimpm)NiMe2 (1) was isolated in good yield (60%). The solid can be further re-crystallized from THF-pentane vapor diffusion in moderate yield (40%) as purple crystals. The crystals of 1(THF) were suitable for X-ray diffraction (XRD) (Fig. 1). The addition of a stoichiometric amount of Cp*2Yb(OEt2) in THF led to the formation of a red suspension after 1 h. After evaporation of the volatiles, the red residue is extracted in Et2O and bright red crystals can be crystallized via slow evaporation at −35 °C of the Et2O solution in moderate yield (51%). Interestingly, instead of forming the expected dimeric Yb-bimpm-Ni anionic complex, a homo-trimetallic neutral Yb-bimpm compound (2) was generated. In 2, the ligand bimpm is coupled as in other N-heteroaromatic ligands13,14,39 including pyrazine23 and pyridazine;22 in exo-position, which is the contrary to the bulkier acridine adduct.40 A third ytterbium metal centre is coordinated in the pocket formed by four of the nitrogen atoms from two bimpm ligands. Only one pentamethylcyclopentadienyl ligand remained on the ytterbium metal centre (Scheme 1a). ORTEP of 1 and 2 and main distances and angles are shown in Fig. 1 and in Table 1.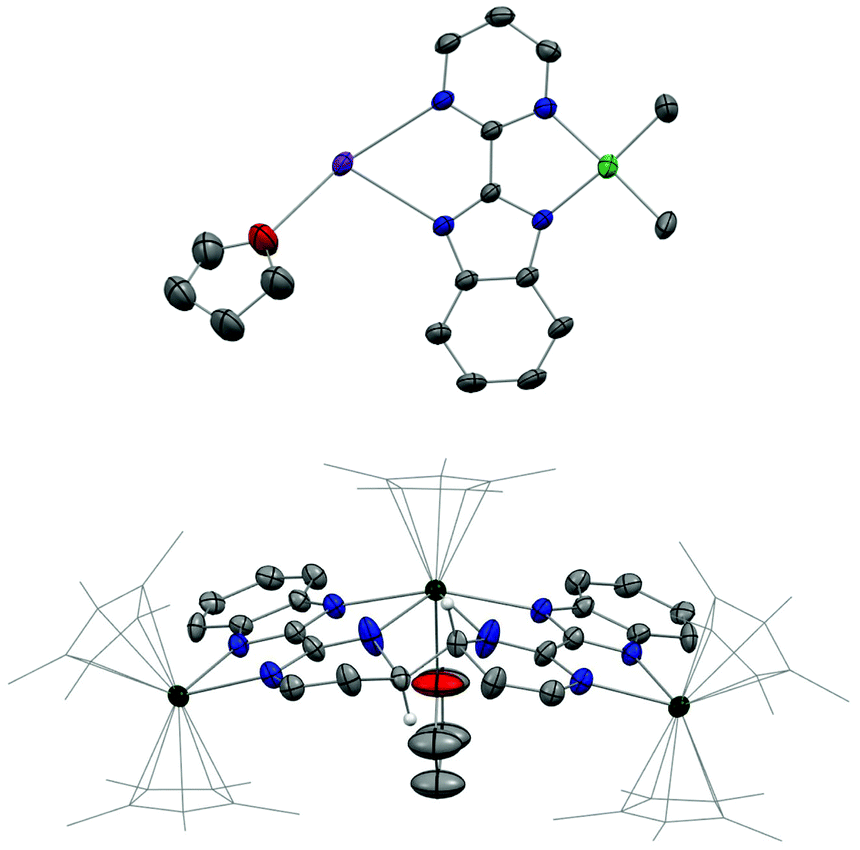 | ||
| Fig. 1 ORTEP of 1(THF) (top) and 2 (bottom). Thermal ellipsoids are at 50% level. Carbon atoms are in grey, hydrogen atoms are in white and have been removed for clarity except the ones on the coupled carbons of the ligands, potassium in purple, nitrogen in blue, nickel in bright green and ytterbium atoms in deep green. Co-recrystallized solvent molecules of Et2O in 2 are omitted. Cp* ligands on ytterbium are presented in wireframe style for clarity. | ||
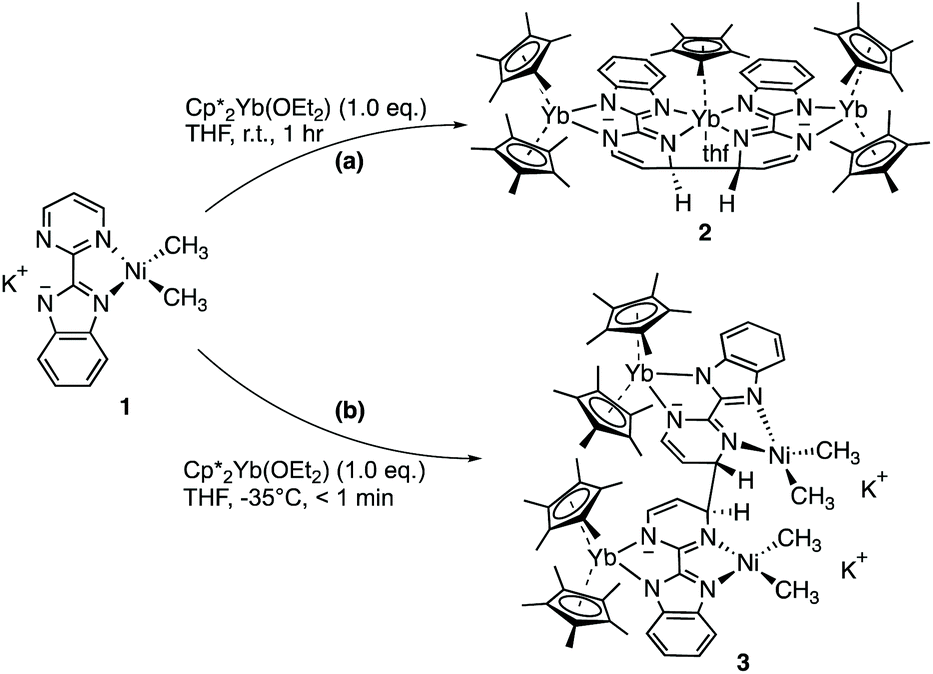 | ||
| Scheme 1 Synthetic routes for 2 and 3. | ||
| 1 | 4 | 2 | 3 | 5 | 6 | |
|---|---|---|---|---|---|---|
| Ni–CH3 | 1.922(5) | 1.92(4) | — | 1.928(5) | 1.92(2) | 1.930(5) |
| Ni–N | 1.97(5) | 1.970(5) | — | 1.987(2) | 2.01(4) | 1.973(5) |
| Yb–N | — | — | 2.364(2) | 2.330(8) | 2.32(2) | — |
| Yb–Cp*centr | — | — | 2.342(2) | 2.33(2) | 2.38(3) | — |
| Bridging C–Cbimpm | 1.451 | 1.45(6) | 1.476 | 1.459(5) | 1.46(3) | 1.46(8) |
| Coupled C–Cbimpm | — | — | 1.520 | 1.56 | 1.56(3) | 1.537 |
| bimpm^NiMe2 | 2 | 12 | — | 31 | 19 | 17 |
In 2, the average distance between Cp* centroid and ytterbium atom is in the range of 2.33–2.34 Å. It is lower than the typical values (2.43–2.50 Å) found in divalent Yb complexes with Cp ligands.41 This indicates the trivalent oxidation state of Yb,8,9,11–13 which means that the Yb-ligand fragment undergoes the transition from YbII(f14, L0) to YbIII(f13, L˙−) before coupling. The C–C σ bond coupling takes place between the same meso-position of pyrimidine cycles on bimpm ligands, causing the dearomatization of pyrimidine and forming a new C(sp3)–C(sp3) bond. This behaviour is explained by the efficient electron transfer from ytterbium fragment to the LUMO of pyrimidine heterocycle, which then undergoes the radical coupling process. The latter has been investigated by Haga et al. on the bimpm ligand system via ab initio molecular orbital calculations.36 Such coupling was already reported with pyridine and pyrimidine moieties.23,39,40 The effective reduction of the ligand also features a longer C–C distance linking the pyrimidine and benzimidazole heterocycles in 2 (1.476 Å) than in 1 (1.451 Å). This is indicative of a loss of good conjugation in the ligand and charge localization on the nitrogen of the pyrimidine.
This surprising compound, 2, obtained in rather good yield could be the result of the easy formation of KCp*, which is soluble in THF and not in Et2O, the solvent of extraction. The formation of the salt could be explained by the ionic bonding nature of the lanthanide to the Cp ring.42 It also demonstrates the relative lability of the nickel dimethyl fragment. As such, 2 is likely to be the thermodynamic product of the reaction by rearrangement of the expected heterobimetallic dimer. In order to get some insights in this singular rearrangement, another reaction was performed in a similar fashion but that was reacted only for several seconds at room temperature before it was dried, dissolved in Et2O and cooled to −35 °C. A brown solution formed without any precipitate. The crystallization of this brown solution yield brown X-ray suitable crystals of an anionic Yb-bimpm-Ni fragment in which two bimpm ligands are coupled (3) and one molecule of KCp* that co-crystallized (see ESI† for more details). The same reaction performed in Et2O, no matter the reaction time, only yielded the red crystalline product of rearrangement (2) (Scheme 1b). ORTEP of 3 and main distances and angle are shown in Fig. 2 and Table 1.
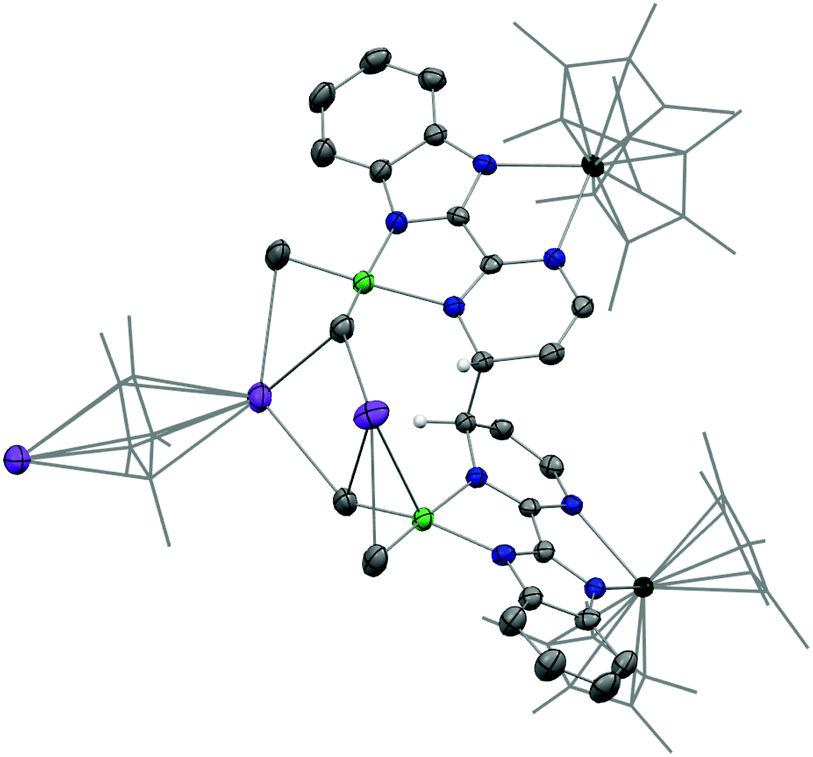 | ||
| Fig. 2 ORTEP of 3. Thermal ellipsoids are at 50% level. Carbon atoms are in grey, hydrogen atoms are in white and have been removed for clarity except the ones on the coupled carbons of the ligands, potassium in purple, nitrogen in blue, nickel in bright green and ytterbium atoms in deep green. Co-recrystallized solvate molecules of Et2O are removed and pentamethylcyclopentadienyl ligands on ytterbium are presented in wireframe style for clarity. | ||
The average Yb–Cp* and Yb–N distances in 3 are 2.33(2) and 2.330(8) Å, respectively, which are slightly shorter than the ones in 2 as 2.342(2) and 2.364(2) Å. The data agree well with a trivalent Yb fragment. A longer bimpm bridging distance of 1.459(5) Å is observed in 3 in comparison to that in 1 (1.451 Å). This follows a similar trend of the ligand reduction. The distance of the coupled σ bond (1.56 Å) between two pyrimidine cycles is much longer than the one in 2 (1.520 Å). It could be due to the less compressed coordination environment in dimeric 3 than in 2. Moreover, in comparison with 1, the average distances in 3 of the Ni–N (1.987(2) Å in 3, 1.97(5) in 1 Å) and Ni–CH3 bonds (1.928(5) Å in 3, 1.922(5) Å in 1) are rather similar. It indicates that 1 and 3 possess a similar divalent coordination system around the nickel centre. The presence of one co-crystallized KCp* molecule is an undoubtedly important part of the dimeric to trimeric transition from 3 to 2, given that the KCp* formation in solution can be regarded as a clear driving force leading to the rearrangement. Another interesting observation in 3 is the noticeable distortion of the nickel fragment (Fig. 3). The nearly planar angle (2°) between the bimpm ligand plane and NiMe2 fragment in 1 became largely bent (31°) in 3. This is clearly indicative of the liable disassociation of the NiMe2 moiety in the coupled ligand system. It is also shown by the facile decomposition of 3 under the inert atmosphere in solid-state at room temperature compared to 2, which is stable in these conditions. This probably indicates that the dimethyl-nickel moiety is ready for disassociation from the bimpm ligand to generate ethane and Ni(0) species (Fig. S11–12†).
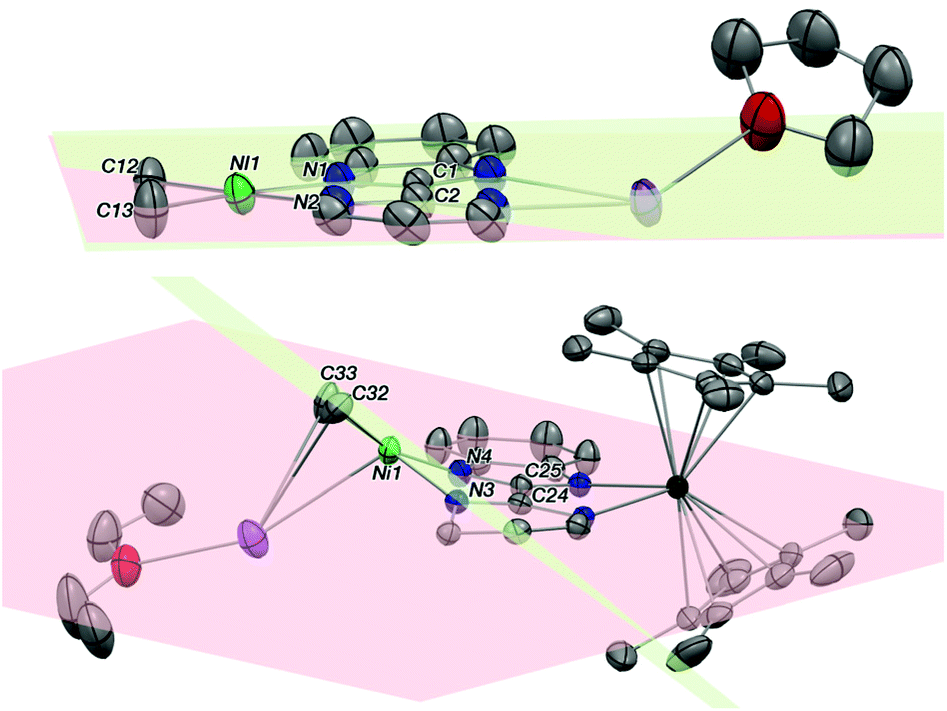 | ||
| Fig. 3 Comparison of distortion of the NiMe2 fragment in 1 (top) and 3 (bottom) presented by ORTEP. N–C–C–N is regarded as the overall ligand plane (red) and atoms are labelled; C–Ni–C is NiMe2 plane (green) and also labelled. Thermal ellipsoids are at 50% level. Carbon atoms are in grey, potassium in purple, nitrogen in blue, nickel in bright green and ytterbium atoms in deep green. Half structure of the coupled 3 is presented. Hydrogen atoms have been removed for clarity. | ||
In the reaction forming 2, the fate of the nickel dimethyl fragment remains uncertain while it is clear that the formation of KCp* helps the rearrangement from 2 to 3. Accordingly, the addition of crown ether (18-crown-6) in solution was considered to cape the active potassium ion.43 The analogous nickel precursor K(18-c-6)(bimpm)NiMe2 (4) was synthesized as deep blue crystalline product in good yield (91%). The combination with Cp*2Yb(OEt2) was performed in both THF and Et2O with different reaction times at room temperature, yielding indifferently an anionic heterobimetallic species containing the Yb-bimpm–Ni fragment, in which two pyrimidine cycles on bimpm ligands are also coupled in exo-position (5). The potassium counter ions are captured by 18-crown-6 (Scheme 2). ORTEP of 4 and 5 are shown in ESI† and Fig. 4, respectively and main distances and angles in Table 1.
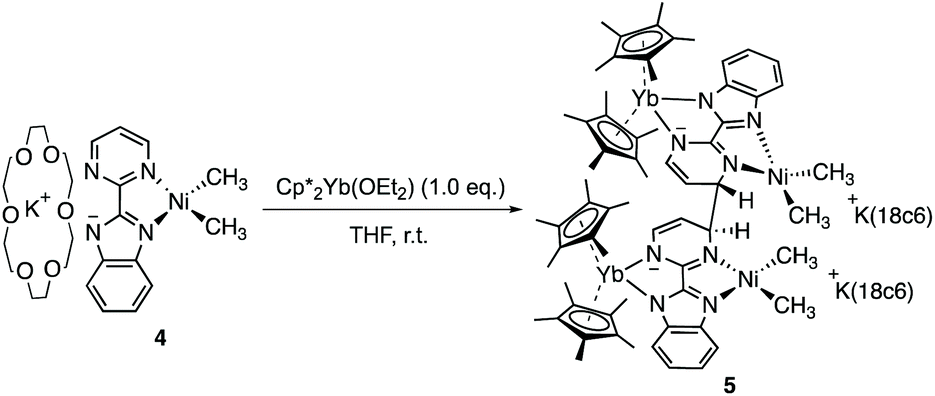 | ||
| Scheme 2 Synthesis of 5 from 4. | ||
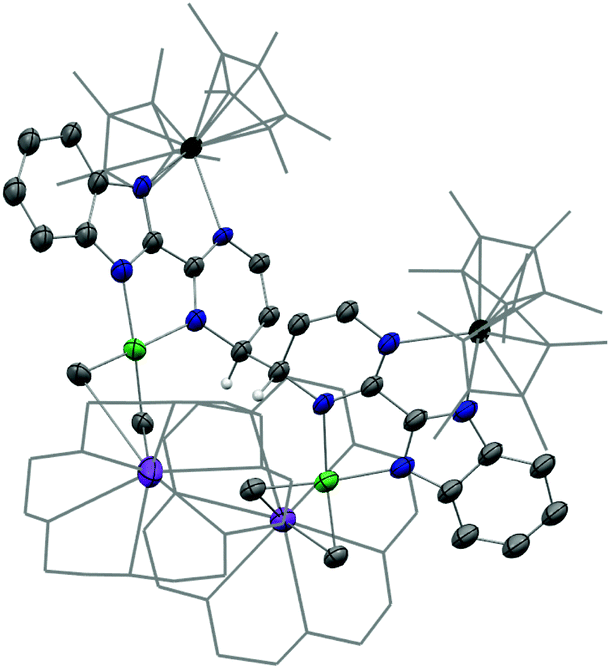 | ||
| Fig. 4 ORTEP of 5. Thermal ellipsoids are at 50% level. Carbon atoms are in grey, hydrogen atoms are in white and have been removed for clarity, except the ones on the coupled carbons of the ligands, potassium in purple, nitrogen in blue, nickel in bright green and ytterbium atoms in deep green. Co-recrystallized solvate molecule of Et2O is removed and pentamethylcyclopentadienyl ligands on ytterbium are presented in wireframe style for clarity. | ||
The average distance Yb–Cpctr in 5 is 2.340(2) Å indicating an YbIII fragment. A reduction has taken place in the ligand, which can be noticed by typical longer ligand linker distance (1.464(5) Å) comparing to the one in 4 as 1.45(6) Å. Similar parameters based on the comparison in Ni–CH3 and Ni–N distances of 4 and 5 indicate that the nickel ion remains at its divalent oxidation state. The distortion angle of bimpm and NiMe2 difference in 4 and 5 is only 7°, indicating a more stable coordination environment, compared to the one found in 3.
As discussed before, it is interesting to compare the angles between the bimpm ligand plane and the dimethyl nickel fragment plane in 3 and 5 (Fig. 5). In 5, the average angle is 19°, which is similar to that of 12° in 4. Thus, the small increase in 5 compare to 1 is likely due to steric hindrance because of the 18-crown-6. Additionally, 5 was found to be stable at room temperature in the solid-state, unlike 3. Thus, the fast decomposition of the nickel fragment in 3 is likely to be the reason for the easy formation of 2 as a thermodynamically stable trimer.
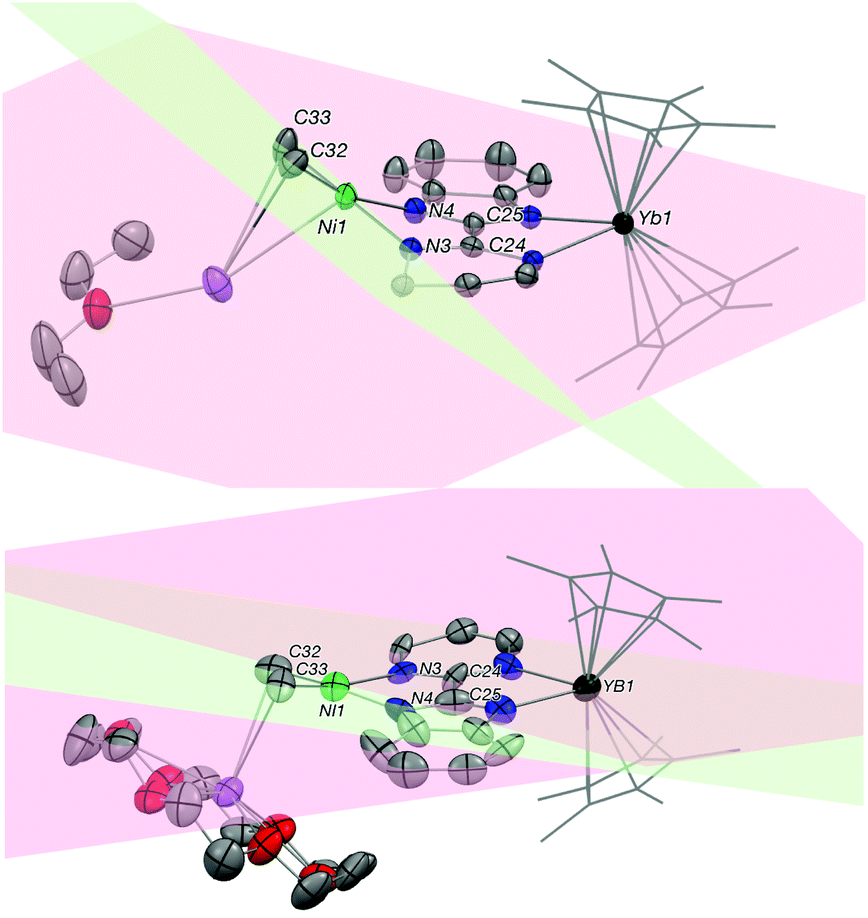 | ||
| Fig. 5 Comparison of the bimpm-NiMe2 distortion angle between 3 and 5. ORTEPs of 3 (top), 5 (bottom). Thermal ellipsoids are at 50% level. Carbon atoms are in grey, potassium in purple, nitrogen in blue, nickel in bright green and ytterbium atoms in deep green. Half structures of 3 and 5 are presented. Hydrogen atoms and co-recrystallized solvate molecules of Et2O in both complexes have been removed and pentamethylcyclopentadienyl ligands on ytterbium are presented in wireframe style for clarity. | ||
In order to estimate the impact of the single-electron transfer system without the lanthanides ion, a reduction was performed on 1 using potassium graphite (in excess) in THF instead of the divalent lanthanide ion reductant. XRD-suitable brown-orange crystals of the reaction product, 6, were isolated via slow diffusion of pentane into THF solution at −35 °C after several days. An ORTEP of 6 and main distances and angle are presented in Fig. 6 and Table 1.
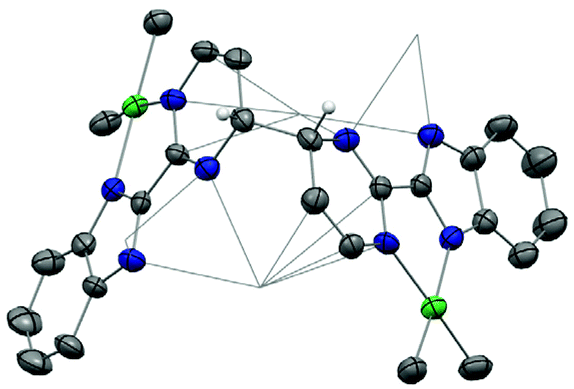 | ||
| Fig. 6 ORTEP of 6. Thermal ellipsoids are at 50% level. Carbon atoms are in grey, hydrogen atoms are in white and have been removed for clarity except the ones on the coupled carbons of bimpm ligands, nitrogen in blue and nickel in bright green. Co-recrystallized solvate molecules of THF have been omitted and potassium atoms are presented in wireframe style for clarity. | ||
In 6, the average distances of the Ni–N and Ni–CH3 bonds are 1.973(5) and 1.930(5) Å, respectively, indicating that the nickel centre remains in a divalent oxidation state. A longer distance of C–C linkage between pyrimidine and benzimidazole of 1.46(8) Å compared to that of 1.451 Å in 1, is indicative of an efficient reduction on the ligand moiety. The torsion angle between the ligand plane and the NiMe2 fragment is 17°, which is similar to the one in 5 (19°). Correspondingly, 6 was found to be thermally stable for several days in solution at room temperature under inert atmosphere. Interestingly, a coupled C–C bond between two pyrimidine rings was also observed but in a different position than that in 2, 3 and 5. In 6, the coupling occurs at the para position of the Ni-coordinated nitrogen atom, while in other structures with the divalent lanthanide, the coupling occurs at the ortho site. The length of the coupled C(sp3)–C(sp3) bond is 1.537 Å, which is similar to the ones in 2, 3 and 5.
This difference in the coupling fashion on bimpm ligand in 3 and 6 is intriguing and it is then plausible that the coordination of the divalent lanthanide ions could subtly modulate the electron density on the redox-active ligand fragment because of strong electron correlation. Additionally, the steric hinderance effect of the Cp* ligands on the lanthanide moiety might also participate in regulating the position at which the coupling occurs. Thus computational investigations were performed at the DFT level.
Theoretical studies
Computational investigations were performed on the anionic bimpm ligand, 1 and the monomeric anion of 3 at the DFT level. The corresponding geometries were optimised using the PBE density functional and Kohn–Sham orbitals are depicted in the ESI†.The optimised geometry of the anionic ligand yields a symmetrical species, in which the LUMO is delocalised equally across the C2 axis of ligand while the density is more localised in the pyrimidine part. The addition of the Ni fragment significantly modifies the electronic density of the LUMO with strong density located on the pyrimidine nitrogen atom that coordinates the nickel ion and on the carbon in para position to the latter. On the other hand, the ortho position to that coordinated nitrogen atom possesses very little density. Thus, an electron transfer that would occur on this species would definitely strongly localise spin density in the para position, which is consistent with the coupling observed in 6.
Now, the addition of the ytterbocene fragment is accompanied by an electron transfer to the ligand and the SOMO is now delocalised on the ligand while the LUMO is principally centred on the pyrimidine moiety. In the SOMO, the spin density has virtually no contribution on the carbon atoms where the coupling is observed in 6 (para to the nickel-coordinated pyrimidine nitrogen atom) while it has little contribution on the carbon atoms where the coupling is observed in 2, 3 and 5. Additionally, the presence of the lanthanide ion may enforce a multiconfigurational energy state allowing the LUMO to contribute significantly11,13 to the spin delocalization and thus explaining the distinct position of the coupling between 6 and 2, 3 and 5. This could be verified by a proper CASSCF computation that was not possible to handle in our hands.
Solution studies
The dissymmetry of the ligand imposes a Cs symmetry in the precursor 1 and 4. 1H NMR of 1 in THF-d8 shows seven resonances in total, two for the methyl (6 protons) at 0.02 and −0.40 ppm, and five for the bimpm ligands (7 protons), found in 8.6–6.8 range. The spectrum of 4 recorded in CD3CN shows six resonances for the bimpm ligands (7 protons), found in 8.8–6.9 ppm range, one signal for 18-crown-6 (24 protons) at 3.55 ppm, as well as two for the methyl (6 protons) at −0.10 and −0.54 ppm. The addition of the ytterbium fragment drastically shifts these protons, in agreement with the presence of a paramagnetic YbIII centre. 2 recorded in THF-d8 at 293 K nicely shows14 signals, integrating for1 proton each and 5 signals, integrating for 15 protons each. This spectrum is in good agreement with a coupled ligand and no symmetry, other than the identity, i.e. a C1 symmetric molecule in solution. The strong paramagnetism of the complex indicates the presence of at least one trivalent ytterbium.The 1H NMR spectra of 3 and 5 (in bracket) present similar spectra in THF-d8 at 293 K, with seven protons at 187.66 (186.25), 181.49 (176.11), 50.43 (53.04), 27.46 (30.55), 19.17 (23.32), 4.91 (−0.03) and −8.27 (−4.50) ppm. Additionally, two non-equivalent Cp* signals are found at 12.28 (7.91) and 2.93 (2.29) ppm and two non-equivalent methyl signals at −4.81 (−0.86) and −18.17 (−14.03) ppm. The presence of only seven signals for the ligand but two different signals for the methyl and Cp* moieties is in agreement with a dimeric species with a C2, an inversion centre at the centre position of the exo C–C bond that was formed or a plane of symmetry cutting the C–C bond that was formed. Thus, the NMR spectra are not informative as to the stereochemical nature of the coupling, but allow identifying its formation. Because both 3 and 5 have very similar coordination environment, their NMR chemical shifts are very close to each other. Again, the paramagnetism of 3 and 5 is indicative of an effective electron transfer from the divalent lanthanide to the redox-active ligand-transition metal fragment with bimpm.
The paramagnetic shift of the signals in 2, 3 and 5 were followed by variable temperature 1H NMR. The chemical shifts of each resonance in these complexes respectively are plotted versus 1/T (see ESI†). A linear δ vs. 1/T plot over the set-up temperature range for each, revealing typical Curie behaviour under this temperature range. This is in good agreement with the recorded solid-state temperature-dependent magnetic data.
The possibility of a reversible C–C transfer was studied. This occurred in multiple occasions after electron transfer from divalent lanthanides.13,14,44 Compared to the relatively stable nature of the Cp*2Yb(phen), 2, 3 and 5 are rather thermosensitive. The eventual dimer-monomer equilibrium was not efficiently observed when dissolving the crystals of 2, 3 and 5 in THF-d8 at room temperature. It might take place at higher temperatures, such as at 60 °C, however, the concomitant decomposition of the dimers occurred when heating up the solutions for extended periods of time, resulting in a number of newly formed, hardly identifiable signals (Fig. S19–24†). Thus, the reversibility of C–C coupling in 2, 3 and 5 remains equivocal.
Magnetism solid-state studies
The organometallic products were analysed by solid-state magnetism using a SQUID (Fig. 7). When the divalent ytterbium is coordinated to the bimpm ligand, one electron is transferred, which allow the coupling to occur. Thus, in 2, 3 and 5, it is expected that Yb centres are at the trivalent state. This was notably indicated by the paramagnetic NMR as well as the Cp*–Yb distance (see previous paragraphs). The solid-state data indicate 300 K χT values of 6.17, 4.45 and 4.45 emu K mol−1 for 2, 3 and 5, respectively. These values agree with the theoretical values of 2.54 emu K mol−1 for uncorrelated 2F7/2 ground-states for each ytterbium centre, that is three ytterbium centres in 2 and two ytterbium centres in 3 and 5. The χT value decreases monotonously up to 6 K with respective values of 3.32, 2.42 and 2.44 emu K mol−1 for 2, 3 and 5. The decrease is due to the depopulation of the higher energy crystal-field states. Note that the very similar decrease of 3 and 5 indicate a very similar crystal field, in agreement with their very similar structural nature. The decrease below 6 K is associated with a mixture of dipolar coupling contributions, while only the ground crystal-field state is populated.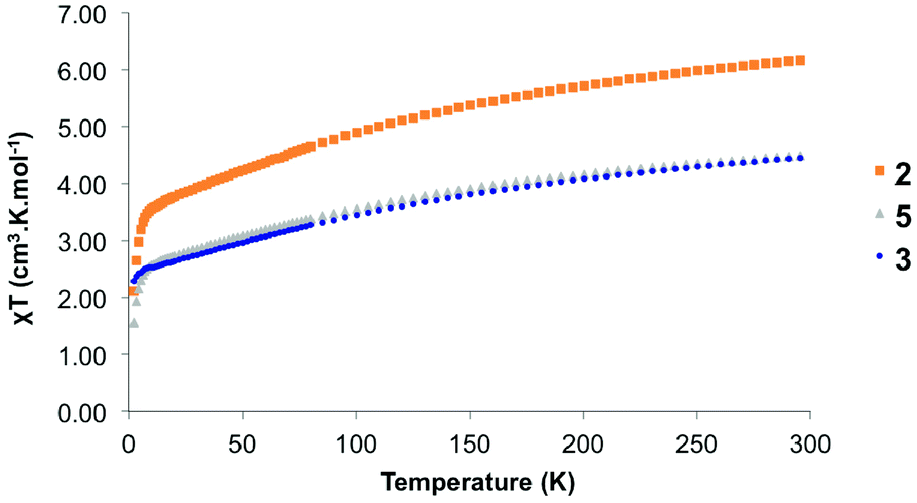 | ||
| Fig. 7 Temperature dependent magnetic data for 2, 3 and 5. | ||
Conclusions
In this article, we have successfully synthesized original redox-active, dissymmetric hetero- or homometallic complexes containing divalent organolanthanides, Cp*2Yb, which allows effective single electron transfer process and leads to the intermolecular C(sp3)–C(sp3) coupling formation between two bridging ligands. The stereochemistry of this ligand coupling is identified as an exo formation, and the dimer-trimer transition is affected by a reactive NiMe2 fragment, which impacts the torsion angle between the bridging ligand and dimethyl functional group. The solid-state explanation of such a rearrangement is provided by the XRD study of the isolated transient exo species. Magnetic measurements show that 2, 3 and 5 possess triplet ground states. This work implements a new redox-active system of divalent lanthanides, in the presence of a dissymmetric ligand, yielding an intermolecular ligand coupling reactivity, yet the reversibility of this bond formation remains ambiguous. This study provides us with a new avenue for the design of our heterometallic architectures with other transition metals using this dissymmetric ligand system, which may lead to different reactivity behaviours compared to the one observed for nickel. The related work is currently under investigation.Experimental section
General considerations
All reactions were performed using standard Schlenk-line techniques or in argon- or nitrogen-filled gloveboxes (MBraun, Garching, Germany). All glassware was dried at 120 °C for at least 12 h prior to use. Tetrahydrofuran (THF), THF-d8, toluene, and toluene-d8 were dried over sodium, degassed; CD3CN was dried over CaH2, degassed. All the solvents were transferred under reduced pressure in a cold flask.1H NMR spectra were recorded in 5 mm tubes adapted with a J. Young valve on Bruker AVANCE II or III-300 MHz (Bruker, Billerica, MA, USA). 1H chemical shifts were expressed relative to TMS (tetramethylsilane) in ppm. Magnetic susceptibility measurements were made for all samples on a SQUID. Diamagnetic corrections were made using Pascal's constants. Temperature dependent magnetic measurements were obtained in sealed quartz tube45 on a SQUID at 0.5 and 2 T. Elemental analyses were obtained from Mikroanalytisches Labor Pascher (Remagen, Germany).
All calculations were performed using the ORCA 4.2.1 software.46 The geometry optimizations were done at the PBE level of theory,47 using scalar relativistic ZORA Hamiltonian with ZORA-def2-TZVP basis set,48 and SARC/J auxiliary basis set for Coulomb fitting.49–51 Each time, dispersion corrections were added to the functional used in the D3 framework proposed by Grimme52 with the addition of the Becke–Johnson damping (D3BJ). Frequencies were calculated numerically to ensure these structures corresponded to energy minima. Single-point energy calculations starting from PBE optimized geometry were then performed in gas phase at the PBE, PBE0,53 TPSSh,54,55 and ωB97X-D3.56
Single crystals of the compounds 1–6 were coated in Paratone-N oil and mounted on a Kapton loop. A BRUKER APEX-II CCD detector and a graphite Mo-Kα monochromator (Nonius, Delft, Netherlands) were used for the data acquisition. All data were measured at 150 K under a nitrogen stream and a refinement method was used for solving the structure. The structure resolution was accomplished using the SHELXS-97 and SHELXT57 program, and the refinement was done with the SHELXL program.58,59 The structure solution and the refinement were achieved with the PLATON60 and Olex2 softwares.61 During the refinement steps, all atoms except hydrogen atoms- were refined anisotropically. The position of the hydrogen atoms was determined using residual electronic densities. Finally, in order to obtain a complete refinement, a weighting step followed by multiples loops of refinement was done. ORTEPs of the compound structures were obtained using the MERCURY software. The structures have been deposited in the CCDC with 2021801–2021805 and 2034256 for 1–5 and 6, respectively.†
(tmeda)NiMe2,62 Cp*2Yb(OEt2),63 and HBimpm36 were synthetized according to published procedures and recrystallized prior use. 18-crown-6 was purchased from Sigma-Aldrich and sublimed before use.
Syntheses
Anal. calcd for C25H37KN4NiO6: C, 51.12; H, 6.35; N, 9.54; found: C, 51.03; H, 6.30; N, 9.06.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The acknowledgements come at the end of an article after the This project has received funding from the H2020 European Research Council (ERC) under the European Union’ Horizon H2020 research program (grant agreement No. 716314). CNRS and Ecole Polytechnique are thanked for funding.Notes and references
- P. J. Chirik and K. Wieghardt, Radical Ligands Confer Nobility on Base-Metal Catalysts, Science, 2010, 327, 794–795 CrossRef CAS PubMed.
- A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Complexes with Nitrogen-Centered Radical Ligands: Classification, Spectroscopic Features, Reactivity, and Catalytic Applications, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef CAS PubMed.
- P. J. Chirik, Preface: Forum on Redox-Active Ligands, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS PubMed.
- V. Lyaskovskyy and B. de Bruin, Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- D. L. J. Broere, L. L. Metz, B. de Bruin, J. N. H. Reek, M. A. Siegler and J. I. van der Vlugt, Redox-Active Ligand-Induced Homolytic Bond Activation, Angew. Chem., Int. Ed., 2015, 54, 1516–1520 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Metal–Ligand Cooperation by Aromatization–Dearomatization: A New Paradigm in Bond Activation and “Green” Catalysis, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Metal–Ligand Cooperation, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- M. Schultz, J. M. Boncella, D. J. Berg, T. D. Tilley and R. A. Andersen, Coordination of 2,2′-Bipyridyl and 1,10-Phenanthroline to Substituted Ytterbocenes: An Experimental Investigation of Spin Coupling in Lanthanide Complexes, Organometallics, 2002, 21, 460–472 CrossRef CAS.
- C. H. Booth, M. D. Walter, D. Kazhdan, Y.-J. Hu, W. W. Lukens, E. D. Bauer, L. Maron, O. Eisenstein and R. A. Andersen, Decamethylytterbocene Complexes of Bipyridines and Diazabutadienes: Multiconfigurational Ground States and Open-Shell Singlet Formation, J. Am. Chem. Soc., 2009, 131, 6480–6491 CrossRef CAS PubMed.
- C. H. Booth, D. Kazhdan, E. L. Werkema, M. D. Walter, W. W. Lukens, E. D. Bauer, Y.-J. Hu, L. Maron, O. Eisenstein, M. Head-Gordon and R. A. Andersen, Intermediate-Valence Tautomerism in Decamethylytterbocene Complexes of Methyl-Substituted Bipyridines, J. Am. Chem. Soc., 2010, 132, 17537–17549 CrossRef CAS PubMed.
- G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Thermal Dihydrogen Elimination from Cp*2Yb(4,5-diazafluorene), Organometallics, 2013, 32, 1150–1158 CrossRef CAS.
- G. Nocton, C. H. Booth, L. Maron, L. Ricard and R. A. Andersen, Carbon–Hydrogen Bond Breaking and Making in the Open-Shell Singlet Molecule Cp*2Yb(4,7-Me2phen), Organometallics, 2014, 33, 6819–6829 CrossRef CAS.
- G. Nocton, W. W. Lukens, C. H. Booth, S. S. Rozenel, S. A. Medling, L. Maron and R. A. Andersen, Reversible Sigma C–C Bond Formation Between Phenanthroline Ligands Activated by (C5Me5)2Yb, J. Am. Chem. Soc., 2014, 136, 8626–8641 CrossRef CAS PubMed.
- G. Nocton and L. Ricard, Reversible C–C coupling in phenanthroline complexes of divalent samarium and thulium, Chem. Commun., 2015, 51, 3578–3581 RSC.
- J. Andrez, G. Bozoklu, G. Nocton, J. Pécaut, R. Scopelliti, L. Dubois and M. Mazzanti, Lanthanide(II) Complexes Supported by N,O-Donor Tripodal Ligands: Synthesis, Structure, and Ligand-Dependent Redox Behavior, Chem. – Eur. J., 2015, 21, 15188–15200 CrossRef CAS PubMed.
- A. Jaoul, C. Clavaguéra and G. Nocton, Electron transfer in tetramethylbiphosphinine complexes of Cp*2Yb and Cp*2Sm, New J. Chem., 2016, 40, 6643–6649 RSC.
- M. Xémard, S. Zimmer, M. Cordier, V. Goudy, L. Ricard, C. Clavaguéra and G. Nocton, Lanthanidocenes: Synthesis, Structure, and Bonding of Linear Sandwich Complexes of Lanthanides, J. Am. Chem. Soc., 2018, 140, 14433–14439 CrossRef PubMed.
- C. A. P. Goodwin, N. F. Chilton, L. S. Natrajan, M.-E. Boulon, J. W. Ziller, W. J. Evans and D. P. Mills, Investigation into the Effects of a Trigonal-Planar Ligand Field on the Electronic Properties of Lanthanide(II) Tris(silylamide) Complexes (Ln = Sm, Eu, Tm, Yb), Inorg. Chem., 2017, 56, 5959–5970 CrossRef CAS PubMed.
- C. J. Kuehl, R. E. D. Re, B. L. Scott, D. E. Morris and K. D. John, Toward new paradigms in mixed-valency: ytterbocene–terpyridine charge-transfer complexes, Chem. Commun., 2003, 2336–2337 RSC.
- F. Jaroschik, A. Momin, A. Martinez, D. Harakat, L. Ricard, X. F. Le Goff and G. Nocton, Synthesis and Characterization of 1,1′-Diphosphaplumbocenes: Oxidative Ligand Transfer Reactions with Divalent Thulium Complexes, Organometallics, 2016, 35, 2032–2038 CrossRef CAS.
- T. P. Gompa, N. Jiang, J. Bacsa and H. S. La Pierre, Synthesis of homoleptic, divalent lanthanide (Sm, Eu) complexes via oxidative transmetallation, Dalton Trans., 2019, 48, 16869–16872 RSC.
- W. J. Evans and D. K. Drummond, Reductive coupling of pyridazine and benzaldehyde azine and reduction of bipyridine by samarium complex (C5Me5)2Sm(THF)2, J. Am. Chem. Soc., 1989, 111, 3329–3335 CrossRef CAS.
- D. J. Berg, J. M. Boncella and R. A. Andersen, Preparation of Coordination Compounds of Cp*2Yb with Heterocyclic Nitrogen Bases: Examples of Antiferromagnetic Exchange Coupling across Bridging Ligands, Organometallics, 2002, 21, 4622–4631 CrossRef CAS.
- P. L. Holland, R. A. Andersen and R. G. Bergman, Synthesis, Characterization, Isomerization, and Reactivity of Dimeric Cyclopentadienylnickel Amido Complexes, J. Am. Chem. Soc., 1996, 118, 1092–1104 CrossRef CAS.
- T. R. Dugan, E. Bill, K. C. MacLeod, G. J. Christian, R. E. Cowley, W. W. Brennessel, S. Ye, F. Neese and P. L. Holland, Reversible C–C Bond Formation between Redox-Active Pyridine Ligands in Iron Complexes, J. Am. Chem. Soc., 2012, 134, 20352–20364 CrossRef CAS PubMed.
- C. C. H. Atienza, C. Milsmann, S. P. Semproni, Z. R. Turner and P. J. Chirik, Reversible Carbon–Carbon Bond Formation Induced by Oxidation and Reduction at a Redox-Active Cobalt Complex, Inorg. Chem., 2013, 52, 5403–5417 CrossRef PubMed.
- V. Goudy, A. Jaoul, M. Cordier, C. Clavaguéra and G. Nocton, Tuning the Stability of Pd(IV) Intermediates Using a Redox Non-innocent Ligand Combined with an Organolanthanide Fragment, J. Am. Chem. Soc., 2017, 139, 10633–10636 CrossRef CAS PubMed.
- D. Wang, J. Moutet, M. Tricoire, M. Cordier and G. Nocton, Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments, Inorganics, 2019, 7, 58 CrossRef CAS PubMed.
- A. Jaoul, M. Tricoire, J. Moutet, M. Cordier, C. Clavaguéra and G. Nocton, Reversible electron transfer in organolanthanide chemistry, Chem. Squared, 2019, 3, 1 Search PubMed.
- R. Ghosh and A. Sarkar, Bidentate P, N−P Ligand for Nickel-Catalyzed Cross-Coupling of Aryl or Benzyl Chlorides with ArMgX, J. Org. Chem., 2010, 75, 8283–8286 CrossRef CAS PubMed.
- J. Magano and J. R. Dunetz, Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- S. Handa, Y. L. N. Mathota Arachchige and L. M. Slaughter, Access to 2′-Substituted Binaphthyl Monoalcohols via Complementary Nickel-Catalyzed Kumada Coupling Reactions under Mild Conditions: Key Role of a P,O Ligand, J. Org. Chem., 2013, 78, 5694–5699 CrossRef CAS PubMed.
- F.-S. Han, Transition-metal-catalyzed Suzuki–Miyaura cross-coupling reactions: a remarkable advance from palladium to nickel catalysts, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- N. Yoshikai, H. Mashima and E. Nakamura, Nickel-Catalyzed Cross-Coupling Reaction of Aryl Fluorides and Chlorides with Grignard Reagents under Nickel/Magnesium Bimetallic Cooperation, J. Am. Chem. Soc., 2005, 127, 17978–17979 CrossRef CAS PubMed.
- H. Xu, J. B. Diccianni, J. Katigbak, C. Hu, Y. Zhang and T. Diao, Bimetallic C–C Bond-Forming Reductive Elimination from Nickel, J. Am. Chem. Soc., 2016, 138, 4779–4786 CrossRef CAS PubMed.
- M. Haga, M. Ishizuya, T. Kanesugi, T. Yutaka, D. Sakiyama, J. Fees and W. Kaim, Synthesis and formation of dinuclear mixed-valent complexes of ruthenium and osmium bridged by 2-(2-pyrimidyl)benzimidazolate, Indian J. Chem. Sect. A, 2003, 42A, 2290–2299 CAS.
- P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Ligand Bite Angle Effects in Metal-catalyzed C−C Bond Formation, Chem. Rev., 2000, 100, 2741–2770 CrossRef CAS PubMed.
- M. Kranenburg, P. C. J. Kamer, P. W. N. M. van Leeuwen, D. Vogt and W. Keim, Effect of the bite angle of diphosphine ligands on activity and selectivity in the nickel-catalysed hydrocyanation of styrene, J. Chem. Soc. Chem. Commun., 1995, 2177–2178 RSC.
- F. Jaroschik, F. Nief, X.-F. Le Goff and L. Ricard, Synthesis and Reactivity of Organometallic Complexes of Divalent Thulium with Cyclopentadienyl and Phospholyl Ligands, Organometallics, 2007, 26, 3552–3558 CrossRef CAS.
- S. Labouille, F. Nief, X.-F. Le Goff, L. Maron, D. R. Kindra, H. L. Houghton, J. W. Ziller and W. J. Evans, Ligand Influence on the Redox Chemistry of Organosamarium Complexes: Experimental and Theoretical Studies of the Reactions of (C5Me5)2Sm(THF)2 and (C4Me4P)2Sm with Pyridine and Acridine, Organometallics, 2012, 31, 5196–5203 CrossRef CAS.
- G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Influence of the Torsion Angle in 3,3′-Dimethyl-2,2′-bipyridine on the Intermediate Valence of Yb in (C5Me5)2 Yb(3,3′-Me 2 -bipy), Organometallics, 2013, 32, 5305–5312 CrossRef CAS.
- J. M. Birmingham and G. Wilkinson, The Cyclopentadienides of Scandium, Yttrium and Some Rare Earth Elements, J. Am. Chem. Soc., 1956, 78, 42–44 CrossRef CAS.
- C. L. Liotta and H. P. Harris, Chemistry of naked anions. I. Reactions of the 18-crown-6 complex of potassium fluoride with organic substrates in aprotic organic solvents, J. Am. Chem. Soc., 1974, 96, 2250–2252 CrossRef CAS.
- T. Cheisson, L. Ricard, F. W. Heinemann, K. Meyer, A. Auffrant and G. Nocton, Synthesis and Reactivity of Low-Valent f-Element Iodide Complexes with Neutral Iminophosphorane Ligands, Inorg. Chem., 2018, 57, 9230–9240 CrossRef CAS PubMed.
- M. D. Walter, M. Schultz and R. A. Andersen, Weak paramagnetism in compounds of the type Cp′2Yb(bipy), New J. Chem., 2006, 30, 238–246 RSC.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- D. A. Pantazis and F. Neese, All-Electron Scalar Relativistic Basis Sets for the Lanthanides, J. Chem. Theory Comput., 2009, 5, 2229–2238 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- O. A. Vydrov and G. E. Scuseria, Assessment of a long-range corrected hybrid functional, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- G. M. Sheldrick, SHELXT– Integrated space–group and crystal–structure determination, Acta Crystallogr. Sect. Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr. A, 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. a. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- W. Kaschube, K. R. Pörschke and G. Wilke, tmeda-Nickel-Komplexe: III. (N,N,N′,N′-Tetramethylethylendiamin)-(dimethyl)nickel(II), J. Organomet. Chem., 1988, 355, 525–532 CrossRef CAS.
- T. D. Tilley, J. M. Boncella, D. J. Berg, C. J. Burns, R. A. Andersen, G. A. Lawless, M. A. Edelman and M. F. Lappert, in Inorganic Syntheses, John Wiley & Sons, Ltd, 2007, pp. 146–150 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data, magnetism measurements, DFT computation data and X-ray crystallography details. CCDC 2021801–20218052034256. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qi00952k |
| This journal is © the Partner Organisations 2021 |